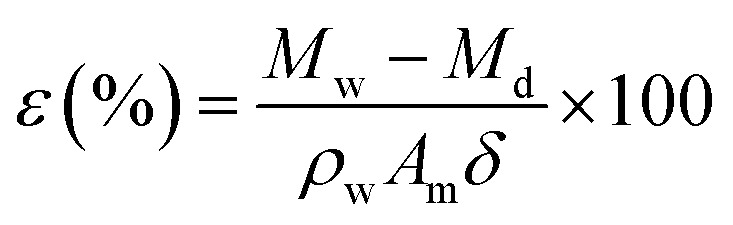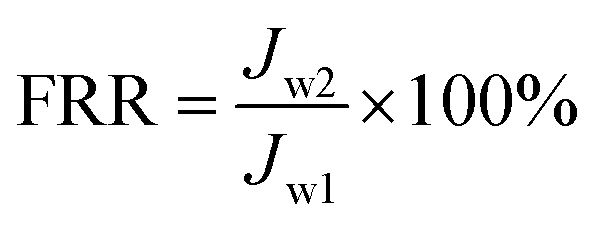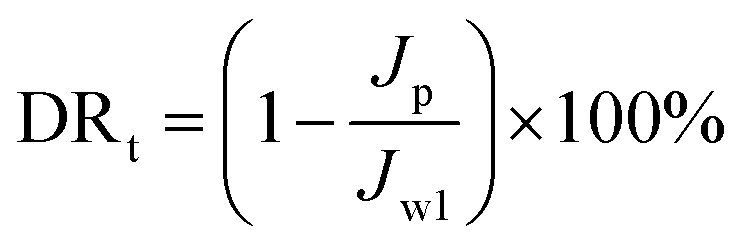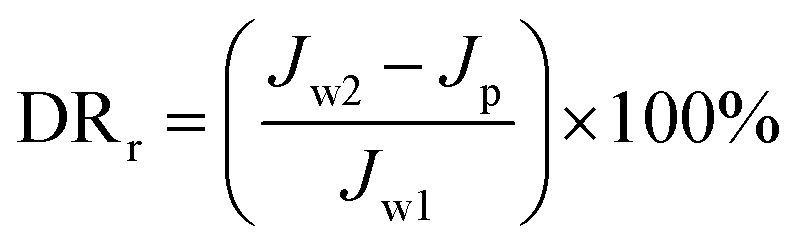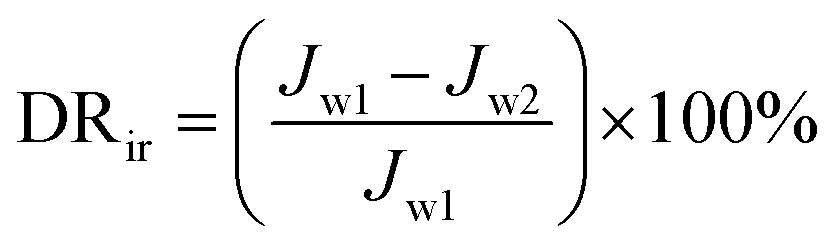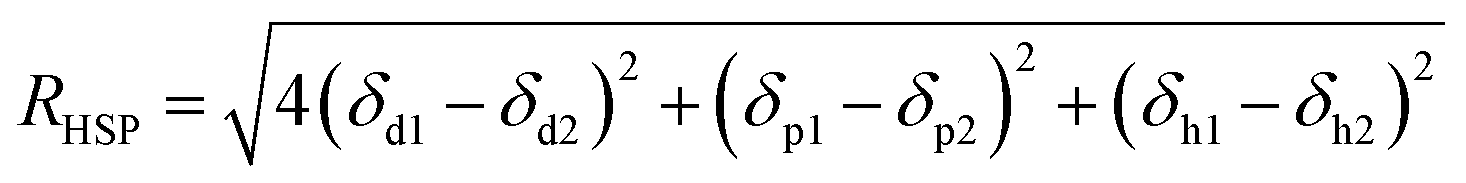DOI:
10.1039/C9RA05454E
(Paper)
RSC Adv., 2019,
9, 34486-34495
Effect of volatile solvent and evaporation time on formation and performance of PVC/PVC-g-PEGMA blended membranes
Received
16th July 2019
, Accepted 21st October 2019
First published on 25th October 2019
1. Introduction
Among the common polymers used to fabricate ultrafiltration membranes, poly(vinyl chloride) (PVC) has received attention owing to its good stiffness, high resistance to bases and acids, and low cost.1 However, PVC membranes exhibit poor antifouling properties due to their hydrophobicity.2 Blending modifications by blending PVC with an amphiphilic copolymer to enhance the antifouling properties of PVC membranes have been reported.3–7 Polymeric membranes can be fabricated via several routes and immersion precipitation (immersion in a non-solvent bath) is the most commonly chosen technique.8 The final membrane morphologies and performances depend on many experimental parameters, including compositions of the casting solution (like polymer concentration, type of solvent and non-solvent, co-solvent/solvent ratio), the solvent evaporation time before immersion in a coagulation bath, and the composition and temperature of the coagulation bath.8–10 Solvents such as N-methyl-2-pyrrolidone (NMP) and N,N-dimethylacetamide (DMAc) were separately used to prepare PVC membranes via the phase inversion method.11–13 Roy et al.12 investigated the effects of molecular weight of additive and solvents (NMP and DMAc) on the performance of PVC-based asymmetric membranes. They found that compared with DMAc, NMP exhibited better interaction with PVC and the membranes prepared using NMP as solvent showed better thermal stability, antifouling properties and mechanical strength.
A volatile solvent like 1,4-dioxane, acetone and tetrahydrofuran (THF) is often chosen as a co-solvent.10 We selected THF based on the following reasons. Firstly, we compared the properties, cost, and environmental impact of the three common volatile solvents. Secondly, THF is a common solvent used for membrane synthesis.10,14–17 Thirdly, during the evaporation before immersion in coagulation bath, THF evaporates fast, which leads to membranes with a tight active layer.15,17 The number of macrovoids underneath the top layer decreased as the evaporation time increased before immersion in coagulation bath.9 Especially, when block copolymers are used to prepare membranes via immersion precipitation, the amount of THF and the evaporation time before precipitation are critical parameters for the formation of self-assemble and ordered structure on the skin layer (Table 1).16,17
Table 1 The parameters of the three common volatile solventsa
| ΔδP/S – polymer/solvent solubility parameter. ΔδS/NS – solvent/non-solvent solubility parameter, non-solvent: water. All information of the hazards of the solvents is according to Regulation (EC) no. 1272/2008. |
| Solvents |
1,4-Dioxane |
THF |
Acetone |
| Vapor pressure (hPa) at 20 °C |
36 |
217.3 |
245.3 |
| Boiling point (°C) |
100 |
66 |
56 |
| Flashing point (°C) |
12 |
−17 |
−17 |
Partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w) Po/w) |
−0.27 |
0.46 |
−0.24 |
| ΔδP/S (MPa1/2) |
6.5 |
7.6 |
7 |
| ΔδS/NS (MPa1/2) |
27.4 |
28.4 |
27.9 |
| Price (500 ml, AR, CNY) |
1623.84 |
678.66 |
554.71 |
| Hazards |
H225, H303, H319, H333, H335, H413, H351 |
H225, H302, H313, H316, H318, H335, H336 |
H225, H319, H336 |
In our previous work,18 we synthesized the amphiphilic copolymer PVC-g-PEGMA and investigated the effect of the dosage of PVC-g-PEGMA on PVC/PVC-g-PEGMA blended membrane properties. We found that PVC-g-PEGMA as an additive could enhance the antifouling properties and pure water flux of PVC ultrafiltration membranes. Especially, when the dosage of PVC-g-PEGMA was 10 wt%, PVC/PVC-g-PEGMA blended membrane exhibited the best comprehensive performance. However, DMAc as the solvent of the membrane casting solutions resulted in large macrovoids at the bottom layer of the membrane. Then Xie et al. used the green solvent dimethyl sulfoxide (DMSO) and traditional solvents as mixing solvents to prepared PVC/PVC-g-PEGMA blended membranes, and the blended membrane showed high permeability and rejection.19
In this work, we chose NMP (a solvent with stronger interaction with PVC but with weaker relative affinity with water compared to DMAc) as solvent and volatile THF as co-solvent according to Hansen solubility parameter and investigated the effects of the addition of THF and evaporation time on the performance of PVC/PVC-g-PEGMA blended membranes. We aimed to decrease the macrovoids at the bottom layer of the membrane and further improve the hydrophilicity and antifouling properties of PVC/PVC-g-PEGMA blended membranes by mixing common solvents. Moreover, we hoped to provide more studies for the blended system of PVC/PVC-g-PEGMA blended membranes.
2. Materials and methods
2.1. Materials
PVC (high molecular weight), PVC-g-PEGMA (synthesized and verified in our previous work18 based on the method previously described20), 1-methyl-2-pyrrolidinone (NMP, anhydrous, 99.5%), sodium chloride (NaCl, ACS reagent, ≥99.0%), sodium alginate (SA, Halal grade) and tetrahydrofuran (THF, anhydrous, ≥99.9%, inhibitor-free) were purchased from MilliporeSigma (St. Louis, MO, USA).
2.2. Membrane preparation
The fabrication of membrane was similar to that of the previous study.21 The membrane casting solution was prepared in 125 mL conical flasks according to Table 2 and stirred at 500 rpm by a stirring hot plate (PC-420D, Corning, USA) at 60 °C for at least 24 h. After the polymers were completely dissolved, the solution was degassed without stirring for at least 3 h until gas bubbles were not completely observed, and then it was cast on a first-grade surface optical glass using a doctor blade set to a thickness of ∼200 μm (Universal blade applicator, Paul N. Gardner Company, Inc., Pompano Beach, FL, USA). Before immersed into a coagulation bath of deionized (DI) water at room temperature for 24 h, the mirror was exposed in air for different time according to Table 2 to allow the solvent to evaporate. Finally, part of membranes immersed in ultrapure water were stored at 4 °C for flux and fouling tests, and the others were air-dried for the characterizations.
Table 2 Compositions of the membrane casting solutionsa
| Membrane ID |
PVC (g) |
PVC-g-PEGMA (g) |
THF (g) |
NMP (g) |
Solvent evaporation time (s) |
PVC-g-PEGMA/PVC wt/wt (%) |
| At 25 °C, the density of THF is 0.889 g mL−1, NMP is 1.028 g mL−1. T/N means the weight/weight ratio of THF to NMP. |
| M1 NMP 60 s |
12 |
1.2 |
— |
86.80 |
60 |
10 |
| M2 T/N = 1/9 0 s |
12 |
1.2 |
8.68 |
78.12 |
0 |
10 |
| M3 T/N = 1/9 30 s |
12 |
1.2 |
8.68 |
78.12 |
30 |
10 |
| M4 T/N = 1/9 60 s |
12 |
1.2 |
8.68 |
78.12 |
60 |
10 |
| M5 T/N = 1/9 90 s |
12 |
1.2 |
8.68 |
78.12 |
90 |
10 |
| M6 T/N = 3/7 60 s |
12 |
1.2 |
26.04 |
60.76 |
60 |
10 |
| M7 T/N = 5/5 60 s |
12 |
1.2 |
43.40 |
43.40 |
60 |
10 |
2.3. Model foulant
Sodium alginate (SA) as a model foulant of extracellular polymeric substances (EPS) to assess the anti-fouling properties of membrane was studied.22–24 A 2 g L−1 SA stock solution was stored at 4 °C. The SA concentration in the filtration test was determined by an UV-vis spectrometer (Thermo Orion Aquamate 8000, USA) at a constant wavelength of 210 nm18
2.4. Membrane characterization
A fourier transform infrared spectroscopy (FTIR) spectrometer with a germanium ATR attachment (FTIR-ATR) (ALPHA, Bruker, Germany) was used to analyze the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the CC stretching band to determine whether PVC-g-PEGMA exist on the blended membrane surfaces. The oxygen, chlorine, and carbon elements on the near surface of all membranes were measured by X-ray photoelectron spectroscopy (XPS) (Axis Ultra, Kratos Analytical Ltd., UK) in the range from 0 to 1100 eV. The morphologies of all blended membranes were imaged using field-emission scanning electron microscopy (FESEM) (JSM-7500F, JEOL Ltd., Tokyo, Japan) at 15 kV accelerating voltage. The hydrophilicity of the membrane surface was characterized by dynamic water contact angle measured using a KRÜSS DSA 25S measuring apparatus (KRÜSS GmbH, Germany). The surface roughness of all blended membranes was determined using atomic force microscopy (AFM) (Multimode 8, Bruker, Germany) at a scan rate of 0.977 Hz and a scan size of 5 μm × 5 μm.
O and the CC stretching band to determine whether PVC-g-PEGMA exist on the blended membrane surfaces. The oxygen, chlorine, and carbon elements on the near surface of all membranes were measured by X-ray photoelectron spectroscopy (XPS) (Axis Ultra, Kratos Analytical Ltd., UK) in the range from 0 to 1100 eV. The morphologies of all blended membranes were imaged using field-emission scanning electron microscopy (FESEM) (JSM-7500F, JEOL Ltd., Tokyo, Japan) at 15 kV accelerating voltage. The hydrophilicity of the membrane surface was characterized by dynamic water contact angle measured using a KRÜSS DSA 25S measuring apparatus (KRÜSS GmbH, Germany). The surface roughness of all blended membranes was determined using atomic force microscopy (AFM) (Multimode 8, Bruker, Germany) at a scan rate of 0.977 Hz and a scan size of 5 μm × 5 μm.
The membrane bulk porosity was measured by its dry and wet weights. First, the membrane stored in DI water was weighed after wiping off the water on the membrane surface with laboratory wipes. Second, the dry weight was measured after the wet membrane was dried at 30 °C for at least 24 h in a vacuum oven. The bulk porosity of the membrane was determined according to the following eqn (1).25
| |
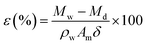 | (1) |
where
ε is the membrane bulk porosity,
Mw (g) is the weight of wet membrane,
Md (g) represents the weight of dry membrane,
Pw (g cm
−3) is the pure water density (4 °C, 1 g cm
−3),
Am (cm
2) represents the area of the wet membrane, and
δ (cm) is the thickness of the wet membrane.
2.5. Ultrafiltration experiments
The membrane filtration test was conducted using a 200 mL dead-end filtration cell (Amicon 8200, Millipore, USA), and the filtration cell was connected to a 5.0 L dispensing vessel which was attached a high-purity nitrogen tank. The effective filtration area was 28.7 cm2. A Pro Balance (AV8101, Ohaus Adventurer, USA) was used to weigh the permeate, and the permeate mass data was recorded every minute using Collect 6.1 software. The transmembrane pressure (TMP) in all flux tests was 20 psi.
The experimental procedures for evaluating the flux performance are described as follows according to previous reports.4,21 First, the pure water flux (Jw1, L m−2 h−1) of the wet membrane was measured by filtering DI water for 2 h. Second, the membrane was conditioned for 2 h using a 10 mM NaCl solution. Then, the fouling experiment was measured for 7 h by filtering a feed solution comprising 20 mgL−1 SA and 10 mM NaCl with stirring at 200 rpm. The flux of the feed solution was denoted as Jp, L m−2 h−1. Finally, the fouled membrane was flushed by DI water for 3 min, and the flux (Jw2, L m−2 h−1) of the cleaned membrane was measured. The membrane filtration tests were repeated at least twice and the average value was reported. The antifouling properties of the membrane were evaluated with some ratios like the flux recovery ratio (FRR), the total flux decline ratio (DRt), reversible flux decline ratio (DRr), and irreversible flux decline ratio (DRir) which were calculated with the following equations from some previous literature.26,27 Higher FRR and lower DRt indicate better antifouling properties of the membrane.28
| |
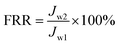 | (2) |
| |
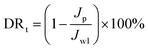 | (3) |
| |
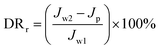 | (4) |
| |
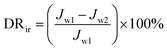 | (5) |
3. Results and discussion
3.1. The effects of solvent and nonsolvent on membranes
The relative affinity of solvent with PVC and water is evaluated using the distance in their Hansen solubility parameter space (RHSP), which was calculated by following eqn (6).29| |
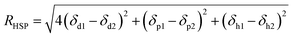 | (6) |
where the “1” represents solvent, “2” represents polymer or water, δd, δp and δh are the dispersion component, polar component and hydrogen bonding component of Hansen solubility parameter (HSP), respectively.
The HSP of PVC, water and solvents, and RHSP values of solvent–PVC pair and solvent–water pair are listed in Table 3. The lower RHSP value of solvent–polymer pair indicates the better dissolving ability of a solvent. A better solvent has a stronger interaction with the polymer.30 RHSP values of NMP–PVC, THF–PVC and DMAc–PVC are 4.9 MPa1/2, 7.5 MPa1/2, and 8.2 MPa1/2, respectively, which indicates that the order of interaction between the three solvents and PVC is NMP > THF > DMAc. Selection of solvent and non-solvent pair is the key factor in preparation of membranes fabricated by non-solvent induced phase separation (NIPS) method.31 The lower RHSP value of water–solvent pair means the better interaction between them, which leads to faster exchange of water and solvent during membrane immersion in water.12 The RHSP value of the THF–water is larger than that of the NMP–water. Therefore, in the current system, THF can theoretically prolong the phase separation, thus suppressing the formation of macrovoids and porous surface layer, which is beneficial to the formation of dense surface layer.32
Table 3 Hansen solubility parameter of solvents, water, PVC and their HSP distance (RHSP)a
| Material |
Hansen solubility parameter (MPa1/2) |
RHSP (MPa1/2) |
| δd |
δp |
δh |
Solvent–PVC |
Solvent–water |
| RHSP is calculated by eqn (6). Hansen solubility parameter of solvents,29 water,29 PVC.33 |
| THF |
16.8 |
5.7 |
8 |
7.5 |
35.91 |
| NMP |
18 |
12.3 |
7.2 |
4.9 |
35.65 |
| DMAc |
16.8 |
11.5 |
10.2 |
8.2 |
32.52 |
| Water |
15.5 |
16 |
42.3 |
— |
— |
| PVC |
18.7 |
10 |
3.1 |
— |
— |
3.2. Analysis of near-surface compositions of all blended membranes
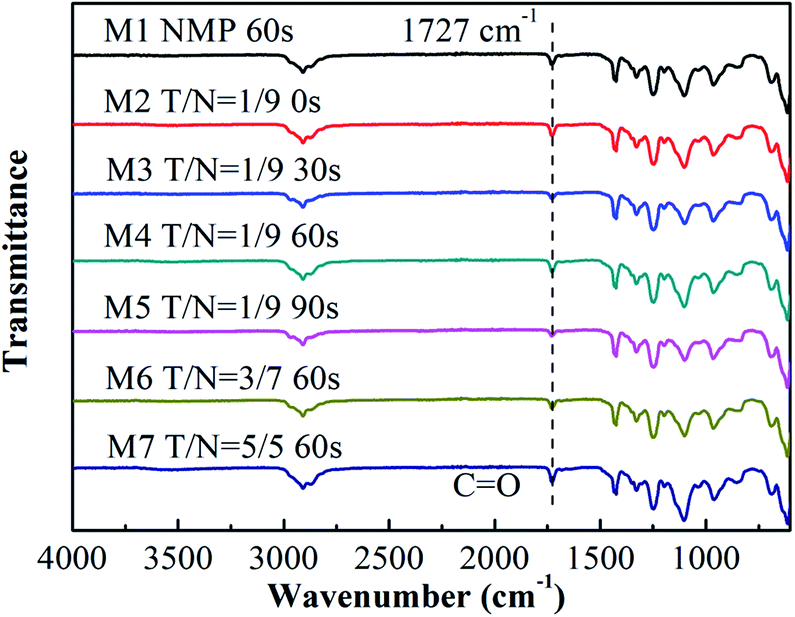
Fig. 1 exhibits FTIR-ATR spectra for the top surface of all blended membranes prepared at different solvents' mixing ratio and varied solvent evaporation time, which is used to prove the presence of PVC-g-PEGMA on the membrane surface. The CO stretching band is represented by the vibrational band at 1727 cm−1,34 and the peak at 1638 cm−1 represents the CC stretching band.35 As shown in Fig. 1, the characteristic peak at 1727 cm−1 but no peak at 1638 cm−1 was observed on all membranes, which indicated the existence of amphiphilic copolymer PVC-g-PEGMA on the surface of all blended membranes.
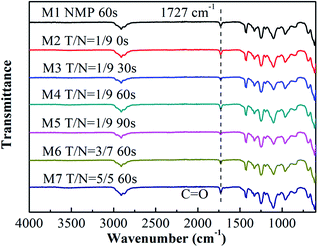 |
| | Fig. 1 FTIR-ATR spectra of the blended membranes at different THF:NMP ratios and different solvent evaporation time. M1, NMP, 60 s; M2, T/N = 1:9, 0 s; M3, T/N = 1:9, 30 s; M4, T/N = 1:9, 60 s; M5, T/N = 1:9, 90 s; M6, T/N = 3:7, 60 s; M7, T/N = 5:5, 60 s. | |
In order to analyze the chemical elements on the near surface of PVC/PVC-g-PEGMA blended ultrafiltration membranes in more detail, the near-surface chemical compositions of the membrane were determined by XPS, shown in Fig. 2. Surface separation commonly occurs in blend systems with amphiphilic polymer during the NIPS process.36 In this work, the PEGMA segments of PVC-g-PEGMA tended to migrate to the membrane surface during the formation of the membrane, being the only source of oxygen element on the membrane surface. Therefore, the near-surface oxygen content of the membrane suggested the migration level of the PEGMA segments. All prepared membranes were blended PVC with the same amount of PVC-g-PEGMA (10 wt%). However, the oxygen content of the membrane surface was different when the amount of THF and solvent evaporation time were varied, which meant both amount of THF and solvent evaporation time had an impact on the migration of PEGMA segments onto the membrane surface.
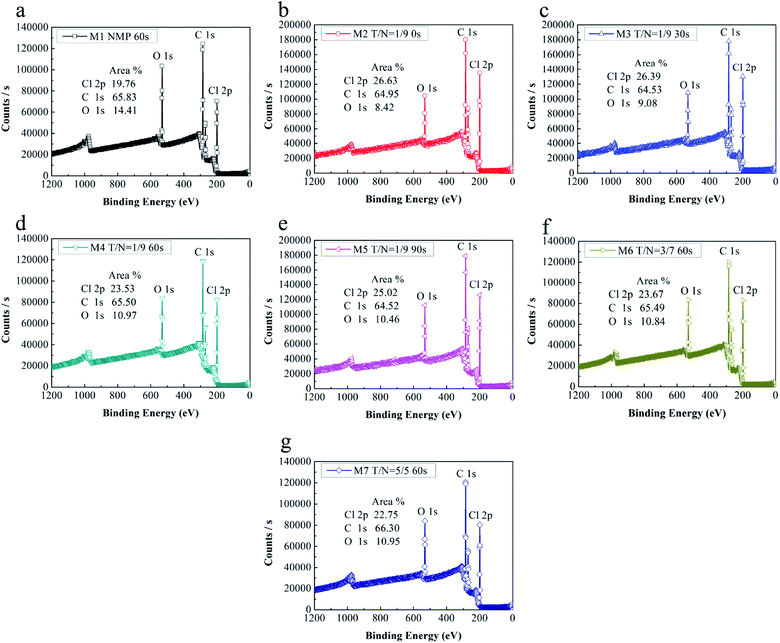 |
| | Fig. 2 XPS spectra of all blended membranes: (a) M1, NMP, 60 s; (b) M2, T/N = 1/9, 0 s; (c) M3, T/N = 1/9, 30 s; (d) M4, T/N = 1/9, 60 s; (e) M5, T/N = 1/9 90 s; (f) M6, T/N = 3/7, 60 s; (g) M7, T/N = 5/5, 60 s. | |
As presented in Fig. 2a, d, f and g, the surface of membrane M1 (prepared with only NMP as solvent) had the most oxygen content of 14.41%. The near-surface oxygen content of membranes M4, M6 and M7 was 10.97%, 10.84% and 10.95%, respectively, which indicated that adding THF as a co-solvent in the casting solution reduced the oxygen content on the membrane surface. However, with increasing the amount of THF, the near-surface oxygen content was basically the same. This phenomenon probably could be explained as follows: THF evaporated selectively during 60 seconds of evaporation, thus forming the nascent top layer,15 which probably prevented the migration of PEGMA segments to the membrane surface. Therefore, when the mixing ratio of THF to NMP was 1:9, the oxygen content on the membrane surface decreased significantly from 14.41% to 10.97%. Meanwhile, the solvent evaporation time before immersion in DI water was the same for 60 s, thus the nascent top layer formed during evaporation had the same hindrance effect on the migration of PEGMA segments. Therefore, even though the THF/NMP mixing ratio increased from 1:9 to 5:5, the oxygen content on the membrane surface was almost unchanged.
Fig. 2b–e shows the XPS spectra of the blended membranes fabricated at different evaporation times under the same THF/NMP ratio of 1:9. The near-surface oxygen content of membranes M2, M3, M4 and M5 was 8.42%, 9.08%, 10.97% and 10.46%, respectively. That was to say, the near-surface oxygen content of the membranes increased with increasing solvent evaporation time from 0 s to 60 s, but when evaporation time continued to increase to 90 s, the oxygen content decreased slightly. This might be because as solvent evaporation time increased, higher polymer concentration was obtained at the surface of the casting membrane before immersion in DI water. Therefore, the exchanging of solvent and DI water was slowed down, which gave PEGMA segments more time to move to the membrane surface.37 However, when solvent evaporation time continued to increase from 60 s to 90 s, the nascent skin layer formed during evaporation hindered the migration of PEGMA segments to the membrane surface, hence the oxygen content decreased slightly. Combined with the analysis of paragraph above, we speculated that solvent evaporation for 60 s initiates the formation of a nascent surface layer, which will hinder the migration of PEGMA segments to the membrane surface during the phase inversion process.
3.3. Morphologies of all blended membranes
Fig. 3 shows the morphologies of all blended membranes observed by the SEM. The surface pore size distribution of the membranes is summarized in Table 4, except for membrane M7, because its surface pore cannot be observed in the SEM image. As presented in Fig. 3a, g, k, m and Table 4, the average diameter (Daverage), pore density and porosity of membrane M1 were the largest. After adding THF, the Daverage, pore density and porosity of membranes M4, M6 and M7 were smaller than those of membrane M1. This was because the rapid evaporation of THF as a co-solvent during the evaporation stage enriched the polymer concentration on the surface of the casting membrane, resulting in a denser or pore-free surface layer.17 Moreover, compared with membranes M4, M6 and M7, it was found that the Daverage, pore density and porosity of the membrane decreased with increasing the amount of THF. The cross-section of the fabricated membrane can be divided into three parts, the dense top layer, the porous finger-like structures, and macrovoids. As shown in Fig. 3b, h, l and n, membranes M1, M4, M6 and M7 all exhibited asymmetric structures and large macrovoids. The heights of the macrovoids of membranes M1, M4, M6, and M7 measured using Image Pro Plus V.7.0 software (Vashaw Scientific Inc.) were 75.442 μm, 67.703 μm, 49.925 μm, 43.178 μm, respectively. This result showed that the large macrovoids became smaller as the amount of THF increased. This result is in accordance with the bulk porosity of the fabricated membranes (Table 5), which is in the order of M7 < M6 < M4 < M1. However, the existence of macrovoids in the cross-section of membranes is usually detrimental, as this will form weak spots within the membrane, especially in the condition with high pressure.31 As mentioned in Section 3.1, THF can increase the time of phase separation due to its worse affinity with water compared to NMP, which can inhibit the formation of macrovoids and porous surface layer.32 The more THF is added, the longer the time of phase separation is, thus the inhibition for the formation of macrovoids is stronger.15 Therefore, the Daverage, pore density and porosity of the membrane decreased with increasing THF/NMP ratio, and the structure of membranes M6 and M7 was denser than that of membranes M1 and M4.
 |
| | Fig. 3 Surface and cross-sectional morphologies of all blended membranes: (a and b) M1, NMP, 60 s; (c and d) M2, T/N = 1/9, 0 s; (e and f) M3, T/N = 1/9, 30 s; (g and h) M4, T/N = 1/9, 60 s; (i and j) M5, T/N = 1/9, 90 s; (k and l) M6, T/N = 3/7, 60 s; (m and n) M7, T/N = 5/5, 60 s. | |
Table 4 The surface pore size distribution of the blended membranes
| Membrane ID |
Daverage (nm) |
Dmax (nm) |
Pore density (m−2) |
Porosity (%) |
| M1 NMP 60 s |
14.79 |
64.53 |
4.03 × 1012 |
0.106 |
| M2 T/N = 1/9 0 s |
14.53 |
50.38 |
10.10 × 1012 |
0.240 |
| M3 T/N = 1/9 30 s |
9.02 |
46.47 |
8.72 × 1012 |
0.077 |
| M4 T/N = 1/9 60 s |
13.89 |
30.98 |
1.64 × 1012 |
0.036 |
| M5 T/N = 1/9 90 s |
7.22 |
19.60 |
1.45 × 1012 |
0.008 |
| M6 T/N = 3/7 60 s |
7.53 |
21.94 |
0.93 × 1012 |
0.005 |
Table 5 The bulk porosity, pure water flux and SA rejection ratio of all blended membranes
| Membrane ID |
Bulk porosity a (%) |
Pure water flux (L m−2 h−1) |
SA rejection ratio (%) |
| Calculated by eqn (1). |
| M1 NMP 60 s |
91.95 ± 0.04 |
226.06 ± 5.71 |
91.05 ± 3.38 |
| M2 T/N = 1/9 0 s |
92.89 ± 0.01 |
254.11 ± 38.68 |
86.10 ± 1.48 |
| M3 T/N = 1/9 30 s |
92.60 ± 1.87 |
247.25 ± 35.19 |
89.16 ± 0.71 |
| M4 T/N = 1/9 60 s |
91.52 ± 2.07 |
141.54 ± 30.18 |
92.12 ± 0.63 |
| M5 T/N = 1/9 90 s |
82.63 ± 1.83 |
138.16 ± 8.92 |
92.21 ± 0.40 |
| M6 T/N = 3/7 60 s |
89.91 ± 0.91 |
73.10 ± 6.70 |
92.47 ± 1.73 |
| M7 T/N = 5/5 60 s |
83.56 ± 2.89 |
24.85 ± 0.45 |
92.16 ± 0.48 |
As shown in Fig. 3c, e, g, i and Table 4 (membranes M2, M3, M4 and M5), when the THF/NMP mixing ratio was 1:9, the Daverage decreased with the increase of evaporation time in general, which was consistent with the result reported in the previous literature.38 The surface pore density and porosity of the membrane also decreased with increasing evaporation time, which was due to the higher polymer concentration at the interface resulted from longer evaporation time. As shown in Fig. 3d, f, h and j, membranes M2, M3, M4 and M5 all exhibited asymmetric structures with macrovoids, but the cross-sectional structure of the membranes did not change much as evaporation time increased from 0 s to 90 s. Hołda et al.9 prepared polysulfone (PSf) solvent resistant nanofiltration membranes with 3/7 of THF/NMP as solvent, distilled water as coagulation bath under 21 wt% of polymer concentration. They found that the number of macrovoids decreased when evaporation time increased, but its shape remained the same. When the solvent evaporated for 120 s, the macrovoids disappeared. This suggested that solvent evaporation could inhibit the formation of macrovoids before the casting solution was immersed in the coagulation bath. However, in the current system, solvent evaporation did not obviously control the macrovoids. There might be three reasons: (1) the polymer concentration was very low, only 13.2%; (2) the amount of THF was very small, and the ratio of THF to NMP was only 1:9; (3) as described in our previous study,18 amphiphilic copolymer PVC-g-PEGMA existed in the blend system, which helped to accelerate the exchange between solvent and non-solvent, thus weakening the inhibition of solvent evaporation on macrovoids formation.
3.4. AFM analysis
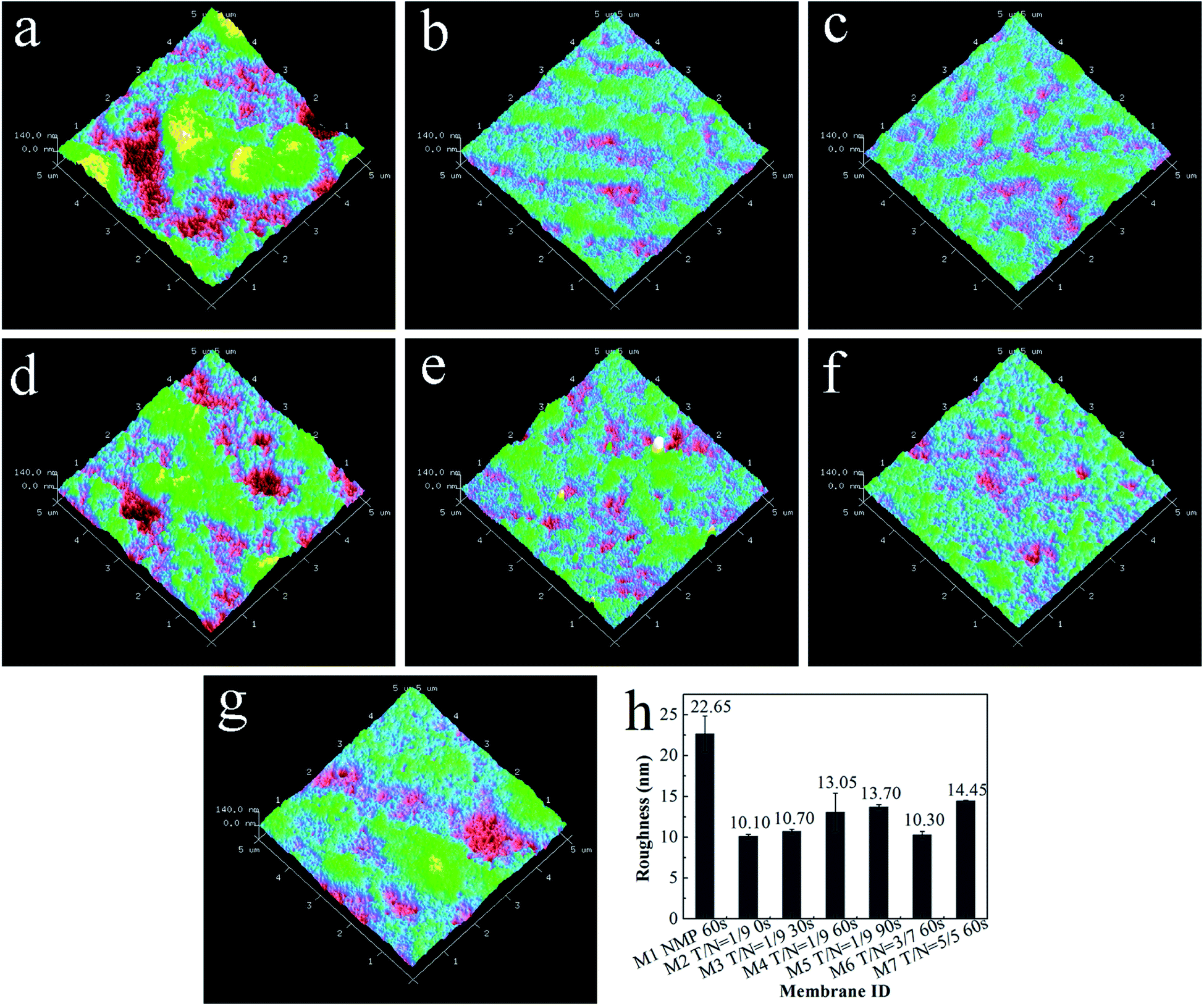
As exhibited in Fig. 4, the root mean square (RMS) roughness of the membrane surface generally decreased with increasing THF/NMP mixing ratio but increased as solvent evaporation time, which was consistent with changes in oxygen content of the membrane surface. Combined with the results in our previous study,18 we ascertained that the more PVC-g-PEGMA on the membrane surface, the rougher the membrane surface. In this work, the addition of THF could significantly lower the surface roughness of the membrane and the casting membrane should not evaporate for too long before immersion in the coagulation bath.
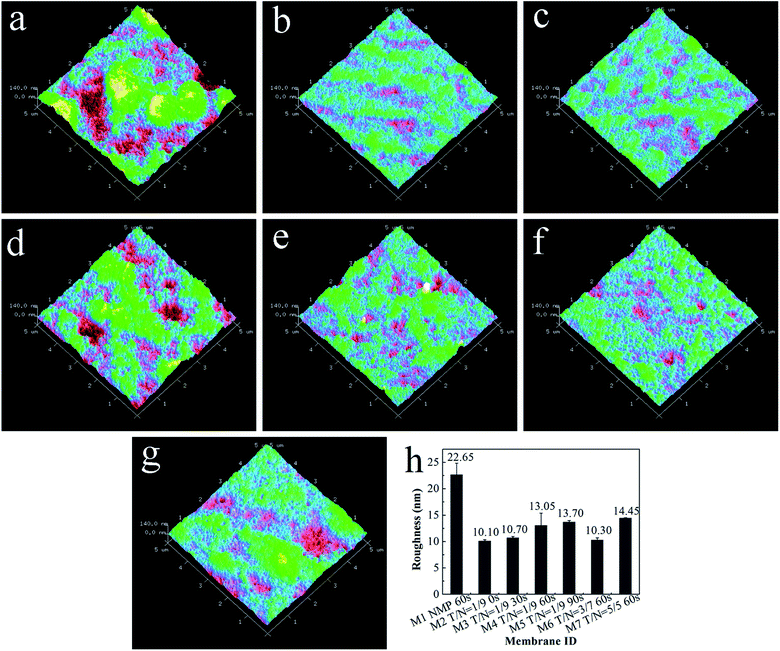 |
| | Fig. 4 AFM images of all blended membranes: (a) M1, NMP, 60 s; (b) M2, T/N = 1/9, 0 s; (c) M3, T/N = 1/9, 30 s; (d) M4, T/N = 1/9, 60 s; (e) M5, T/N = 1/9, 90 s; (f) M6, T/N = 3/7, 60 s; (g) M7, T/N = 5/5, 60 s; (h) the RMS roughness of all blended membranes. | |
3.5. Hydrophilicity analysis of the blended membranes
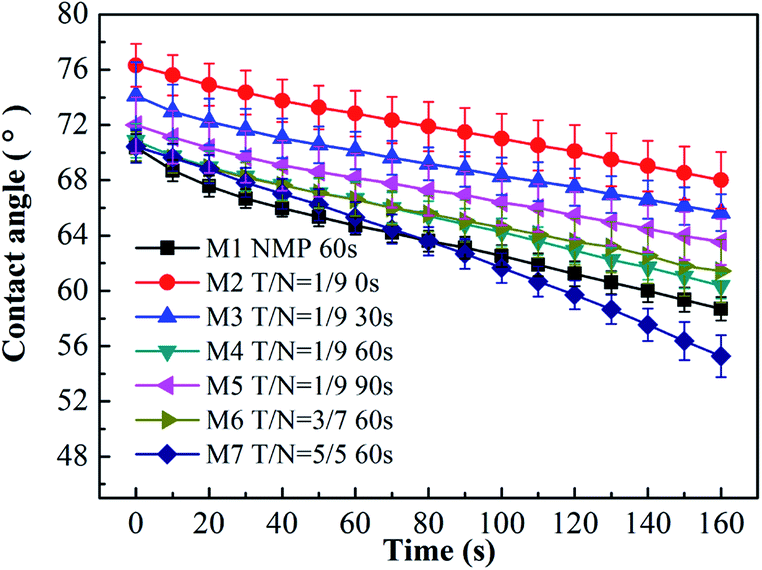
As presented in Fig. 5, the water contact angle of all blended membranes declined with time. For the case of increasing THF/NMP mixing ratio, the initial water contact angle of membranes M1, M4, M6 and M7 was 70.38 ± 0.98°, 70.87 ± 1.24°, 70.46 ± 1.09° and 70.45 ± 1.19°, respectively. In other words, the water contact angle was similar for the membrane M1 and the membranes M4, M6, M7, which was probably due to the synergistic effect of hydrophilic PEGMA segments on the membrane surface and the surface roughness of the membrane. Membrane M1 had the most hydrophilic PEGMA segments on the surface, but simultaneously, its surface roughness was higher than that of membranes M4, M6, M7. The hydrophilicity and the roughness of the membrane surface can impact the change of contact angle.39,40 The surface oxygen content of membranes M4, M6 and M7 was almost the same, and their surface roughness was similar. Therefore, the initial water contact angles of membranes M4, M6 and M7 were similar.
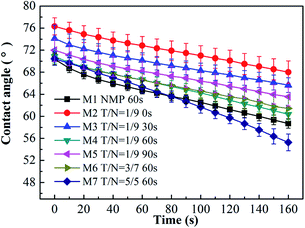 |
| | Fig. 5 Change of water contact angle for all blended membranes at different THF/NMP ratios and different solvent evaporation time. M1, NMP, 60 s; M2, T/N = 1:9, 0 s; M3, T/N = 1:9, 30 s; M4, T/N = 1:9, 60 s; M5, T/N = 1:9, 90 s; M6, T/N = 3:7, 60 s; M7, T/N = 5:5, 60 s. | |
When the THF/NMP mixing ratio was 1:9, the initial water contact angle of the blended membranes first declined from 76.32 ± 1.55° to 70.82 ± 1.24° with increasing solvent evaporation time from 0 s to 60 s and then increased to 72.00 ± 2.03° when evaporation time increased to 90 s. This result suggested that increasing evaporation time properly could enrich the hydrophilic PEGMA segments on the membrane surface, thus enhancing the hydrophilicity of the membrane.
3.6. Filtration performance
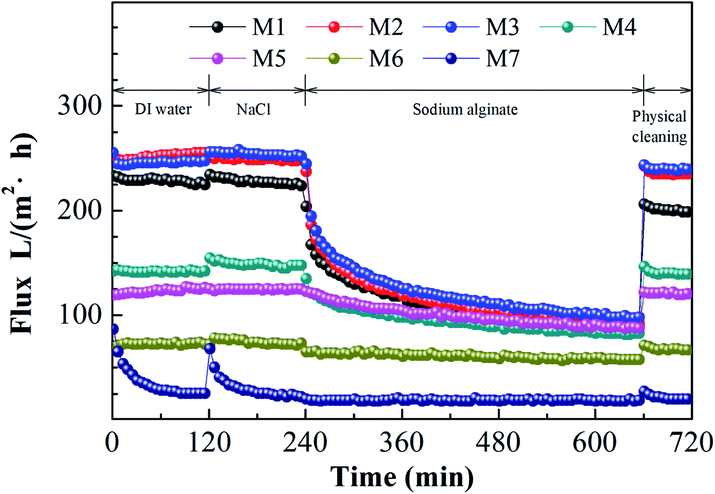
Fig. 6 shows the flux performance of the blended membranes under a constant pressure of 20 psi at room temperature (∼25 °C). As shown in Fig. 6 and Table 5, both the amount of THF and the solvent evaporation time could significantly affect the pure water fluxes of the membranes. For the case of different THF/NMP mixing ratio, membrane M1 exhibited the highest pure water flux of 226.06 ± 5.71 L m−2 h−1. The pure water flux decreased to 24.85 ± 0.45 L m−2 h−1 with increasing THF:NMP mixing ratio to 5:5. The pure water flux of the membrane is affected by hydrophilicity, pore size, porosity and transmembrane pressure. Surface porosity, Daverage (shown in Table 4) and bulk porosity (shown in Table 5) of the membrane decreased with increasing the amount of THF, which caused a decrease in the pure water flux of the membrane. Moreover, the SA rejection ratios of the blended membranes increased as THF/NMP mixing ratio increased, which was probably due to the decreased Daverage and maximum diameter (Dmax) of the membrane. For the case of changing evaporation time, the water pure flux of the membrane declined from 254.11 ± 38.68 L m−2 h−1 to 138.16 ± 8.92 L m−2 h−1 as the solvent evaporation time increased, which was probably because of the decreased surface porosity, Daverage and bulk porosity of the membrane. In addition, the decrease of maximum diameter (Dmax) resulted in higher SA rejection ratio for membranes M2, M3, M4, M5.
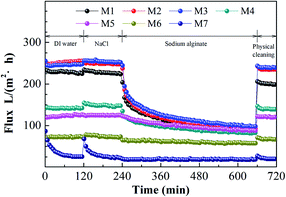 |
| | Fig. 6 Flux as a function of time using DI water or SA as feed solution of the PVC/PVC-g-PEGMA membranes fabricated at different THF/NMP ratio and solvent evaporation time. M1: NMP, 60 s; M2, T/N = 1:9, 0 s; M3, T/N = 1:9, 30 s; M4, T/N = 1:9, 60 s; M5, T/N = 1:9, 90 s; M6, T/N = 3:7, 60 s; M7, T/N = 5:5, 60 s. | |
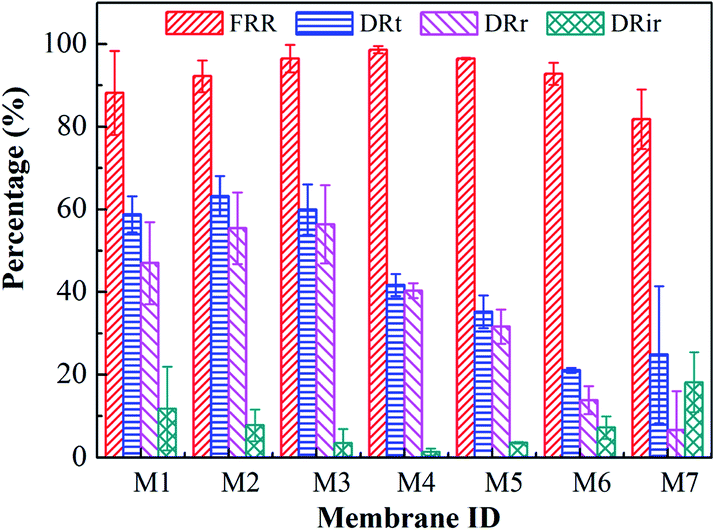
Characterization of the antifouling properties for all blended membranes are shown in Fig. 7. For the case of increasing THF/NMP mixing ratio, the flux recovery ratio (FRR) of membranes M1, M4, M6 and M7 was 88.21 ± 10.13%, 98.65 ± 0.85%, 92.79 ± 2.71%, and 81.81 ± 7.22%, respectively. The FRR of membrane M1 was lower than that of membranes M4 and M6, which was probably due to larger surface pore size41 and higher surface roughness42 of membrane M1 comparing with membranes M4 and M6. The FRR of membrane M7 was lower than that of membranes M4 and M6, which probably was because the surface roughness of membrane M7 was larger than that of membranes M4 and M6. For the case of varied solvent evaporation time, the FRR of membranes M2, M3, M4, and M5 was 92.20 ± 3.83%, 96.48 ± 3.35%, 98.65 ± 0.85%, 96.44 ± 0.19%, and the DRt of them was 63.21 ± 4.86%, 59.91 ± 6.11%, 41.67 ± 2.65%, and 35.21 ± 3.91%, respectively. The FRR generally increased and the DRt decreased as solvent evaporation time increased, which was due to the increase of hydrophilicity.43 Overall, membrane M4 exhibited the best antifouling properties with high pure water flux and SA rejection rate simultaneously.
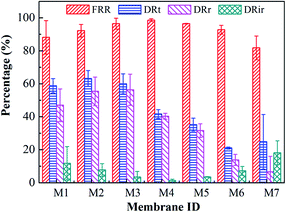 |
| | Fig. 7 Characterization of the membrane antifouling properties: the flux recovery ratio (FRR), total flux decline ratio (DRt), reversible flux decline ratio (DRr), and irreversible flux decline ratio (DRir). M1, NMP, 60 s; M2, T/N = 1:9, 0 s; M3, T/N = 1:9, 30 s; M4, T/N = 1:9, 60 s; M5, T/N = 1:9, 90 s; M6, T/N = 3:7, 60 s; M7, T/N = 5:5, 60 s. | |
4. Conclusion
In this work, we investigated the effect of THF as co-solvent and the solvent evaporation time on the performances of PVC/PVC-g-PEGMA blended membranes. When the THF/NMP mixing ratio was up to 3:7, the macrovoids at the bottom layer could become smaller. The addition of THF could effectively reduce the surface roughness of PVC/PVC-g-PEGMA membranes, thus improving its antifouling properties. Increasing the dose of THF or solvents evaporation time both could decrease the surface pore size of the membrane, causing the increase of SA rejection ratio. But it was worth noting that too much THF and too long solvent evaporation time reduced the membrane flux. Therefore, based on the performances of the membrane including hydrophilicity, flux performance, SA rejection ratio, the surface roughness, and the flux recovery ratio, the blended membrane prepared under the THF/NMP mixing ratio of 1:9 with solvent evaporation for 60 s showed optimal performance.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51678377), the State Key Laboratory of Separation Membranes and Membrane Processes (Tianjin Polytechnic University) (M2-201809), and the Fundamental Research Funds for the Central Universities. We thank Yi He at Analytical & Testing Center, Sichuan University for his kind help with SEM measurements. The views and ideas expressed herein are solely those of the authors and do not represent the ideas of the funding agencies in any form.
References
- J. Xu and Z.-L. Xu, J. Membr. Sci., 2002, 208, 203–212 CrossRef CAS.
- X. Fan, Y. Su, X. Zhao, Y. Li, R. Zhang, J. Zhao, Z. Jiang, J. Zhu, Y. Ma and Y. Liu, J. Membr. Sci., 2014, 464, 100–109 CrossRef CAS.
- L.-F. Fang, B.-K. Zhu, L.-P. Zhu, H. Matsuyama and S. Zhao, J. Membr. Sci., 2017, 524, 235–244 CrossRef CAS.
- B. Liu, C. Chen, W. Zhang, J. Crittenden and Y. Chen, Desalination, 2012, 307, 26–33 CrossRef CAS.
- W.-d. Liu, Y.-h. Zhang, L.-f. Fang, B.-k. Zhu and L.-p. Zhu, Chin. J. Polym. Sci., 2012, 30, 568–577 CrossRef CAS.
- Y. Zhang, X. Tong, B. Zhang, C. Zhang, H. Zhang and Y. Chen, J. Membr. Sci., 2018, 548, 32–41 CrossRef CAS.
- Z. Zhou, S. Rajabzadeh, A. R. Shaikh, Y. Kakihana, W. Ma and H. Matsuyama, J. Membr. Sci., 2016, 514, 537–546 CrossRef CAS.
- I. Soroko, M. P. Lopes and A. Livingston, J. Membr. Sci., 2011, 381, 152–162 CrossRef CAS.
- A. K. Hołda, B. Aernouts, W. Saeys and I. F. J. Vankelecom, J. Membr. Sci., 2013, 442, 196–205 CrossRef.
- I. Soroko, M. Makowski, F. Spill and A. Livingston, J. Membr. Sci., 2011, 381, 163–171 CrossRef CAS.
- H. Rabiee, M. H. D. A. Farahani and V. Vatanpour, J. Membr. Sci., 2014, 472, 185–193 CrossRef CAS.
- K. J. Roy, T. V. Anjali and A. Sujith, J. Mater. Sci., 2017, 52, 5708–5725 CrossRef CAS.
- H. Zhang, X. Hu, Y. Chen, B. Cheng, D. Liu and Y. Zhang, J. Mater. Sci., 2015, 50, 4371–4378 CrossRef CAS.
- W. Qiu, L. Liu and W. J. Koros, J. Membr. Sci., 2017, 529, 150–158 CrossRef CAS.
- P. Vandezande, X. Li, L. E. M. Gevers and I. F. J. Vankelecom, J. Membr. Sci., 2009, 330, 307–318 CrossRef CAS.
- S. P. Nunes, R. Sougrat, B. Hooghan, D. H. Anjum, A. R. Behzad, L. Zhao, N. Pradeep, I. Pinnau, U. Vainio and K.-V. Peinemann, Macromolecules, 2010, 43, 8079–8085 CrossRef CAS.
- K.-V. Peinemann, V. Abetz and P. F. W. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS.
- H. Wu, T. Li, B. Liu, C. Chen, S. Wang and J. C. Crittenden, Appl. Surf. Sci., 2018, 455, 987–996 CrossRef CAS.
- W. Xie, T. Li, C. Chen, H. Wu, S. Liang, H. Chang, B. Liu, E. Drioli, Q. Wang and J. C. Crittenden, Ind. Eng. Chem. Res., 2019, 58(16), 6413–6423 CrossRef CAS.
- S. H. Ahn, J. A. Seo, J. H. Kim, Y. Ko and S. U. Hong, J. Membr. Sci., 2009, 345, 128–133 CrossRef CAS.
- B. Liu, C. Chen, T. Li, J. Crittenden and Y. Chen, J. Membr. Sci., 2013, 445, 66–75 CrossRef CAS.
- K. Katsoufidou, S. G. Yiantsios and A. J. Karabelas, Desalination, 2008, 220, 214–227 CrossRef CAS.
- Y. Ye, P. L. Clech, V. Chen and A. G. Fane, J. Membr. Sci., 2005, 264, 190–199 CrossRef CAS.
- Y. Ye, p. L. Clech, V. Chen, A. G. Fane and B. Jefferson, Desalination, 2005, 175, 7–20 CrossRef CAS.
- J.-F. Li, Z.-L. Xu, H. Yang, L.-Y. Yu and M. Liu, Appl. Surf. Sci., 2009, 255, 4725–4732 CrossRef CAS.
- X. Zhao, Y. Su, W. Chen, J. Peng and Z. Jiang, J. Membr. Sci., 2012, 415–416, 824–834 CrossRef CAS.
- X. Zhao, Y. Su, Y. Li, R. Zhang, J. Zhao and Z. Jiang, J. Membr. Sci., 2014, 450, 111–123 CrossRef CAS.
- G. Rong, D. Zhou, X. Han and J. Pang, Appl. Surf. Sci., 2018, 427, 1065–1075 CrossRef CAS.
- C. M. Hansen, Hansen solubility parameters: a users hand book, CRC Press, Taylor & Francis Group, Florida, 2nd edn, 2007 Search PubMed.
- R. Guan, H. Dai, C. Li, J. Liu and J. Xu, J. Membr. Sci., 2006, 277, 148–156 CrossRef CAS.
- M. Mulder, Basic Principles of Membrane Technology, Kluwer Academic Publishers, 2rd edn, 1996 Search PubMed.
- E. Bagheripour, A. R. Moghadassi and S. M. Hosseini, Arabian J. Chem., 2017, 10, S3375–S3380 CrossRef CAS.
- J. Brandrup and E. H. Immergut, Polymer handbook, Wiley, New York, 3rd edn, 1989 Search PubMed.
- N. A. Hashim, F. Liu and K. Li, J. Membr. Sci., 2009, 345, 134–141 CrossRef CAS.
- R. Patel, M. Patel, S. H. Ahn, Y. K. Sung, H.-K. Lee, J. H. Kim and J.-S. Sung, Mater. Sci. Eng., C, 2013, 33, 1662–1670 CrossRef CAS.
- X. Ma, Y. Su, Q. Sun, Y. Wang and Z. Jiang, J. Membr. Sci., 2007, 292, 116–124 CrossRef CAS.
- C. Chen, L. Tang, B. Liu, X. Zhang, J. Crittenden, K. L. Chen and Y. Chen, J. Membr. Sci., 2015, 487, 1–11 CrossRef CAS.
- Y. Gu, R. M. Dorin and U. Wiesner, Nano Lett., 2013, 13, 5323–5328 CrossRef CAS.
- H. Guo and M. Ulbricht, J. Membr. Sci., 2011, 372, 331–339 CrossRef CAS.
- Q. Zhang, J. Jiang, F. Gao, G. Zhang, X. Zhan and F. Chen, Chem. Eng. J., 2017, 321, 412–423 CrossRef CAS.
- X. Fu, T. Maruyama, T. Sotani and H. Matsuyama, J. Membr. Sci., 2008, 320, 483–491 CrossRef CAS.
- S. H. Woo, J. Park and B. R. Min, Sep. Purif. Technol., 2015, 146, 187–191 CrossRef CAS.
- Z. Xu, J. Liao, H. Tang and N. Li, J. Membr. Sci., 2018, 548, 481–489 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Songmiao Liangc,
Haiqing Changa and
Baicang Liu
*b,
Songmiao Liangc,
Haiqing Changa and
Baicang Liu