
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-doped graphene–TiOxNy nanocomposite electrode for highly efficient capacitive deionization†
Yuchen Wuab,
Gaopeng Jiang b,
Qian Lib,
Zisheng Zhang*a and
Zhongwei Chen*b
b,
Qian Lib,
Zisheng Zhang*a and
Zhongwei Chen*b
aDepartment of Chemical and Biological Engineering, University of Ottawa, 161 Louis Pasteur Private, Ottawa, Ontario K1N 6N5, Canada. E-mail: Jason.zhang@uottawa.ca
bDepartment of Chemical Engineering, University of Waterloo, 200 University Ave W, Waterloo, Ontario N2L 3G1, Canada. E-mail: zhwchen@uwaterloo.ca
First published on 9th September 2019
Abstract
In this work, the first-ever reported nanocomposite electrode of nitrogen-doped graphene–titanium oxynitride (NG–TiOxNy) for capacitive deionization (CDI) was synthesized via hydrothermal reaction and a high-temperature nitridation process. The physiochemical characterizations revealed that the nitrogen was doped in the graphene structure mainly in the form of graphitic nitrogen and the TiOxNy was successfully formed via TiO2 nitridation process. The layered NG nanosheets facilitated the diffusion of ions in saline water and formed electrical double layer on the surface of the electrode material, while the presence of TiOxNy enhanced the electrochemical performance by increasing surface area and generating surface vacancies via nitridation. The CDI cell employed NG–TiOxNy electrode delivered a breakthrough salt adsorption capacity of 26.1 mg g−1 in 500 mg L−1 saline water, and retained over 90% of its initial salt removal efficacy after 12 regeneration cycles. Such high CDI performance exhibits the promising application of NG–TiOxNy as a novel CDI electrode candidate.
1 Introduction
Obtaining fresh and clean water is one of the most urgent environmental problems in the 21st century.1 An effective solution for increasing the fresh water supply is to develop sustainable desalination technology, which can treat brackish and saline water.2 However, traditional water desalination techniques including distillation, reverse osmosis, and vacuum evaporation are approaching their limitations as they require high operating temperature, huge energy consumption, and large capital cost.3 Alternatively, capacitive deionization (CDI) has become one of the most promising solutions for water desalination,4 which works by separating and adsorbing ions on each electrode. It has the advantageous nature of high efficiency, low cost, low energy-consumption, ambient atmosphere operation, and environmental-benignity.5 The electrode material in a CDI cell plays a pivotal role in determining the desalination performance.6 Currently, most reported and commercialized CDI electrode materials are based on various kinds of novel porous carbon electrodes.7–14Graphene, as a novel two-dimensional carbon nanomaterial, has been widely used as the electrode materials in batteries and supercapacitors due to its high electrochemical performance and large surface area.15–20 Its heteroatom-doped derivative, especially nitrogen-doped graphene (NG), has also been investigated as electrode materials to achieve even better electrochemical performance in batteries and supercapacitors as opposed to graphene due to nitrogen-doped active sites and enhanced electrical conductivity.21,22 It is also worth to notice that it is reported that by nitrogen doping, the NG achieved significantly high adsorption capacity in CDI test.23–28 On the other hand, titanium oxynitride (TiOxNy, where x and y stand for the unspecific atom ratios of oxygen and nitrogen to titanium, respectively) and/or titanium nitride (TiN) can be fabricated into nanomaterials and used as electrodes materials in batteries and supercapacitors with enhanced electrochemical performance and wettability.29–31 Additionally, with various morphologies, various Ti species and excellent electrical performance, TiN and/or TiOxNy can be used as additives to enhance the performance of graphene nanosheets by forming nanocomposites.32,33 Moreover, our previous research disclosed that the porous TiN/TiOxNy had shown excellent performance as the CDI electrode material.34 However, there is no reported nanocomposites of NG and TiOxNy that have been synthesized and used as CDI electrode materials. Therefore, it is highly promising to prepare this kind of hybrid material and investigate its potential performance in CDI devices for water desalination.
In this study, a first-ever reported nanocomposite electrode of nitrogen-doped graphene–titanium oxynitride (NG–TiOxNy) was synthesized via the hydrothermal reaction and high-temperature nitridation process and then tested for CDI. The physiochemical characterization results demonstrated the successful formation of nitrogen-doped-sites on the graphene nanosheets and the TiOxNy nanoparticles embedded on the graphene nanosheets. The multi-layered graphene structure and TiOxNy nanoparticles allowed the saline water stream to pass through and facilitates a fast ion diffusion process to form the electrical double layer (EDL) at the electrode interface of both the graphene layers and TiOxNy nanoparticles. As a result, the NG–TiOxNy electrode delivered a significantly higher salt removal efficiency than the commercial AC, pure rGO and rGO–TiO2 electrodes in single-pass CDI test (Fig. 1). Furthermore, the electrode reached a maximum salt adsorption capacity of 26.1 mg g−1 in 500 mg L−1 feed in saline water. The electrode material also exhibited great regeneration capacity and long-life cycling stability. The overall superior results demonstrated that the NG–TiOxNy could be an ideal CDI electrode material candidate.
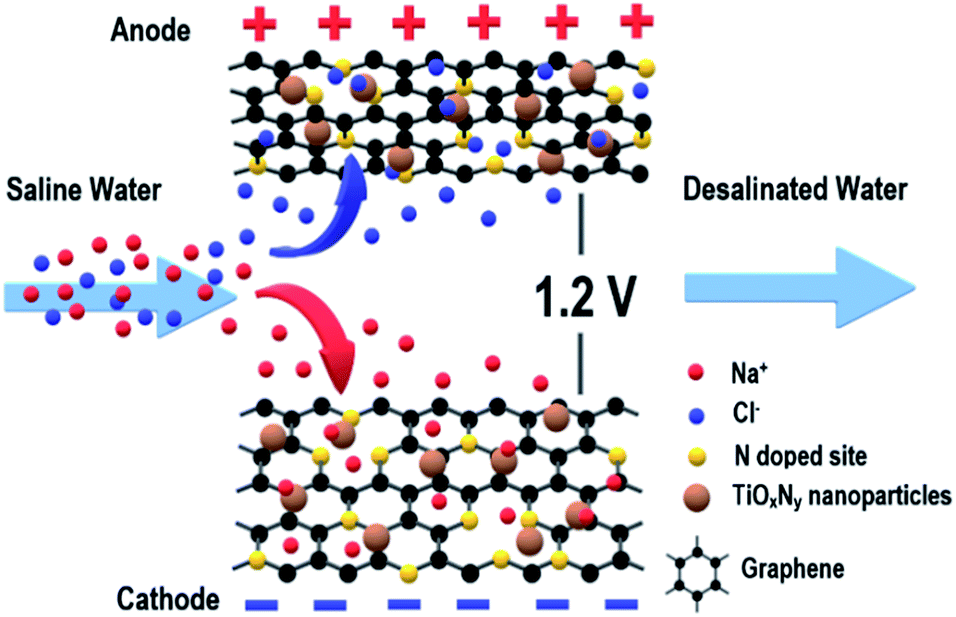 | ||
| Fig. 1 Schematic of a flow-by CDI cell using NG–TiOxNy electrodes. | ||
2 Experimental
2.1 Synthesis of TiO2 nanoparticles
The synthesis procedure of the polystyrene (PS) nanosphere is adapted from the literature.35 First, 2.5 g polyvinylpyrrolidone (PVP, M.W. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was charged in a three-neck flask that contains 200 mL boiled distilled deionized water (oxygen-free DDI). After PVP was fully dissolved, the flask was placed in an oil bath with N2 gas purging under the surface of the solution to expel the oxygen in the system. As the temperature was elevated to 70 °C for 15 min, styrene monomer (24 mL) was gradually added into the flask for 20 min with vigorous stirring and refluxing. Then, the potassium persulfate (40 mL, 5 mg mL−1) was dropwise added into the mixture, and the emulsion polymerization was continued under an air-free atmosphere at 70 °C for 24 h. The PS emulsion was centrifuged and washed with DDI water for several times. Then, after 3 days' freeze-drying, the PS nanospheres were obtained.
000) was charged in a three-neck flask that contains 200 mL boiled distilled deionized water (oxygen-free DDI). After PVP was fully dissolved, the flask was placed in an oil bath with N2 gas purging under the surface of the solution to expel the oxygen in the system. As the temperature was elevated to 70 °C for 15 min, styrene monomer (24 mL) was gradually added into the flask for 20 min with vigorous stirring and refluxing. Then, the potassium persulfate (40 mL, 5 mg mL−1) was dropwise added into the mixture, and the emulsion polymerization was continued under an air-free atmosphere at 70 °C for 24 h. The PS emulsion was centrifuged and washed with DDI water for several times. Then, after 3 days' freeze-drying, the PS nanospheres were obtained.
Titanium(IV) butoxide (TBOT, 97%, 10 mL), ethanol (>99%, 10 mL), and hydrochloric acid (37%, 2 mL) were mixed with a volume ratio of 5:5:1 and stirred for at least 30 min. Meanwhile, 2 grams of the PS nanosphere was sonicated and re-dispersed in 20 mL ethanol. The PS nanosphere ethanol dispersion was then added into the TBOT solution and the mixture was sonicated until it turns light yellow after 30 min36–38 After drying the sol–gel in ambient conditions overnight, the composite sample was loaded into a tube furnace and subjected to sequential heat treatments at 300 °C for 1 h and 540 °C for 30 min under argon atmosphere to form the TiO2 nanoparticles.
2.2 Synthesis of NG–TiOxNy
In this study, the GO and TiO2 were mixed in designed mass ratios (1:1, 2:1, and 3:1) in a beaker with 400 mL DDI water and thoroughly stirred for over 24 hours. Then, the mixed solution was sealed in an autoclave and hydrothermally treated for 12 h at 180 °C. The hydrotreated product was then centrifuged at 2500 rpm and separated from the solution. Subsequently, the product was washed by DDI water and centrifuged several times. The obtained product was then oven dried at 60 °C for 12 h, and then transferred to a vacuum oven and fully dried. Thus, in this step, GO was reduced to rGO39 and the obtained composite is denoted as rGO–TiO2 for comparison.
The dried composite was transferred to a ceramic boat and was subjected to heat treatment from room temperature to 800 °C with a ramp rate of 1 °C min−1 under argon flow. Then, the atmosphere was switched from argon to ammonia after the temperature reached 800 °C and was kept at 800 °C for 1 h to proceed the nitridation. Then, the sample was cooled down until room temperature in argon.
2.3 Physicochemical and electrochemical characterization
The crystal structure property of the electrode material was characterized by X-ray diffraction (XRD, RIETVELDXRG 3000) and the chemical composition was characterized by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha spectrometer) at University of Waterloo. Sample morphology was examined using scanning electron microscopy (SEM, ZEISS ULTRA PLUS), and transmission electron microscopy (TEM, Carl Zeiss Libra 200MC STEM).The crystallite size was calculated according to the Scherrer equation, given in eqn (1):
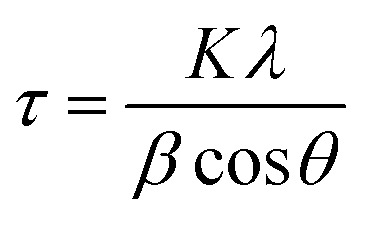 | (1) |
Cyclic voltammetry (CV) measurements were employed for evaluating the electrochemical performance of the CDI electrode and its adsorption/desorption capacity. The CV experiment was performed in a three-electrode system, which employed a carbon electrode as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode in the NaCl electrolyte solution. Reversible Hydrogen Electrode (RHE) were used to calibrate the SCE electrode in a pH = 7 NaCl solution, as shown in Fig. S1 in the ESI.† The actual potential was calibrated by add 0.5 V to the original value, where the potential range from −1 V to 0 V is corresponded to the actual potential range of −0.5 V to 0.5 V, representing both the cathode and anode adsorption processes. The experiment was conducted using an EC-lab SP-300 working station under the sweep rate range of 5–50 mV s−1 in 0.1–1 mol L−1 NaCl solution. The value of specific capacity of the prepared materials was calculated by integrating the area of the CV curve, using eqn (2):
 | (2) |
2.4 CDI test
The synthesized NG–TiOxNy electrode material with different mass ratios was mixed with PTFE and conductivity carbon (Super P) with a mass ratio of 8:1:1. The electrode material was dried in a vacuum oven and pressed on metal mesh with copper current collectors. Each plate had an effective surface area of 4 cm2 and was separated by glass fiber with 5 mm space. Saline water with the salinity of 100–500 mg L−1 was fed into the cell by a peristaltic pump (Baoding Lange) and passed between two electrodes under the flowrate from 10–30 mL min−1, and the potential from 0.8–1.2 V applied on each side. The salinity of the outflow water was measured by a CON6+ conductivity meter, and the raw data was converted into salinity using the calibration curve in Fig. S2.† The salt removal efficiency is calculated using the following eqn (3):
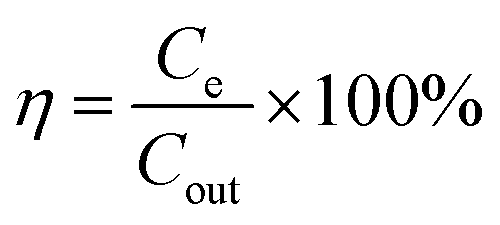 | (3) |
The salt adsorption capacity (SAC) was obtained using eqn (4):
 | (4) |
The salt adsorption rate (SAR) was calculated using eqn (5):
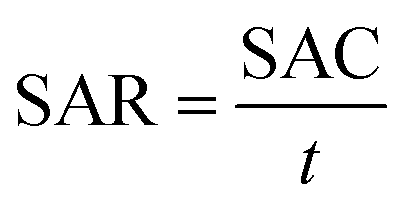 | (5) |
3 Result and discussion
The XRD patterns of the synthesized NG–TiOxNy, rGO–TiO2, and rGO are shown in Fig. 2a. For both NG–TiOxNy and rGO–TiO2, the main (101) peak at 25° and minor (200), (215) peaks at 48° and 75° can be assigned to the anatase-phase TiO2 (JCPDS no. 21-1272), respectively. The peaks at 31°, 54°, 56°, can be assigned crystal planes (121), (211), (123) to the rutile-phase TiO2 (JCPDS no. 46-1238). Both the presence of anatase-TiO2 and rutile-TiO2 are confirmed in the rGO–TiO2 synthesized via hydrothermal method. New peaks appeared at 36°, 42°, and 62° indicated the successful nitridation of rGO–TiO2 after ammonia annealing. The upshift of the characteristic peaks in the XRD pattern of NG–TiOxNy as opposed to peaks for crystal planes (111), (200), (220) in standard TiN XRD profile (JCPDS: 38-1420, Fig. S3†) indicated the presence of the oxynitride. The value of N/Ti ratio (y) was calculated to be 0.61 (Table S1†), further proving the low degree of nitridation.40–42 The XRD results prove the presence of various Ti species, including both the anatase-phase and rutile TiO2 and the existence of TiOxNy crystal phase. Based on previous reports, both anatase and rutile phase TiO2 can have a positive effect on increasing the ion adsorption by increasing the wettability and forming a more cohesive EDL.38,43 The calculated average size of the TiOxNy nanoparticles was 43 nm. Such small size of nanoparticles can potentially provided more active sites for ion absorbing and forming the EDL on the surface of the nanoparticles.43,44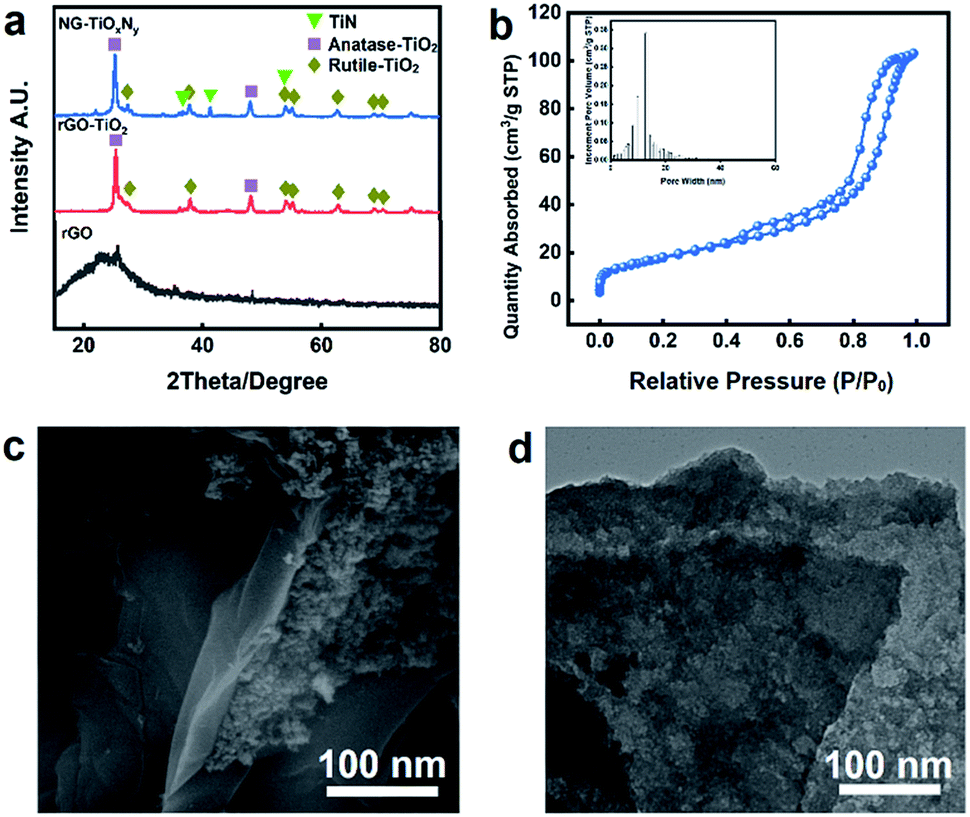 | ||
| Fig. 2 (a) XRD patterns of NG–TiOxNy, rGO–TiO2 and rGO; (b) N2 adsorption–desorption isotherm and associated pore size distribution of NG–TiOxNy; (c) SEM image and (d) TEM image of NG–TiOxNy. | ||
The BET analysis (Fig. 2b) shows a typical type IV isotherm curve, further confirmed the existence of the mesoporous structure with the pore distribution around 18 nm. Compare with the rGO–TiO2 (Table 1). The nitrogen doping process dramatically increased the BET surface area and the pore volume. The increase of the BET surface area is attributed to the loaded TiOxNy nanoparticles with small crystal size and could be further explained by the surface vacancies generated during the ammonia heat treatment, which may result in a positive effect on ion absorbing.45,46
| BET surface area (m2 g−1) | BET pore volume (cm3 g−1) | |
|---|---|---|
| NG–TiOxNy | 64.22 | 0.391 |
| rGO–TiO2 | 24.17 | 0.049 |
The SEM images in Fig. S4† at 1 μm magnitude indicates the layered structure of the graphene nanosheet. The layered nanosheets provide a relatively large surface area for interaction between liquid and solid as well as enough space for the saline water to pass through and assisted the ion diffusion process. Fig. 2c shows the TiOxNy nanoparticles attached to the graphene nanosheet layer. The TiOxNy nanoparticles provides the space for the formation of cohesive electrical double layer for ion absorbing. Fig. 2d is the TEM image of the synthesized NG–TiOxNy, which indicates the layered nanosheets of the graphene. Furthermore, the TiOxNy nanoparticles embedded on graphene the layer further allows the absorption of saline water into the electrode structure, which extended the electrode–electrolyte interface into NG–TiOxNy and minimized the distance of ion diffusion between the electrode surface and the saline water during the electrosorption process.
The elemental mapping (Fig. 3) suggestes the homogenous distribution of the elements of the NG–TiOxNy at 5 μm magnitude. The EDX mapping spectra reveales the mass ratio generally follows the rGO:TiO2 = 2:1 as initial. Nitrogen accounts for 13.40% atomic percentage (Fig. 3b), demonstrating the nitrogen-enriched doped surface, which furthers the surface absorbing capacity.46,47 To further study the chemical composition of the NG–TiOxNy electrode material. X-ray photoelectron spectroscopy (XPS) was utilized, as shown in Fig. 4. The wide survey of the NG–TiOxNy, as shown in Fig. S5,† exhibits several main peaks assigned as Ti 2s, O 1s, Ti 2p, N 1s, C 1s, Ti 3s, and Ti 3p, respectively. The high resolution XPS spectrum of O 1s in Fig. 4a can be deconvoluted to Ti–O, C–O, –OH and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks, respectively. The formation of hydrogen bonds with water further enhanced the wettability of the electrode and had a positive effect on the ion storage process in the electrode material.48,49 The XPS spectrum of C 1s scan in Fig. 4b is deconvoluted to the peaks of sp2 CC at 284.8 eV, sp3 C–C peak at 286.8 eV, they are assigned to the CC/C–C bond on the graphene aromatic ring. The peak at 287.4 eV and 289.2 eV are assigned to CN and C–N bonds. Both demonstrate the successful nitrogen doping on the aromatic rings of graphene layer. The nitrogen configuration of the graphite layer can possibly result in a stronger binding energy with the ions in the electrolyte and enhancing the surface ion retention capacity.50 Additionally, it is also reported the NG contributed to better wettability of the electrode material.51 Thus, the presence of nitrogen doping graphene can be possible contributing to a higher salt adsorption capacity.52 The carbon–oxygen peak at 291.3 eV is significantly diminished compared with the reported GO C 1s spectra,53 which can be attributed to the reduction of oxygen on the GO nanosheets during the hydrothermal treatment. The nitrogen-doped graphite potentially contributes extra salt adsorption capacity during the CDI process by creating the surface vacancies.54 The XPS spectrum of N 1s scan is presented in Fig. 4c, where the main peak at 396.3 eV and the peak at 397.4 eV assigned to Ti–N and Ti–O–N confirm the successful nitridation process of the Ti species. Such formation of the Ti–N and Ti–O–N bonds resulted in various Ti species, providing possibility for cation intercalation within the chemistry bonds and increased its adsorption capacity.33,55 The peaks at 398.6 eV and 400.8 eV further confirm the pyridine and graphite nitrogen doping sites. Fig. 4d illustrates the XPS spectrum of Ti 2p scan. The peaks at 458.8 eV and 456.8 eV are assigned to the Ti–O and the N intercalation into TiO2 lattice or the formation of TiOxNy, respectively.56 Both the N 1s scan and the Ti 2p scan confirmed the existing of oxygen and nitrogen species and the formation of oxynitride group in the TiOxNy, which is corresponded with the results in XRD analysis. The various Ti states of Ti potentially facilitate fast oxidation/reduction surface faradaic reactions during the electrochemical charge and discharge process, which provide an opportunity to effectively improve the specific capacitance and ion adsorption capacity.57 Through the analysis of atomic percentages of different Ti species from the Ti 2p scan (Fig. S6†), the value of O/Ti ratio (x) is calculated as 0.66 (Table S2†). Thus, the composition of titanium compounds in this NG–TiOxNy nanocomposite (rGO:TiOxNy = 2:1 (w/w)) can be estimated as TiO0.66N0.61.
O peaks, respectively. The formation of hydrogen bonds with water further enhanced the wettability of the electrode and had a positive effect on the ion storage process in the electrode material.48,49 The XPS spectrum of C 1s scan in Fig. 4b is deconvoluted to the peaks of sp2 CC at 284.8 eV, sp3 C–C peak at 286.8 eV, they are assigned to the CC/C–C bond on the graphene aromatic ring. The peak at 287.4 eV and 289.2 eV are assigned to CN and C–N bonds. Both demonstrate the successful nitrogen doping on the aromatic rings of graphene layer. The nitrogen configuration of the graphite layer can possibly result in a stronger binding energy with the ions in the electrolyte and enhancing the surface ion retention capacity.50 Additionally, it is also reported the NG contributed to better wettability of the electrode material.51 Thus, the presence of nitrogen doping graphene can be possible contributing to a higher salt adsorption capacity.52 The carbon–oxygen peak at 291.3 eV is significantly diminished compared with the reported GO C 1s spectra,53 which can be attributed to the reduction of oxygen on the GO nanosheets during the hydrothermal treatment. The nitrogen-doped graphite potentially contributes extra salt adsorption capacity during the CDI process by creating the surface vacancies.54 The XPS spectrum of N 1s scan is presented in Fig. 4c, where the main peak at 396.3 eV and the peak at 397.4 eV assigned to Ti–N and Ti–O–N confirm the successful nitridation process of the Ti species. Such formation of the Ti–N and Ti–O–N bonds resulted in various Ti species, providing possibility for cation intercalation within the chemistry bonds and increased its adsorption capacity.33,55 The peaks at 398.6 eV and 400.8 eV further confirm the pyridine and graphite nitrogen doping sites. Fig. 4d illustrates the XPS spectrum of Ti 2p scan. The peaks at 458.8 eV and 456.8 eV are assigned to the Ti–O and the N intercalation into TiO2 lattice or the formation of TiOxNy, respectively.56 Both the N 1s scan and the Ti 2p scan confirmed the existing of oxygen and nitrogen species and the formation of oxynitride group in the TiOxNy, which is corresponded with the results in XRD analysis. The various Ti states of Ti potentially facilitate fast oxidation/reduction surface faradaic reactions during the electrochemical charge and discharge process, which provide an opportunity to effectively improve the specific capacitance and ion adsorption capacity.57 Through the analysis of atomic percentages of different Ti species from the Ti 2p scan (Fig. S6†), the value of O/Ti ratio (x) is calculated as 0.66 (Table S2†). Thus, the composition of titanium compounds in this NG–TiOxNy nanocomposite (rGO:TiOxNy = 2:1 (w/w)) can be estimated as TiO0.66N0.61.
 | ||
| Fig. 3 (a) SEM images of NG–TiOxNy; (b) EDX spectrum of the NG–TiOxNy associated with (a); elemental mapping of (c) C; (d) O; (e) N and (f) Ti. | ||
 | ||
| Fig. 4 High resolution XPS spectra of NG–TiOxNy (a) C 1s scan; (b) O 1s scan; (c) N 1s scan; (d) Ti 2p scan. | ||
The CV curves were obtained at a wide range of scan rates (Fig. 5a) and in NaCl electrolyte solution with 3 different concentrations (Fig. 5c). All the CV curves of NG–TiOxNy are symmetric with respect to the x-axis and no significant oxidation/redox peaks were observed, indicating the reversibility of the EDL formation process dominated the electrosorption processes on the surface of the fabricated electrodes.58 As calibrated in Fig. S1,† the symmetric CV curve shape of the CDI electrode is ranged from −0.5 V to 0.5 V in actual, which indicates the electrode has the adsorption capacity for both Na+ and Cl−. To evaluate the electrochemical performance, the specific capacitance was calculated from the I–V plots, as shown in Fig. 5b. The specific capacitance ranges from 84.3–66.5 F g−1 as the scan rate increased from 10 to 50 mV s−1. The decrease in the specific capacitance is possibly due to the charge-resistive behavior when the scan rate increased.59 The mass ratio of N-doped graphene:TiOxNy is approximately 2:1, which results in a significant decrease of specific area and electrochemical active area as opposed to sole N-doped graphene and a further decrease of specific capacitance.46,60,61 Fig. 5c shows the I–V curves for electrolyte solutions with concentrations of 0.1 mol L−1, 0.5 mol L−1, and 1 mol L−1. Higher concentration of the electrolyte solution increased the mobility of the ions, while facilitated the formation of EDL on the electrode surface more easily.62 Thus, the specific capacitance, ranging from 66.5–28.5 F g−1, increased as the electrolyte concentration increased. In Fig. 5d after 5000 CV cycles with at the scan rate of 50 mV s−1, the NG–TiOxNy electrode retained 90% to its initial capacitance, demonstrating its excellent electrochemical stability. Fig. 5e presents the comparison of NG–TiOxNy with commercial AC electrode, pure rGO, and rGO–TiO2 (initial mass ratio 2:1). As can be seen from the figure, NG–TiOxNy showes higher specific capacitance at the scan rate of 50 mV s−1 than the AC, rGO and rGO–TiO2 electrodes. The higher capacitance can be attributed to the nitrogen-doped sites on the graphene layer and the surface vacancies generated during the Ti species nitridation.46,63 Electrochemical Impedance Spectroscopy (EIS) analysis, which is usually used for evaluating the electrical conductivity of electrode materials, was performed to further investigate the electrochemical properties of the synthesized NG–TiOxNy. The Nyquist plots for the electrode material are shown in Fig. 5f, where the typical semicircles can be observed, representing low resistance of the electrode/electrolyte interface and its high capacitance. The slope after the semicircle indicates the formation of the electrical double layer on the surface of the electrode material.49 Compared with AC, rGO, and rGO–TiO2 electrodes, the higher slope and smaller semicircle of NG–TiOxNy indicates its higher diffusion coefficient and smaller diffusion resistance for the adsorbed ions.64 Such phenomena can be explained by the formation of the surface oxygen vacancies and the capacitance from oxygen vacancies ion adsorption during ammonia heat treatment.65
 | ||
| Fig. 5 (a) CV curves of NG–TiOxNy electrode with scan rates of 10, 20, 30, 40, and 50 mV s−1; (b) specific capacitance of NG–TiOxNy vs. scan rate. (c) CV curves of NG–TiOxNy with 0.1 mol L−1, 0.5 mol L−1, and 1 mol L−1 aqueous NaCl solutions; (d) cycling stability test for 5000 cycles; (e) CV curves and specific capacitance of NG–TiOxNy, AC, rGO and rGO–TiO2; (f) EIS Nyquist plots of NG–TiOxNy, AC, rGO and rGO–TiO2. | ||
The whole CDI process can be seen in Fig. S6.† The mass ratio graphite and Ti species were optimized by preliminary desalination experiments using different rGO:TiO2 mass ratios as shown in Fig. 6a. Compared to the pristine rGO, rGO–TiO2, and AC, the NG–TiOxNy material exhibited significantly lower salt retention rate, which is less than 50% at 25 min. The higher performance of NG–TiOxNy can be attributed to the enhanced ion absorbing capacity due to the nitrogen doping process with higher binding capacity between nitrogen doping sites and the ions,33,46 which is also corresponded with the better electrochemical performance. Furthermore, the higher CDI performance of the electrodes after nitrogen doping can be explained enhanced ions storage capacity in the surface vacancies formed during the ammonia heat treatment.54 The optimized initial mass ratio of rGO:TiO2 = 2:1 was chosen for further study. Compared with rGO–TiO2 (initial mass ratio 2:1), the n-doping and nitridation process significantly enhanced the salt adsorption capacity of the NG–TiOxNy CDI electrode material. The dramatically high salt efficiency of NG–TiOxNy can be attributed the multi-layered graphene nanosheets facilitated the rapid liquid diffusion; higher specific capacitance and the surface adsorption and ion intercalation within the Ti–O–N lattice.57,66
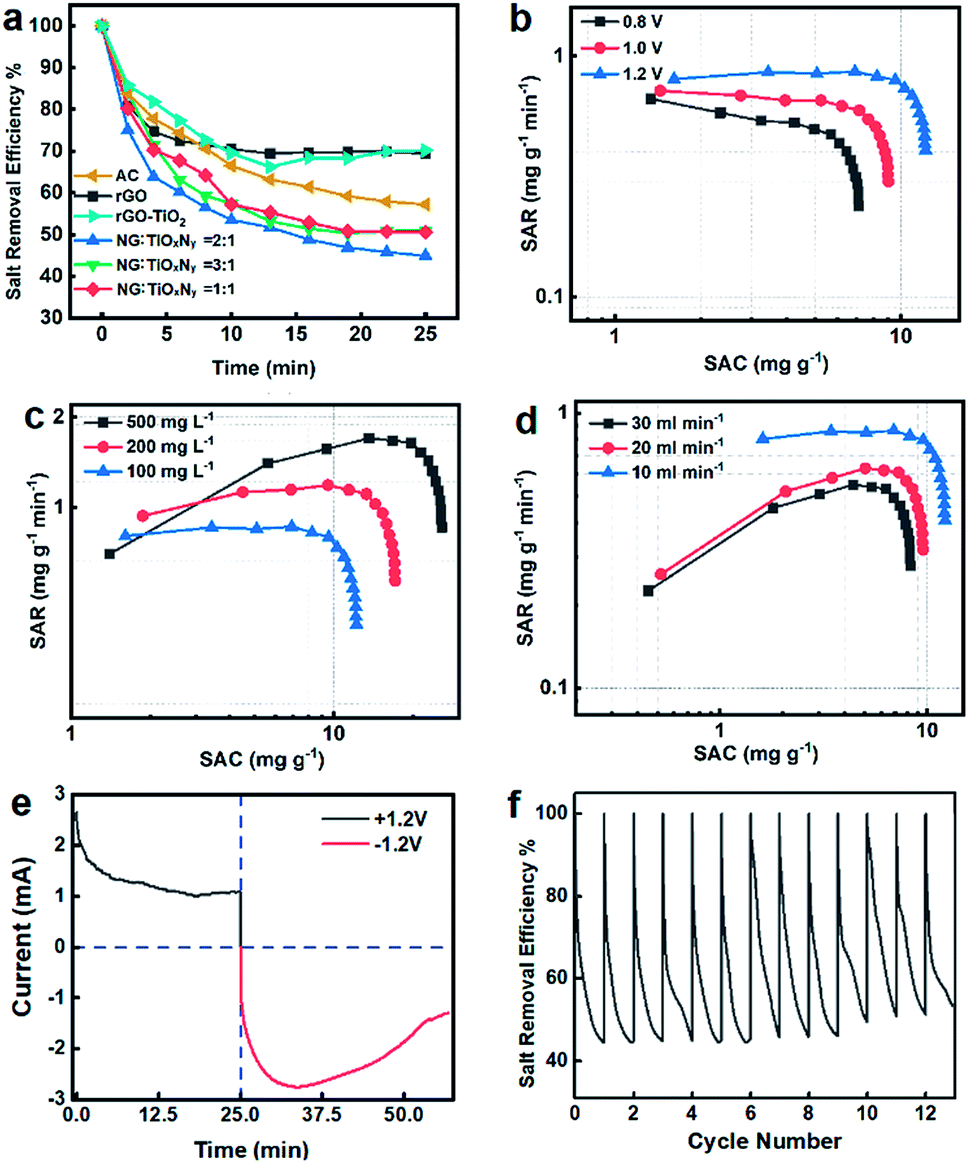 | ||
| Fig. 6 (a) Comparison of salt retention curves of electrodes NG:TiOxNy = 1:1 to 3:1, rGO, and AC; Ragone plots of NG–TiOxNy at: (b) potential of 0.8 V, 1.0 V and 1.2 V; (c) water salinity of 500 mg L−1, 200 mg L−1, and 100 mg L−1; (d) flow rates of 30 mL min−1, 20 mL min−1 and 10 mL min−1; (e) current vs. time plot; (f) cycling stability for over 12 cycles. | ||
Fig. 6b–d shows the Ragone plots of SAC vs. SAR under different working conditions.67 In all the three tests, the SAC increases rapidly during the first 5 min and gradually reached saturated after 25 min, while the SAR follows the same pattern. Fig. 6b shows the batch-mode CDI test was performed at 10 mL min−1 flow rate with 100 mg L−1 NaCl solution. The SACs reaches equilibrium approximately 8.1 mg g−1, 9.6 mg g−1, and 12.8 mg g−1 with cell voltages of 1.2–0.8 V at near saturated adsorption, respectively. Meanwhile, the SAR of each group at initial stage reached 0.7–0.95 mg g−1 min−1. The increase in the SACs and SARs indicated that the salt adsorption capacity was strongly depended on potential applied on the cell, which will result in a more cohesive electrical double layer and enhance the ion absorbing capacity.68 In Fig. 6c, the SACs were obtained with 10 mL min−1 flow rate and 1.2 V while the salinity of the pumped-in water concentrations ranges from 100–500 mg L−1. Despite the increase in saline water concentration, the electrode maintained its salt adsorption capacity, demonstrating the possibility of its application in high salt concentrations treatment. At the salt concentration of 500 mg L−1, the salt adsorption capacity reached a breakthrough SAC of 26.1 mg g−1 at its near saturation adsorption. It is also worth to mention the SAR of 500 mg L−1 saline water group reached as high as 1.8 mg g−1 min−1, which is considerable high SAR compared with previous reports.67 Fig. 6d shows the SACs for different flow rates (10–30 mL min−1) at 1.2 V and salt concentration of 100 mg L−1. The SAC curves have a similar trend as the previous experiments, and the SACs were 12.6–7.0 mg g−1 while the SARs were 0.95–0.45 mg g−1 min−1 at near saturated adsorption, respectively. The relatively smaller SAC declines when performing the CDI test in higher flowrates enabled its potential in fast water desalination. Fig. 6e shows the current-elapsed time curve, which directly corresponded to the conductivity and salt removal efficiency curve by measuring the current at the outflow stream. The potential applied to the CDI cell shifted from 1.0 V to −1.0 V at 25 minutes. The current curve quickly dropped in the first 10 minutes, and then reached the lowest current at 25 minutes. The conductivity of the outflow water recovered slightly and reached the saturation point of ions adsorption. The potential was shifted at 25 minutes, resulting in a rapid increase in reverse current, showing the release of ions from the porous electrode. The negative current then decreased after a few minutes, which indicated that the reverse adsorption process occurred. The rapid charge and discharge performance of the NG–TiOxNy electrode demonstrated its great potential in ad/desorption potential. In Fig. 6f, the regeneration test was performed using NaCl saline water with 10 mL min−1 flow rate and 100 mg L−1 salt concentration and repeated charge–discharge cycles for 30 minutes. The salt removal efficiency remained at 90% of its original capacity for over 12 cycles which is superior than most reported titanium-based CDI electrodes.43,69 The stable cycling stability demonstrated that the synthesized NG–TiOxNy has an excellent potential for longtime-continuous desalination application. Compared with several reported porous carbon and titanium-carbon electrode materials (Table 2), the NG–TiOxNy material in this work exhibites significantly higher salt adsorption capacity.
| Electrode | Cell voltage V | Salinity mg L−1 | SAC mg g−1 | Ref |
|---|---|---|---|---|
| Commercial AC | 1.2 | 292–1170 | 10.9–13.0 | 70 |
| Carbon aerogel | 1.2 | 100–1000 | 5–15 | 71 |
| Graphene and derivatives | 0.8–1.2 | 100–1000 | 5–20 | 72 |
| rGO–TiO2 | 1.2 | 300 | 9.2–13.2 | 73 |
| CNT–TiO2 | 1.2 | 500 | ∼4.3 | 74 |
| TiO2–AC | 1.2 | 500 | ∼2.7 | 75 |
| TiO2 NPs/AC | 1.2 | 100 | ∼8.04 | 69 |
| Ti3C2–mxene | 1.2 | 100 | 13 | 76 |
| Ti3C2Tx–mxene | 1.2 | 500 | 20–24 | 77 |
| N-doped graphene | 1.2 | 500 | >17.8 | 78 |
| N-doped carbon sphere | 1.2 | 1000 | 14.91 | 79 |
| N-doped TiO2/ZrO2 | 1.2 | 50 | 3.98 | 80 |
| 3DOM–TiN | 1.2 | 500 | 25.3 | 34 |
| NG–TiOxNy | 1.2 | 500 | 26.1 | This work |
4 Conclusions
In this work, we successfully synthesized a novel type of nitrogen-doped graphene–titanium oxynitride (NG–TiOxNy) nanocomposite electrode via hydrothermal reaction and high-temperature nitridation process and then tested for CDI. The physical and chemicalcharacterization revealed the nitrogen was successfully doped onto the graphite layer and formed various species of nitrogen-doped carbon. Additionally, the TiOxNy nanoparticles were also formed and embedded on the graphene nanosheets. Further study revealed the ammonia treatment and the loaded TiOxNy nanoparticles gave the electrode better electrochemical performance and extra ion absorbing capacity compared with graphene and TiO2 based CDI electrodes. Consequently, the multi-layered graphene nanosheets allowed the saline water stream to pass through the electrodes and absorbing ions in both the surface of nitrogen-doped graphene and TiOxNy nanoparticles. In the batch mode CDI test, the NG–TiOxNy electrode-based CDI device exhibited a significantly high salt adsorption capacity of 26.1 mg g−1 at its near saturated adsorption using 500 mg L−1 feed-in saline water. In the cycling stability test, the NG–TiOxNy electrode lasted for over 12 cycles while retaining over 90% of its original salt removal capacity, furthers its great potential in long-time continuous CDI application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely acknowledge the Department of Chemical Engineering at University of Waterloo, the Department of Chemical and Biological Engineering at University of Ottawa and the China Scholarship Council.References
- M. Catley-Carlson, Nature, 2011, 473, 27 CrossRef CAS
.
- F. A. AlMarzooqi, A. A. Al Ghaferi, I. Saadat and N. Hilal, Desalination, 2014, 342, 3–15 CrossRef CAS
- Y. H. Teow and A. W. Mohammad, Desalination, 2019, 451, 2–17 CrossRef CAS
- S. Santangelo, Appl. Sci., 2019, 9, 1049 CrossRef
- Y. Oren, Desalination, 2008, 228, 10–29 CrossRef CAS
- J. Oladunni, J. H. Zain, A. Hai, F. Banat, G. Bharath and E. Alhseinat, Sep. Purif. Technol., 2018, 207, 291–320 CrossRef CAS
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS
- K. Shi and I. Zhitomirsky, ChemElectroChem, 2015, 2, 396–403 CrossRef CAS
- K. Tang, J. Chang, H. Cao, C. Su, Y. Li, Z. Zhang and Y. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 11324–11333 CrossRef CAS
- F. Duan, X. Du, Y. Li, H. Cao and Y. Zhang, Desalination, 2015, 376, 17–24 CrossRef CAS
- K. Shi, X. Yang, E. D. Cranston and I. Zhitomirsky, Adv. Funct. Mater., 2016, 26, 6437–6445 CrossRef CAS
- G. Divyapriya, K. K. Vijayakumar and I. Nambi, Desalination, 2019, 451, 102–110 CrossRef CAS
- Z. U. Khan, T. Yan, L. Shi and D. Zhang, Environ. Sci.: Nano, 2018, 5, 980–991 RSC
- J. Han, L. Shi, T. Yan, J. Zhang and D. Zhang, Environ. Sci.: Nano, 2018, 5, 2337–2345 RSC
- H.-J. Choi, S.-M. Jung, J.-M. Seo, D. W. Chang, L. Dai and J.-B. Baek, Nano Energy, 2012, 1, 534–551 CrossRef CAS
- J. H. Chang, A. Huzayyin, K. Lian and F. Dawson, Appl. Phys. Lett., 2015, 107, 193902 CrossRef
- Z. Luan, Y. Tian, L. Gai, H. Jiang, X. Guo and Y. Yang, J. Alloys Compd., 2017, 729, 9–18 CrossRef CAS
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174–182 CrossRef CAS
- A. Abdelkader and D. Fray, Nanoscale, 2017, 9, 14548–14557 RSC
- W. Dianbudiyanto and S.-H. Liu, Desalination, 2019, 468, 114069 CrossRef CAS
- H. Tian, N. Wang, F. Xu, P. Zhang, D. Hou, Y. Mai and X. Feng, J. Mater. Chem. A, 2018, 6, 10354–10360 RSC
- H. Gao, J. Li and K. Lian, RSC Adv., 2014, 4, 21332–21339 RSC
- X. Xu, Z. Sun, D. H. Chua and L. Pan, Sci. Rep., 2015, 5, 11225 CrossRef CAS PubMed
- W. Niu and Y. Yang, ACS Energy Lett., 2018, 3, 2796–2815 CrossRef CAS
- W. Niu and Y. Yang, ACS Appl. Energy Mater., 2018, 1, 2440–2445 CrossRef CAS
- W. Lei, J. Guo, Z. Wu, C. Xuan, W. Xiao and D. Wang, Science Bulletin, 2017, 62, 1011–1017 CrossRef CAS
- W. Lei, L. Han, C. Xuan, R. Lin, H. Liu, H. L. Xin and D. Wang, Electrochim. Acta, 2016, 210, 130–137 CrossRef CAS
- X. Cao, G. Tian, Y. Chen, J. Zhou, W. Zhou, C. Tian and H. Fu, J. Mater. Chem. A, 2014, 2, 4366–4374 RSC
- J. Kim, W.-H. Khoh, B.-H. Wee and J.-D. Hong, RSC Adv., 2015, 5, 9904–9911 RSC
- Y.-J. B. Ting, K. Lian and N. Kherani, ECS Trans., 2011, 35, 133–139 CrossRef CAS
- G. Lui, G. Li, X. Wang, G. Jiang, E. Lin, M. Fowler, A. Yu and Z. Chen, Nano Energy, 2016, 24, 72–77 CrossRef CAS
- L. Chen, H. Dai, Y. Zhou, Y. Hu, T. Yu, J. Liu and Z. Zou, Chem. Commun., 2014, 50, 14321–14324 RSC
- A. Achour, M. Chaker, H. Achour, A. Arman, M. Islam, M. Mardani, M. Boujtita, L. Le Brizoual, M. Djouadi and T. Brousse, J. Power Sources, 2017, 359, 349–354 CrossRef CAS
- Y. Wu, G. Jiang, G. Liu, G. Lui, Z. Cano, Q. Li, Z. Zhang, A. Yu, J. Zhang and Z. Chen, J. Mater. Chem. A, 2019, 7, 15633–15639 RSC
- Z. Liu, Z. Jin, X. Liu, Y. Fu and G. Liu, J. Sol-Gel Sci. Technol., 2006, 38, 73–78 CrossRef CAS
- S. Sung, S. Park, W.-J. Lee, J. Son, C.-H. Kim, Y. Kim, D. Y. Noh and M.-H. Yoon, ACS Appl. Mater. Interfaces, 2015, 7, 7456–7461 CrossRef CAS
- O. Wiranwetchayan, S. Promnopas, T. Thongtem, A. Chaipanich and S. Thongtem, Surf. Coat. Technol., 2017, 326, 310–315 CrossRef CAS
- C. Kim, J. Lee, S. Kim and J. Yoon, Desalination, 2014, 342, 70–74 CrossRef CAS
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS
- I. G. Morozov, O. Belousova, O. Belyakov, I. Parkin, S. Sathasivam and M. Kuznetcov, J. Alloys Compd., 2016, 675, 266–276 CrossRef CAS
- R. A. Sait and R. B. M. Cross, Appl. Surf. Sci., 2017, 424, 290–298 CrossRef CAS
- C.-L. Lee, C. Kim and I.-D. Kim, RSC Adv., 2017, 7, 44804–44808 RSC
- M. Ding, S. Fan, S. Huang, M. E. Pam, L. Guo, Y. Shi and H. Y. Yang, ACS Appl. Energy Mater., 2019, 2, 1812–1822 CrossRef CAS
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed
- G. Liu, J. Li, J. Fu, G. Jiang, G. Lui, D. Luo, Y. P. Deng, J. Zhang, Z. P. Cano and A. Yu, Adv. Mater., 2018, 1806761 CrossRef PubMed
- P. Bharathidasan, S. Sridhar, P. V. Vardhan, S. Sivakkumar, D.-W. Kim and S. Devaraj, J. Mater. Sci.: Mater. Electron., 2018, 29, 7661–7667 CrossRef CAS
- Y. Liu, T. Chen, T. Lu, Z. Sun, D. H. C. Chua and L. Pan, Electrochim. Acta, 2015, 158, 403–409 CrossRef CAS
- M. Hashemi, M. S. Rahmanifar, M. F. El-Kady, A. Noori, M. F. Mousavi and R. B. Kaner, Nano energy, 2018, 44, 489–498 CrossRef CAS
- A. S. Yasin, H. O. Mohamed, I. M. Mohamed, H. M. Mousa and N. A. Barakat, Sep. Purif. Technol., 2016, 171, 34–43 CrossRef CAS
- L.-F. Chen, X.-D. Zhang, H.-W. Liang, M. Kong, Q.-F. Guan, P. Chen, Z.-Y. Wu and S.-H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS
- W. Xiong, M. Liu, L. Gan, Y. Lv, Z. Xu, Z. Hao and L. Chen, Colloids Surf., A, 2012, 411, 34–39 CrossRef CAS
- K. Wang, L. Li, T. Zhang and Z. Liu, Energy, 2014, 70, 612–617 CrossRef CAS
- B. Yu, X. Wang, X. Qian, W. Xing, H. Yang, L. Ma, Y. Lin, S. Jiang, L. Song and Y. Hu, RSC Adv., 2014, 4, 31782–31794 RSC
- W. Lei, W. Xiao, J. Li, G. Li, Z. Wu, C. Xuan, D. Luo, Y.-P. Deng, D. Wang and Z. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 28604–28611 CrossRef CAS PubMed
- Z. Lei, N. Christov and X. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 RSC
- M. S. Akple, J. Low, Z. Qin, S. Wageh, A. A. Al-Ghamdi, J. Yu and S. Liu, Chin. J. Catal., 2015, 36, 2127–2134 CrossRef CAS
- G. Hasegawa, A. Kitada, S. Kawasaki, K. Kanamori, K. Nakanishi, Y. Kobayashi, H. Kageyama and T. Abe, J. Electrochem. Soc., 2015, 162, A77–A85 CrossRef CAS
- G. Trefalt, S. H. Behrens and M. Borkovec, Langmuir, 2015, 32, 380–400 CrossRef PubMed
- S. Karade, D. Dubal and B. Sankapal, MoS2 ultrathin nanoflakes for high performance supercapacitors: Room temperature chemical bath deposition (CBD), 2016 Search PubMed
- C. Xiang, M. Li, M. Zhi, A. Manivannan and N. Wu, J. Mater. Chem., 2012, 22, 19161–19167 RSC
- J. Lazarte, R. Dipasupil, G. Pasco, R. Eusebio, A. Orbecido, R.-a. Doong and L. Bautista-Patacsil, Nanomaterials, 2018, 8, 934 CrossRef PubMed
- K.-C. Tsay, L. Zhang and J. Zhang, Electrochim. Acta, 2012, 60, 428–436 CrossRef CAS
- A. Achour, J. Ducros, R. Porto, M. Boujtita, E. Gautron, L. Le Brizoual, M. Djouadi and T. Brousse, Nano Energy, 2014, 7, 104–113 CrossRef CAS
- Z. Xing, G. Li, S. Sy and Z. Chen, Nano Energy, 2018, 54, 1–9 CrossRef CAS
- M. Inagaki, M. Toyoda, Y. Soneda and T. Morishita, Carbon, 2018, 132, 104–140 CrossRef CAS
- C. Zhu, P. Yang, D. Chao, X. Wang, X. Zhang, S. Chen, B. K. Tay, H. Huang, H. Zhang and W. Mai, Adv. Mater., 2015, 27, 4566–4571 CrossRef CAS PubMed
- T. Kim and J. Yoon, RSC Adv., 2015, 5, 1456–1461 RSC
- J. Zhang, J. Fang, J. Han, T. Yan, L. Shi and D. Zhang, J. Mater. Chem. A, 2018, 6, 15245–15252 RSC
- P.-I. Liu, L.-C. Chung, H. Shao, T.-M. Liang, R.-Y. Horng, C.-C. M. Ma and M.-C. Chang, Electrochim. Acta, 2013, 96, 173–179 CrossRef CAS
- R. Zhao, P. M. Biesheuvel, H. Miedema, H. Bruning and A. van der Wal, J. Phys. Chem. Lett., 2010, 1, 205–210 CrossRef CAS
- A. Thamilselvan, A. Nesaraj and M. Noel, Int. J. Environ. Sci. Technol., 2016, 13, 2961–2976 CrossRef CAS
- H. Yin, S. Zhao, J. Wan, H. Tang, L. Chang, L. He, H. Zhao, Y. Gao and Z. Tang, Adv. Mat., 2013, 25, 6270–6276 CrossRef CAS PubMed
- A. G. El-Deen, J.-H. Choi, C. S. Kim, K. A. Khalil, A. A. Almajid and N. A. Barakat, Desalination, 2015, 361, 53–64 CrossRef CAS
- H. Li, Y. Ma and R. Niu, Separation and Purification Technology, 2016 Search PubMed
- P. Srimuk, M. Zeiger, N. Jäckel, A. Tolosa, B. Krüner, S. Fleischmann, I. Grobelsek, M. Aslan, B. Shvartsev and M. E. Suss, Electrochim. Acta, 2017, 224, 314–328 CrossRef CAS
- P. Srimuk, F. Kaasik, B. Krüner, A. Tolosa, S. Fleischmann, N. Jäckel, M. C. Tekeli, M. Aslan, M. E. Suss and V. Presser, J. Mater. Chem. A, 2016, 4, 18265–18271 RSC
- W. Bao, X. Tang, X. Guo, S. Choi, C. Wang, Y. Gogotsi and G. Wang, Joule, 2018, 2, 778–787 CrossRef CAS
- M. Mi, X. Liu, W. Kong, Y. Ge, W. Dang and J. Hu, Desalination, 2019, 464, 18–24 CrossRef CAS
- Y. Liu, T. Chen, T. Lu, Z. Sun, D. H. Chua and L. Pan, Electrochim. Acta, 2015, 158, 403–409 CrossRef CAS
- A. S. Yasin, I. M. Mohamed, H. M. Mousa, C. H. Park and C. S. Kim, Sci. Rep., 2018, 8, 541 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05380h |
| This journal is © The Royal Society of Chemistry 2019 |