
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePPEGMEMA-based cationic copolymers designed for layer-by-layer assembly†
Tao Jiang ,
Saeed Zajforoushan Moghaddam and
Esben Thormann*
,
Saeed Zajforoushan Moghaddam and
Esben Thormann*
Department of Chemistry, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark. E-mail: esth@kemi.dtu.dk; Tel: +45 4525-2439
First published on 28th August 2019
Abstract
We have synthesized three PPEGMEMA-based cationic copolymers with similar amine contents but with systematic variation in the average length of the PEG side chains. The positively charged copolymers were paired with alginate to fabricate layer-by-layer assembled multilayered films. It was demonstrated that the polymeric design, in terms of the systematic variation in the average length of the PEG units, affects the polyelectrolyte multilayer growth mechanism and can be used to tune the structural properties and the water content of the layers. In addition, by partial cross-linking of the amine groups present in the copolymer backbone, disintegration of the film induced by pH changes was prevented. Finally, it was demonstrated how the cross-linked multilayered film can exhibit cationic, zwitterionic and anionic properties depending on the pH value and how these changes are associated with swelling, layer contraction and changes in water content.
1. Introduction
Layer-by-layer (LbL) assembly has drawn great attention as a convenient and adaptable method to fabricate functional polymer-coated surfaces.1–7 The designated polymeric components are sequentially adsorbed onto a solid substrate, driven by different intermolecular forces, such as electrostatic attraction,2,3,8–10 hydrogen bonding,11–14 and covalent bonding.15,16 The electrostatic LbL assembly method has been extensively investigated due to its simplicity and high efficiency.1 Accordingly, polyelectrolyte multilayered films can be readily prepared by alternate immersion of a charged substrate into solutions of oppositely charged polyelectrolytes with rinsing steps in between. To this end, the thicknesses and composition of the layer are controlled by the number of deposited layers, pH,10,17,18 ionic strength,10,19,20 temperature,21 adsorption time,22 and polymer molecular weight.23The need for ionizable groups in the backbone of the polymers for electrostatic LbL assembly effectively hinders the incorporation of uncharged polymers into the polyelectrolyte multilayered films. It is, however, feasible to accommodate desired functionalities into the backbone of charged polymers by chemical modification. Several authors have implemented such an approach to tune the functionality of multilayered films obtained by electrostatic LbL assembly.24–26 For instance, Sun and coworkers modified polyethyleneimine (PEI) with polyethylene glycol (PEG) and fabricated a multilayered film based on PEGylated-PEI and hyaluronic acid. The obtained film demonstrated antifouling properties resulting from the grafted PEG units.24 In another relevant work, Neoh and coworkers custom synthesized poly(isobutylene-alt-maleic acid) (PIAMA) derivatives bearing either alkyl- or PEG-carboxyl side chains, and LbL assembled the obtained polyanions with PEI.25 It was shown that varying the side chains in the polyanion affects the wettability of the multilayered film; hence, such a film could be used to tune the protein adsorption and cell adhesion properties of the surface. In both the works mentioned above, the functional units (PEG or alkyl chains) are grafted onto a polyelectrolyte backbone, which has limitations in terms of tuning the properties of the polyion such as charge density and molecular architecture. A more versatile approach is to copolymerize neutral and charged monomers instead, where the former unit provides functional properties and the latter is responsible for the electrostatic interaction needed for LbL assembly. For instance, Schlenoff and coworkers copolymerized N-isopropylacrylamide (NIPAM, neutral monomer) with allylamine (cationic comonomer) as well as styrene sulfonic acid (anionic comonomer).27 The synthesized PNIPAM-based copolymers were then paired to fabricate a thermoresponsive multilayered film. Based on this approach, one can thus control not only the chemical composition but also the charge density, molecular weight and chain architecture of the components in the multilayered film.
Poly(polyethylene glycol methyl ether methacrylate) (PPEGMEMA) is a methacrylate derivative of polyethylene glycol that has been vastly studied as a potential coating material due to its hydrophilic nature and high water content, which leads to lubricating28,29 and protein-repellent properties.30–33 A common method to fabricate PPEGMEMA-coated surfaces is surface-initiated polymerization.34–37 However, this method has several disadvantages that can restrict its applicability. It is generally challenging to achieve a thick coating using surface-initiated polymerization, which is a less versatile and scalable method that requires special substrate treatment and reaction control. In contrast, LbL assembly can be conducted through various methods, e.g., dip-coating, spray-coating and spin-coating, which not only covers a broader range of substrates but can also render considerably thicker coatings.7 Therefore, an alternative way to fabricate PEGMEMA-based coatings is to incorporate PEGMEMA units into charged chains so that electrostatic LbL assembly can be utilized.
Adopting this approach, we have synthesized a PPEGMEMA-based cationic copolymer in which PEGMEMA (neutral monomer) was randomly copolymerized with aminoethyl methacrylate (cationic comonomer). Three PPEGMEMA copolymer samples with comparable degrees of polymerization and amine content, but with different average lengths for the PEG side chains were prepared and then paired with alginate as a reference anionic polymer. This approach provided a method to study how systematic variation of the polymer architecture affects the multilayer buildup process and the multilayer structure. For this purpose, the assembly of the multilayered films was simultaneously monitored with a quartz crystal microbalance with dissipation monitoring (QCM-D) and spectroscopic ellipsometry. Accordingly, information for the thickness, conformation and water content of the films was extracted. Finally, the film structure was stabilized by cross-linking the amine groups, which preserved the pH-responsivity of the film while preventing its charge-induced disintegration.
2. Experimental section
2.1. Materials
Di(ethylene glycol) methyl ether methacrylate (DEGMEMA, 95%), poly(ethylene glycol) methyl ether methacrylate (PEGMEMA) with number average molecular weights (Mn) of 300 and PEGMEMA with Mn of 500 g mol−1 were purchased from Sigma Aldrich. All three monomers were treated with neutral alumina column chromatography to remove the inhibitors before use. Sodium alginate (NaAlg, medium viscosity ≥2.000 cP, 2% (25 °C)), (2-Boc-amino)ethyl methacrylate (BocAMA, 99%), diethyl meso-2,5-dibromoadipate (98%), glutaraldehyde (50% aqueous solution), trifluoroacetic acid (TFA, 99%), (3-aminopropyl)triethoxysilane (APTES, 99%) and copper(I) chloride (CuCl, >99%, washed sequentially with acetic acid and ethanol before use) was purchased from Sigma Aldrich and used as received unless otherwise stated. Tris(2-dimethylaminoethyl)amine (Me6TREN, 99%) was purchased from Alfa Aesar, USA. All the solvents used in this work were obtained from Sigma Aldrich and of HPLC grade. A pH 3 citric acid/sodium phosphate buffer (1.62 g citric acid, 0.56 g sodium phosphate dibasic dehydrate in 500 mL water) was used for the preparation of the polymer solutions as well as for rinsing in between each deposition step. Solutions of 100 ppm (w/v) PPEGMEMA and alginate were prepared for LbL assembly and filtered through 0.44 μm regenerative cellulose syringe filters to remove possible aggregates and dust. Ultra-pure water (Milli-Q Plus system, resistivity of 18.2 MΩ-cm, TOC value less than 5 ppb.) was used to prepare all the solutions.2.2. Synthesis of poly(poly(ethylene glycol) methyl ether methacrylate)-stat-poly(aminoethyl methacrylate) (P(PEGMEMA-stat-AMA))
Atom transfer radical polymerization (ATRP) is a well-developed living radical polymerization (LRP) technique, which has been widely utilized in preparation of polymers with narrow molecular weight distribution and macromolecular engineering.38 PPEGMEMA copolymers with randomly distributed primary amine groups were synthesized by ATRP with a protection-deprotection strategy according to previous reports.14,39,40 ATRP in the presence of primary amine groups (the cationic group) is challenging due to possible complex formation between the amine groups and Cu(I), which may readily inactivate the catalyst; thus, a two-step protocol was employed to overcome this problem. First, PEGMEMA was copolymerized with protected BocAMA, yielding a P(PEGMEMA-stat-BocAMA) statistical copolymer. In the second step, Boc groups were hydrolyzed under acidic condition (TFA/DCM), yielding the targeted P(PEGMEMA-stat-AMA) copolymer (Scheme 1).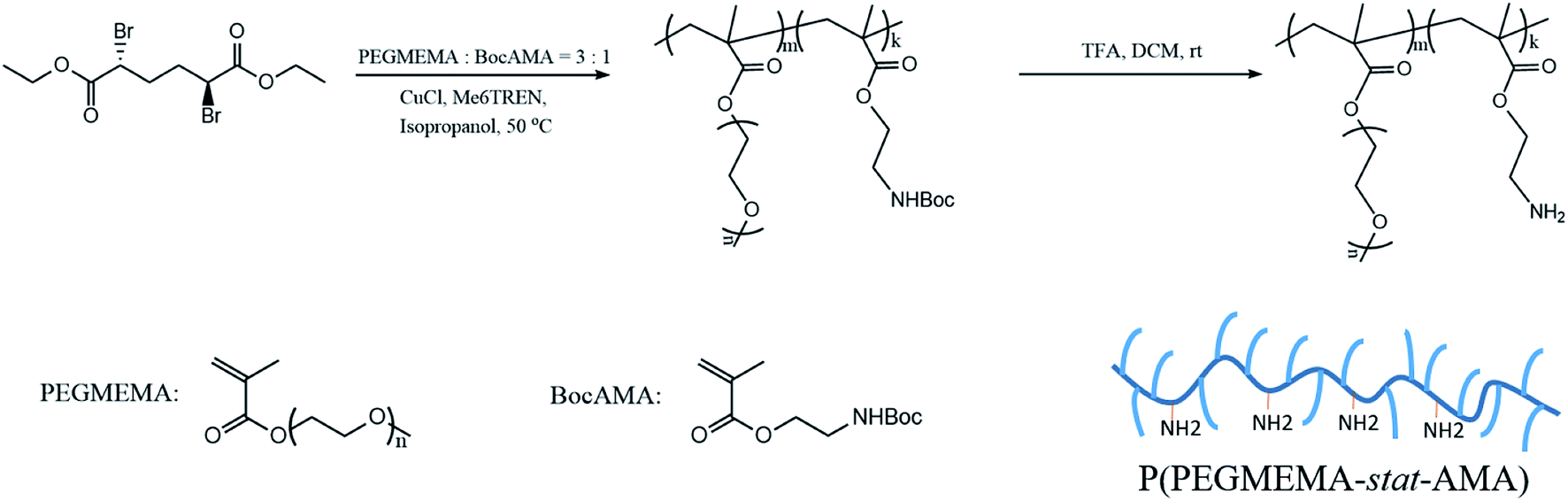 | ||
| Scheme 1 Synthesis procedure used for the P(PEGMEMA-stat-AMA) polymer. | ||
The detailed ATRP experimental procedure is as follows. For the synthesis of the protected P(PEGMEMA-stat-BocAMA), diethyl meso-2,5-dibromoadipate (initiator, 7.2 mg, 0.02 mmol), PEGMEMA (monomer, 3 mmol), (2-Boc-amino)ethyl methacrylate (monomer, 229.27 mg, 1 mmol) and Me6TREN (ligand, 9.2 mg, 0.04 mmol) were dissolved in 5 mL isopropanol. Dimethylformamide (0.1 mL) was added to the reaction mixture as an internal standard for subsequent calculation of the monomer conversion. The reaction mixture was bubbled with nitrogen gas for 30 min, after which the prewashed copper(I) chloride (1.23 mg, 0.0125 mmol) was added. The polymerization was triggered by immersing the reaction flask into a 50 °C water bath. After 16 hours, the reaction was quenched by adding 5 mL of water into the solution and by exposure to air. A small volume of the solution (0.1 mL) was collected for 1H NMR measurement to determine the reaction conversion. The polymer solution was then purified via dialysis (regenerative cellulose tubing with MWCO 6–8 kDa, Spectrum Laboratories, Inc.) against water for 3 days. Finally, residual water was removed via lyophilization to obtain the pure P(PEGMEMA-stat-BocAMA) copolymers. Fig. 1 presents the 1H NMR spectra for one PPEGMEMA-based copolymer before and after the deprotection process. Accordingly, the peak corresponding to t-butyl (1.43 ppm) in the Boc protecting group is eliminated after the deprotection process.
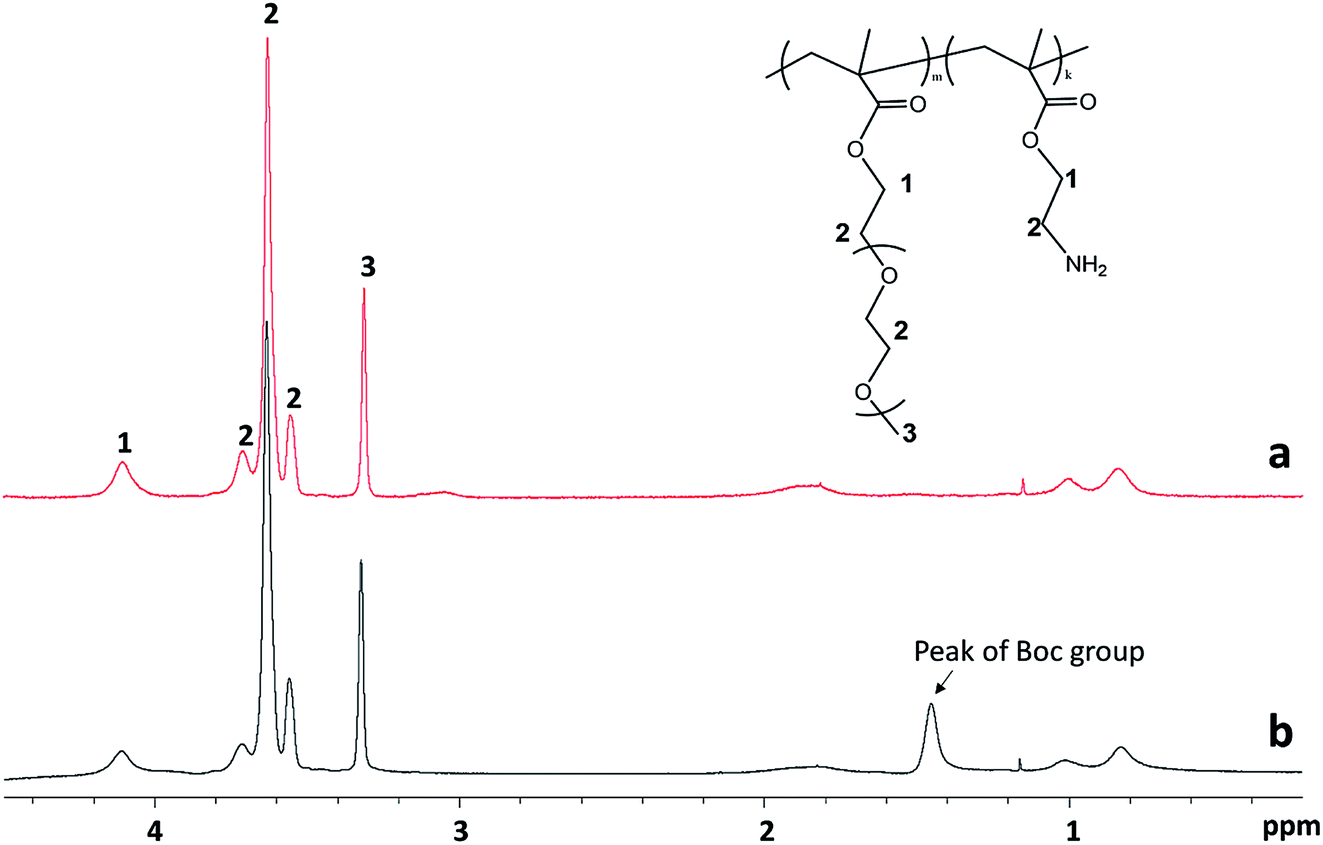 | ||
| Fig. 1 NMR spectra for m-PPEGMEMA (a) after and (b) before deprotection to remove Boc groups. | ||
In the deprotection process, the obtained P(PEGMEMA-stat-BocAMA) copolymers (300 mg) were dissolved in 4 mL dichloromethane followed by the addition of 0.5 mL of trifluoroacetic acid. The reaction mixture was stirred at room temperature for 3 hours, after which dichloromethane was removed by evaporation. The viscous polymer residue was redissolved in 5 mL of water, and the pH was adjusted to 7 with 1 M NaOH solution. The polymer was purified by dialysis and subsequent lyophilization, yielding a light-yellow viscous liquid.
All the P(PEGMEMA-stat-AMA) copolymers in this work were synthesized with the same procedure described above. In order to obtain polymers with a similar degree of polymerization (DP), the monomer to initiator ratio is fixed at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for all polymerizations, and the reaction time is set to be the same value of 16 hours to achieve high conversion (>80%) for all three polymerization processes. To determine the DPs of the polymers obtained, samples of the reaction mixture were collected before and after the polymerization process, and 1H NMR spectra (Brucker 400 MHz NMR spectrometer) for both samples were taken using deuterated DMSO as the solvent. The integral ratios of δ 6.0 ppm (monomer double bond (vinyl))/δ 7.9 ppm (DMF) before (r1) and after (r2) the polymerization were used to obtain the monomer conversion (c) via c = 1 − r2/r1. The DP values were calculated accordingly and shown in Table 1.
:1 for all polymerizations, and the reaction time is set to be the same value of 16 hours to achieve high conversion (>80%) for all three polymerization processes. To determine the DPs of the polymers obtained, samples of the reaction mixture were collected before and after the polymerization process, and 1H NMR spectra (Brucker 400 MHz NMR spectrometer) for both samples were taken using deuterated DMSO as the solvent. The integral ratios of δ 6.0 ppm (monomer double bond (vinyl))/δ 7.9 ppm (DMF) before (r1) and after (r2) the polymerization were used to obtain the monomer conversion (c) via c = 1 − r2/r1. The DP values were calculated accordingly and shown in Table 1.
The average number of ethylene oxide units in PEGMEMA monomers were tuned, in order to obtain P(PEGMEMA-stat-AMA) copolymers with different PEG side chain lengths. Overall three P(PEGMEMA-stat-AMA) copolymers, namely P((EO)2.5MEMA-stat-AMA), P((EO)4.5MEMA-stat-AMA) and P((EO)9MEMA-stat-AMA), with average PEGMEMA monomer molecular weights of 211, 300 and 500, corresponding to an average 2.5, 4.5 and 9 ethylene oxide units in the PEG chains in each monomer were synthesized, respectively. To this end, PEGMEMA with average molecular weights of Mn = 300 and Mn = 500, were used for the synthesis of P((EO)4.5MEMA-stat-AMA) and P((EO)9MEMA-stat-AMA), respectively, while a mixture of PEGMEMA with average molecular weights of Mn = 188 and Mn = 300 in the ratio of 4:1 was used for the synthesis of P((EO)2.5MEMA-stat-AMA). For convenience, from now we will refer to these three copolymers as s-PPEGMEMA (for the shortest PEG units), m-PPEGMEMA (for the medium PEG units) and l-PPEGMEMA (for the longest PEG units), with s, m and l referring to short, medium and longer PEG side chains, respectively.
To determine the number average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity index (PDI) of the copolymers, asymmetric flow field-flow fractionation (AF4) was performed using a Wyatt Eclipse instrument with UV (Agilent 1230 infinity, Agilent), refractive index (Optilab rex, 633 nm, Wyatt) and multi-angle light scattering (MALS) (Dawn Heleos-II, 662 nm, Wyatt) detectors. A frit-inlet channel equipped with a regenerated cellulose membrane (MWCO 5 kDa, Millipore) and a W350 spacer was used as the separation channel. The samples were analyzed with a constant detector flow of 0.5 mL min−1, and a cross flow that decreased exponentially from 3 mL min−1 to 0 mL min−1 in 20 min. A 50 mM PBS buffer at pH 7.4 was prepared and filtered with a 0.1 μm membrane (Millipore) immediately before use as the eluent. The samples were dissolved in the PBS buffer with a concentration of 5 mg mL−1, and the injection volume was set to be 100 μL. The refractive index increment (dn/dc) values for the copolymers were determined with an Optilab rex detector. For the molecular weight calculation, dn/dc values of 0.135, 0.120 and 0.118 were measured for s-PPEGMEMA, m-PPEGMEMA, and l-PPEGMEMA, respectively. Astra software (Wyatt, version 7.1.3.15) was utilized for data analysis and determination of the molecular weight from Debye plots.41
2.3. Quartz crystal microbalance with dissipation monitoring (QCM-D)
LbL assembly and the pH-responsiveness of PPEGMEMA/alginate multilayered films was monitored with QCM-D (Q-Sense E1, Biolin Scientific, Gothenburg, Sweden) using silica-coated sensors (QSX 335, Biolin Scientific). In a typical QCM-D experiment, an alternating voltage is applied to a quartz crystal sensor, which gives rise to oscillation of the sensor at its fundamental resonance frequency (F). The resonance frequency is related to the mass of the oscillating sensor as well as any coupled mass. Additionally, the decay in the sensor oscillation is monitored upon repeated stopping of the drive generator output. Accordingly, the dissipation factor (D) is determined as the ratio of the dissipated energy to the total stored energy.The sensor was first aminated with APTES before the LbL assembly.42 To do so, the sensor was rinsed with copious amounts of ethanol and water, dried, and then plasma-treated (PDC-32G plasma cleaner, Harrick Plasma) in water vapor with a constant pressure of 0.5 Torr for 1 min. Afterwards, the sensor was exposed to APTES and toluene vapor for 18 hours by placing it in a vacuumed desiccator with a 50% (v/v) APTES/Toluene solution. Hereafter, the sensor was rinsed with copious amounts of toluene and ethanol and dried with compressed air. The sensor was then immediately mounted into the QCM-D module (QSense ellipsometry module, QELM 401, Biolin Scientific, Gothenburg, Sweden), and the measurement was started at 23 °C under a 150 μL min−1 flow rate of citric acid/phosphate buffer with pH 3. After obtaining a stable baseline for all the harmonics, alginate and PEGMEMA solutions were alternately loaded and rinsed with buffer to obtain 7 bilayers in total. Glutaraldehyde solution (1%, w/w) was then loaded for 2 hours to cross-link the multilayered film, after which the chamber was rinsed with pH 3 buffer to remove the unbounded glutaraldehyde. To test the pH stability, 30 mM NaCl solutions with pH 3 and 5.6 were loaded, following which three consecutive pH cycles were conducted. Finally, the pH-responsiveness of the film was examined through titration of NaCl solutions with pH values ranging between 2 to 9. In all cases, the pH was adjusted with HCl/NaOH and NaCl was the only added electrolyte.
For thin, uniform and rigid films, the frequency shifts show a linear dependence to the adsorbed mass per unit area according to the Sauerbrey equation.43 The Sauerbrey equation, however, only provides a valid estimation of the ratio if dissipation and normalized frequency shifts ((ΔDn)/(−Δfn/n)) are significantly smaller than 4 × 10−7 Hz−1; otherwise, the adsorbed mass will be underestimated.44 For soft and highly hydrated polymeric films, as in the present case, the measured shifts (for different harmonics) in the resonance frequency and dissipation factor can be related to the thickness and viscoelastic properties of the film through modeling.44 Here, the viscoelastic Voigt model, where the adhered layer is represented by a film of uniform thickness and density with distinct viscous and elastic components can provide a better estimation of the film properties. The measured frequency and dissipation shifts are then related to the properties of the film and medium by:45
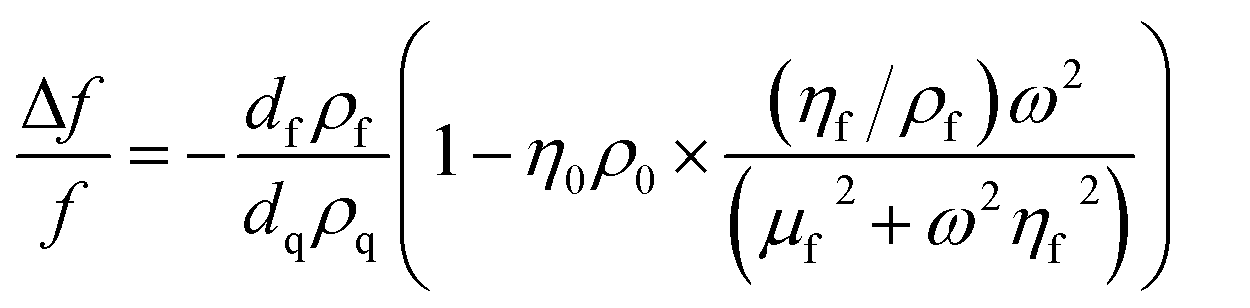
where ω is the angular frequency of oscillation, η0 and ρ0 are the viscosity and density of the medium, respectively, and dq and ρq are the thickness and density of the quartz crystal resonator, respectively. The density and viscosity of the medium together with the density of the hydrated film are estimated and treated as fixed parameters to avoid overparameterization. The thickness (df), viscosity (ηf) and shear modulus (μf) of the polymeric film are thus obtained from Voigt modeling. The instrument software (Dfind, Biolin Scientific) was employed for analysis of the data. The density of the hydrated film (ρf) was estimated to be 1050 kg m−3 (see ESI, Section S2†). The density and viscosity of water at 23 °C (from the software library) was used for the medium.
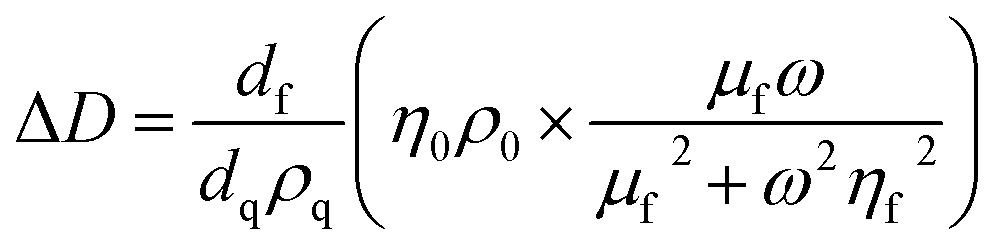
2.4. Spectroscopic ellipsometry
As mentioned above, the QCM-D studies of the LbL assembly and tests of the pH responsiveness of the films were conducted in an ellipsometry module allowing for simultaneous QCM-D and ellipsometry measurement. Spectroscopic ellipsometry (M2000, JA Woollam Co., USA) is an optical method in which changes in the polarization state of light upon interaction with a sample are measured. Two parameters, i.e., ψ (amplitude ratio) and Δ (phase shift) are used to quantify the polarization change. The measured parameters correlate with the optical properties and thickness of the sample components:46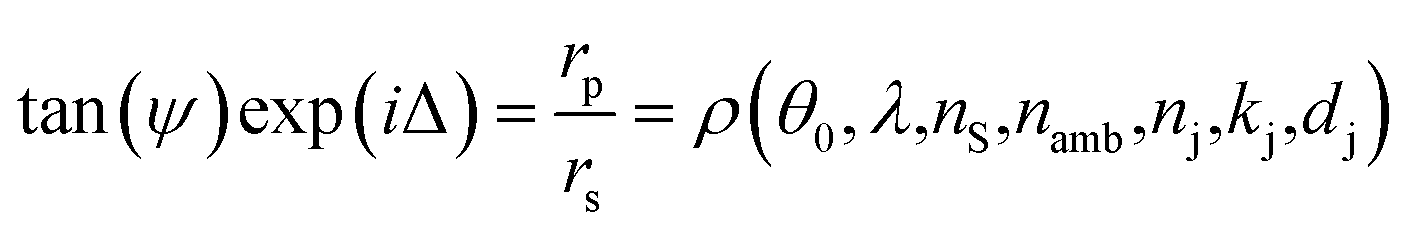
Here, the subscripts p and s refer to the parallel and perpendicular directions with respect to the plane of incidence, respectively, and rp and rs represent the reflection coefficients. The rP to rS ratio is a complex function of the angle of incidence (θ0), wavelength (λ), optical functions of the substrate (ns) and the ambient (namb), as well as the optical functions (nj, kj) and thickness (dj) of the film.
The QSense ellipsometry module (QELM 401, Biolin Scientific, Gothenburg, Sweden) allows in situ ellipsometry measurements on the QCM-D sensor through two optical windows. Accordingly, ψ and Δ spectra were collected (wavelength range: 250–1000 nm, angle of incidence of 70°) before adsorption of the first layer (bare sensor data) and at the end of each rinsing step. The instrument software (CompleteEASE, JA Woollam Co., USA) was employed to model the ψ and Δ data. The optical model employed was composed of three layers representing a uniform hydrated polymer film, a silica coating, and a thick optically opaque titanium substrate. First, the bare sensor was modeled as a pseudosubstrate, and the fitted parameters were fixed in the model (see ESI, Section S3†). Considering the low ionic strength of the buffer solutions, the optical data for pure water at 23 °C from the software library were used for the medium.47 The multilayered film was regarded as a transparent and homogeneous layer with no adsorption (k = 0); then, the refractive index (n) was described by the Cauchy equation:48
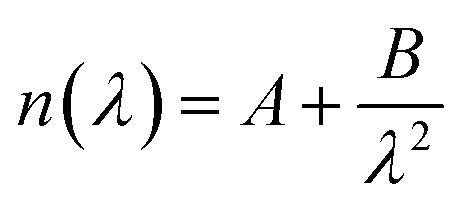
In the first modeling approach, the hydrated multilayered film was considered as a single-component film characterized by a thickness and two effective optical constants (Aeff, Beff). In the second modeling approach, the multilayered film was considered as a two-component layer consisting of a dry polymer (A = 1.5 and B = 0.005)49,50 and water. In addition to film thickness, the volume fraction of water (fw) was then estimated according to the Bruggeman Effective Medium Approximation (BEMA):51
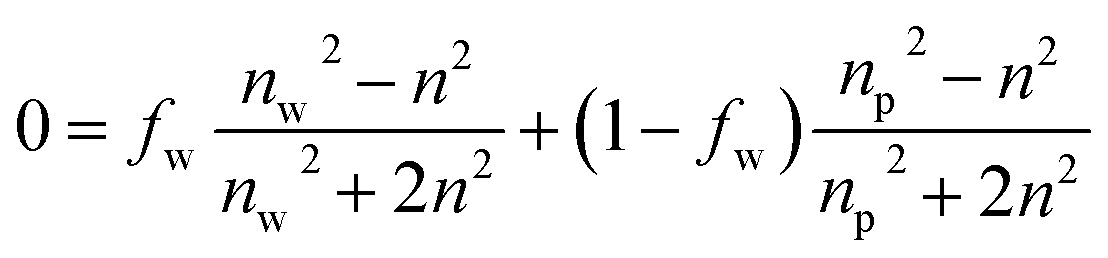
3. Results and discussion
3.1. P(PEGMEMA-stat-AMA) cationic copolymers for multilayer assembly
As described in the experimental section, three PEGMEMA-based cationic copolymers, with varying average PEG side chain lengths were prepared by ATRP, as shown in Scheme 1; a summary of the properties of the synthesized copolymers is provided in Table 1. The conversion of each polymerization was determined from 1H NMR data, based on which the actual degree of polymerization of each monomer was obtained. Overall, the degree of polymerization for all three copolymers was comparable, with a slight variation observed due to a minor difference in the conversion. The number average molecular weight and polydispersity of the copolymers was characterized by AF4 measurement (see Table 1). Here, it is seen that the s-PPEGMEMA has a relatively narrow molecular weight distribution, while the PDI value was observed to increase as the average PEG chain length increased. The latter observation indicates less control of the polymerization for longer PEG side chains.3.2. LbL assembly
Fig. 2 demonstrates the QCM-D data in terms of the shifts in the oscillation frequency (F) and dissipation factor (D) resulting from LbL assembly of PPEGMEMA/alginate multilayered films. The negative shift in oscillation frequency correlates to the effective mass (polymer film plus hydrodynamic water content) coupled to the sensor. The dissipation factor, on the other hand, represents the ability of the adhered film to dissipate the oscillatory energy; consequently, it is considered as a semiquantitative measure of the film conformation and viscoelasticity. Accordingly, a soft and hydrated film is characterized by a relatively large positive shift in the dissipation factor, while a rigid and compact film produces relatively small shifts in the dissipation factor.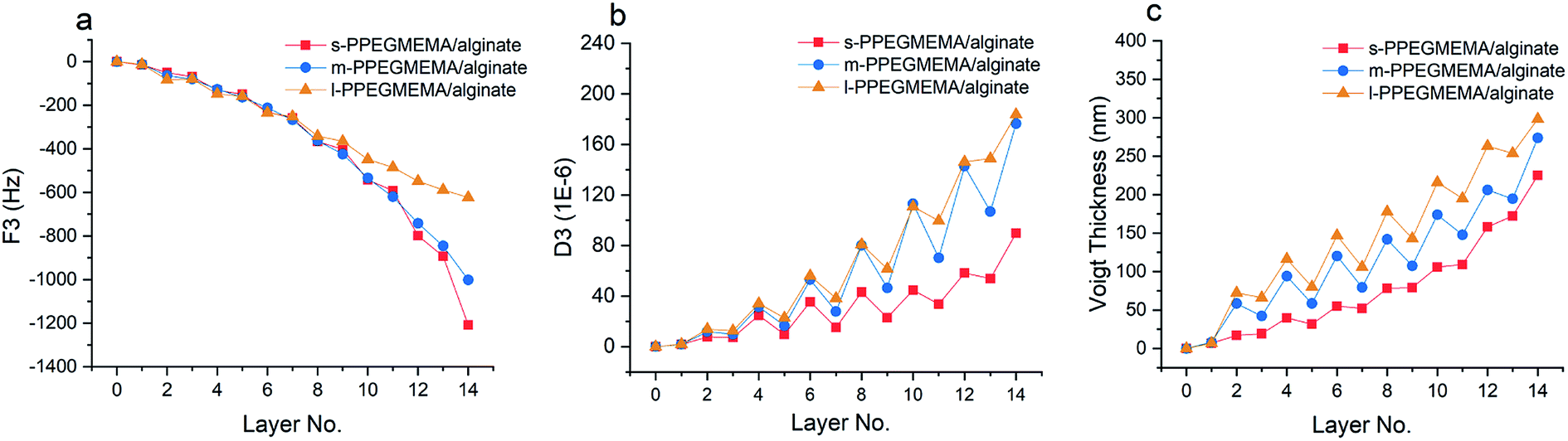 | ||
| Fig. 2 QCM-D frequency shift (a), dissipation shift (b) and the corresponding Voigt thickness (c) for LbL assembly of s-PEGMEMA(red square), m-PEGMEMA(blue circle) and l-PEGMEMA (yellow triangle) with alginate in pH 3 citric acid/phosphate buffer. | ||
According to Fig. 2, the measured dissipation shifts for 14 layers (7 bilayers) for s-PPEGMEMA, m-PPEGMEMA and l-PPEGMEMA samples are approximately 90 × 10−6, 180 × 10−6 and 183 × 10−6, respectively. Such large dissipation values suggest that the multilayered films are all swollen and highly hydrated. On the other hand, the frequency shifts for 14 layers for the s-PPEGMEMA, m-PPEGMEMA, and l-PPEGMEMA samples are around −1200, −1000 and −620 Hz, respectively. Accordingly, increasing the average length of the PEG side chains leads to a relatively larger dissipation shift (more viscoelastic film) and a relatively smaller frequency shift (smaller coupled mass). The model was then fitted to the frequency and dissipation data to estimate the Voigt thickness of each PPEGMEMA/alginate multilayered film as a function of the deposition number (see Fig. 2c and Table 2).
| polymer | Voigt Thickness (nm) | Cauchy Thickness (nm) | BEMA Thicknessa (nm) | BEMA Water Contenta (%) | Dry Thicknessb (nm) |
|---|---|---|---|---|---|
| a Fitted with the BEMA method, Bruggeman model.b Fitted with the Cauchy method, with mean A, B and C parameters for PPEGMEMA and alginate Cauchy parameters. | |||||
| s-PPEGMEMA | 225.1 | 200.7 ± 0.3 | 198.9 ± 0.3 | 77.3 ± 0.1 | 48.9 ± 0.1 |
| m-PPEGMEMA | 273.8 | 180.6 ± 0.6 | 177.1 ± 0.6 | 83.3 ± 0.1 | 29.4 ± 0.1 |
| l-PPEGMEMA | 297.6 | 163.3 ± 2.7 | 156.1 ± 2.6 | 91.5 ± 0.2 | 17.6 ± 0.1 |
According to Fig. 2c, it appears that the LbL assembly of s-PPEGMEMA/alginate multilayered films follows a nonlinear growth mechanism. However, the multilayer growth seems to be inclined towards a linear growth mechanism when increasing the length of the PEG side chains in the copolymers. It has been suggested that the diffusion of polyelectrolyte chains “in and out” of the film during the LbL buildup can give rise to nonlinear growth.1,52–54 Accordingly, one could speculate that the bulkiness of the PEG side chains can hinder segmental diffusion within the film, and, as a consequence, promote a linear growth mechanism.
In addition, while the frequency data show a monotonic gain, the dissipation data demonstrate a zigzag trend. Accordingly, adsorption of PPEGMEMA copolymers gives rise to a relatively large increment in dissipation, whereas the deposition of alginate produces a considerable decrement in dissipation. This can be interpreted based on the conformation of the interfacial top layer that has been previously observed.55 When the PPEGMEMA copolymer is the top layer, a highly swollen and hydrated conformation is expected for the interfacial layer, which thus explains the large dissipation gain. On the other hand, adsorption of alginate seems to promote partial collapse of the sublayer, subsequently producing a relatively rigid and less hydrated interfacial layer.
Spectroscopic ellipsometry data can provide further information for the structural features of the multilayered films. Fig. 3 illustrates the ψ and Δ spectra for the bare sensor as well as polymer-coated sensor after each deposition step. For s-PPEGMEMA, the LbL deposition is characterized by marked spectral oscillations in both the ψ and Δ spectra. These changes are more pronounced specifically after the 10th deposited layer, where a secondary peak evolves in the UV range of the ψ spectra and a significant shift in the Δ spectra is found. This observation is in line with the QCM-D data, suggesting a non-monotonic growth in thickness. For m-PPEGMEMA, the changes in the ψ and Δ spectra are relatively less pronounced, which can imply a relatively smaller thickness and (or) higher water content in the film. For l-PPEGMEMA, the evolution of the ψ and Δ spectra is even less significant as no trace of a secondary peak is found in the ψ spectra and a relatively smaller shift in the Δ spectra is demonstrated, which together suggests a thinner and (or) more hydrated layer compared to the layers obtained with s-PPEGMEMA and m-PPEGMEMA. In addition, the shifts in ψ and Δ following LbL deposition indicate a monotonic trend, which suggests a linear growth mechanism.
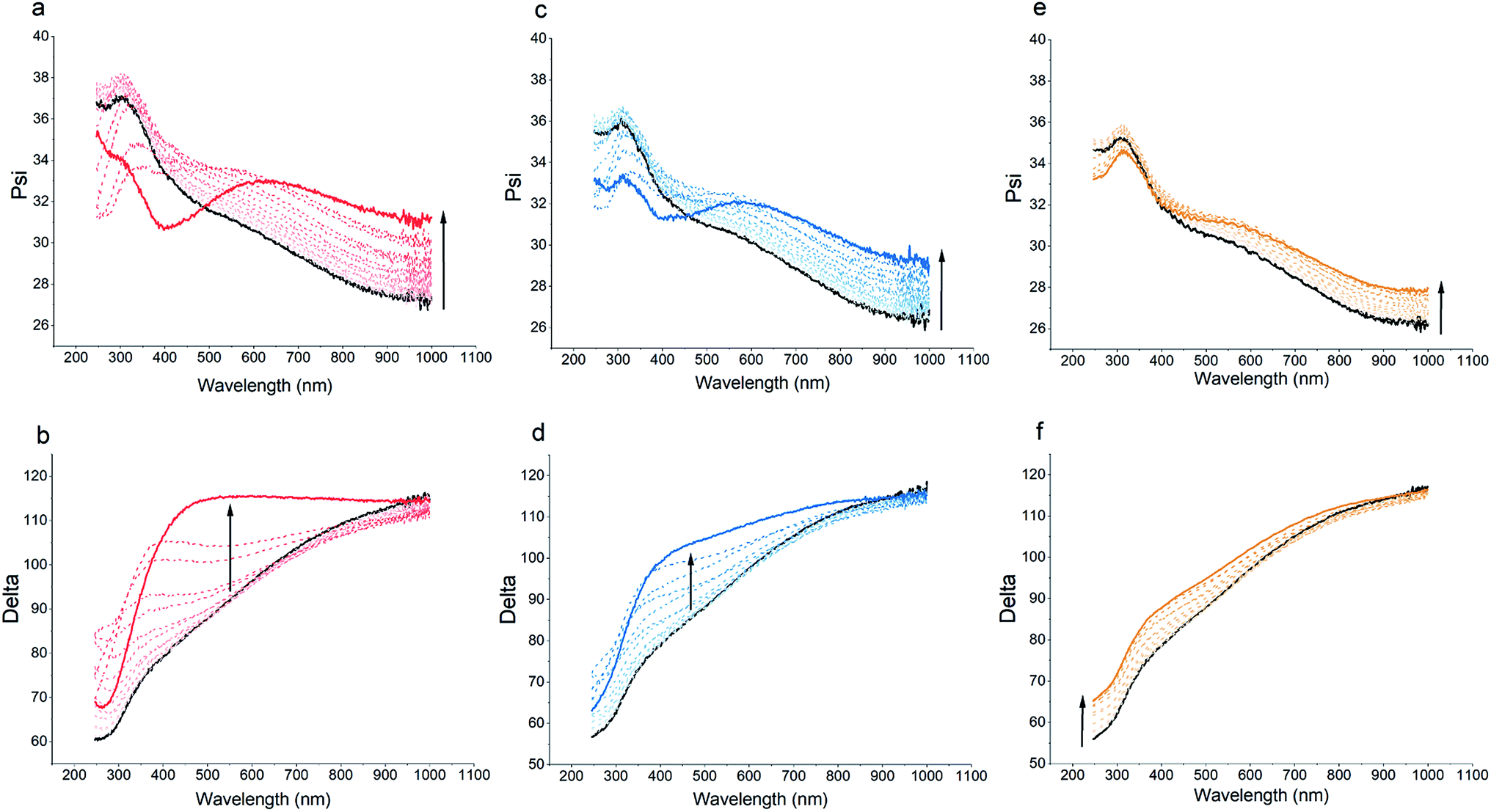 | ||
| Fig. 3 Ellipsometric Psi (top row) and Delta (bottom row) for LbL assembly of s-PPEGMEMA (a and b), m-PPEGMEMA (c and d) and l-PPEGMEMA (e and f) with alginate in pH 3 citric acid/phosphate buffer (the black solid curve represents the bare sensor and the arrows hint at LbL deposition). | ||
To obtain more quantitative information for the structure of the films, the spectroscopic ellipsometry data were examined using a single-component Cauchy and a two-component BEMA model. According to both modeling approaches, increasing the average length of the PEG side chains leads to a decrement in the total hydrated film thickness from approximately 200 nm for the s-PPEGMEMA/alginate multilayered film to approximately 160 nm for the l-PPEGMEMA/alginate multilayered film, while the water content within the film increases roughly from 77% for the s-PPEGMEMA/alginate multilayered film to 91% for the l-PPEGMEMA/alginate multilayered film. This difference in the water content of the films is reflected in the refractive index dispersion of the hydrated layers (see Fig. 4). As a general observation, the refractive indices for all three layers are closer to that of water than of the dry polymer, suggesting a large amount of water in the films. In addition, increasing the average PEG side chain length gives rise to a decrement in the refractive index, suggesting that the water content is relatively large. The increase in water content obtained upon increasing the average PEG side chain length can also explain the larger QCM-D dissipation shift for this layer. In addition, the dry thicknesses of the s-PPEGMEMA, m-PPEGMEMA, and l-PPEGMEMA samples are found to be approximately 49, 29 and 18 nm, respectively. Accordingly, it can be concluded that increasing the average length of the PEG side chains decreases the amount of adsorbed polymer chains, which can be attributed to a larger steric hindrance for LbL deposition.
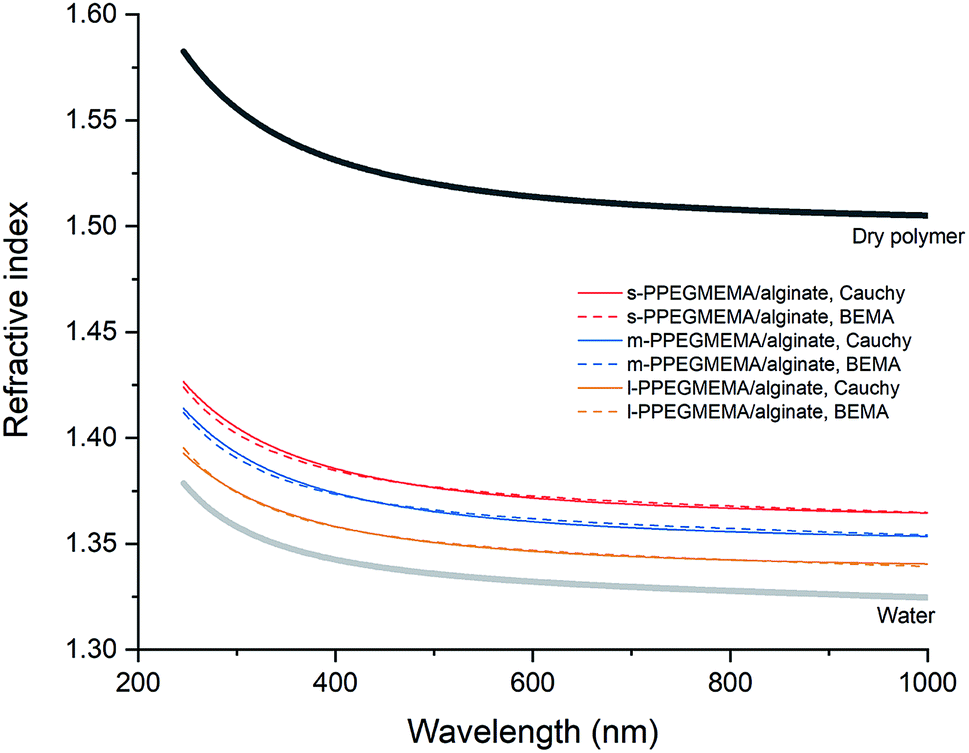 | ||
| Fig. 4 Refractive indices obtained from single-component Cauchy (solid line) and two-component BEMA (dashed line) models for 7 bilayers of s-PPEGMEMA/alginate (red), m-PPEGMEMA/alginate (blue) and l-PPEGMEMA/alginate (yellow) multilayered films. Refractive indices for water (bold gray) and dry polymer (bold black) are provided for comparison. | ||
It is noteworthy that a discrepancy in film thickness was observed between Voigt modeling and the optical thickness (Table 2). We attribute this discrepancy to the inhomogeneous nature of the PPEGMEMA/alginate multilayered film. In particular, while the inner layers of the obtained film are relatively impact, the outer PPEGMEMA layer is highly swollen and hydrated. The sparsely distributed PPEGMEMA chains in the outermost layer of the film significantly affect the dissipation value and add to the overestimation of the Voigt thickness. This effect is much less significant to ellipsometry modeling, where an outer layer with low polymer density will contribute with a low weight to the optical thickness.
3.3. Stabilization of the PPEGMEMA/alginate film by cross-linking
As electrostatic interactions are the driving force for the multilayer buildup, the pH at which the multilayered film is fabricated should roughly correspond to a state of charge neutrality. Accordingly, a subsequent change in pH will create a charge imbalance that will cause excessive swelling and disintegration of the film (see ESI, Fig. S17†). Chemical cross-linking, on the other hand, is an effective method to enhance the pH-stability of polyelectrolyte multilayered films. Glutaraldehyde, as a common cross-linker for proteins and polysaccharides, can form a dynamic covalent bond between free amine groups on the PPEGMEMA copolymer through the Schiff base reaction.56–58 Herein, we selected the m-PPEGMEMA/alginate multilayered film to investigate the effect of cross-linking and pH-stability.Fig. 5 demonstrates the structural changes in the multilayered film resulting from the cross-linking process. With respect to the QCM-D data (Fig. 5a), a significant decrement in the dissipation of around −80 × 10−6 accompanied by a small frequency shift of around −70 Hz, is observed. While the change in frequency can be considered insignificant compared to the total frequency shift during the layer buildup (see Fig. 2a), the relatively large decrease in dissipation suggests structural collapse and enhanced rigidity of the film after cross-linking.
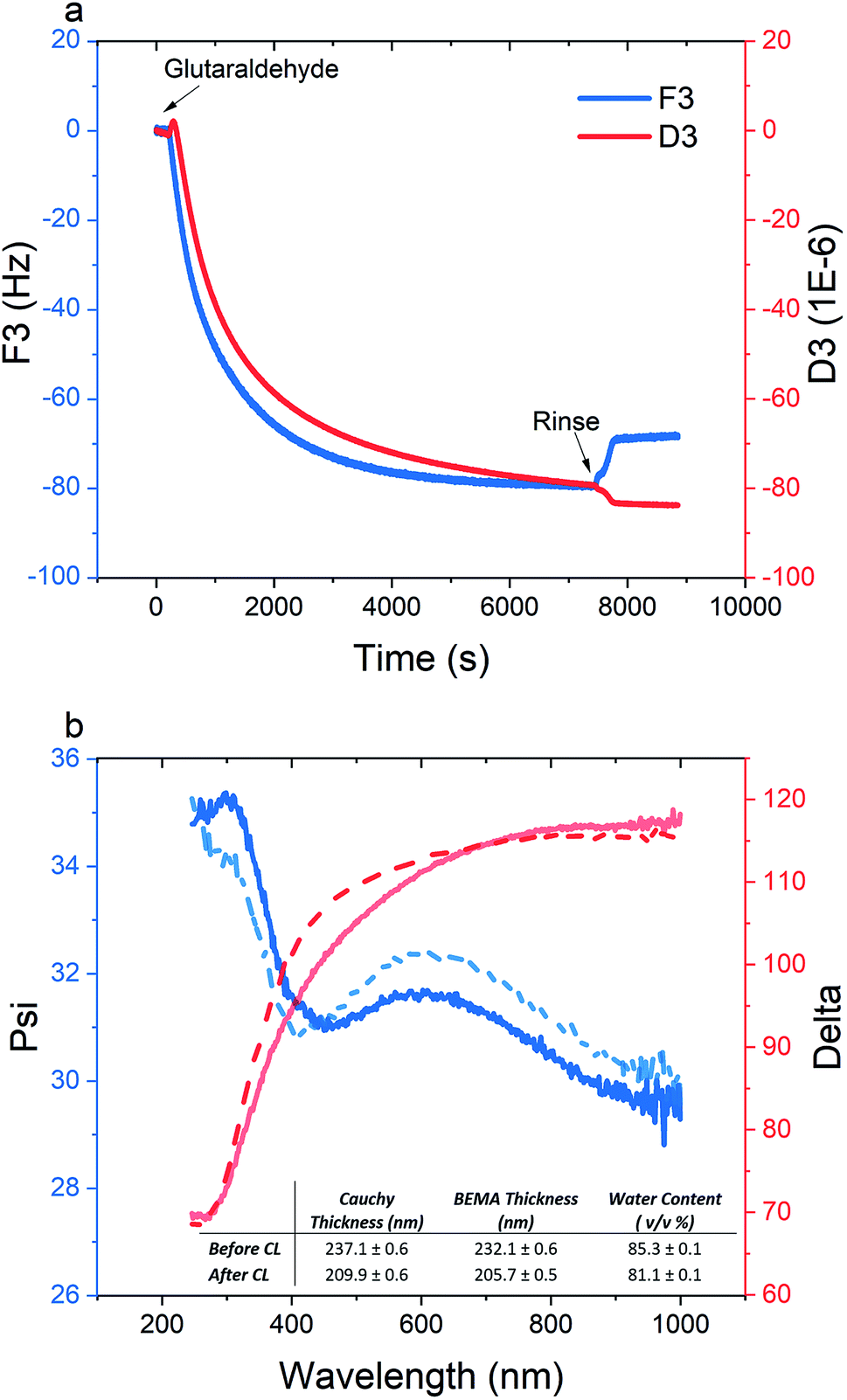 | ||
| Fig. 5 (a) Frequency (blue) and dissipation (red) shifts for the cross-linking step of a m-PPEGMEMA/alginate multilayered film with 1% glutaraldehyde; (b) Psi (blue) and Delta (red) before (dashed line) and after (solid line) cross-linking. The inset shows the modeling result obtained from the Cauchy and BEMA methods. | ||
According to the ellipsometry data, chemical cross-linking is accompanied by a decrease in the optical thickness from 237 nm to 210 nm, confirming the film shrinkage, as inferred by the dissipation data. In the same line, a decrease in water content from 85% to 81% is demonstrated. The film shrinkage could be attributed to the consumption of a portion of the amine groups, and, hence, charge regulation within the film, as well as the conformational immobilization caused by the chemical cross-links.
3.4. pH-responsive behavior of the m-PPEGMEMA/alginate multilayered film
Fig. 6 demonstrates the stability of the multilayered film upon three consecutive pH cycles between 3 and 5.6. According to the QCM-D data (Fig. 6a), increasing the pH from 3 to 5.6 leads to a gain in dissipation and a decline in frequency, which both imply swelling and hydration of the polymer film. By lowering the pH back to 3, the frequency and dissipation shifts demonstrate conformational collapse and dehydration of the film. During the three pH cycles, no significant layer disintegration and mass loss was observed, indicating enhanced pH stability of the film after cross-linking. It is, however, worth mentioning that a slight structural hysteresis (irreversible frequency and dissipation shifts) is found after each pH cycle, which could be attributed to conformational restructuring within the film that can also cause irreversible ionization of amine and carboxyl groups.59–61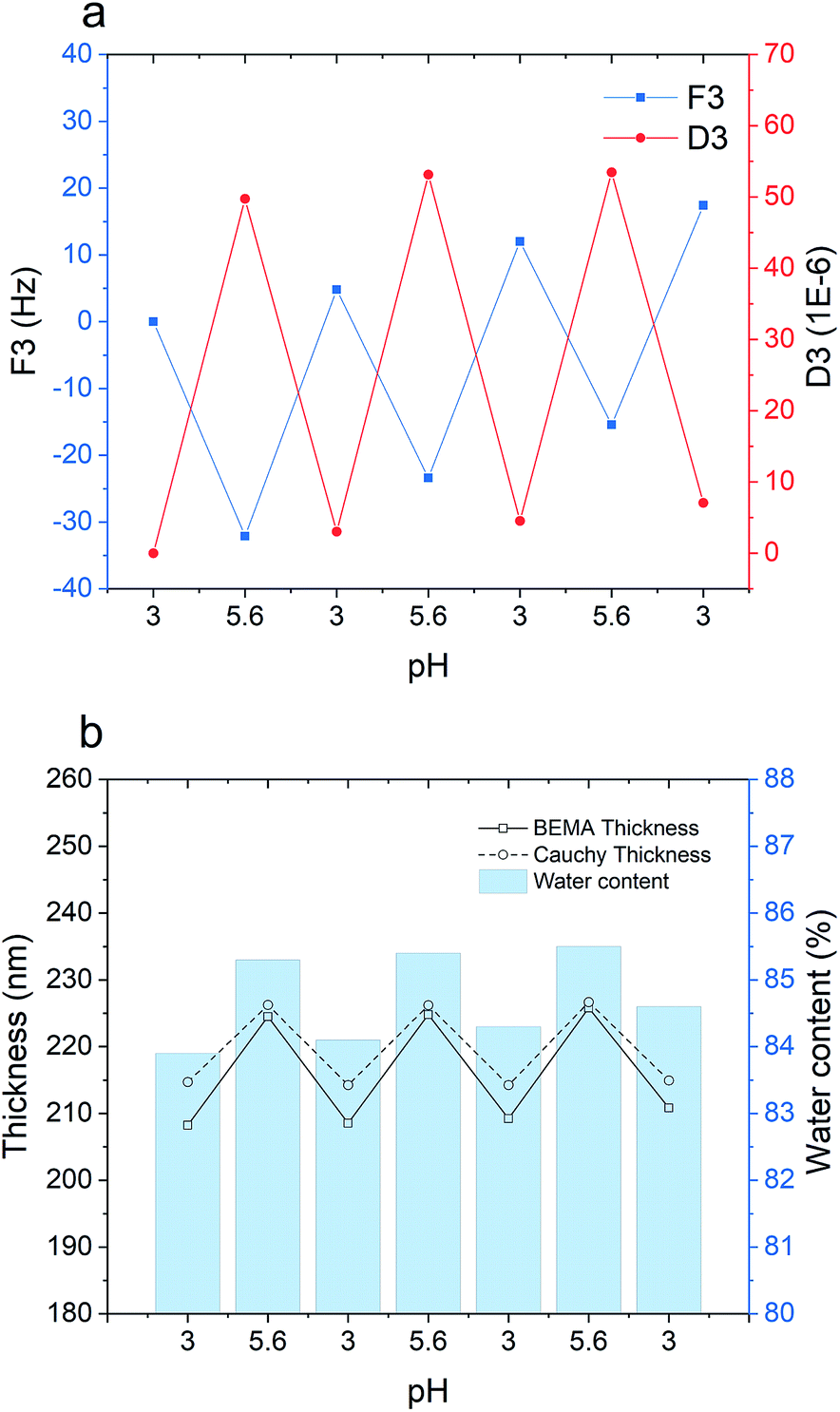 | ||
| Fig. 6 m-PPEGMEMA/alginate multilayered film stability as a function of pH during repeated changes between pH 3 and 5.6: (a) frequency and dissipation changes monitored with QCM-D; (b) Cauchy (dashed line, circle) and BEMA thicknesses (solid line, square) as well as water content (violet bar) obtained via the BEMA model from ellipsometry. | ||
Ellipsometry results (Fig. 6b) represent the same effect as concluded from the QCM-D data. During pH cycling, the Cauchy thickness exhibited a zig-zag trend in agreement with the frequency and dissipation shifts. Accordingly, the optical thickness obtained with the BEMA model fluctuates between approximately 210 nm at pH 3 and 225 nm at pH 6, while the estimated water content at the same time also varies systematically.
To further investigate the pH-responsiveness of the PPEGMEMA/alginate multilayered film, a pH titration was performed (see Fig. 7). For the QCM-D data presented in Fig. 7a, it can be seen that increasing the pH from 2 to 4 results in a relatively large dissipation shift of around −50 × 10−6, suggesting shrinkage of the film. In contrast, further increasing the pH from 4 to 9 is characterized by a relatively large gain in the dissipation of approximately 140 × 10−6, indicating a reswelling of the film. For the frequency shift, a similar variation is observed although the trend is less clear.
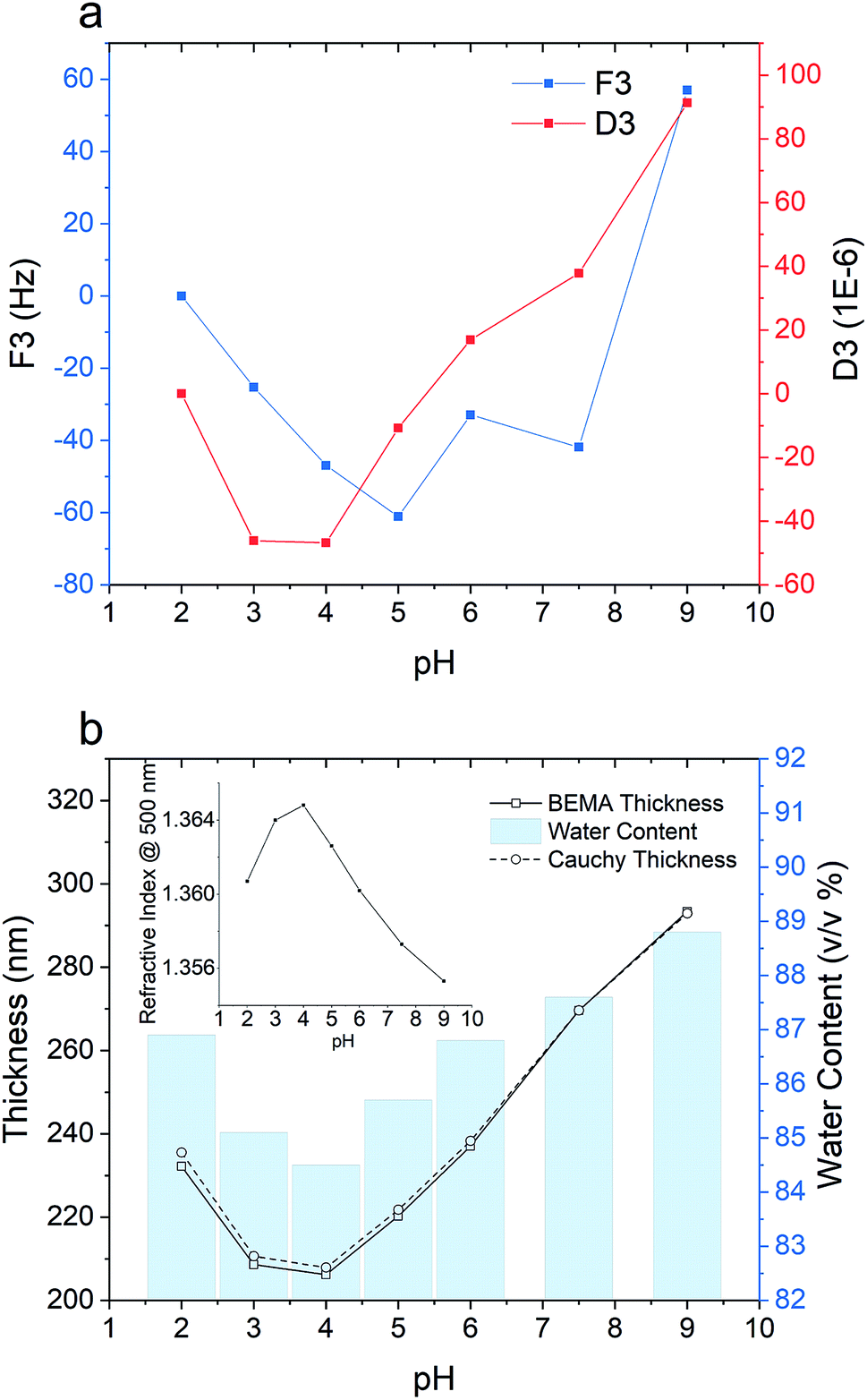 | ||
| Fig. 7 pH responsiveness of a m-PPEGMEMA/alginate multilayered film; (a) frequency and dissipation changes obtained from QCM-D; (b) Cauchy thickness (circle, dashed line), BEMA thickness (square, solid line) and water content (blue bar). The inset shows how the refractive index changes with the pH variation). | ||
In the ellipsometry data (see Fig. 7b), increasing the pH from 2 to 4 is associated with a decline in the BEMA thickness from 235 nm to 208 nm, while further increasing the pH results in a gain in thickness from 208 to 290 nm. The estimated water content followed the same trend as the film thickness, i.e., decreasing from 87 to 85% when the pH is changed from 2 to 4 and increasing from 85 to 89% when the pH is changed from 4 to 9.
The swollen-collapsed-swollen conformational change of the multilayered film when the pH is first changed from 2 to 4 and then subsequently changed to 9 can be explained based on pH-dependent charge regulation within the multilayered film. The collapsed state in the pH range of 3–4 could be attributed to the charge neutral state within the multilayered film. Under such conditions, the number of charged amine and carboxyl groups within the film are roughly comparable. Decreasing the pH to 2 is associated with protonation of the amine groups on the PPEGMEMA copolymer and the carboxylate groups on the alginate, creating a net positive charge within the film. This leads to the accumulation of counterions (Cl−) within the film that can produce an osmotic pressure difference, and, hence, swelling of the film. On the other hand, increasing the pH to 9 is accompanied by deprotonation of the carboxyl groups and amine groups, which produces a net negative charge in the film. Accumulation of Na+ counterions then results in an osmotic swelling. Thus, the swollen-collapsed-swollen conformational change is due to the cross-linked PPEGMEMA/alginate multilayered film exhibiting cationic, zwitterionic and anionic states as a function of pH. To this end, we note that the charging state of the multilayered film, as well as the possibility to change between different charging states, is of high importance for the film functionality and for possible applications. While both the hydration level and the sign of the surface potential will be decisive for the films ability to either attract or repel specific biomolecules or cells,62 the responsive nature of the film can be used for control release of encapsulated drugs,63 for response membranes64 or in sensor technologies.65 For these three different states, it must, however, be considered that the swelling capacity of the PPEGMEMA/alginate multilayered film is governed by the balance between the osmotic pressure of the solution and the entropic penalty of stretching the polymer chains between the cross-linking points.
4. Conclusion
In this work, three PPEGMEMA-based cationic copolymers with different average PEG side chain lengths were paired with alginate via electrostatic LbL assembly. In situ QCM-D and ellipsometry measurements were used to follow the multilayer buildup process for the three different systems, and the result shows that the thickness and structure of the multilayered film is closely correlated to the average PEG side chain length in the PPEGMEMA-based cationic copolymers. Specifically, as the average PEG side chain length increases, two main observations are noted. First, it is found that the multilayer builds up less effectively with a reduced increase in added mass during the alternating deposition of polymers. Second, the film is found to become more dissipative and exhibit a higher water content. Both of these effects are interpreted as being directly related to the average PEG side chain length, which, due to the high hydrophilicity of the PEG units, leads to a highly hydrated layer and to steric hindrance, which limits the amount of polymer adsorbed in each deposition step.After buildup, the obtained PPEGMEMA/alginate multilayered films were cross-linked with glutaraldehyde to exhibit pH responsiveness without disintegration of the layer due to charge imbalance in the layer. It was further demonstrated how the PPEGMEMA/alginate multilayered films can be in a cationic, zwitterionic or anionic state depending on the pH, and how the transitions between these states lead to structural changes in the layer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the financial support from the Independent Research Fund Denmark (grant # 6111-00102B).References
- J. Borges and J. F. Mano, Molecular Interactions Driving the Layer-by-Layer Assembly of Multilayers, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS PubMed.
- G. Decher, Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites, Science, 1997, 277, 1232–1237 CrossRef CAS.
- K. Ariga, Y. Lvov and T. Kunitake, Assembling Alternate Dye-Polyion Molecular Films by Electrostatic Layer-by-Layer Adsorption, J. Am. Chem. Soc., 1997, 119, 2224–2231 CrossRef CAS.
- J. Huang, S. Zajforoushan Moghaddam and E. Thormann, Structural Investigation of a Self-Cross-Linked Chitosan/Alginate Dialdehyde Multilayered Film with in Situ QCM-D and Spectroscopic Ellipsometry, ACS Omega, 2019, 4, 2019–2029 CrossRef CAS.
- J. D. Delgado, R. L. Surmaitis, S. Abou Shaheen and J. B. Schlenoff, Engineering Thiolated Surfaces with Polyelectrolyte Multilayers, ACS Appl. Mater. Interfaces, 2019, 11, 3524–3535 CrossRef CAS PubMed.
- P. Gentile, I. Carmagnola, T. Nardo and V. Chiono, Layer-by-Layer Assembly for Biomedical Applications in the Last Decade, Nanotechnology, 2015, 26, 422001 CrossRef CAS PubMed.
- J. J. Richardson, M. Björnmalm and F. Caruso, Technology-Driven Layer-by-Layer Assembly of Nanofilms, Science, 2015, 348, aaa2491 CrossRef.
- Y. Liu, M. L. Bruening, D. E. Bergbreiter and R. M. Crooks, Multilayer Dendrimer-Polyanhydride Composite Films on Glass, Silicon, and Gold Wafers, Angew. Chem., Int. Ed. Engl., 1997, 36, 2114–2116 CrossRef CAS.
- C. Liu, E. Thormann, P. M. Claesson and E. Tyrode, Surface Grafted Chitosan Gels. Part I. Molecular Insight into the Formation of Chitosan and Poly(Acrylic Acid) Multilayers, Langmuir, 2014, 30, 8866–8877 CrossRef CAS.
- M. Lundin, F. Solaqa, E. Thormann, L. MacAkova and E. Blomberg, Layer-by-Layer Assemblies of Chitosan and Heparin: Effect of Solution Ionic Strength and PH, Langmuir, 2011, 27, 7537–7548 CrossRef CAS.
- W. B. Stockton and M. F. Rubner, Molecular-Level Processing of Conjugated Polymers. 4. Layer-by-Layer Manipulation of Polyaniline via Hydrogen-Bonding Interactions, Macromolecules, 1997, 30, 2717–2725 CrossRef CAS.
- L. Wang, Z. Wang, X. Zhang, J. Shen, L. Chi and H. Fuchs, A New Approach for the Fabrication of an Alternating Multilayer Film of Poly(4-Vinylpyridine) and Poly(Acrylic Acid) Based on Hydrogen Bonding, Macromol. Rapid Commun., 1997, 18, 509–514 CrossRef CAS.
- G. K. Such, A. P. R. Johnston and F. Caruso, Engineered Hydrogen-Bonded Polymer Multilayers: From Assembly to Biomedical Applications, Chem. Soc. Rev., 2011, 40, 19–29 RSC.
- X. Liang, V. Kozlovskaya, Y. Chen, O. Zavgorodnya and E. Kharlampieva, Thermosensitive Multilayer Hydrogels of Poly(N-Vinylcaprolactam) as Nanothin Films and Shaped Capsules, Chem. Mater., 2012, 24, 3707–3719 CrossRef CAS PubMed.
- D. E. Bergbreiter and K.-S. Liao, Covalent Layer-by-Layer Assembly—an Effective, Forgiving Way to Construct Functional Robust Ultrathin Films and Nanocomposites, Soft Matter, 2009, 5, 23–28 RSC.
- P. Kohli and G. J. Blanchard, Applying Polymer Chemistry to Interfaces: Layer-by-Layer and Spontaneous Growth of Covalently Bound Multilayers, Langmuir, 2000, 16, 4655–4661 CrossRef CAS.
- M. Schönhoff and P. Bieker, Linear and Exponential Growth Regimes of Multilayers of Weak Polyelectrolytes in Dependence on PH, Macromolecules, 2010, 43, 5052–5059 CrossRef.
- S. S. Shiratori and M. F. Rubner, PH-Dependent Thickness Behavior of Sequentially Adsorbed Layers of Weak Polyelectrolytes, Macromolecules, 2000, 33, 4213–4219 CrossRef CAS.
- S. T. Dubas and J. B. Schlenoff, Swelling and Smoothing of Polyelectrolyte Multilayers by Salt, Langmuir, 2001, 17, 7725–7727 CrossRef CAS.
- R. A. McAloney, M. Sinyor, V. Dudnik and M. Cynthia Goh, Atomic Force Microscopy Studies of Salt Effects on Polyelectrolyte Multilayer Film Morphology, Langmuir, 2001, 17, 6655–6663 CrossRef CAS.
- H. L. Tan, M. J. McMurdo, G. Pan and P. G. Van Patten, Temperature Dependence of Polyelectrolyte Multilayer Assembly, Langmuir, 2003, 19, 9311–9314 CrossRef CAS.
- Y. H. Yang, F. A. Malek and J. C. Grunlan, Influence of Deposition Time on Layer-by-Layer Growth of Clay-Based Thin Films, Ind. Eng. Chem. Res., 2010, 49, 8501–8509 CrossRef CAS.
- P. Kujawa, P. Moraille, J. Sanchez, A. Badia and F. M. Winnik, Effect of Molecular Weight on the Exponential Growth and Morphology of Hyaluronan/Chitosan Multilayers: A Surface Plasmon Resonance Spectroscopy and Atomic Force Microscopy Investigation, J. Am. Chem. Soc., 2005, 127, 9224–9234 CrossRef CAS.
- D. Chen, M. Wu, B. Li, K. Ren, Z. Cheng, J. Ji, Y. Li and J. Sun, Layer-by-Layer-Assembled Healable Antifouling Films, Adv. Mater., 2015, 27, 5882–5888 CrossRef CAS PubMed.
- S. Guo, X. Zhu, M. Li, L. Shi, J. L. T. Ong, D. Jańczewski and K. G. Neoh, Parallel Control over Surface Charge and Wettability Using Polyelectrolyte Architecture: Effect on Protein Adsorption and Cell Adhesion, ACS Appl. Mater. Interfaces, 2016, 8, 30552–30563 CrossRef CAS PubMed.
- P. Esmaeilzadeh, A. Köwitsch, F. Heyroth, G. Schmidt, S. Fischer, K. Richter and T. Groth, Synthesis of Thiolated Polysaccharides for Formation of Polyelectrolyte Multilayers with Improved Cellular Adhesion, Carbohydr. Polym., 2017, 157, 1205–1214 CrossRef CAS PubMed.
- J. A. Jaber and J. B. Schlenoff, Polyelectrolyte Multilayers with Reversible Thermal Responsivity, Macromolecules, 2005, 38, 1300–1306 CrossRef CAS.
- X. Liu, E. Thormann, A. Dedinaite, M. Rutland, C. Visnevskij, R. Makuska and P. M. Claesson, Low Friction and High Load Bearing Capacity Layers Formed by Cationic-Block-Non-Ionic Bottle-Brush Copolymers in Aqueous Media, Soft Matter, 2013, 9, 5361 RSC.
- T. Pettersson, A. Naderi, R. Makuška and P. M. Claesson, Lubrication Properties of Bottle-Brush Polyelectrolytes: An AFM Study on the Effect of Side Chain and Charge Density, Langmuir, 2008, 24, 3336–3347 CrossRef CAS PubMed.
- A. Dworak, A. Utrata-Wesołek, D. Szweda, A. Kowalczuk, B. Trzebicka, J. Anioł, A. L. Sieroń, A. Klama-Baryła and M. Kawecki, Poly[Tri(Ethylene Glycol) Ethyl Ether Methacrylate]-Coated Surfaces for Controlled Fibroblasts Culturing, ACS Appl. Mater. Interfaces, 2013, 5, 2197–2207 CrossRef CAS PubMed.
- W. L. Chen, R. Cordero, H. Tran and C. K. Ober, 50th Anniversary Perspective: Polymer Brushes: Novel Surfaces for Future Materials, Macromolecules, 2017, 50, 4089–4113 CrossRef CAS.
- J.-F. Lutz, Thermo-Switchable Materials Prepared Using the OEGMA-Platform, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- X. Deng, N. M. B. Smeets, C. Sicard, J. Wang, J. D. Brennan, C. D. M. Filipe and T. Hoare, Poly(Oligoethylene Glycol Methacrylate) Dip-Coating: Turning Cellulose Paper into a Protein-Repellent Platform for Biosensors, J. Am. Chem. Soc., 2014, 136, 12852–12855 CrossRef CAS.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Surface-Initiated Controlled Radical Polymerization: State-of-the-Art, Opportunities, and Challenges in Surface and Interface Engineering with Polymer Brushes, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- M. C. R. Tria, C. D. T. Grande, R. R. Ponnapati and R. C. Advincula, Electrochemical Deposition and Surface-Initiated RAFT Polymerization: Protein and Cell-Resistant PPEGMEMA Polymer Brushes, Biomacromolecules, 2010, 11, 3422–3431 CrossRef CAS.
- Y. Liu, V. Klep and I. Luzinov, To Patterned Binary Polymer Brushes via Capillary Force Lithography and Surface-Initiated Polymerization, J. Am. Chem. Soc., 2006, 128, 8106–8107 CrossRef CAS PubMed.
- H. Ma, J. Hyun, P. Stiller and a. Chilkoti, “Non-Fouling” Oligo(Ethylene Glycol)-Functionalized Polymer Brushes Synthesized by Surface-Initiated Atom Transfer Radical Polymerization, Adv. Mater., 2004, 16, 338–341 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Macromolecular Engineering by Atom Transfer Radical Polymerization, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- M. H. Dufresne and J. C. Leroux, Study of the Micellization Behavior of Different Order Amino Block Copolymers with Heparin, Pharm. Res., 2004, 21, 160–169 CrossRef CAS PubMed.
- E. k. P. Kumar, L. N. Feldborg, K. Almdal and T. L. Andresen, Synthesis and Characterization of a Micelle-Based PH Nanosensor with an Unprecedented Broad Measurement Range, Chem. Mater., 2013, 25, 1496–1501 CrossRef.
- B. H. Zimm, The Scattering of Light and the Radial Distribution Function of High Polymer Solutions, J. Chem. Phys., 1948, 16, 1093–1099 CrossRef CAS.
- F. Zhang, K. Sautter, A. M. Larsen, D. A. Findley, R. C. Davis, H. Samha and M. R. Linford, Chemical Vapor Deposition of Three Aminosilanes on Silicon Dioxide: Surface Characterization, Stability, Effects of Silane Concentration, and Cyanine Dye Adsorption, Langmuir, 2010, 26, 14648–14654 CrossRef CAS PubMed.
- G. Sauerbrey, Use of a Quartz Vibrator for Weighing Thin Films on a Microbalance, Zeitschrift für Physik, 1959, 155, 206–222 CrossRef CAS.
- I. Reviakine, D. Johannsmann and R. P. Richter, Hearing What You Cannot See and Visualizing What You Hear: Interpreting Quartz Crystal Microbalance Data from Solvated Interfaces, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Viscoelastic Acoustic Response of Layered Polymer Films at Fluid-Solid Interfaces: Continuum Mechanics Approach, Phys. Scr., 1999, 59, 391–396 CrossRef CAS.
- J. A. Woollam, P. G. Snyder and M. C. Rost, Variable Angle Spectroscopic Ellipsometry: A Non-Destructive Characterization Technique for Ultrathin and Multilayer Materials, Thin Solid Films, 1988, 166, 317–323 CrossRef CAS.
- C. Laguerie, M. Aubry and J. P. Couderc, Some Physicochemical Data on Monohydrate Citric Acid Solutions in Water: Solubility, Density, Viscosity, Diffusivity, PH of Standard Solution, and Refractive Index, J. Chem. Eng. Data, 1976, 21, 85–87 CrossRef CAS.
- J. A. Woollam, B. D. Johs, C. M. Herzinger, J. N. Hilfiker, R. A. Synowicki and C. L. Bungay, Overview of Variable-Angle Spectroscopic Ellipsometry (VASE): I. Basic Theory and Typical Applications, Proc. SPIE, 1999, 10294, 1029402 CrossRef.
- J. J. I. Ramos and S. E. Moya, Water Content of Hydrated Polymer Brushes Measured by an in Situ Combination of a Quartz Crystal Microbalance with Dissipation Monitoring and Spectroscopic Ellipsometry, Macromol. Rapid Commun., 2011, 32, 1972–1978 CrossRef CAS PubMed.
- J. J. Iturri Ramos, S. Stahl, R. P. Richter and S. E. Moya, Water Content and Buildup of Poly(Diallyldimethylammonium Chloride)/Poly(Sodium 4-Styrenesulfonate) and Poly(Allylamine Hydrochloride)/Poly(Sodium 4-Styrenesulfonate) Polyelectrolyte Multilayers Studied by an in Situ Combination of a Quartz Crystal Microb, Macromolecules, 2010, 43, 9063–9070 CrossRef CAS.
- D. E. Aspnes, J. B. Theeten and F. Hottier, Investigation of Effective-Medium Models of Microscopic Surface Roughness by Spectroscopic Ellipsometry, Phys. Rev. B, 1979, 20, 3292–3302 CrossRef CAS.
- J. Ruths, F. Essler, G. Decher and H. Riegler, Polyelectrolytes. I: Polyanion/Polycation Multilayers at the Air/Monolayer/Water Interface as Elements for Quantitative Polymer Adsorption Studies and Preparation of Hetero-Superlattices on Solid Surfaces, Langmuir, 2000, 16, 8871–8878 CrossRef CAS.
- C. Picart, J. Mutterer, L. Richert, Y. Luo, G. D. Prestwich, P. Schaaf, J.-C. Voegel and P. Lavalle, Molecular Basis for the Explanation of the Exponential Growth of Polyelectrolyte Multilayers, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12531–12535 CrossRef CAS PubMed.
- D. Volodkin and R. Von Klitzing, Competing Mechanisms in Polyelectrolyte Multilayer Formation and Swelling: Polycation-Polyanion Pairing vs. Polyelectrolyte-Ion Pairing, Curr. Opin. Colloid Interface Sci., 2014, 19, 25–31 CrossRef CAS.
- R. A. Ghostine, M. Z. Markarian and J. B. Schlenoff, Asymmetric Growth in Polyelectrolyte Multilayers, J. Am. Chem. Soc., 2013, 135, 7636–7646 CrossRef CAS PubMed.
- A. Bigi, G. Cojazzi, S. Panzavolta, K. Rubini and N. Roveri, Mechanical and Thermal Properties of Gelatin Films at Different Degrees of Glutaraldehyde Crosslinking, Biomaterials, 2001, 22, 763–768 CrossRef CAS.
- S. Avrameas and T. Ternynck, The Cross-Linking of Proteins with Glutaraldehyde and Its Use for the Preparation of Immunoadsorbents, Immunochemistry, 1969, 6, 53–66 CrossRef CAS PubMed.
- C. Liu, E. Thormann, P. M. Claesson and E. Tyrode, Surface Grafted Chitosan Gels. Part II. Gel Formation and Characterization, Langmuir, 2014, 30, 8878–8888 CrossRef CAS PubMed.
- S. C. Howard, V. S. J. Craig, P. A. Fitzgerald and E. J. Wanless, Swelling and Collapse of an Adsorbed PH-Responsive Film-Forming Microgel Measured by Optical Reflectometry and QCM, Langmuir, 2010, 26, 14615–14623 CrossRef CAS PubMed.
- D. Lee, A. J. Nolte, A. L. Kunz, M. F. Rubner and R. E. Cohen, PH-Induced Hysteretic Gating of Track-Etched Polycarbonate Membranes: Swelling/Deswelling Behavior of Polyelectrolyte Multilayers in Confined Geometry, J. Am. Chem. Soc., 2006, 128, 8521–8529 CrossRef CAS PubMed.
- K. Itano, J. Choi and M. F. Rubner, Mechanism of the PH-Induced Discontinuous Swelling/Deswelling Transitions of Poly(Allylamine Hydrochloride)-Containing Polyelectrolyte Multilayer Films, Macromolecules, 2005, 38, 3450–3460 CrossRef CAS.
- D. S. Salloum and J. B. Schlenoff, Protein Adsorption Modalities of Polyelectrolyte Multilayers, Biomacromolecules, 2004, 5, 1089–1096 CrossRef CAS PubMed.
- J. Wu and M. J. Sailor, Chitosan Hydrogel-Capped Porous SiO2 as a PH Responsive Nano-Valve for Triggered Release of Insulin, Adv. Funct. Mater., 2009, 19, 733–741 CrossRef CAS.
- X. Jin and Y. Lo. Hsieh, PH-Responsive Swelling Behavior of Poly(Vinyl Alcohol)/Poly(Acrylic Acid) Bi-Component Fibrous Hydrogel Membranes, Polymer, 2005, 46, 5149–5160 CrossRef CAS.
- J. Goicoechea, C. R. Zamarreño, I. R. Matías and F. J. Arregui, Optical Fiber PH Sensors Based on Layer-by-Layer Electrostatic Self-Assembled Neutral Red, Sens. Actuators, B, 2008, 132, 305–311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05464b |
| This journal is © The Royal Society of Chemistry 2019 |