
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and thermal stability of ZrO2@SiO2 core–shell submicron particles†
Maik Finsel a,
Maria Hemmea,
Sebastian Döring‡
a,
Jil S. V. Rütera,
Gregor T. Dahla,
Tobias Krekelerb,
Andreas Kornowskia,
Martin Ritterb,
Horst Wellera and
Tobias Vossmeyer*a
a,
Maria Hemmea,
Sebastian Döring‡
a,
Jil S. V. Rütera,
Gregor T. Dahla,
Tobias Krekelerb,
Andreas Kornowskia,
Martin Ritterb,
Horst Wellera and
Tobias Vossmeyer*a
aInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, D-20146 Hamburg, Germany. E-mail: tobias.vossmeyer@chemie.uni-hamburg.de
bElectron Microscopy Unit, Hamburg University of Technology, Eißendorfer Straße 42, D-21073 Hamburg, Germany
First published on 28th August 2019
Abstract
ZrO2@SiO2 core–shell submicron particles are promising candidates for the development of advanced optical materials. Here, submicron zirconia particles were synthesized using a modified sol–gel method and pre-calcined at 400 °C. Silica shells were grown on these particles (average size: ∼270 nm) with well-defined thicknesses (26 to 61 nm) using a seeded-growth Stöber approach. To study the thermal stability of bare ZrO2 cores and ZrO2@SiO2 core–shell particles they were calcined at 450 to 1200 °C. After heat treatments, the particles were characterized by SEM, TEM, STEM, cross-sectional EDX mapping, and XRD. The non-encapsulated, bare ZrO2 particles predominantly transitioned to the tetragonal phase after pre-calcination at 400 °C. Increasing the temperature to 600 °C transformed them to monoclinic. Finally, grain coarsening destroyed the spheroidal particle shape after heating to 800 °C. In striking contrast, SiO2-encapsulation significantly inhibited grain growth and the t → m transition progressed considerably only after heating to 1000 °C, whereupon the particle shape, with a smooth silica shell, remained stable. Particle disintegration was observed after heating to 1200 °C. Thus, ZrO2@SiO2 core–shell particles are suited for high-temperature applications up to ∼1000 °C. Different mechanisms are considered to explain the markedly enhanced stability of ZrO2@SiO2 core–shell particles.
Introduction
Core–shell nano- and microparticles represent a novel class of functional materials with numerous prospective applications. For example, they are well-suited for applications in catalysis,1–7 high performance liquid chromatography,8–10 as well as biomedical diagnosis and therapy.6,7,11–15 Furthermore, they are highly promising candidates for the development of advanced optical materials, which are needed, e.g., for thermophotovoltaic (TPV) systems,16,17 photonic displays,18 and structural coloration.19–21 Among the broad variety of core–shell particles studied to date, especially those consisting of ceramic materials are very robust and suited for applications in hostile chemical environments, or at elevated processing and operating temperatures.Inorganic cores encapsulated by silica shells are probably one of the most widely studied types of core–shell particles. Silica coatings can be grown on various inorganic materials, including metals,15,22 semiconductors,12 and high band gap metal oxides.23–25 Usually, the Stöber method is adapted to grow silica shells with well-controlled thickness. In general, silica encapsulation decreases the particles' initial polydispersity and surface roughness. Furthermore, silica is optically transparent and has good mechanical, chemical, and thermal stability. Using well-established silane coupling chemistry, the silica shells can easily be functionalized and bioconjugated, as has been shown, e.g., for fluorescent nanodiamonds and magnetic nanoparticles.11,12 In other examples, gold nanoparticles have been coated with differently thick silica shells to tune the optical properties of thin films assembled from these particles.26 Further, photonic display pixels have been fabricated using silica encapsulated iron oxide nanoparticles.27
Zirconia is an interesting core material. Ceramics based on ZrO2 have excellent mechanical properties, are chemically inert, and have low thermal conductivity. In addition, they are heat-resistant and used, e.g., as thermal barrier coatings (TBCs) in jet engines. Furthermore, zirconia has a high refractive index (>2 in visible and NIR range) and is well-suited for various optical applications. For example, zirconia-based microparticles have been used for the fabrication of photonic glasses, which may find applications in advanced TBCs.28–31 Another study proposed the application of zirconia microparticles for the design of novel NIR absorbers/emitters to enhance the efficiency of TPV systems.32 Furthermore, zirconia-based microceramics are of interest for the fabrication of heat-resistant structural coloration.19,33
The sol–gel synthesis of ZrO2@SiO2 core–shell nanoparticles has been reported in previous works by Bai et al.23 and Yang et al.24 They proposed applications of these particles for the self-assembly-based fabrication of functional optical devices and coatings with adjustable refractive index. In another study, zirconia nanoparticles were silica coated to enable their surface functionalization via silane coupling chemistry.34 The modified zirconia particles were then used to provide quartz fiber reinforced composites with radiopacity. ZrO2@SiO2 core–shell particles are also highly promising, e.g., for structural coloration and advanced optical coatings. It is also expected that they could be well-suited for processing at elevated temperatures and for various high-temperature applications, as already mentioned above. Thus, there is a need to study the thermal stability of ZrO2@SiO2 core–shell particles.
When heated to several hundred degree centigrade sol–gel derived zirconia submicron particles shrink significantly and transition, first, from the amorphous (a) to the metastable tetragonal (t) phase and, later, to the stable monoclinic (m) phase.28,35,36 These transitions are associated with significant grain coarsening and the t → m transition is accompanied by a 5% volume increase of formed crystallites.37 As a result, submicron zirconia particles disintegrate after calcining at 850 °C.28 However, since earlier studies have shown improved phase stability in sol–gel derived ZrO2–SiO2 mixed oxides,38,39 it is conceivable that silica encapsulation may significantly improve the thermal stability of zirconia submicron particles.
Thus, the objective of our present study was to characterize the thermal stability of ZrO2@SiO2 core–shell submicron particles. To this end, ZrO2@SiO2 core–shell particles were synthesized using an improved seeded growth protocol, which enables the well-controlled deposition of homogeneous silica shells onto pre-calcined zirconia cores without using additional organic coupling agents. The thermal stability of synthesized core–shell particles was explored by calcining them at temperatures ranging from 450 to 1200 °C. Thermally induced morphological changes were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Phase transitions and grain growth were studied by ex situ X-ray diffraction (XRD). Furthermore, cross-sectional TEM lamellae were prepared via focused ion beam (FIB) technique and analyzed by high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) in combination with energy-dispersive X-ray spectroscopy (EDX) mapping.
Experimental
Chemicals
Anhydrous ethanol (99.5%) was purchased from Acros, demineralized water (ASC reagent), eicosanoic acid (99.0%), and hydroxypropyl cellulose (HPC, average Mw ∼ 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mn = 10000, 99%) were purchased from Sigma-Aldrich, and zirconium(IV) n-propoxide (70% in n-propanol) was purchased from Alfa Aesar. These chemicals were used for the synthesis of zirconia cores. Denatured ethanol (96%) from Grüssing was used for purification.
000, Mn = 10000, 99%) were purchased from Sigma-Aldrich, and zirconium(IV) n-propoxide (70% in n-propanol) was purchased from Alfa Aesar. These chemicals were used for the synthesis of zirconia cores. Denatured ethanol (96%) from Grüssing was used for purification.
Anhydrous tetrahydrofuran (THF, ≥99.7%) was purchased from VWR, anhydrous ethanol (99.5%) from Acros, demineralized water (ASC reagent) from Sigma-Aldrich, ammonium hydroxide solution (28% NH3 in H2O) from VWR, and tetraethyl orthosilicate (TEOS, 99%) from Alfa Aesar. These chemicals were used for growing silica shells on zirconia cores. Demineralized water (Millipore Simplicity System, 18.2 MΩ cm) and denatured ethanol (96%) from Grüssing were used for purification.
Materials and methods
Synthesis of submicron zirconia particles: 21 mg (0.067 mmol) eicosanoic acid and 30 mg HPC were dissolved in 45 mL anhydrous ethanol using a 100 mL reactor vessel. Prior to use, ethanol was dried over 0.4 nm molecular sieves and filtered through a polyethersulfone (PES) syringe filter (0.1 μm pore size) to remove dust of the drying agent. The clear solution was heated to 60 °C under stirring (550 rpm). Meanwhile, 1.655 g (5.54 mmol) zirconium n-propoxide were sonicated (Badelin, Sonorex Super RK 106) for 10 min. After adding 200 μL of demineralized water to the ethanolic solution of stabilizers, the undiluted zirconia precursor was added quickly by using a syringe (Vivomed, 2.00 × 120 mm). Some seconds afterwards, the solution turned white (induction time) and the suspension was stirred continuously (550 rpm) for 3 h. For purification, the obtained suspension was added to 50 mL ice-cooled ethanol and then centrifuged (−5 °C, 5 min, 6000 × g) to separate the particles from the supernatant. In three additional washing steps (each step ∼35 mL ethanol), the centrifugation speed was gradually reduced by 500 × g down to 4500 × g. Finally, the zirconia particles were resuspended by sonication for 5 min in 15 mL ethanol.
Results and discussion
Synthesis of ZrO2@SiO2 core–shell particles
Fig. 1 shows the strategy we used for the sol–gel synthesis of ZrO2@SiO2 core–shell particles. We were especially interested in exploring the synthesis and properties of submicron core–shell particles with core sizes ranging from 100 to 400 nm. Such particles are of significant interest for photonic applications in the visible range, such as structural colors,19 and the design of optical materials with adjustable refractive index.24 Furthermore, sol–gel derived zirconia particles exhibit a phase transition from the metastable tetragonal (t) phase to the monoclinic (m) phase upon thermal treatment.35,36,42–44 As this t → m transition is associated with a volume expansion of 5% (ref. 37) it leads to stress-induced destabilization of the particles. Because grain coarsening and the t → m transition proceed in smaller submicron particles at lower temperatures than in larger micrometer-sized particles,28 they are especially suited to study possible grain growth attenuation and phase stabilization via silica encapsulation. | ||
| Fig. 1 Sol–gel synthesis of ZrO2@SiO2 core–shell particles: first, primary particles are formed by hydrolysis of zirconium n-propoxide and subsequent polycondensation. The primary particles aggregate and form ZrO2 submicron particles, which are calcined at 400 °C. Next, SiO2 seeds are deposited onto the particles by treatment with TEOS/H2O in THF. After removing primary silica particles, closed SiO2 shells are deposited according to a seeded growth process by successive addition of TEOS/H2O in ethanol. | ||
Synthesis of ZrO2 cores
Over the past three decades, various protocols have been elaborated for the wet-chemical synthesis of zirconia particles. Based on some of these previous works35,42,45–47 we reported a facile sol–gel method for the synthesis of submicron zirconia particles with diameters ranging from 0.4 to 0.9 μm.28 In that approach, zirconia particles were formed by the hydrolysis of zirconium alkoxide precursors in the presence of carboxylic acid stabilizers. In order to further decrease the particle size for our present study we modified our previous protocol by adjusting various parameters, including the stabilizer composition and the amount of water added, as detailed in the Experimental section.The application of hydroxypropyl cellulose (HPC) as an efficient stabilizer in the sol–gel synthesis of powders and nanoparticles (e.g., for TiO2,48 ZrO2,49 PbS,50 and ZnS nanoparticles51) has already been established. As the addition of HPC seems to favor the stabilization of relatively small particles52 we used HPC (in addition to previously used eicosanoic acid) as stabilizer for the synthesis of ZrO2 particles with diameters below 0.4 μm. This approach yielded spheroidal particles with a mean diameter of ∼270 nm (after calcination at 400 °C for 3 h) and quite narrow size distributions (7–12% standard deviation). Fig. 2(a) shows a representative TEM image of prepared zirconia cores. Details on sizing statistics can be found in the ESI (Fig. S1†). The TEM image captured at larger magnification (Fig. 2(a), inset) reveals a quite rough particle surface with occasionally attached ∼10–30 nm sized granular structures. This morphology is attributed to the formation mechanism of the particles, in which primary particles with sizes in the 10–20 nm range are initially formed, which then aggregate to form the final particles, as indicated in Fig. 1.35,45,53
 | ||
| Fig. 2 TEM images of calcined (400 °C, 3 h) zirconia core particles (a), pre-encapsulated and dried (80 °C, 4 h) ZrO2@SiO2-seeds particles (b), and dried (80 °C, 4 h) ZrO2@SiO2 core–shell particles after deposition of a ∼38 nm thick silica shell (c). HAADF-STEM images of ZrO2@SiO2 particles with initial shell thicknesses of ∼26 (d), ∼38 (e), and ∼61 nm (f). The particles shown in (d)–(f) were calcined at 450 °C for 3 h before TEM characterization. Scale bars in (a) and in the inset of (a) also apply to figure parts (b and c) and all insets, respectively. Scale bar in (d) also applies to figure parts (e and f). | ||
Growth of SiO2 shells
So far, only very few studies have been published regarding the synthesis of ZrO2@SiO2 core–shell particles. As pointed out by Bai et al.23 growing a homogeneous silica shell onto zirconia cores is challenging. They reported that pre-adsorption of PVP improved the colloidal stability of zirconia cores and enabled the deposition of homogeneous silica shells via hydrolysis of TEOS. However, a polymeric adhesion layer may compromise the thermal stability of such core–shell particles. A different approach, which does not require the need of a polymeric interfacial layer, was reported by Yang et al.24 In order to initiate the growth of homogeneous silica shells, without inducing particle aggregation, they first adsorbed citric acid onto the particles' surface. This step decreased the negative zeta potential of the particles and, thus, enhanced the colloidal stability. Further, it was proposed that the adsorbed citric acid molecules served as a surface coupled catalyst for the hydrolysis of TEOS. As a result, they obtained non-aggregated ZrO2@SiO2 particles with adjustable shell thicknesses.Here, we developed a facile seeded-growth approach for the growth of dense and homogeneous silica shells on zirconia cores without the need of any additional pre-adsorbed organic material. In the first step, silica seeds were deposited onto the zirconia cores (pre-calcined at 400 °C for 3 h), as indicated in Fig. 1. This was achieved by the hydrolysis of TEOS in THF in the presence of zirconia cores at elevated temperature (60 °C). The particles with silica seeds attached (ZrO2@SiO2-seeds particles) were then separated from non-bound silica clusters via centrifugation. The TEM image in the inset in Fig. 2(b) indicates that, after silica seed deposition and drying at 80 °C for 4 h, the surface roughness of the particles was somewhat more pronounced. However, in order to clearly prove the deposition of silica, the ZrO2@SiO2-seeds particles were characterized using EDX analysis. Fig. 3 shows an EDX line scan analysis across a single, pre-encapsulated zirconia particle. The Si signal clearly proves the successful deposition of silica at the particle's surface.
 | ||
| Fig. 3 EDX line scan analysis of a pre-encapsulated ZrO2@SiO2-seeds particle. The silicon sum line profile (10 lines, green) confirms successful deposition of silica on the particle's surface (left). The green lines shown in the STEM image of the particle indicate the positions of the 10 line scans used to produce the sum line profile (right). | ||
In the second step, silica shells were grown on the seed covered core particles. To this end, the seeded cores were redispersed in ethanol and, after addition of ammonium hydroxide solution, TEOS and water were added under mild heating (30 °C). In order to avoid secondary nucleation and growth of silica particles without zirconia cores, it was necessary to keep the TEOS concentration low. Therefore, TEOS and water were added gradually in portions and not at once. Finally, the obtained core–shell particles were separated from the remaining reaction mixture by centrifugation, resuspended in ethanol, and stored in suspension at room temperature until further use. In general, the thickness of the silica shells could be adjusted in the range from 10 to 180 nm by increasing the total amount of gradually added TEOS. However, for our present study, we prepared three samples of core–shell particles with initial silica shell thicknesses of ∼26, ∼38, and ∼61 nm (determined after drying at 80 °C for 4 hours, see the ESI, Fig. S2†). Fig. 2, parts (c)–(f), show representative TEM and HAADF-STEM images of these ZrO2@SiO2 particles after drying at 80 °C for 4 h (c) and calcination at 450 °C for 3 h (d)–(f). These images reveal a smooth, compact and homogeneous shell covering the surface of the zirconia cores. With increasing shell thickness some narrowing of the particle size distribution was observed, as reported previously.23,24 Further, the HAADF-STEM images in Fig. 2(d)–(f) reveal that calcining the ZrO2@SiO2 particles at 450 °C for 3 h decreased the initial silica shell thicknesses to ∼10, ∼30, and ∼50 nm, respectively. We attribute this finding to the loss of residual water and organic components of the silica precursor, similar as reported previously for sol–gel derived zirconia microparticles. This interpretation was confirmed by TGA measurements (see TGA-DSC data provided in the ESI, Fig. S3†).28
It is to emphasize that for successfully growing homogeneous silica shells it was mandatory to first deposit the silica seed particles and then to separate the obtained seeded zirconia particles from the reaction mixture, before growing continuous silica shells. In a first set of experiments, we tried to grow continuous silica shells directly onto the core particles by adding relatively high amounts of TEOS. These attempts only yielded SiO2 particles loosely attached to the zirconia cores, similar as reported by Bai et al.23 According to the model of LaMer,54 too high TEOS concentrations lead to homonucleation and subsequent growth of pure silica particles, competing with silica shell growth. As a result, very heterogeneous samples were obtained, initially.
Thermal stability
 | ||
| Fig. 4 SEM images of bare ZrO2 cores, ZrO2@SiO2 core–shell particles (initial shell thickness: ∼38 nm), and pre-encapsulated ZrO2@SiO2-seeds particles after calcination at 450, 800, 1000, and 1200 °C for 3 h (heating rate: 5 °C min−1). Scale bars in the top left SEM image and the inset (ZrO2, 450 °C) apply to all other SEM images and insets, respectively. | ||
In striking contrast, the core–shell particles were stable and shape persistent, even after calcination at 1000 °C. After heating to 450 °C, the shell surface appears rather smooth. Overall, this morphology did not change after increasing the temperature to 800 and 1000 °C. Finally, after calcination at 1200 °C also the core–shell particles disintegrated completely. The SEM image suggests that at such high temperature a molten mass of silica was formed which, upon cooling, solidified with enclosed fractured zirconia cores.
The ZrO2@SiO2 particles with initial shell thicknesses of ∼26 and ∼61 nm showed similar shape stability upon calcination. SEM images of these particles after calcination at various temperatures are included in Fig. S5 of the ESI.†
Interestingly, the spheroidal shape of the pre-encapsulated ZrO2@SiO2-seeds particles was also preserved after calcination at 1000 °C (see Fig. 4). However, in contrast to the ZrO2@SiO2 core–shell particles, progression of grain coarsening with increasing temperature was clearly observable. After calcination at 1200 °C, the ZrO2@SiO2-seeds particles also lost their spheroidal shape, but the formed grains were obviously smaller than in case of the bare zirconia particles.
 | ||
| Fig. 5 XRD data of ZrO2 submicron core particles (a), and ZrO2@SiO2 core–shell submicron particles with an initial shell thickness of 38 nm (b). XRD data of the bare ZrO2 cores and the core–shell particles with ∼26, ∼38, and ∼61 nm thick silica shells (c). The samples were calcined for 3 h at different temperatures, as indicated (heating rate: 5 °C min−1). The bare zirconia cores transitioned almost completely to the monoclinic phase after heating above 600 °C, while the core–shell particles still exhibited significant tetragonal phase fractions after heating to 1000 °C. | ||
It is well known that sol–gel derived amorphous zirconia first transitions from the amorphous state to the metastable tetragonal phase and, upon further increasing the temperature, to the stable monoclinic phase.35,36,43,44,55–62 The initial transition to the tetragonal phase is attributed to the local coordination environment and short-range order in the amorphous phase that is more similar to the tetragonal rather than the monoclinic polymorph.36,56,59,62–64 Thus, the initial crystallization yields the tetragonal phase. The thermodynamically stable monoclinic phase is formed only after heating to higher temperatures.
The phase transitions presented in Fig. 5(a) are very similar to those reported earlier for somewhat larger submicron zirconia particles (diameter: ∼0.75 μm, after calcination at 450 °C).28 However, in our present study, the particles started to transition to the monoclinic phase already at lower temperature. In general, tetragonal grain growth is less inhibited in smaller particles.28,65 Thus, in the case of presently studied smaller particles the critical tetragonal grain size, at which the phase transition occurs (see below), is already reached at lower calcination temperature.
The XRD data shown in Fig. 5(b) reveal a striking influence of silica encapsulation on the phase stability. The data show the phase transitions of the ZrO2@SiO2 core–shell particles with an initial shell thickness of ∼38 nm. While the bare zirconia cores had transitioned almost completely to the monoclinic phase after calcination at 600 °C, the initial phase composition of the silica encapsulated cores (>80 wt% tetragonal, <20 wt% monocline, after 450 °C) changed only marginally after calcination at temperatures of up to 800 °C (∼70% tetragonal, ∼30% monoclinic). The main phase transition to the monoclinic phase was observed after heating to 1000 °C. Finally, after calcination at 1200 °C, the t → m transition was finished (≥94% monocline) and the core–shell structure of the particles was lost (see Fig. 4).
The ZrO2@SiO2 particles with the initial shell thicknesses of ∼26 and ∼61 nm, showed very similar phase transitions as those shown in Fig. 5(b). For comparison, Fig. 5(c) presents XRD data of core–shell particles with the three different shell thicknesses after calcination at 800, 1000, and 1200 °C. In all three samples the t → m transition occurred mainly when the samples were heated to 1000 °C. The data reveal only minor differences in phase stability.
In contrast, a clear difference in phase stability was observed for the pre-encapsulated ZrO2@SiO2-seeds particles. The XRD data shown in Fig. 6 clearly indicate that the initial treatment of the core particles with the silica precursor (TEOS) already led to some stabilization of the tetragonal phase. However, the effect was much less pronounced than in the case of the core–shell particles. A major fraction of the silica seeded particles transitioned already after calcination at 600 °C to the monoclinic phase and after calcination at 1000 °C the t → m transformation was nearly finished.
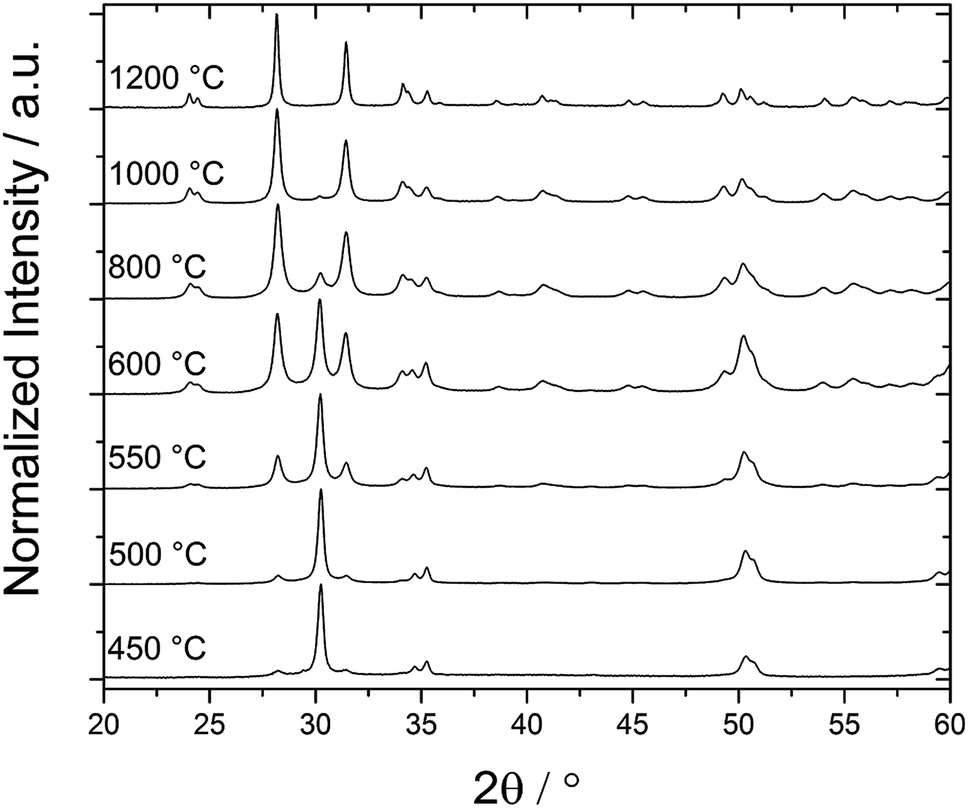 | ||
| Fig. 6 XRD data of pre-encapsulated ZrO2@SiO2-seeds particles. The samples were calcined for 3 h at different temperatures, as indicated (heating rate: 5 °C min−1). The major fraction of the initial tetragonal phase had transitioned to the monoclinic phase after heating to 600 °C. | ||
It is to note that within the considered temperature range of up to 1200 °C our XRD data did not indicate the formation of zircon (ZrSiO4). This finding is in agreement with the study of Monte et al.,38 who explored phase transitions in ZrO2–SiO2 binary oxides at similar temperatures. However, Aguilar et al.,39 who also studied the crystallization of zirconia in ZrO2–SiO2 binary oxides, reported the formation of zircon after calcination at 1200 °C (and higher temperatures). In a silica rich mixture they also observed the formation of crystobalite after calcination at 1300 °C, which was absent in our present work.
In order to gain more insight into the microstructural changes after calcination, the grain sizes were determined after each thermal treatment (see the ESI† for details). As shown in Fig. 7 the tetragonal crystallite size of the bare zirconia cores was ∼45 nm after calcination at 450 °C. This size is similar to the critical grain size reported by Shukla and Seal36 for submicron-sized (500–600 nm) zirconia particles. As they pointed out, for isolated zirconia nanocrystals with sizes up to 10 nm the tetragonal structure is more stable as it provides lower surface energy than the monoclinic phase. For larger isolated nanocrystals the volume free energy becomes the dominating parameter and forces the transition to the monoclinic phase. Thus, isolated tetragonal zirconia nanocrystals grow until they reach a size of ∼10 nm before they transition to the monoclinic phase. However, in contrast to isolated nanocrystals, the surface energy of tetragonal crystallites in aggregated structures of zirconia nanocrystals is significantly lower. Furthermore, due to the spatial confinement of the crystallites within aggregated structures the hydrostatic strain energy has to be taken into consideration. Because the martensitic tetragonal-to-monoclinic phase transition is associated with a 5% (ref. 37) volume increase, the resulting strain energy makes this transition less favorable. Taking into account these contributions, Shukla and Seal36 calculated a critical tetragonal grain size of 41 nm in aggregated zirconia particles, which is similar to the maximum tetragonal crystallite sizes observed in our current study (see Fig. 7).
 | ||
| Fig. 7 Crystallite sizes (grain sizes) of bare ZrO2 cores (black), pre-encapsulated ZrO2@SiO2-seeds particles (brown), and completely encapsulated ZrO2@SiO2 core–shell particles (red, blue, green) after calcination at different temperatures (squares: tetragonal phase; circles: monoclinic phase). Crystallite sizes were determined from XRD data using the Scherrer equation. Due to instrumental broadening, crystallite sizes above ∼80 nm can only be considered as rough estimates of the actual crystallite size. | ||
After calcination at 500 °C, the apparent tetragonal crystallite size of the bare zirconia particles decreased to ∼37 nm. This finding is attributed to the predominant phase transition of larger tetragonal grains, leaving behind a population of somewhat smaller crystallites. The formed monoclinic crystallites had an average size of only ∼30 nm. This observation may suggest that the phase transition is associated with twinning of crystallites to relieve stresses, as reported previously.66–69
After calcination at 550 °C some remaining smaller (∼22 nm) tetragonal crystallites are still observed while the majority of larger grains had transitioned to the monoclinic phase with a crystallite size of ∼35 nm. Upon further increasing the calcination temperature, significant grain growth is observed leading to monoclinic crystallite sizes >80 nm after heating to 1200 °C. The grain size of ∼95 nm indicated for the calcination temperature of 1200 °C (see Fig. 7) must be considered with caution. Due to instrumental broadening, such large crystallite sizes cannot be determined accurately with our XRD equipment.
In striking contrast to the bare zirconia particles, the grain growth of the encapsulated ZrO2@SiO2 particles was strongly inhibited. After calcining at 450 °C the tetragonal grain size was ∼29 nm. After heating to 500 and 550 °C, the grain size apparently decreased somewhat. We attribute this observation to the formation of smaller tetragonal crystallites from residual amorphous zirconia, producing a smaller average grain size. Upon increasing the calcination temperature from 550 to 800 °C the tetragonal crystallites grew to sizes of ∼30, ∼32, and ∼34 nm for particles with the initial shell thicknesses of ∼61, ∼38, and ∼26 nm, respectively. At 1000 °C the t → m transition occurred to somewhat different extent (see Fig. 5) for the three samples, leaving behind differently sized tetragonal crystallites. However, as a general trend it was observed that, for each calcination temperature, the tetragonal crystallite size was smaller the thicker the silica shell was. Thus, the attenuation of grain growth became somewhat more effective with increasing shell thickness.
The monoclinic crystallites, which were formed after calcination at 1000 °C, had a size of only ∼20 nm. Similar to the t → m transition of the bare zirconia cores, these crystallites were much smaller than the remaining tetragonal grains. As already mentioned, we attribute this observation to the relief of stress via twinning.66–69 After sintering at 1200 °C the monoclinic crystallites grew to sizes of 27 to 32 nm.
Compared to the bare zirconia cores and the fully encapsulated ZrO2@SiO2 particles, the pre-encapsulated ZrO2@SiO2-seeds particles showed an intermediate progression in monoclinic grain growth after calcination at 1000 °C and 1200 °C, see Fig. 7. These findings are in general agreement with grain sizes and the shape stability observed in the SEM images of Fig. 4.
In summary, the deposition of silica shells on zirconia cores enabled a significant stabilization of the tetragonal phase. This is evidenced by a pronounced shift of the calcination temperature, from 500 to 1000 °C, at which the t → m transition is observed. In general agreement with previous studies, this stabilization is associated with a strong inhibition of the tetragonal grain growth.
In order to explain these findings it is useful to take into consideration the results of previous studies on ZrO2–SiO2 binary oxides.38,39 These studies showed that already small amounts (1–10 mol%) of silica stabilize the tetragonal zirconia phase effectively. This stabilization has been attributed to constraint imposed on zirconia crystallites by the surrounding amorphous silica matrix as well as hindered mass transport through the silica matrix.70 Thereby, crystallite growth is blocked and the tetragonal phase is stabilized by keeping the crystallite size below the critical grain size.38,71,72 Additionally, it has been proposed that the high-melting, covalently bonded silica matrix with a low expansion coefficient impedes the martensitic transition to the monoclinic phase as it requires a volume increase of 5%.37
We propose that similar mechanisms are responsible for the observed stabilization of the tetragonal phase of ZrO2@SiO2 core–shell particles. In our previous study,28 we observed that initially formed tetragonal crystallites in submicron zirconia particles are significantly larger than those found in larger micrometer-sized particles. Thus, grain growth seems to be enhanced in particles with a larger surface-to-volume ratio. This finding indicates that crystallite growth is favored at the particle's surface, most likely because the crystallites are spatially less confined, mass transport at the surface is enhanced, and the overall surface energy decreases as the initially rough surface is smoothened due to consumption of material during grain growth. In line with this explanation and in general agreement with the constraint effect proposed for the phase stabilization in ZrO2–SiO2 mixed oxides, the silica shell attenuates grain growth at the particle's surface because it enforces spatial confinement and inhibits mass transport. Thus, in order to grow the tetragonal crystallites, which are covered by a silica shell, to the critical grain size, the calcination temperature has to be increased significantly.
Another mechanism that may play a crucial role in the t → m transition is related to chemisorption of oxygen. Livage et al.64 reported that tetragonal zirconia is much more stable when heated in an oxygen-free atmosphere than under air. Srinivasan et al.62 also observed that the phase transition was accomplished more readily under air than under inert gas. They proposed that chemisorption of oxygen at surface defects (oxygen vacancies) upon cooling below 300 °C initiates the t → m transition. Similarly, Collins and Bowman73 suggested that incorporation of oxygen from the atmosphere eliminates oxygen vacancies and, thereby, facilitates the transition to the monoclinic phase. Furthermore, it has been demonstrated by Penner et al.74 that in inert atmospheres, the persisting structural defectivity leads to a high stability of tetragonal ZrO2 up to 1000 °C. As the solubility of oxygen in amorphous silica is quite low,75 and the high Si–O bond energy predicts a low concentration of oxygen vacancies and a low diffusion coefficient of oxygen in silica,75–77 a silica shell is expected to provide an efficient diffusion barrier for oxygen. Thus, chemisorption of oxygen at oxygen vacancies of zirconia is impeded and the tetragonal phase is stabilized.
 | ||
| Fig. 8 High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and EDX mapping data (Zr: magenta, Si: green, O: red) of cross-sectional lamellae prepared from ZrO2@SiO2 core–shell particles after calcination at 450 °C (a), 800 °C (b), and 1000 °C (c). The lamellae were prepared from ZrO2@SiO2 core–shell particles with an initial shell thickness of 38 nm using FIB technique. | ||
Fig. 8(a) shows the HAADF-STEM image of a core–shell particle that was heat-treated at 450 °C. The apparent diameter of the zirconia core (∼240 nm) is significantly smaller than the average core diameter of ∼270 nm, suggesting that the FIB cut through this particle was off-center. Thus, the images of this particle represent the projection of a cap of the particle through the FIB lamella (thickness < 100 nm) and provide insight into the ZrO2/SiO2 interfacial structure. First, the HAADF-STEM image and the Zr map clearly show that the ZrO2 core has a fine-grained structure with distinct surface roughness. Second, the Si map suggests a closed silica shell covering the ZrO2 core. On its inner side the amorphous silica shell adapts well to the rough ZrO2 surface and penetrates deep into pores. This finding confirms the good wettability of zirconia by silica. Third, the O map shows a quite homogenous distribution of oxygen. Only few oxygen deficient spots are observed, which match dark spots seen in the HAADF-STEM image, and indicate the presence of pores, which have not been filled (completely) by silica.
The particle that was calcined at 800 °C (Fig. 8(b)) had an apparent core size of ∼270 nm, which matches the average core diameter. Thus, this FIB lamella was most likely cut close to the center of the particle. The HAADF-STEM image and the Zr and O maps show that the porosity of the core increased significantly. Furthermore, the Si map shows a smooth densified silica shell with a thickness of 25–30 nm, in agreement with the HAADF-STEM images shown in Fig. 2(e). At the ZrO2/SiO2 interface, the two materials appear quite sharply demarcated, though some silica still extends into cavities at the core's surface. Although the Si map indicates no silica at the center of the core, the integrated EDX signal reveals a Si fraction of 1–2 wt% (see the ESI, Fig. S9 and Table S1†). This finding suggests that a small fraction of silicon may have diffused into the inner core region.
The HAADF-STEM image and the elemental maps of the particle, which was calcined at 1000 °C (Fig. 8(c)), indicates somewhat more pronounced coarsening of the porous core structure. In contrast, the silica shell as well as the ZrO2/SiO2 interfacial region did not indicate any significant morphological changes. The EDX analysis at the center of the core suggests an unchanged Si fraction of ∼1–2 wt% (see the ESI, Fig. S9 and Table S1†).
In general, the EDX analyses of the core–shell particles support our explanation for the observed attenuation of grain growth and phase stabilization. The zirconia cores are enclosed by a homogeneous, dense silica shell with a conformal ZrO2/SiO2 interface, at which amorphous silica penetrates deep into the porous core structure. Thus, the silica shell is expected to effectively block mass transport at the core's surface70 and to enforce the spatial confinement of crystallites. The closed silica shell can also block chemisorption of oxygen at oxygen vacancies and, thereby, suppress the t → m transition. In addition, a small fraction of Si was detected within the core after calcination at 800 and 1000 °C. This finding may suggest that doping by Si may also contribute to the observed phase stabilization. In the literature, the possibility of Si-doping in zirconia has been discussed controversially. On the one hand, Kajihara et al.70 and Ikuhara et al.78 suggested that grain growth and mass transport in zirconia are inhibited by Si ions dissolved in the tetragonal zirconia lattice near grain boundaries and by Si segregation across grain boundaries. On the other hand, Guo et al.79 and Gremillard et al.80 found that silica forms vitreous pockets of glassy phase at the triple grain junctions.
The HAADF-STEM images shown in Fig. 8 do not provide information on the polycrystalline character of the zirconia cores. Therefore, the FIB lamellae were also characterized by bright field (BF-) TEM and high resolution (HR-) TEM. The obtained images are presented in the ESI (Fig. S7 and S8†). They clearly confirm the polycrystalline nature of the zirconia cores. Also, they show that, after calcination at 450 °C, the zirconia cores still contained amorphous material, besides nanocrystalline domains. Furthermore, the images confirm the formation of a conformal SiO2/ZrO2 interface, as already discussed above.
Conclusions
In summary, spheroidal submicron ZrO2 particles were synthesized according to a modified sol–gel method using a mixture of eicosanoic acid and hydroxypropyl cellulose (HPC) as stabilizers. Subsequently, the core particles were calcined at 400 °C for 3 h. The deposition of silica shells was done in two steps without using any additional organic coupling agents. First, the zirconia core particles were coated with silica seeds in a pre-encapsulation step. Second, silica shells were grown stepwise with thicknesses of ∼26, ∼38, and ∼61 nm by adding TEOS in a modified Stöber approach. The resulting core–shell structure was confirmed by HAADF-STEM imaging.The bare zirconia cores showed pronounced grain coarsening after calcination at 600 °C and transitioned almost completely to the monoclinic phase. After calcination at 1000 °C the initial spheroidal particle shape was completely lost. In striking contrast, SEM images of the silica encapsulated zirconia particles did not show obvious morphological changes after calcination at 1000 °C. Further, compared to the bare cores, grain growth was strongly inhibited. However, after calcination at 1000 °C the encapsulated cores transitioned partially to the monoclinic phase, and after calcination at 1200 °C this transition was complete. After this heat treatment the core–shell structure was completely disintegrated. Interestingly, the grain growth of the pre-encapsulated ZrO2@SiO2-seeds particles showed an intermediate progression compared to the bare zirconia cores and the fully encapsulated ZrO2@SiO2 particles. Although SEM images clearly revealed significant grain coarsening after heating to 1000 °C, the initial spheroidal shape of the pre-encapsulated particles was still preserved.
EDX analyses of cross-sectional lamellae confirmed a smooth, closed shell of amorphous silica encapsulating the zirconia cores. On the inner side, the shell adapted to the rough surface of the core and silica penetrated deep into pores.
Attenuation of grain growth and stabilization of the tetragonal phase was also observed for the pre-encapsulated ZrO2@SiO2-seeds particles, but the effect was smaller than for the ZrO2@SiO2 particles with a closed silica shell.
In accordance with previous studies on ZrO2–SiO2 binary oxides,38,39 it is concluded that the silica shell hinders mass transport at the particle's surface and spatially confines the crystallites. Thereby, grain growth is blocked and the tetragonal phase is stabilized. Furthermore, previous studies indicated that chemisorption of oxygen at oxygen vacancies can initiate the t → m transition.62,64,73 Thus, the shielding effect of the silica shell, which impedes oxygen diffusion from the surrounding atmosphere to the core, may play an additional role in stabilizing the tetragonal phase. Finally, the Si fraction of 1–2%, which was detected within the zirconia core, may cause lattice distortion and/or the formation of intergranular diffusion barriers and, thereby, contribute to phase stabilization.38,39,70,78–80
In view of potential high-temperature processing and applications, our results demonstrate that submicron zirconia particles, which have been encapsulated by a thin, continuous silica shell, are temperature-resistant up to ∼1000 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 192346071 – SFB 986 (Projects C6, Z3, A1). The authors thank Robert Schön for particle characterization by HR-SEM and EDX measurements, Kaline P. Furlan and Anja Borchert for TGA-DSC measurements, Stefan Werner for TEM measurements, and Almut Barck for XRD measurements.References
- C. Xiao, R. V. Maligal-Ganesh, T. Li, Z. Qi, Z. Guo, K. T. Brashler, S. Goes, X. Li, T. W. Goh, R. E. Winans and W. Huang, ChemSusChem, 2013, 6, 1915–1922 CrossRef CAS PubMed.
- K. A. Dahlberg and J. W. Schwank, Chem. Mater., 2012, 24, 2635–2644 CrossRef CAS.
- V. R. Calderone, J. Schütz-Widoniak, G. L. Bezemer, G. Bakker, C. Steurs and A. P. Philipse, Catal. Lett., 2010, 137, 132–140 CrossRef CAS.
- J. C. Park, J. U. Bang, J. Lee, C. H. Ko and H. Song, J. Mater. Chem., 2010, 20, 1239–1246 RSC.
- Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef CAS.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS.
- J. F. Li, Y. J. Zhang, S. Y. Ding, R. Panneerselvam and Z. Q. Tian, Chem. Rev., 2017, 117, 5002–5069 CrossRef CAS.
- A. Ahmed, K. Skinley, S. Herodotou and H. Zhang, J. Sep. Sci., 2018, 41, 99–124 CrossRef CAS.
- R. Hayes, A. Ahmed, T. Edge and H. Zhang, J. Chromatogr. A, 2014, 1357, 36–52 CrossRef CAS.
- M. Gumustas, P. Zalewski, S. A. Ozkan and B. Uslu, Chromatographia, 2019, 82, 17–48 CrossRef CAS.
- A. Bumb, S. K. Sarkar, N. Billington, M. W. Brechbiel and K. C. Neuman, J. Am. Chem. Soc., 2013, 135, 7815–7818 CrossRef CAS PubMed.
- R. He, X. You, J. Shao, F. Gao, B. Pan and D. Cui, Nanotechnology, 2007, 18, 315601 CrossRef.
- K. Y. Yoon, C. Kotsmar, D. R. Ingram, C. Huh, S. L. Bryant, T. E. Milner and K. P. Johnston, Langmuir, 2011, 27, 10962–10969 CrossRef CAS PubMed.
- F. Wang, X. Chen, Z. Zhao, S. Tang, X. Huang, C. Lin, C. Cai and N. Zheng, J. Mater. Chem., 2011, 21, 11244–11252 RSC.
- Y. Kobayashi, H. Inose, T. Nakagawa, K. Gonda, M. Takeda, N. Ohuchi and A. Kasuya, J. Colloid Interface Sci., 2011, 358, 329–333 CrossRef CAS.
- P. N. Dyachenko, A. Y. Petrov and M. Eich, Appl. Phys. Lett., 2013, 103, 211105 CrossRef.
- A. Petrov, H. Lehmann, M. Finsel, C. Klinke, H. Weller and T. Vossmeyer, Langmuir, 2016, 32, 848–857 CrossRef CAS.
- L. Shi, Y. Zhang, B. Dong, T. Zhan, X. Liu and J. Zi, Adv. Mater., 2013, 25, 5314–5320 CrossRef CAS PubMed.
- G. Shang, L. Maiwald, H. Renner, D. Jalas, M. Dosta, S. Heinrich, A. Petrov and M. Eich, Sci. Rep., 2018, 8, 1–9 CrossRef CAS PubMed.
- J. G. Park, S. H. Kim, S. Magkiriadou, T. M. Choi, Y. S. Kim and V. N. Manoharan, Angew. Chem., Int. Ed., 2014, 53, 2899–2903 CrossRef CAS.
- J. Wang, Y. Zhang, S. Wang, Y. Song and L. E. I. Jiang, Acc. Chem. Res., 2011, 405–415 CrossRef.
- J. Ye, B. Van de Broek, R. De Palma, W. Libaers, K. Clays, W. Van Roy, G. Borghs and G. Maes, Colloids Surf., A, 2008, 322, 225–233 CrossRef CAS.
- A. Bai, H. Song, G. He, Q. Li, C. Yang, L. Tang and Y. Yu, Ceram. Int., 2016, 42, 7583–7592 CrossRef CAS.
- X. Yang, N. Zhao, Q. Zhou, C. Cai, X. Zhang and J. Xu, J. Mater. Chem. C, 2013, 1, 3359–3366 RSC.
- V. Srdic, B. Mojic, M. Nikolic and S. Ognjanovic, Process. Appl. Ceram., 2013, 7, 45–62 CrossRef CAS.
- T. Ung, L. M. Liz-Marzán and P. Mulvaney, J. Phys. Chem. B, 2002, 105, 3441–3452 CrossRef.
- I. Lee, D. Kim, J. Kal, H. Baek, D. Kwak, D. Go, E. Kim, C. Kang, J. Chung, Y. Jang, S. Ji, J. Joo and Y. Kang, Adv. Mater., 2010, 22, 4973–4977 CrossRef CAS.
- E. W. Leib, U. Vainio, R. M. Pasquarelli, J. Kus, C. Czaschke, N. Walter, R. Janssen, M. Müller, A. Schreyer, H. Weller and T. Vossmeyer, J. Colloid Interface Sci., 2015, 448, 582–592 CrossRef CAS.
- E. W. Leib, R. M. Pasquarelli, J. J. do Rosário, P. N. Dyachenko, S. Döring, A. Puchert, A. Y. Petrov, M. Eich, G. A. Schneider, R. Janssen, H. Weller and T. Vossmeyer, J. Mater. Chem. C, 2015, 4, 62–74 RSC.
- E. W. Leib, R. M. Pasquarelli, M. Blankenburg, M. Müller, A. Schreyer, R. Janssen, H. Weller and T. Vossmeyer, Part. Part. Syst. Charact., 2016, 33, 645–655 CrossRef CAS.
- P. N. Dyachenko, J. J. Do Rosário, E. W. Leib, A. Y. Petrov, R. Kubrin, G. A. Schneider, H. Weller, T. Vossmeyer and M. Eich, ACS Photonics, 2014, 1, 1127–1133 CrossRef CAS.
- P. N. Dyachenko, J. J. do Rosário, E. W. Leib, A. Y. Petrov, M. Störmer, H. Weller, T. Vossmeyer, G. A. Schneider and M. Eich, Opt. Express, 2015, 23, A1236–A1244 CrossRef CAS.
- Y. Häntsch, G. Shang, A. Petrov, M. Eich and G. A. Schneider, Adv. Opt. Mater., 2019, 1900428 CrossRef.
- H. Wang, X. Jiang, L. Qian, W. Chen and M. Zhu, Adv. Funct. Mater., 2015, 723–730 CAS.
- J. Widoniak, S. Eiden-Assmann and G. Maret, Eur. J. Inorg. Chem., 2005, 3149–3155 CrossRef CAS.
- S. Shukla and S. Seal, J. Phys. Chem. B, 2004, 108, 3395–3399 CrossRef CAS.
- S. Shukla and S. Seal, Int. Mater. Rev., 2005, 50, 45–64 CrossRef CAS.
- F. Monte, W. Larsen and J. D. Mackenzie, J. Am. Ceram. Soc., 2000, 83, 628–634 CrossRef.
- D. H. Aguilar, L. C. Torres-Gonzalez and L. M. Torres-Martinez, J. Solid State Chem., 2000, 158, 349–357 CrossRef.
- L. Lutterotti and P. Scardi, J. Appl. Crystallogr., 1990, 23, 246–252 CrossRef CAS.
- C. J. Howard, R. J. Hill and B. E. Reichert, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 44, 116–120 CrossRef.
- L. Lerot, F. Legrand and P. De Bruycker, J. Mater. Sci., 1991, 26, 2353–2358 CrossRef CAS.
- Y. T. Moon, H. K. Park, D. K. Kim, C. H. Kim and I.-S. Seog, J. Am. Ceram. Soc., 1995, 78, 2690–2694 CrossRef CAS.
- M. A. Blesa, A. J. G. Maroto, S. I. Passaggio, N. E. Figliolia and G. Rigotti, J. Mater. Sci., 1985, 20, 4601–4609 CrossRef CAS.
- T. Ogihara, N. Mizutani and M. Kato, J. Am. Ceram. Soc., 1989, 72, 421–426 CrossRef CAS.
- H. Kumazawa, Y. Hori and E. Sada, Chem. Eng. J., 1993, 51, 129–133 CrossRef CAS.
- B. Yan, C. V. McNeff, F. Chen, P. W. Carr and A. V. McCormick, J. Am. Ceram. Soc., 2001, 84, 1721–1727 CrossRef CAS.
- T. Halamus, P. Wojciechowski and I. Bobowska, Polym. Adv. Technol., 2007, 19, 807–811 CrossRef.
- J. Y. Choi, C. H. Kim and D. K. Kim, J. Am. Ceram. Soc., 2010, 81, 1353–1356 CrossRef.
- S. Lu, U. Sohling, T. Krajewski, M. Mennig and H. Schmidt, J. Mater. Sci. Lett., 1998, 17, 2071–2073 CrossRef CAS.
- T. Cai, Z. Hu, B. Ponder, J. St. John and D. Moro, Macromolecules, 2003, 36, 6559–6564 CrossRef CAS.
- S. Shukla, S. Seal, R. Vij and S. Bandyopadhyay, J. Nanopart. Res., 2002, 4, 553–559 CrossRef CAS.
- K. Uchiyama, T. Ogihara, T. Ikemoto, N. Mizutani and M. Kato, J. Mater. Sci., 1987, 22, 4343–4347 CrossRef CAS.
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- R. Srinivasan, B. H. Davis, O. B. Cavin and C. R. Hubbard, J. Am. Ceram. Soc., 1992, 75, 1217–1222 CrossRef CAS.
- V. G. Keramidas and W. B. White, J. Am. Ceram. Soc., 1974, 57, 22–24 CrossRef CAS.
- H. Uchiyama, K. Takagi and H. Kozuka, Colloids Surf., A, 2012, 403, 121–128 CrossRef CAS.
- M. Y. Ghotbi, V. Nasiri and M. Rafiee, J. Colloid Interface Sci., 2013, 389, 121–125 CrossRef CAS PubMed.
- F. Zhang, P. J. Chupas, S. L. A. Lui, J. C. Hanson, W. A. Caliebe, P. L. Lee and S. W. Chan, Chem. Mater., 2007, 19, 3118–3126 CrossRef CAS.
- E. Tani, M. Yoshimura and S. Somiya, J. Am. Ceram. Soc., 1983, 66, 11–14 CrossRef CAS.
- R. C. Garvie and M. F. Goss, J. Mater. Sci., 1986, 21, 1253–1257 CrossRef CAS.
- R. Srinivasan, R. J. De Angelis, G. Ice and B. H. Davis, J. Mater. Res., 1991, 6, 1287–1292 CrossRef CAS.
- T. Chraska, A. H. King and C. C. Berndt, Mater. Sci. Eng., A, 2000, 286, 169–178 CrossRef.
- J. Livage, K. Doi and C. Mazieres, J. Am. Ceram. Soc., 1968, 51, 349–353 CrossRef CAS.
- S. Shukla, S. Seal, R. Vij and S. Bandyopadhyay, Nano Lett., 2003, 3, 397–401 CrossRef CAS.
- G. K. Bansal and A. H. Heuer, Acta Metall., 1974, 22, 409–417 CrossRef CAS.
- V. P. Dravid, M. R. Notis and C. E. Lyman, J. Am. Ceram. Soc., 1988, 71, C-219–C-221 CrossRef CAS.
- J. E. Bailey, Proc. R. Soc. London, Ser. A, 1964, 279, 395–412 CrossRef.
- Y. C. Wu and Y. T. Chiang, J. Am. Ceram. Soc., 2011, 94, 2200–2212 CrossRef CAS.
- K. Kajihara, Y. Yoshizawa and T. Sakuma, Acta Metall. Mater., 1995, 43, 1235–1242 CrossRef CAS.
- T. Ono, M. Kagawa and Y. Syono, J. Mater. Sci., 1985, 20, 2483–2487 CrossRef CAS.
- V. S. Nagarajan and K. J. Rao, J. Mater. Sci., 1989, 24, 2140–2146 CrossRef CAS.
- D. E. Collins and K. J. Bowman, J. Mater. Res., 1998, 13, 1230–1237 CrossRef CAS.
- E. Köck, M. Kogler, T. Götsch, L. Schlicker, M. F. Bekheet, A. Doran, A. Gurlo, B. Klötzer, B. Petermüller, D. Schildhammer and S. Penner, Dalton Trans., 2017, 4554–4570 RSC.
- T. Bakos, S. N. Rashkeev and S. T. Pantelides, Phys. Rev. Lett., 2002, 88, 1–4 CrossRef PubMed.
- R. H. Doremus, J. Phys. Chem., 1976, 80, 1773–1775 CrossRef CAS.
- J. J. Perez-Bueno, R. Ramirez-Bon, Y. V. Vorobiev, F. Espinoza-Beltran and J. Gonzalez-Hernandez, Thin Solid Films, 2000, 379, 57–63 CrossRef CAS.
- Y. Ikuhara, P. Thavorniti and T. Sakuma, Acta Mater., 1997, 45, 5275–5284 CrossRef CAS.
- X. Guo, W. Sigle, J. U. Fleig and J. Maier, Solid State Ionics, 2002, 154–155, 555–561 CrossRef CAS.
- L. Gremillard, T. Epicier, J. Chevalier and G. Fantozzi, Acta Mater., 2000, 48, 4647–4652 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05078g |
| ‡ Present address: AB-Analytik Dr A. Berg GmbH, Ruhrstraβe 49, D-22761 Hamburg, Germany. |
| This journal is © The Royal Society of Chemistry 2019 |