
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptidoglycan pathways: there are still more!
Ahmed M. Helal
a,
Ahmed M. Sayed
a,
Mariam Omara
a,
Mohamed M. Elsebaei
 a and
Abdelrahman S. Mayhoub
*ab
a and
Abdelrahman S. Mayhoub
*ab
aDepartment of Pharmaceutical Organic Chemistry, College of Pharmacy, Al-Azhar University, Cairo 11884, Egypt. E-mail: amayhoub@azhar.edu.eg
bUniversity of Science and Technology, Zewail City of Science and Technology, Giza, Egypt
First published on 9th September 2019
Abstract
The discovery of 3rd and 4th generations of currently existing classes of antibiotics has not hindered bacterial resistance, which is escalating at an alarming global level. This review follows WHO recommendations through implementing new criteria for newly discovered antibiotics. These recommendations focus on abandoning old scaffolds and hitting new targets. In light of these recommendations, this review discusses seven bacterial proteins that no commercial antibiotics have targeted yet, alongside their reported chemical scaffolds.
 Ahmed M. Helal | Ahmed Helal (MSc) is a PhD candidate at Dr Mayhoub's laboratory. His thesis focuses on the development of new therapeutics for the treatment of MDR-pathogens. Before joining Dr Mayhoub's lab, he obtained his MSc in Organic Chemistry from Al-Azhar University in 2016. His research interests focus on heterocyclic synthesis. |
 Ahmed M. Sayed | Ahmed M. Sayed is currently working as an R&D specialist in an Egyptian local pharmaceutical company, where he is following his passion for learning every day. He graduated from Al-Azhar University with a BSc in pharmaceutical sciences in 2017. He joined Dr Mayhoub's lab as a researcher in 2016 while he was an undergraduate. He feels like he is still not content with his level of education and has to go further in pursuit of knowledge. |
 Mariam Omara | Mariam Omara is a new MSc student in Dr Mayhoub's lab; however, she joined the lab early while she was a sophomore undergraduate. She obtained her BSc in Pharmaceutical Sciences from the College of Pharmacy, Al-Azhar University, obtaining the highest Honors degree, in June 2019. |
 Mohamed M. Elsebaei | Mohamed M. Elsebaei (PhD) is a lecturer of Organic Chemistry at Al-Azhar University, College of Pharmacy. He obtained his MSc and PhD in Organic Chemistry, under the supervision of Dr Mayhoub, from the same University in 2015 and 2018, respectively. His research interests focus on cross-coupling reactions, metal-catalyzed reactions, and heterocyclic scaffold design. |
 Abdelrahman S. Mayhoub | Dr Abdelrahman S. Mayhoub holds a PhD degree (2012) in Medicinal Chemistry from Purdue University, USA. He was a postdoctoral fellow at the University of Michigan (2013) and currently he is an associate professor at Al-Azhar University and Zewail City for Science and Technology (Egypt). His main contributions in the field of Medicinal Chemistry include the development of several cancer chemopreventatives, aromatase inhibitors, and antiflaviviral agents (Dengue and West-Nile viruses). He discovered phenylthiazoles as a new antibacterial class of compounds for the treatment of notorious bacterial strains such as MRSA, VRSA, and VRE. Dr Mayhoub places a special focus on drug-likeness and controlling the pharmacokinetic profiles of bioactive molecules. |
1. Introduction
It is apparent that currently approved antimicrobials are losing the anti-resistance battle against multidrug-resistant pathogens (MDRP). Microbial resistance has extended to include reagents once deemed to be last resorts, such as vancomycin,1 linezolid,2 and even colistin.3To avoid the rapid development of microbial resistance against upcoming antibiotics, the World Health Organization (WHO)4 has set innovation criteria to maintain antibiotic clinical efficacy over an extended period.4 These criteria mainly include hitting new microbial pathways through novel chemical scaffolds.
This review sheds light on genuine bacterial pathways that have not been targeted with any commercial antibiotics, and provides examples of those potential antibacterial leads through a structure-based approach.
2. Targeting peptidoglycan
2.1. Biological importance
Peptidoglycan (PG) is a single macromolecule; however, it surrounds bacterial bodies to grant them integrity along with protection, resulting in it being a key player in the survival of all prokaryotic pathogens.5 Consequently, focusing on the PG biosynthetic pathway is favorable for medicinal chemists, since the peptidoglycan bacterial cell wall does not exist in mammalian cells. Moreover, it is multilayered and thicker in Gram-positive bacteria, weighing about half of the dry weight of some types of Gram-positive bacteria.5 On the other hand, PG is thinner in Gram-negative bacteria, which are protected by an outer cell wall.6 The differences between Gram-positive and Gram-negative bacteria in addition to their results in relation to inhibition and resistance will be discussed below, respectively.2.2. Structure
Chemically, PG includes short chains of glycans and alternative N-acetylmuramic acid (MurNAc) and β-1,4-N-acetylglucosamine (GlcNAc) components, connected via pentapeptides (MurNAc-GlcNAc-pentapeptide).7 The detailed biosynthetic pathway has been covered in many reviews and articles previously.8–12 In Fig. 1, we simplified the PG biosynthetic pathway with a focus on the lipid carrier as a promising target for future antibiotic development.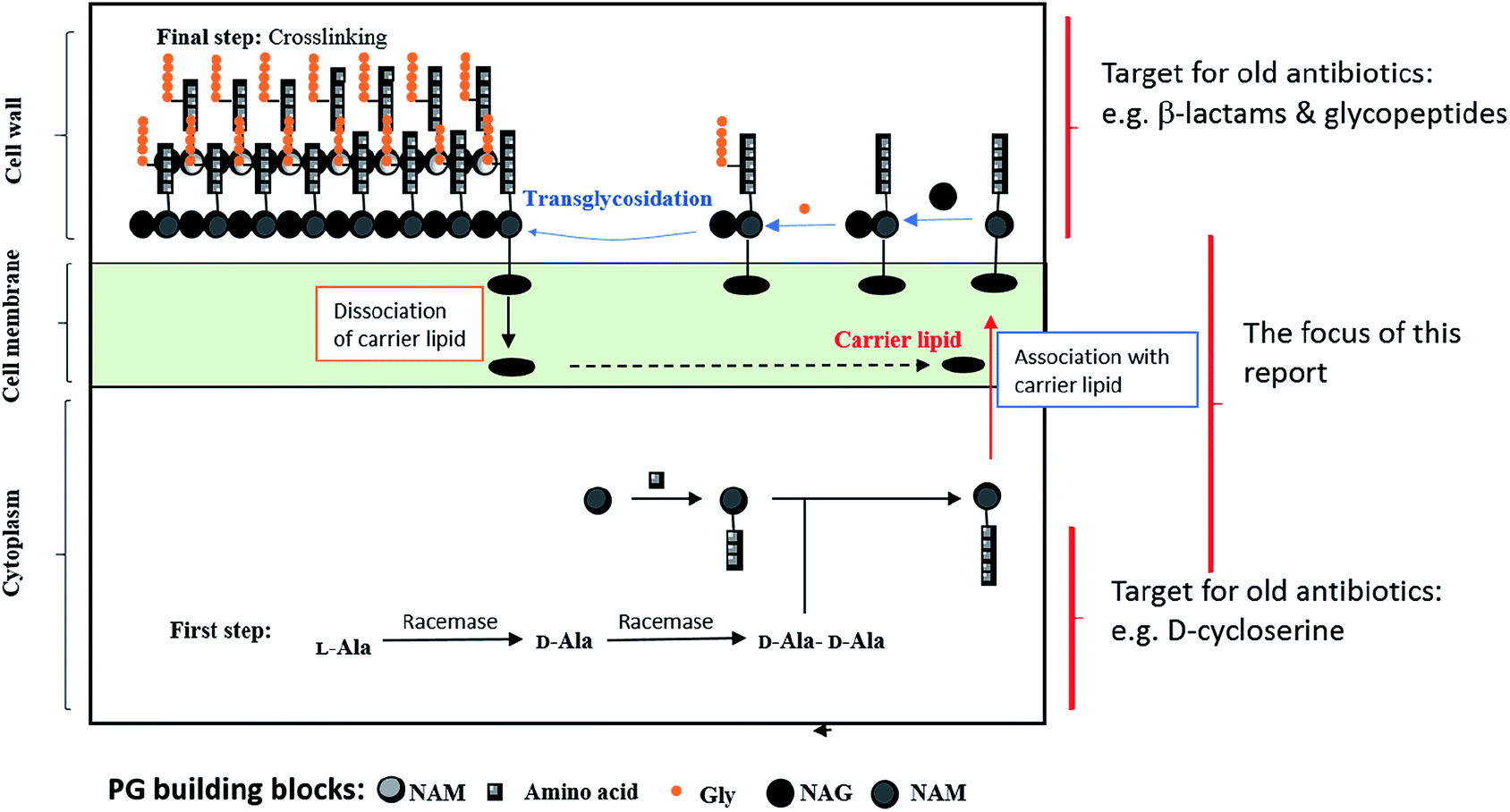 | ||
| Fig. 1 A diagram showing PG biosynthesis. | ||
Remarkably, there are different steps in the PG biosynthetic pathway take place at different cellular levels. The process starts in the cytosolic region via the synthesis of sugar-pentapeptide units, which then relocate through the cytoplasmic membrane to polymerize with other sugars and finally cross-link together to generate the PG molecule (Fig. 1). Nevertheless, all existing antibiotics, perturbing the cell wall synthesis, target either a cytosolic step13 or the growing pro-PG.14 Except for the locally acting antibiotic bacitracin,15 which interferes with the carrier lipid, this step of PG biosynthesis has somehow been overlooked by medicinal chemists for decades. In the following section, we will discuss targets within the carrier lipid that bode well for developing novel antibiotics.
2.3. Extracytoplasmic steps

 | ||
| Fig. 2 A schematic illustration of some key steps of PG biosynthesis. Reprinted with permission from Mohammad H., et al., J. Med. Chem., 2017, 2425; copyright:121 American Chemical Society. | ||
Moenomycin 1 is a natural product that inhibits the GT;22 therefore, it is considered one of the most active antibiotics (MIC = 0.05 μg ml−1 for S. aureus). Bacteria have shown no resistance against moenomycin,23,24 despite its use as a growth promoter in animal feed, which was supposed to provoke resistance.25,26 Moreover, resistance induction in vitro showed slow rates of resistance development,27 the absence of cross resistance28 and a lack of transferable resistance.29 Nevertheless, it is not used as an antibiotic as a result of its poor pharmacokinetics profile by virtue of its lipophilicity leading to adverse effects.10,30 Recent approaches for targeting the GT include:
• Moenomycin analogues that can bind to the donor part of the receptor, but with better pharmacokinetic profiles. As moenomycin is a pentasaccharide compound linked to a polypernyl chain through a phosphoglycerate linkage, it mimics lipid II through binding competitively to the GT. Hence, there have been several attempts to build moenomycin-like structures.
The first try included simplifying the polysaccharide part of moenomycin via building a disaccharide derivative. This approach resulted in some compounds with good IC50 values (the concentration of an inhibitor where the response (or binding) is reduced by half) on an in vitro scale; however, they did not show in vivo activity, such as in the case of compound 2.21,31–34
The second try was to directly mimic lipid II35 or ring F of the moenomycin,36 which resulted in low to moderate activity compounds like compound 3 with an MIC (minimum inhibitory concentration) of 60 μM against Bacillus cereus.35

Additionally, several articles relied on HTS (high throughput screening) or in silico protocols to generate new leads. Despite advances in obtaining novel leads with apparently potent in vitro activities, none of the tested new structures were efficient in in vivo models.37–40
Furthermore, monosaccharide-based scaffolds (e.g. compounds 4 and 5) maintain the essential features of moenomycin with better physicochemical properties and antimicrobial efficacies (MIC values of around 2 μg ml−1 against Gram-positive pathogens).21
• A recent approach was to create a lipid II analogue in an attempt to imitate the transition state of the GT catalyzed reaction; hence, the compound could bind to the acceptor site of the receptor.34
• Researchers managed to synthesize a pseudo-disaccharide lipid II analogue, 6, to inhibit 70% of the GT enzyme activity, advancing bacterial cell lysis.30
2.3.2.1. The biological importance of MraY. Bristol–Myers Squibb has shown that mraY (mra: murein synthesis gene cluster-A) is an essential gene in Streptococcus pneumoniae via a gene knockout approach.41 Moreover, investigations through genetic and biochemical methods have substantiated that mraY and murG gene products in E. coli are essential for cell growth and survival.42,43 This discovery originated from an influx of data about translocase I, a lipid I precursor, dating from the year 1965. However, a leap in knowledge occurred in 1991 when Ikeda et al.44 were able to solve the mystery of the MraY gene sequence, which was officially attached to lipid I production 20 years later.20,45
Ultimately, MraY has gained its importance as a result of it not only being a prime molecular enzyme intervening in the prokaryotic cellular envelope, but also because it has no twin in mammalian cells.46 Therefore, it is deemed to be an attractive target for antibiotic development. Here, we focus on related discoveries, as well as the implications and challenges related to developing such inhibitors.
2.3.2.2. MraY inhibitors. So far, only a few classes of MraY inhibitors have been studied, due to difficulties in administering antibacterial compounds through membranes.45 Interestingly, these inhibitors can be classified, according to their chemical nature, into nucleosides, peptides and non-nucleoside-non-peptides.
Nucleoside MraY inhibitors. These arrays of small molecules have effectively inhibited MraY, for example mureidomycin A,47 the first-in-class, muraymycin, tunicamycin and mureidomycin.48–53 Chemically, these peptidyl-nucleoside compounds share a common structural feature: a uridine or dihydrouridine skeleton. This particular sub-structure is a key element in their MraY inhibitory properties, which are backed by their ability to recognize and competitively bind to UDP-Mpp binding sites on MraY.48,52,54,55 For further information, the SAR details of this group have been reviewed by T. D. Bugg et al.56 Unfortunately, the poor physicochemical properties are a perennial limitation that has prevented any further clinical development.
Peptide MraY inhibitors. Amphomycin is a lipopeptide antibiotic that was isolated following the fermentation of Streptomyces canus in 1953. It is a non-competitive inhibitor of MraY, forming a complex with the undecaprenyl phosphate in the presence of calcium ions; hence it prevents the enzymatic reaction.57 Notably, amphomycin is active against Gram-positive cocci with MIC50 values ranging between 2 and 4 μg ml−1 against MSSA and MRSA.
The second member of this family is protein E, which is produced by the bacteriophage φX-174, and amassed mainly from lipophilic amino acids.58,59 Both peptide inhibitors have no clinical value as a result of their unsuitable physicochemical and pharmacokinetic behaviors.
Others (non-nucleoside-non-peptide MraY inhibitors). Unlike nucleoside and peptide inhibitors, articles reporting small molecule MraY inhibitors are very scarce. Bristol–Myers Squibb published the results of a program it launched, intending to screen a large internal library against purified MraY and MruG.60 The results, in general, were not encouraging enough to escalate this project to the pre-clinical phase. Briefly, two compounds, BMS-185937 and BMS-187979, displayed antimicrobial potency but with considerable cytotoxicity, which disqualified them from further consideration. Additionally, a third lead, BMS-190134, was an analogue to the undecaprenyl phosphate substrate of MraY; however, its lack of antibacterial activity in vitro led to the termination of this lead as well. Furthermore, the natural product isolate BMS-304245 also suffered from the same drawback; i.e. a lack of sufficient antimicrobial activity.
2.3.2.3. Remarks. Notably, the MraY enzyme belongs to a superfamily of proteins called PNPTs (polyribonucleotide nucleotidyltransferases), which also includes a mammalian protein (called GPT), which is used as a post-translational glycosylation catalyst and shares some structural similarities with MraY. Therefore, cross-toxicity against both bacterial and host cells is highly expected with all MraY inhibitors. Consequently, tunicamycin is a useless antibiotic because of its powerful inhibitory activity against human GPT.47,61 Likewise, amphomycin can complex with eukaryotic lipid carriers.62,63 Thus, differences in binding features, as established by G. Brändén's group, have to be taken into consideration.64
2.4. Targeting cytoplasmic steps
2.4.2.1. Natural product leads. • Cnicin 7 is isolated from artichoke and blessed thistle.66 While it exerts its activity through covalently binding to the thiol group of Cys115, it is thought that the unsaturated ester side chain is essential to its activity from a structure–activity relationship (SAR) study.67

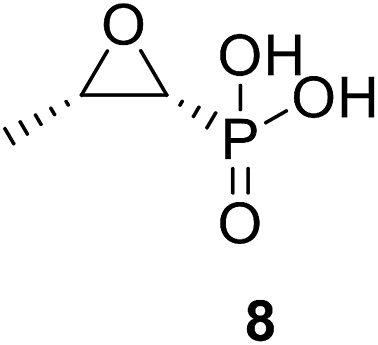
• Fosfomycin 8 is a phosphoenolpyruvate analogue produced by Streptomyces fradiae; moreover, it alkylates the active site Cys115 of MurA, causing bactericidal activity.68
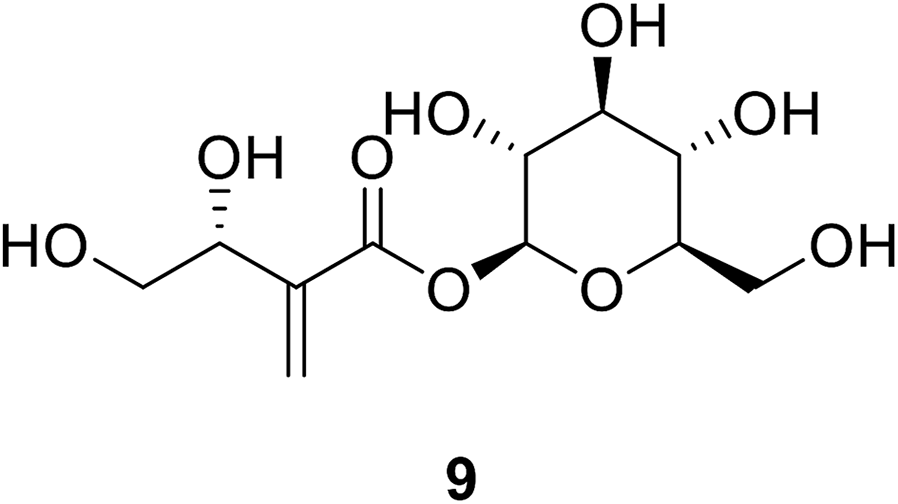
Tuliposide B 9 is a secondary metabolite occurring in tulip anthers. The hydroxyl group of these compounds was found to be crucial for the anti-bacterial activities of tulipaline B and its analogues.69
2.4.2.2. Structure-based design. • By screening a chemical library from the Johnson Pharmaceutical Research Institute, a pyrazolopyrimidine, (RWJ-110192) 10, and a purine analogue, (RWJ-140998) 11, were found to be active against Escherichia coli, with IC50 values ranging from 0.2 μM to 0.9 μM, and showed lower inhibitory activity against Staphylococcus aureus.70

• 5-Sulfonoxy-anthranilic acid derivatives were obtained using HTS. T6361 12 and T6362 13 are known as competitive inhibitors of MurA, with a Ki value of 16 μM.71

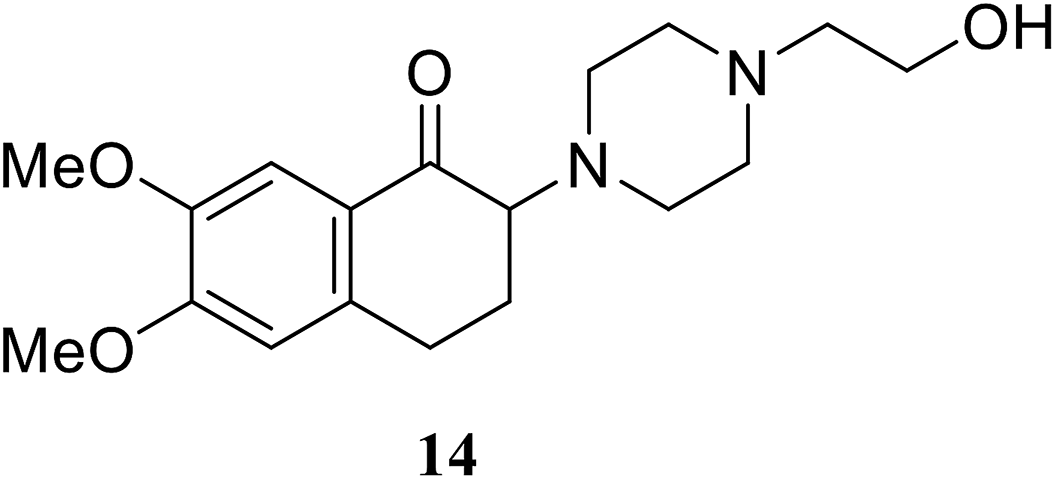
• The 2-aminotetralone 14 demonstrated antibacterial activity against Staphylococcus aureus and Escherichia coli, with MICs of 17 and 3.13 μg ml−1, respectively. α-Aminoketone is responsible for the inhibitory activity and evidence has been provided to support its covalent mode of action, involving the C115 thiol group of MurA/MurZ.72
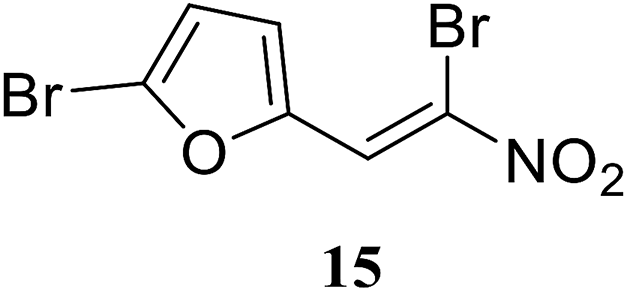
• The di-bromo substituted nitrovinylfuran 15 has broad-spectrum antibacterial activity, via the inhibition of MurA in E. coli, P. aeruginosa and S. aureus, in the low micromolar concentration range.73
• The benzothioxalone series was found to have potent MurA inhibitory activity, with IC50 values between 0.25 μM and 18.54 μM. Compound 16 has an IC50 value towards MurA of E. coli of 0.28 μM. This inhibition of MurA was irreversible.74

2.4.2.3. Remarks. It should be noted that, because they have one copy of the MurA gene, Gram-negative bacteria are more susceptible to MurA inhibitors, unlike Gram-positive bacteria, which may show resistance as a result of gene duplication.
2.4.3.1. Structure-based design. • The imidazolinone 17 showed antibacterial activity against S. aureus.75

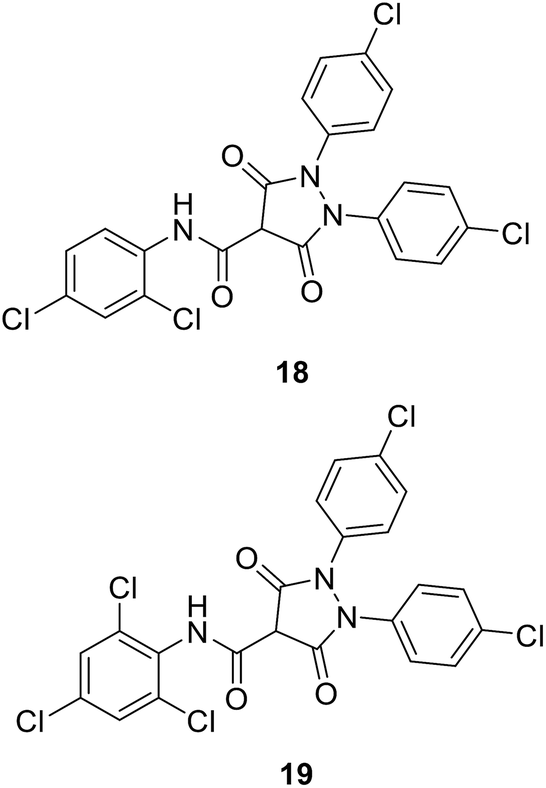
• 3,5-Dioxopyrazolidine inhibitors of MurB were obtained (18 and 19) with IC50 values of 4–10 μM. Antibacterial activities were shown by these compounds against Gram-positive bacteria, including methicillin-resistant S. aureus, vancomycin-resistant Enterococcus faecalis, and penicillin-resistant Streptococcus pneumonia.76
• A 4-thiazolidinone derivative, 20, was synthesized and evaluated for its ability to inhibit the bacterial enzyme MurB. Compound 20 has an IC50 value of 7.7 μM.77

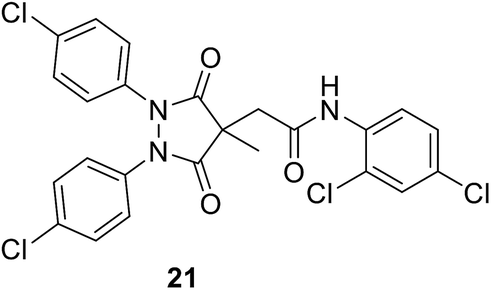
• A series of pyrazolidine-3,5-dione (21) and 5-hydroxy-1H-pyrazol-3(2H)-one inhibitors of E. coli MurB have been synthesized. The 5-hydroxy-1H-pyrazol-3(2H)-ones showed low micromolar IC50 values versus E. coli MurB and sub-micromolar minimum inhibitory concentrations (MICs) against Staphylococcus aureus, Enterococcus faecalis, Streptococcus pneumoniae, and E. coli. Docking studies produced several binding orientations for these molecules at the MurB active site. Improvements in MIC values towards MurB are correlated with the increasing lipophilicity of the C-4 substituent of the 5-hydroxy-1H-pyrazol-3(2H)-one core.78
• Phenylthiazolyl urea and carbamate derivatives were synthesized for evaluation as new inhibitors of bacterial cell-wall biosynthesis. Many of them demonstrated good activity against MurA and MurB in Gram-positive bacteria such as MRSA, VRE and PRSP. 3,4-Difluorophenyl 5-cyanothiazolylurea 22 with a clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value of 2.64 demonstrated antibacterial activity against both Gram-positive and Gram-negative bacteria.77
P value of 2.64 demonstrated antibacterial activity against both Gram-positive and Gram-negative bacteria.77

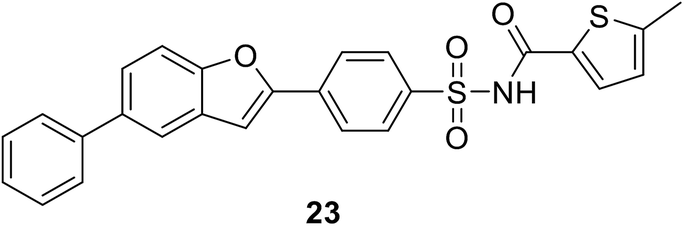
2.4.4.1. Results of high-throughput screening (HTS). • A series of benzofuran acyl-sulfonamides was identified as a potential lead. Compound 23 inhibited Escherichia coli MurC with an IC50 value of 2.3 μM, exhibiting time-dependent partially reversible inhibition.79
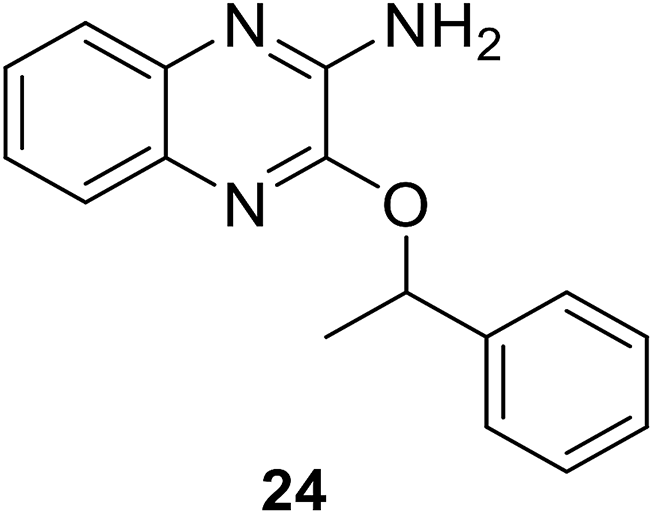
• The quinoxaline inhibitor 24 competes with ATP; experiments indicated that it was a competitive inhibitor of ATP, binding to the MurC enzyme.80
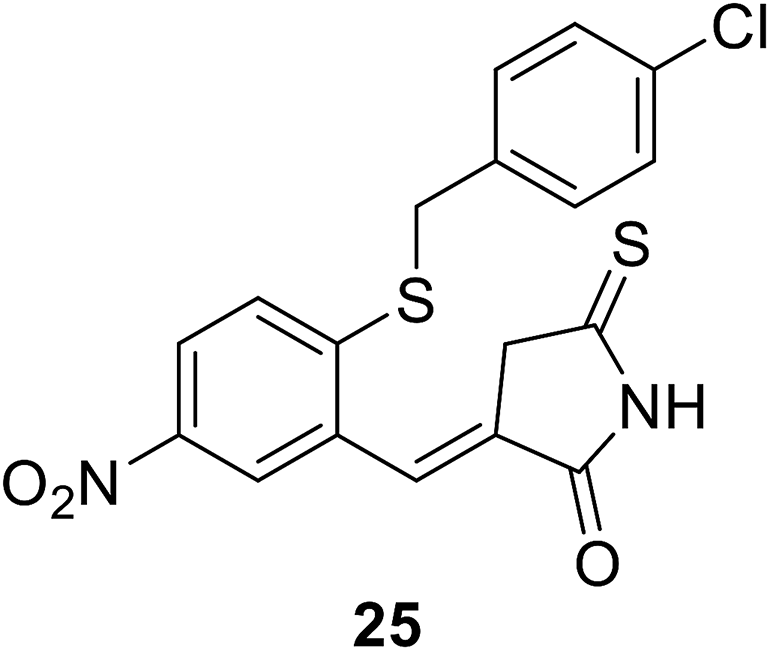
• Benzylidene rhodanines inhibit MurC with IC50 values ranging from 12 μM to 27 μM, with whole-cell activity against Gram-positive MRSA but not against Gram-negative E. coli. Compound 25 showed the best MIC value against MRSA of 31 μM.81
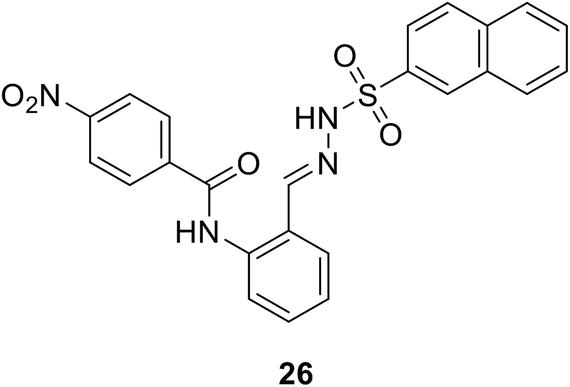
• N-Benzylidenesulfonohydrazide-based compounds, e.g. compound 26, possess inhibitory activity against both MurC and MurD enzymes, with IC50 values of 27 μM and 43 μM.82
• Compounds with an N-acylhydrazone scaffold were characterized as a new class of inhibitors of MurC and MurD from E. coli. Compound 27 has IC50 values of 123 μM and 230 μM against MurC and MurD, respectively, and 28 is selective against MurC with an IC50 value of 32 μM.83


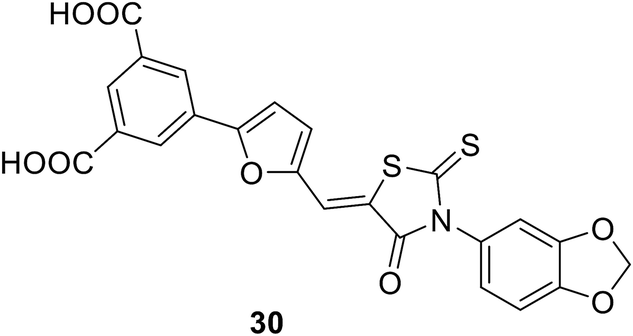
• Glutamic acid surrogate-benzene-1,3-dicarboxylic acid derivatives were discovered through virtual screening. Compound 30 is the most potent, with an IC50 value of 270 μM.85
• Naphthalene-N-sulfonyl-D-glutamic acid derivatives were synthesized and screened against MurD from E. coli because of the high binding affinity of D-Glu toward MurD. They displayed IC50 values ranging from 80 to 600 mM.86
• 5-Benzylidenethiazolidin-4-ones (31 and 32) with low-micromolar affinity against MurD have been discovered. Remarkably, the thiazolidine-2,4-dione heterocyclic ring binds at the site where the uracil ring of the natural substrate UDP-MurNAc-Ala binds.87

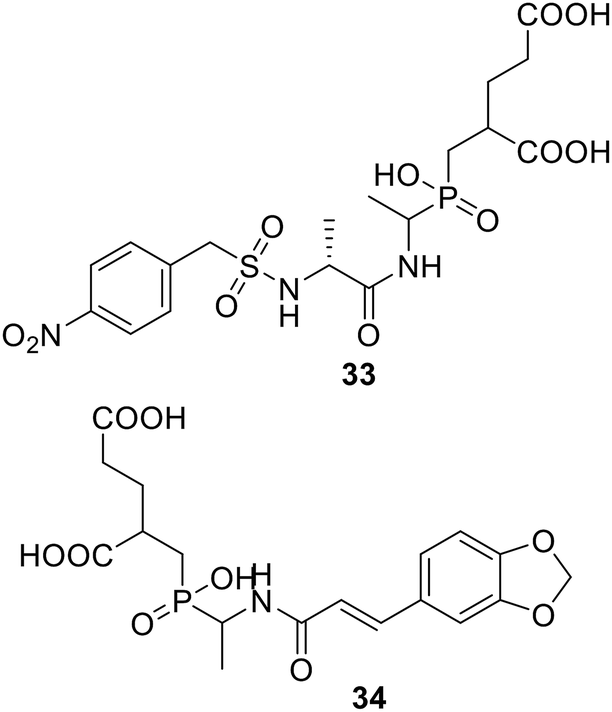
• Phosphinate compounds were designed and synthesized as inhibitors of the D-glutamic acid-adding enzyme MurD from Escherichia coli. Compounds 33 and 34 both had IC50 values near 100 μM.88

• Phosphinates also could inhibit MurE via MurD, with an IC50 value of 1.1 μM.90
• Peptide inhibitors were discovered; the protein MurEp1 could inhibit MurE with an IC50 value of 500 μM. This inhibition proved to be time-dependent and was reversed via the addition of UDP-MurNAc-Ala-Glu during the pre-incubation step.91
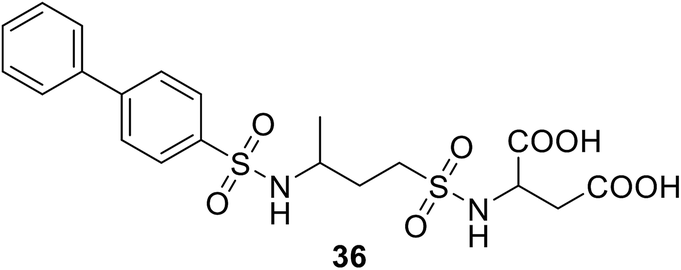
• Sulfonamides that are similar to inhibitors of MurD can inhibit MurE; e.g., compound 36 has an IC50 value of 181 μM.
2.4.6.1. Remarks. There is a variation between Gram-positive and Gram-negative bacteria regarding the MurE substrate. In most Gram-positive bacteria, the MurE substrate should contain L-lysine as the third amino acid in the peptide, while it could be meso-A2pm in Gram-negative bacteria.92,93 As MurE is highly sensitive to the amino acid, this difference between the two types of bacteria will change the nature of MurE inhibitors regarding how each bacteria is targeted.
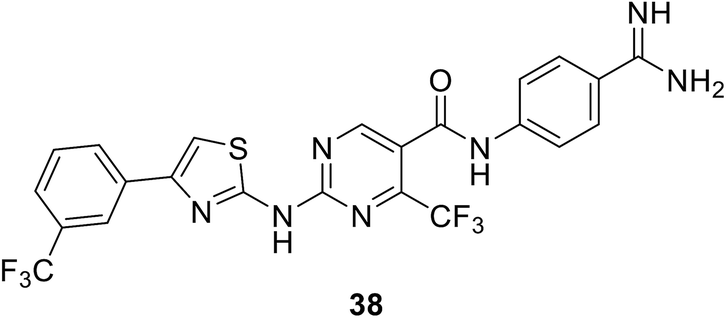
• A thiazolylaminopyrimidine series of MurF inhibitors from E. coli was identified in 2006 by Johnson & Johnson, with the use of an Mpl-based in vitro assay. The most potent compound from this series was compound 38, with an IC50 value of 2.5 μM.95
2.4.8.1. The biological importance of UppP. Unlike Gram-negative bacteria, undecaprenyl pyrophosphatase phosphate (UppP) is the sole enzyme attributed to the biosynthesis of undecaprenyl phosphate (Up OR C55-P) in Gram-positive bacteria, except in Bacillus subtills where the protein BcrC shares this function.96 Unfortunately, in Gram-negative bacteria, it is not just UppP but 3 other genes that are the chief architects of Up biosynthesis: pgpB, ybgG and lpxT. This makes UppP a less potent target for Gram-negative bacteria.97 Up is deemed a vital lipid carrier, as with MraY and MurG, which produce lipid I and lipid II, respectively.
Interestingly, the phosphatase enzyme is essential for the two paths of Up production, synthesis and recycling, which suggests that phosphatase is a necessity for cell viability.98 To generate the monophosphate form of Und-P, the cell implements the following system: de novo synthesis through the condensation of 8 molecules of isopentenyl pyrophosphate and farnesyl pyrophosphate using the undecaprenyl pyrophosphate synthetase to form undecaprenyl pyrophosphate. This is followed by the cell using the phosphatase enzyme to generate the monophosphate Up.99 Moreover, Upp released during vital processes in the cell is recycled to Up using the phosphatase enzyme. Hence, UppP is one of the key enzymes in peptidoglycan biosynthesis, as shown in Fig. 3.
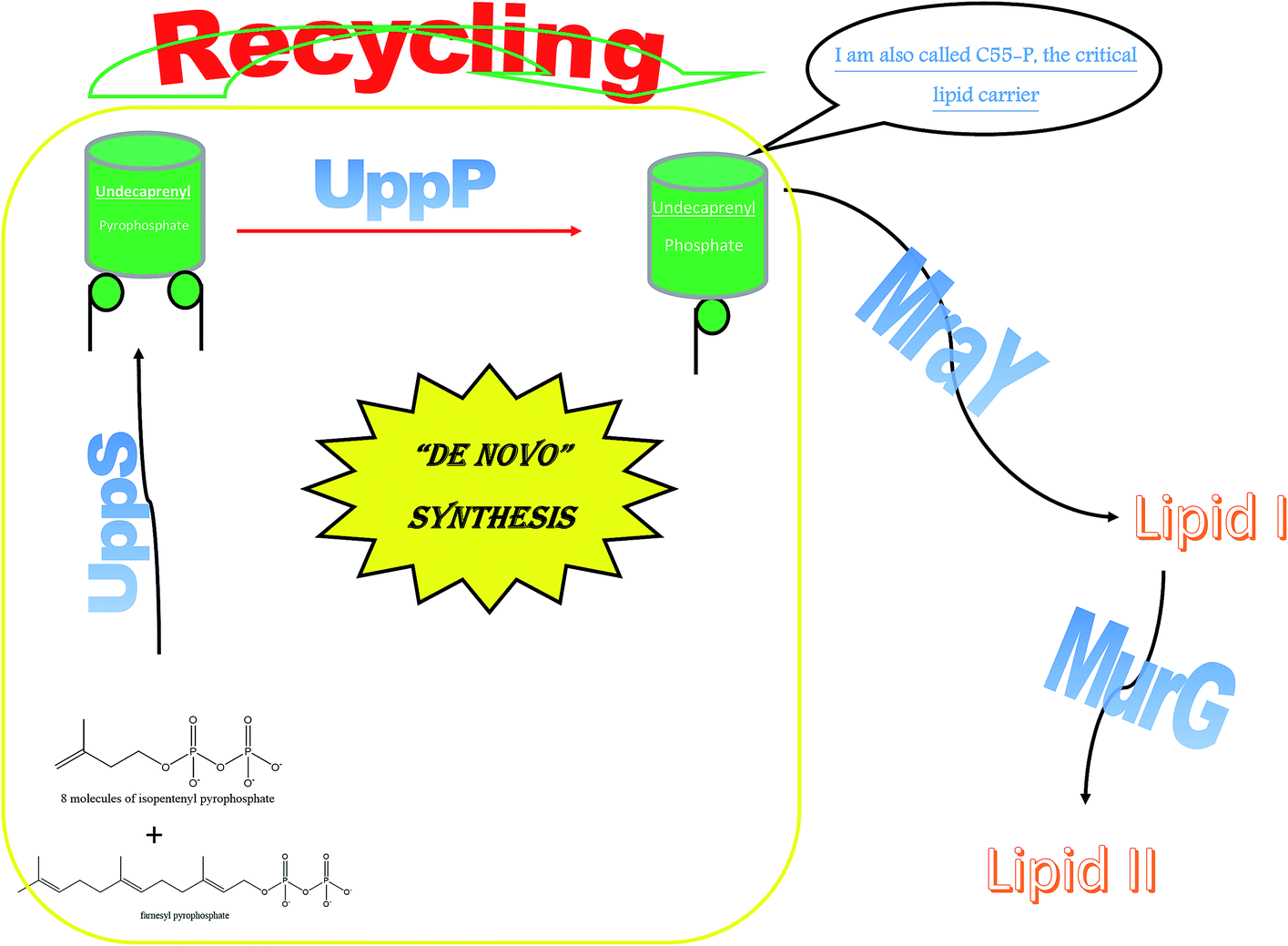 | ||
| Fig. 3 A diagram emphasizing the essential role of UppP. | ||
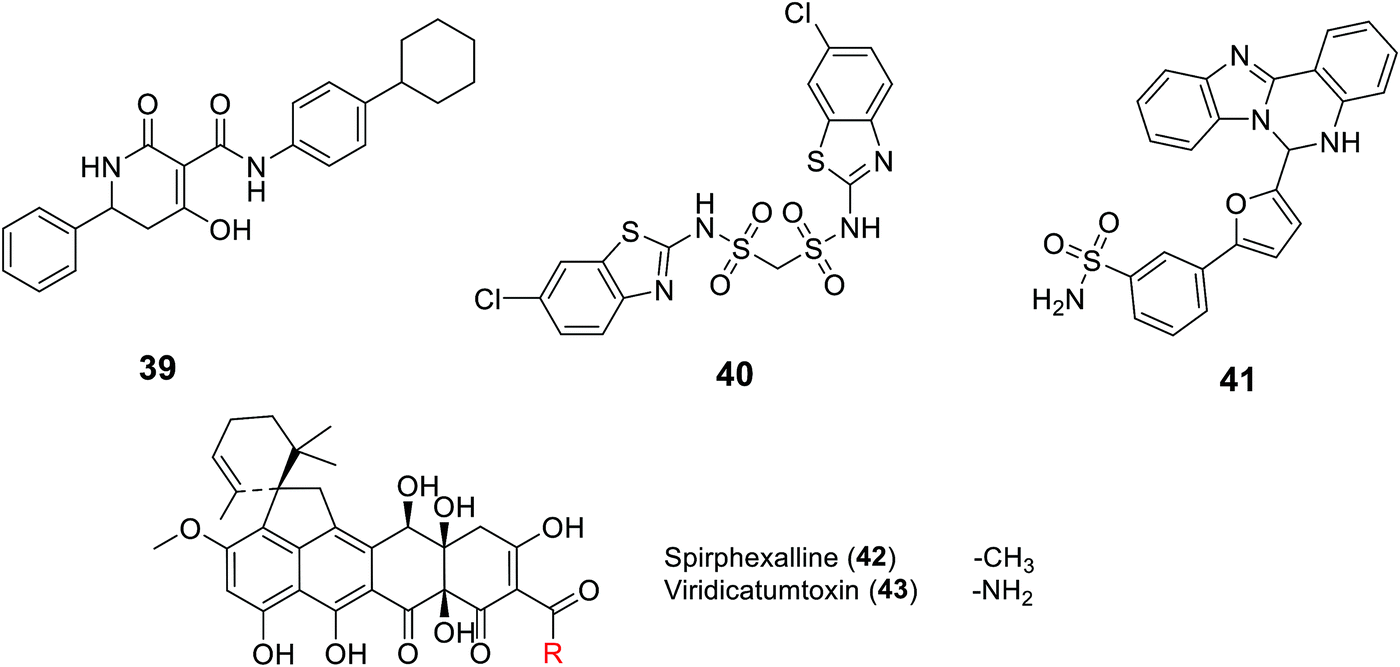
2.4.8.2. UppP inhibitors. Recently, UppP became a new potential target for antibiotic development, including a focus on the multi-drug resistant S. aureus. This was started by GlaxoSmithKline who, for the first time, discovered UppP inhibitors via utilizing an enzyme-based assay system that measured the inorganic phosphate (Pi) released in the enzymatic reaction of the UppP system, and a cell-based assay which analyzed the incorporation of 14C isopentenyl pyrophosphate (IPP). However, their active structures were undisclosed.100 The Novartis group studied a pharmacophore model of a co-crystal structure of UppP with a natural substrate at the active site of the enzyme, which successfully led to the discovery of the tetramic acid derivative 39 as a potent inhibitor of Staphylococcus pneumonia Upp synthase.101 Kuo C. et al.102 also conducted virtual screening based on the crystal structure of Helicobacter pylori Upp synthase and discovered the bis-sulfonyl-containing compound BTB06061 40, a potent and selective inhibitor of H. pylori Upp synthase.102 Furthermore, Durrant et al. studied a docking model of substrates/inhibitors of Upp synthase based on the X-ray structures of Upp synthase-substrate (for example, farnesyl diphosphate)/inhibitor (such as bisphosphonate drugs) complexes, which led to the development of the non-bisphosphonate derivative HTS04781 41, which has potent inhibitory activity against S. aureus Upp synthase.103 Recently, the same group reported two new structurally related compounds named spirohexaline (42) and viridicatumtoxin (43) as UppP inhibitors isolated from the culture broth of Penicillium brasilianum FKI-3368.103,104 These compounds have a hexacycline structure, with a tetracyclic ring fused with a spirobicyclic ring.
Others have reported the discovery of several inhibitors of UppS, comprising bisphosphonates such as BPH-629 44105 and tetramic acids such as 45,101 as well as diketoacids such as 46,106 and benzoic acids such as 47.107 Based on HTS,107 Durrant et al. produced a series of benzoic (48–51), phosphonic acid (52), and diketoacid (53) compounds with activity against UppS. Furthermore, they discovered several potent cationic inhibitors 54–56, which was unexpected from both a computational and experimental perspective, as these compounds do not mimic the (anionic) FPP substrate and the UppP mechanism is not thought to involve carbocation intermediates.108 Therefore, they tried to explain how these inhibitors could bind to the UppP target through obtaining crystal structures of 47–54 and 56 bound to Escherichia coli UppP.

2.4.8.3. Phenylthiazoles as a novel class of dual UppP and UppS inhibitors. Phenylthiazoles were reported as a new scaffold with wide antimicrobial activity against multidrug-resistant Gram-positive strains, including MRSA, VRSA and VRE.109 This class of antimicrobials exhibited a selective advantage over vancomycin in terms of the rate of elimination of MRSA cells.109,110 This criteria is clinically important, as it would affect the size of the dosing regimen necessary for patients.111,112
Through the more than 400 published phenylthiazole derivatives,110,113–126 the SAR of this class of compounds has become well defined (Fig. 4). In brief, the presence of a lipophilic tail and a cationic nitrogenous head is essential for the antimicrobial activities.
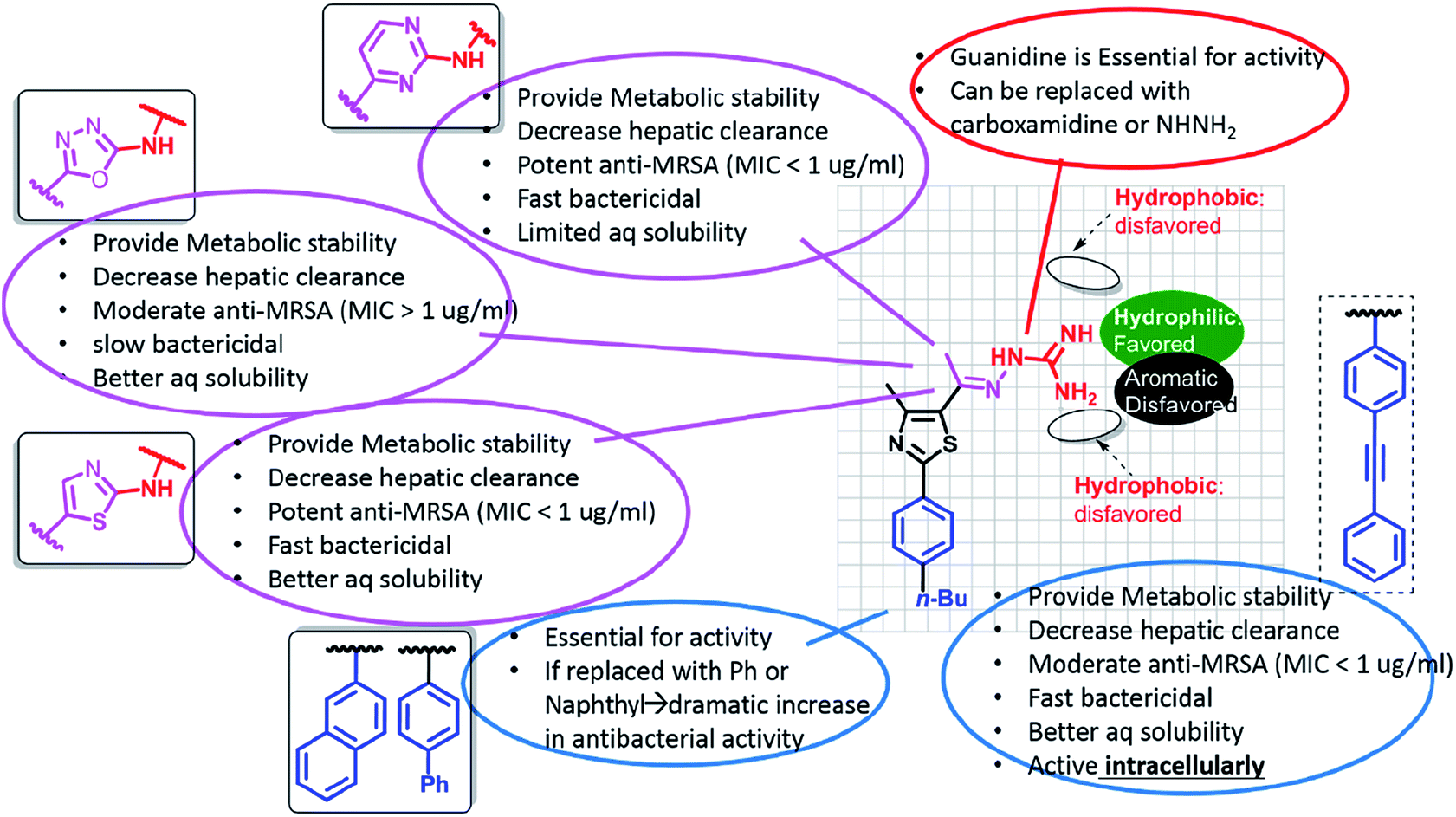 | ||
| Fig. 4 The SAR of phenylthiazole antibiotics. | ||
The main drawbacks of the earliest discovered phenylthiazoles were their ultra-short half-lives,118 which did not exceed the limit of 30 min, and rapid hepatic elimination rates. Incorporating the imine bond of 1st generation phentylthiazoles within a pyrimidine ring yielded 2nd generation analogues with enhanced metabolic stability (t1/2 > 8 h).124 So far, the promising antibacterial potencies of 1st and 2nd generation phentylthiazoles has been offset by their limited activity against intracellular bacterial pathogens, similar to vancomycin and linezolid. Fine tuning the size and polar surface area of the linking heteroaromatic ring provided 3rd generation PTs with balanced properties that allow them to cross cell barriers and accumulate intracellularly in sufficiently lethal concentrations, while maintaining the metabolic stability and rapid bactericidal attributes (Fig. 5).113
 | ||
| Fig. 5 Different generations of phenylthiazole antibiotics with their associated antimicrobial, physicochemical and pharmacokinetic properties. | ||
Later on, a detailed metabolite analysis was completed and this indicated the presence of an additional metabolic soft spot at the butyl benzylic carbon.125 Addressing this limitation, replacing the methylene soft spot with oxygen125 or acetylenyl114 moieties or undergoing replacement with t-butyl,117 has yielded phenylthiazoles with even more pronounced stability with respect to the hepatic metabolism (Fig. 6).
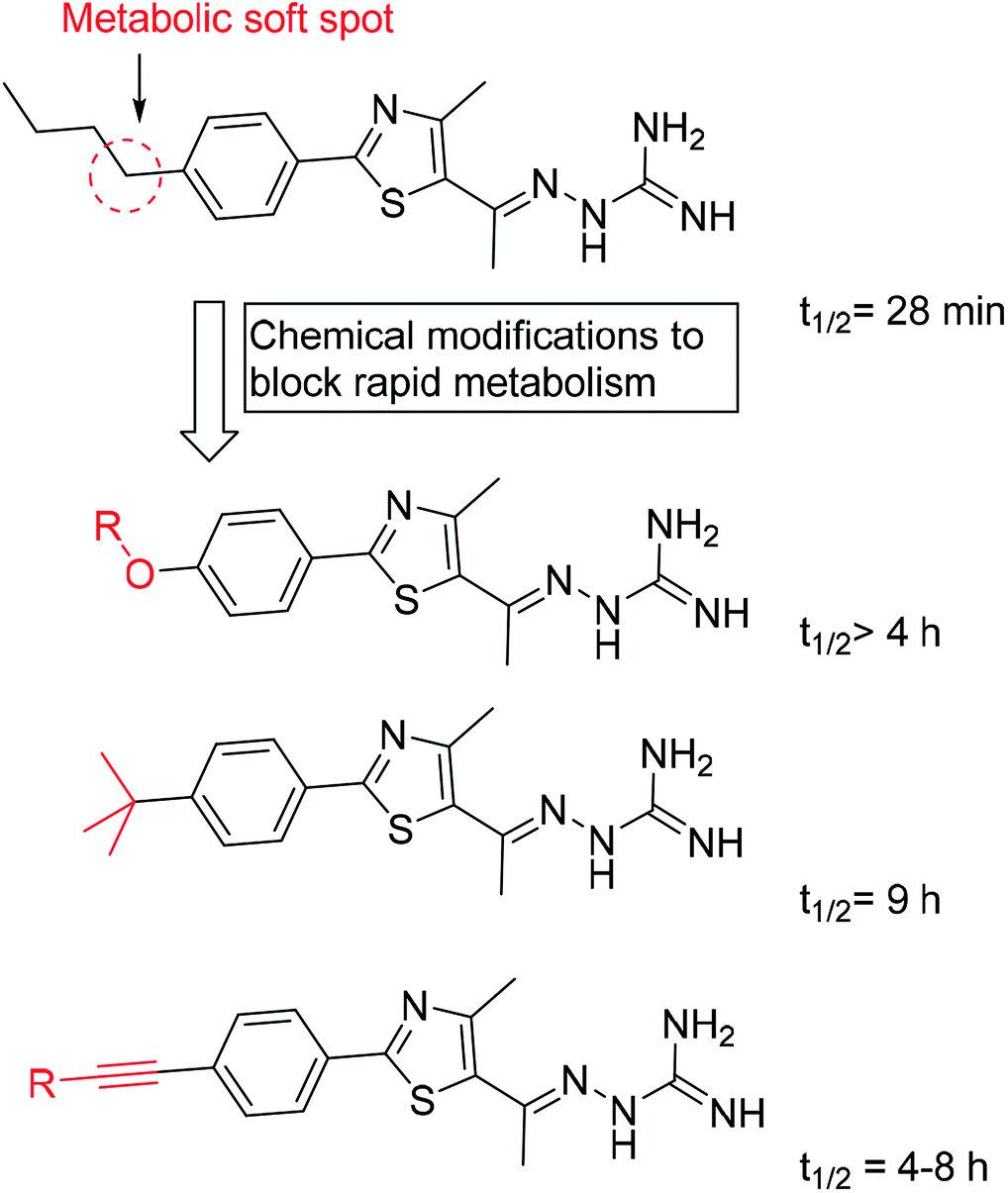 | ||
| Fig. 6 The half-lives of different classes of phenylthiazole antibiotics. | ||
Interestingly, at the time of the first discovery of phenylthiazoles, the antibacterial mechanism of action was unknown. Later on, transposon mutagenesis studies conducted by our group suggested three possible antibacterial targets: YubA, YubB (undecaprenyl diphosphate phosphatase (UppP)) and YubD. Using a direct biochemical assay, the lead phenylthiazole inhibited UppP with an IC50 value of 6 μM. It also inhibited another enzyme involved in PG biosynthesis called undecaprenyl diphosphate synthase (IC50 = 19 μM).121 More importantly, the inhibitory activity of the phenylthiazole lead compound was tested against human FPPS (HsFPPS) and there was no inhibition of HsFPPS.
2.4.8.4. Remarks. The multitarget inhibition mode of action exhibited by phenylthiazoles for both UppP and UppS, the two adjacent proteins in the cell wall biosynthetic pathway (Fig. 3), may contribute to the inability of MDR-bacterial cells to develop rapid resistance to many members of the phenylthiazole family, even after 14 passages.110,117,126
3. Conclusions
The pharmaceutical industry is still relying on hitting the same bacterial targets over and over. This strategy has strengthened the resistance pathways of pathogenic bacteria. Therefore, in order to limit bacterial resistance and increase the survival rates of newly developed antibiotics, the WHO recommends hitting new bacterial pathways using novel chemical cores that are not recognized yet by any resistance machinery of microorganisms. This review illuminates seven potential targets, all participating in peptidoglycan biosynthesis, that may be considered as highly potent and selective targets for future antibiotics in the global fight against increasing microbial resistance. Following WHO criteria, we recommend that medicinal chemists and medical research centers should discard old scaffolds and targets that are already used in the commercial sphere. Additionally, we urge researchers to investigate more novel microbial targets to enable humanity to face microbial resistance waves.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Science & Technology Development Funds (STDF), Grant #5334.References
- K. Hiramatsu, Lancet Infect. Dis., 2001, 1, 147–155 CrossRef CAS.
- P. Wilson, J. Andrews, R. Charlesworth, R. Walesby, M. Singer, D. Farrell and M. Robbins, J. Antimicrob. Chemother., 2003, 51, 186–188 CrossRef CAS PubMed.
- A. Jayol, P. Nordmann, A. Brink, M.-V. Villegas, V. Dubois and L. Poirel, Antimicrob. Agents Chemother., 2017, 61, e01423-17 CrossRef PubMed.
- WHO, Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline, including tuberculosis, World Health Organization, Geneva, 2017, WHO/EMP/IAU/2017.11 Search PubMed.
- W. Vollmer, D. Blanot and M. A. de Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS PubMed.
- L. Gan, S. Chen and G. J. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18953–18957 CrossRef CAS PubMed.
- P. Giesbrecht, T. Kersten, H. Maidhof and J. Wecke, Microbiol. Mol. Biol. Rev., 1998, 62, 1371–1414 CAS.
- A. Tomasz, Rev. Infect. Dis., 1986, 8, S260–S278 CrossRef CAS PubMed.
- W. P. Hammes and F. C. Neuhaus, Antimicrob. Agents Chemother., 1974, 6, 722–728 CrossRef CAS PubMed.
- B. Ostash and S. Walker, Nat. Prod. Rep., 2010, 27, 1594–1617 RSC.
- N. A. Caveney, F. K. Li and N. C. Strynadka, Curr. Opin. Struct. Biol., 2018, 53, 45–58 CrossRef CAS PubMed.
- K. J. Stone and J. L. Strominger, Proc. Natl. Acad. Sci. U.S.A., 1971, 68, 3223–3227 CrossRef CAS PubMed.
- S. Batson, C. de Chiara, V. Majce, A. J. Lloyd, S. Gobec, D. Rea, V. Fulop, C. W. Thoroughgood, K. J. Simmons, C. G. Dowson, C. W. G. Fishwick, L. P. S. de Carvalho and D. I. Roper, Nat. Commun., 2017, 8, 1939 CrossRef PubMed.
- T. D. Bugg, D. Braddick, C. G. Dowson and D. I. Roper, Trends Biotechnol., 2011, 29, 167–173 CrossRef CAS PubMed.
- G. Siewert and J. L. Strominger, Proc. Natl. Acad. Sci. U.S.A., 1967, 57, 767–773 CrossRef CAS PubMed.
- A. Derouaux, E. Sauvage and M. Terrak, Front. Immunol., 2013, 4, 78 Search PubMed.
- A. Bouhss, A. E. Trunkfield, T. D. Bugg and D. Mengin-Lecreulx, FEMS Microbiol. Rev., 2007, 32, 208–233 CrossRef PubMed.
- N. F. Galley, A. M. O'Reilly and D. I. Roper, Bioorg. Chem., 2014, 55, 16–26 CrossRef CAS PubMed.
- C. A. Arias and B. E. Murray, Nat. Rev. Microbiol., 2012, 10, 266–278 CrossRef CAS PubMed.
- J. S. Anderson, M. Matsuhashi, M. A. Haskin and J. L. Strominger, Proc. Natl. Acad. Sci. U.S.A., 1965, 53, 881–889 CrossRef CAS PubMed.
- J. Zuegg, C. Muldoon, G. Adamson, D. McKeveney, G. Le Thanh, R. Premraj, B. Becker, M. Cheng, A. G. Elliott and J. X. Huang, Nat. Commun., 2015, 6, 7719 CrossRef CAS PubMed.
- P. Welzel, Chem. Rev., 2005, 105, 4610–4660 CrossRef CAS PubMed.
- P. Butaye, L. A. Devriese and F. Haesebrouck, Antimicrob. Agents Chemother., 2001, 45, 1374–1378 CrossRef CAS PubMed.
- S. Hentschel, D. Kusch and H. Sinell, Zentralbl. Bakteriol., Parasitenkd., Infektionskrankh. Hyg., Abt. 1: Orig., Reihe B, 1979, 168, 546–561 CAS.
- R. C. Goldman and D. Gange, Curr. Med. Chem., 2000, 7, 801–820 CrossRef CAS PubMed.
- M. D. Barton, Nutr. Res. Rev., 2000, 13, 279–299 CrossRef CAS PubMed.
- Y. Rebets, T. Lupoli, Y. Qiao, K. Schirner, R. Villet, D. Hooper, D. Kahne and S. Walker, ACS Chem. Biol., 2013, 9, 459–467 CrossRef PubMed.
- M. A. Pfaller, Diagn. Microbiol. Infect. Dis., 2006, 56, 115–121 CrossRef CAS PubMed.
- G. Huber, in Mechanism of Action of Antibacterial Agents, Springer, 1979, pp. 135–153 Search PubMed.
- X. Wang, L. Krasnova, K. B. Wu, W. S. Wu, T. J. Cheng and C. H. Wong, Bioorg. Med. Chem. Lett., 2018, 28(16), 2708–2712 CrossRef CAS PubMed.
- M. J. Sofia, N. Allanson, N. T. Hatzenbuhler, R. Jain, R. Kakarla, N. Kogan, R. Liang, D. Liu, D. J. Silva and H. Wang, J. Med. Chem., 1999, 42, 3193–3198 CrossRef CAS PubMed.
- E. R. Baizman, A. A. Branstrom, C. B. Longley, N. Allanson, M. J. Sofia, D. Gange and R. C. Goldman, Microbiology, 2000, 146, 3129–3140 CrossRef CAS PubMed.
- J. Halliday, D. McKeveney, C. Muldoon, P. Rajaratnam and W. Meutermans, Biochem. Pharmacol., 2006, 71, 957–967 CrossRef CAS PubMed.
- H. W. Shih, Y. F. Chang, W. J. Li, F. C. Meng, C. Y. Huang, C. Ma, T. J. R. Cheng, C. H. Wong and W. C. Cheng, Angew. Chem., Int. Ed., 2012, 51, 10123–10126 CrossRef CAS PubMed.
- H. Kuhn, D. Gutelius, E. Black, C. Nadolny, A. Basu and C. Reid, MedChemComm, 2014, 5, 1213–1217 RSC.
- S. Dumbre, A. Derouaux, E. Lescrinier, A. Piette, B. Joris, M. Terrak and P. Herdewijn, J. Am. Chem. Soc., 2012, 134, 9343–9351 CrossRef CAS PubMed.
- T.-J. R. Cheng, Y.-T. Wu, S.-T. Yang, K.-H. Lo, S.-K. Chen, Y.-H. Chen, W.-I. Huang, C.-H. Yuan, C.-W. Guo and L.-Y. Huang, Bioorg. Med. Chem., 2010, 18, 8512–8529 CrossRef CAS PubMed.
- S.-H. Huang, W.-S. Wu, L.-Y. Huang, W.-F. Huang, W.-C. Fu, P.-T. Chen, J.-M. Fang, W.-C. Cheng, T.-J. R. Cheng and C.-H. Wong, J. Am. Chem. Soc., 2013, 135, 17078–17089 CrossRef CAS PubMed.
- C. M. Gampe, H. Tsukamoto, E. H. Doud, S. Walker and D. Kahne, J. Am. Chem. Soc., 2013, 135, 3776–3779 CrossRef CAS PubMed.
- Y. Wang, F. Y. Chan, N. Sun, H. K. Lui, P. K. So, S. C. Yan, K. F. Chan, J. Chiou, S. Chen and R. Abagyan, Chem. Biol. Drug Des., 2014, 84, 685–696 CrossRef CAS PubMed.
- J. A. Thanassi, S. L. Hartman-Neumann, T. J. Dougherty, B. A. Dougherty and M. J. Pucci, Nucleic Acids Res., 2002, 30, 3152–3162 CrossRef CAS PubMed.
- D. S. Boyle and W. D. Donachie, J. Bacteriol., 1998, 180, 6429–6432 CAS.
- D. Mengin-Lecreulx, L. Texier, M. Rousseau and J. van Heijenoort, J. Bacteriol., 1991, 173, 4625–4636 CrossRef CAS PubMed.
- M. Ikeda, M. Wachi, H. Jung, F. Ishino and M. Matsuhashi, J. Bacteriol., 1991, 173, 1021–1026 CrossRef CAS PubMed.
- Y. Liu and E. Breukink, Antibiotics, 2016, 5, 28 CrossRef PubMed.
- T. Mohammadi, A. Karczmarek, M. Crouvoisier, A. Bouhss, D. Mengin-Lecreulx and T. Den Blaauwen, Mol. Microbiol., 2007, 65, 1106–1121 CrossRef CAS PubMed.
- F. Isono and M. Inukai, Antimicrob. Agents Chemother., 1991, 35, 234–236 CrossRef CAS PubMed.
- T. D. Bugg, A. J. Lloyd and D. I. Roper, Infect. Disord. - Drug Targets, 2006, 6, 85–106 CrossRef CAS.
- P. E. Brandish, K.-i. Kimura, M. Inukai, R. Southgate, J. T. Lonsdale and T. Bugg, Antimicrob. Agents Chemother., 1996, 40, 1640–1644 CrossRef CAS.
- P. E. Brandish, M. K. Burnham, J. T. Lonsdale, R. Southgate, M. Inukai and T. D. Bugg, J. Biol. Chem., 1996, 271, 7609–7614 CrossRef CAS PubMed.
- A. J. Lloyd, P. E. Brandish, A. M. Gilbey and T. D. Bugg, J. Bacteriol., 2004, 186, 1747–1757 CrossRef CAS PubMed.
- M. T. Rodolis, A. Mihalyi, C. Ducho, K. Eitel, B. Gust, R. J. Goss and T. D. Bugg, Chem. Commun., 2014, 50, 13023–13025 RSC.
- M. T. Rodolis, A. Mihalyi, A. O'Reilly, J. Slikas, D. I. Roper, R. E. Hancock and T. D. Bugg, ChemBioChem, 2014, 15, 1300–1308 CrossRef CAS PubMed.
- C. Dini, P. Collette, N. Drochon, J. Guillot, G. Lemoine, P. Mauvais and J. Aszodi, Bioorg. Med. Chem. Lett., 2000, 10, 1839–1843 CrossRef CAS PubMed.
- T. Tanino, B. Al-Dabbagh, D. Mengin-Lecreulx, A. Bouhss, H. Oyama, S. Ichikawa and A. Matsuda, J. Med. Chem., 2011, 54, 8421–8439 CrossRef CAS PubMed.
- T. D. Bugg, M. T. Rodolis, A. Mihalyi and S. Jamshidi, Bioorg. Med. Chem., 2016, 24, 6340–6347 CrossRef CAS PubMed.
- D. K. Banerjee, J. Biol. Chem., 1989, 264, 2024–2028 CAS.
- T. G. Bernhardt, W. D. Roof and R. Young, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 4297–4302 CrossRef CAS PubMed.
- T. G. Bernhardt, D. K. Struck and R. Young, J. Biol. Chem., 2001, 276, 6093–6097 CrossRef CAS PubMed.
- L. E. Zawadzke, P. Wu, L. Cook, L. Fan, M. Casperson, M. Kishnani, D. Calambur, S. J. Hofstead and R. Padmanabha, Anal. Biochem., 2003, 314, 243–252 CrossRef CAS PubMed.
- J. Yoo, E. H. Mashalidis, A. C. Kuk, K. Yamamoto, B. Kaeser, S. Ichikawa and S.-Y. Lee, Nat. Struct. Mol. Biol., 2018, 25, 217 CrossRef CAS PubMed.
- M. Kang, J. P. Spencer and A. D. Elbein, Biochem. Biophys. Res. Commun., 1978, 82, 568–574 CrossRef CAS.
- M. S. Kang, J. P. Spencer and A. D. Elbein, J. Biol. Chem., 1978, 253, 8860–8866 CAS.
- J. Hering, E. Dunevall, M. Ek and G. Brändén, Drug Discovery Today, 2018, 1426–1435 CrossRef CAS PubMed.
- W. Du, J. R. Brown, D. R. Sylvester, J. Huang, A. F. Chalker, C. Y. So, D. J. Holmes, D. J. Payne and N. G. Wallis, J. Bacteriol., 2000, 182, 4146–4152 CrossRef CAS.
- H. Mizuno and T. Usuki, ChemistrySelect, 2018, 3, 1781–1786 CrossRef CAS.
- A. Bachelier, R. Mayer and C. D. Klein, Bioorg. Med. Chem. Lett., 2006, 16, 5605–5609 CrossRef CAS.
- T. D. Bugg, Compr. Nat. Prod. Chem., 1999, 3, 241–294 CAS.
- K. Shigetomi, K. Shoji, S. Mitsuhashi and M. Ubukata, Phytochemistry, 2010, 71, 312–324 CrossRef CAS PubMed.
- E. Z. Baum, D. A. Montenegro, L. Licata, I. Turchi, G. C. Webb, B. D. Foleno and K. Bush, Antimicrob. Agents Chemother., 2001, 45, 3182–3188 CrossRef CAS PubMed.
- S. Eschenburg, M. A. Priestman, F. A. Abdul-Latif, C. Delachaume, F. Fassy and E. Schönbrunn, J. Biol. Chem., 2005, 280, 14070–14075 CrossRef CAS PubMed.
- C. J. Dunsmore, K. Miller, K. L. Blake, S. G. Patching, P. J. F. Henderson, J. A. Garnett, W. J. Stubbings, S. E. V. Phillips, D. J. Palestrant, J. D. L. Angeles, J. A. Leeds, I. Chopra and C. W. G. Fishwick, Bioorg. Med. Chem. Lett., 2008, 18, 1730–1734 CrossRef CAS PubMed.
- T. Scholz, C. L. Heyl, D. Bernardi, S. Zimmermann, L. Kattner and C. D. Klein, Bioorg. Med. Chem., 2013, 21, 795–804 CrossRef CAS.
- K. Miller, C. J. Dunsmore, J. A. Leeds, S. G. Patching, M. Sachdeva, K. L. Blake, W. J. Stubbings, K. J. Simmons, P. J. Henderson and J. De Los Angeles, J. Antimicrob. Chemother., 2010, 65, 2566–2573 CrossRef CAS.
- J. J. Bronson, K. L. DenBleyker, P. J. Falk, R. A. Mate, H.-T. Ho, M. J. Pucci and L. B. Snyder, Bioorg. Med. Chem. Lett., 2003, 13, 873–875 CrossRef CAS.
- Y. Yang, A. Severin, R. Chopra, G. Krishnamurthy, G. Singh, W. Hu, D. Keeney, K. Svenson, P. J. Petersen, P. Labthavikul, D. M. Shlaes, B. A. Rasmussen, A. A. Failli, J. S. Shumsky, K. M. K. Kutterer, A. Gilbert and T. S. Mansour, Antimicrob. Agents Chemother., 2006, 50, 556–564 CrossRef CAS.
- C. J. Andres, J. J. Bronson, S. V. D'Andrea, M. S. Deshpande, P. J. Falk, K. A. Grant-Young, W. E. Harte, H.-T. Ho, P. F. Misco, J. G. Robertson, D. Stock, Y. Sun and A. W. Walsh, Bioorg. Med. Chem. Lett., 2000, 10, 715–717 CrossRef CAS.
- A. M. Gilbert, A. Failli, J. Shumsky, Y. Yang, A. Severin, G. Singh, W. Hu, D. Keeney, P. J. Petersen and A. H. Katz, J. Med. Chem., 2006, 49, 6027–6036 CrossRef CAS PubMed.
- D. E. Ehmann, J. E. Demeritt, K. G. Hull and S. L. Fisher, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1698, 167–174 CrossRef CAS PubMed.
- L. E. Zawadzke, M. Norcia, C. R. Desbonnet, H. Wang, K. Freeman-Cook and T. J. Dougherty, Assay Drug Dev. Technol., 2008, 6, 95–103 CrossRef CAS PubMed.
- T. Tomasic and L. P. Masic, Curr. Med. Chem., 2009, 16, 1596–1629 CrossRef CAS PubMed.
- R. Frlan, A. Kovač, D. Blanot, S. Gobec, S. Pečar and A. Obreza, Molecules, 2008, 13, 11 CrossRef CAS.
- R. Šink, A. Kovač, T. Tomašić, V. Rupnik, A. Boniface, J. Bostock, I. Chopra, D. Blanot, L. P. Mašič and S. Gobec, ChemMedChem, 2008, 3, 1362–1370 CrossRef PubMed.
- S. Turk, A. Kovač, A. Boniface, J. M. Bostock, I. Chopra, D. Blanot and S. Gobec, Bioorg. Med. Chem., 2009, 17, 1884–1889 CrossRef CAS PubMed.
- A. Perdih, A. Kovač, G. Wolber, D. Blanot, S. Gobec and T. Solmajer, Bioorg. Med. Chem. Lett., 2009, 19, 2668–2673 CrossRef CAS PubMed.
- J. Humljan, M. Kotnik, C. Contreras-Martel, D. Blanot, U. Urleb, A. Dessen, T. Šolmajer and S. Gobec, J. Med. Chem., 2008, 51, 7486–7494 CrossRef CAS PubMed.
- N. Zidar, T. Tomašić, R. Šink, V. Rupnik, A. Kovač, S. Turk, D. Patin, D. Blanot, C. Contreras Martel, A. Dessen, M. Müller Premru, A. Zega, S. Gobec, L. Peterlin Mašič and D. Kikelj, J. Med. Chem., 2010, 53, 6584–6594 CrossRef CAS.
- K. Štrancar, D. Blanot and S. Gobec, Bioorg. Med. Chem. Lett., 2006, 16, 343–348 CrossRef.
- J. D. Guzman, A. Gupta, D. Evangelopoulos, C. Basavannacharya, L. C. Pabon, E. A. Plazas, D. R. Munoz, W. A. Delgado, L. E. Cuca and W. Ribon, J. Antimicrob. Chemother., 2010, 65, 2101–2107 CrossRef CAS PubMed.
- K. Štrancar, A. Boniface, D. Blanot and S. Gobec, Arch. Pharm., 2007, 340, 127–134 CrossRef.
- C. Paradis-Bleau, A. Lloyd, F. Sanschagrin, H. Maaroufi, T. Clarke, A. Blewett, C. Dowson, D. I. Roper, T. D. H. Bugg and R. C. Levesque, Biochem. J., 2009, 421, 263–272 CrossRef CAS PubMed.
- D. Mengin-Lecreulx, T. Falla, D. Blanot, J. van Heijenoort, D. J. Adams and I. Chopra, J. Bacteriol., 1999, 181, 5909–5914 CAS.
- K. H. Schleifer and O. Kandler, Bacteriol. Rev., 1972, 36, 407–477 CAS.
- I. Sosic, B. Stefane and A. Kovac, Heterocycles, 2010, 81, 91–115 CrossRef CAS.
- E. Z. Baum, S. M. Crespo-Carbone, D. Abbanat, B. Foleno, A. Maden, R. Goldschmidt and K. Bush, Antimicrob. Agents Chemother., 2006, 50, 230–236 CrossRef CAS PubMed.
- R. Bernard, M. El Ghachi, D. Mengin-Lecreulx, M. Chippaux and F. Denizot, J. Biol. Chem., 2005, 280, 28852–28857 CrossRef CAS PubMed.
- Y. Li and X. Tian, Immunogenetics, 2016, 1, 2 Search PubMed.
- M. El Ghachi, A. Bouhss, D. Blanot and D. Mengin-Lecreulx, J. Biol. Chem., 2004, 279, 30106–30113 CrossRef CAS.
- M. Jukic, K. Rozman and S. Gobec, Curr. Med. Chem., 2016, 23, 464–482 CrossRef CAS.
- H. Li, J. Huang, X. Jiang, M. Seefeld, M. McQueney and R. Macarron, J. Biomol. Screening, 2003, 8, 712–715 CrossRef CAS.
- S. Peukert, Y. Sun, R. Zhang, B. Hurley, M. Sabio, X. Shen, C. Gray, J. Dzink-Fox, J. Tao and R. Cebula, Bioorg. Med. Chem. Lett., 2008, 18, 1840–1844 CrossRef CAS PubMed.
- C.-J. Kuo, R.-T. Guo, I.-L. Lu, H.-G. Liu, S.-Y. Wu, T.-P. Ko, A. H.-J. Wang and P.-H. Liang, BioMed Res. Int., 2008, 2008, 841312 Search PubMed.
- W. Sinko, C. de Oliveira, S. Williams, A. Van Wynsberghe, J. D. Durrant, R. Cao, E. Oldfield and J. A. McCammon, Chem. Biol. Drug Des., 2011, 77, 412–420 CrossRef CAS.
- J. Inokoshi, Y. Nakamura, Z. Hongbin, R. Uchida, K.-i. Nonaka, R. Masuma and H. Tomoda, J. Antibiot., 2013, 66, 37–41 CrossRef CAS.
- R.-T. Guo, R. Cao, P.-H. Liang, T.-P. Ko, T.-H. Chang, M. P. Hudock, W.-Y. Jeng, C. K.-M. Chen, Y. Zhang and Y. Song, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 10022–10027 CrossRef CAS PubMed.
- Y. Zhang, F.-Y. Lin, K. Li, W. Zhu, Y.-L. Liu, R. Cao, R. Pang, E. Lee, J. Axelson and M. Hensler, ACS Med. Chem. Lett., 2012, 3, 402–406 CrossRef CAS PubMed.
- J. D. Durrant, R. Cao, A. A. Gorfe, W. Zhu, J. Li, A. Sankovsky, E. Oldfield and J. A. McCammon, Chem. Biol. Drug Des., 2011, 78, 323–332 CrossRef CAS.
- Y.-P. Lu, H.-G. Liu and P.-H. Liang, Biochem. Biophys. Res. Commun., 2009, 379, 351–355 CrossRef CAS.
- H. Mohammad, A. S. Mayhoub, A. Ghafoor, M. Soofi, R. A. Alajlouni, M. Cushman and M. N. Seleem, J. Med. Chem., 2014, 57, 1609–1615 CrossRef CAS PubMed.
- H. Mohammad, A. S. Mayhoub, M. Cushman and M. N. Seleem, J. Antibiot., 2015, 68, 259–266 CrossRef CAS.
- G. Pankey and L. Sabath, Clin. Infect. Dis., 2004, 38, 864–870 CrossRef CAS.
- G. French, J. Antimicrob. Chemother., 2006, 58, 1107–1117 CrossRef CAS.
- I. Eid, M. M. Elsebaei, H. Mohammad, M. Hagras, C. E. Peters, Y. A. Hegazy, B. Cooper, J. Pogliano, K. Pogliano, H. S. Abulkhair, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2017, 139, 665–673 CrossRef CAS PubMed.
- M. M. Elsebaei, H. Mohammad, M. Abouf, N. S. Abutaleb, Y. A. Hegazy, A. Ghiaty, L. Chen, J. Zhang, S. R. Malwal, E. Oldfield, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 148, 195–209 CrossRef CAS.
- M. Hagras, Y. A. Hegazy, A. H. Elkabbany, H. Mohammad, A. Ghiaty, T. M. Abdelghany, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 143, 1448–1456 CrossRef CAS.
- M. Hagras, H. Mohammad, M. S. Mandour, Y. A. Hegazy, A. Ghiaty, M. N. Seleem and A. S. Mayhoub, J. Med. Chem., 2017, 60, 4074–4085 CrossRef CAS PubMed.
- A. Kotb, N. S. Abutaleb, M. A. Seleem, M. Hagras, H. Mohammad, A. Bayoumi, A. Ghiaty, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 151, 110–120 CrossRef CAS PubMed.
- H. Mohammad, A. S. Mayhoub, A. Ghafoor, M. Soofi, R. A. Alajlouni, M. Cushman and M. N. Seleem, J. Med. Chem., 2014, 57, 1609–1615 CrossRef CAS PubMed.
- H. Mohammad, P. V. Reddy, D. Monteleone, A. S. Mayhoub, M. Cushman, G. K. Hammac and M. N. Seleem, PLoS One, 2015, 10, e0130385 CrossRef PubMed.
- H. Mohammad, P. V. Reddy, D. Monteleone, A. S. Mayhoub, M. Cushman and M. N. Seleem, Eur. J. Med. Chem., 2015, 94, 306–316 CrossRef CAS PubMed.
- H. Mohammad, W. Younis, L. Chen, C. E. Peters, J. Pogliano, K. Pogliano, B. Cooper, J. Zhang, A. Mayhoub, E. Oldfield, M. Cushman and M. N. Seleem, J. Med. Chem., 2017, 60, 2425–2438 CrossRef CAS PubMed.
- H. Mohammad, W. Younis, H. G. Ezzat, C. E. Peters, A. AbdelKhalek, B. Cooper, K. Pogliano, J. Pogliano, A. S. Mayhoub and M. N. Seleem, PLoS One, 2017, 12, e0182821 CrossRef PubMed.
- H. X. Ngo, K. D. Green, C. S. Gajadeera, M. J. Willby, S. Y. Holbrook, C. Hou, A. Garzan, A. S. Mayhoub, J. E. Posey and O. V. Tsodikov, ACS Infect. Dis., 2018, 1030–1040 CrossRef CAS PubMed.
- M. A. Seleem, A. M. Disouky, H. Mohammad, T. M. Abdelghany, A. S. Mancy, S. A. Bayoumi, A. Elshafeey, A. El-Morsy, M. N. Seleem and A. S. Mayhoub, J. Med. Chem., 2016, 59, 4900–4912 CrossRef CAS PubMed.
- E. Yahia, H. Mohammad, T. M. Abdelghany, E. Fayed, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2017, 126, 604–613 CrossRef CAS PubMed.
- M. ElAwamy, H. Mohammad, A. Hussien, N. S. Abutaleb, M. Hagras, R. A. T. Serya, A. T. Taher, K. A. Abouzid, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 152, 318–328 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |