
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew α-pyrones from an endophytic fungus, Hypoxylon investiens J2†
Chao Yuan a,
Hong-Xia Yangb,
Yu-Hua Guoc,
Lin Fand,
Ying-Bo Zhanga and
Gang Li*b
a,
Hong-Xia Yangb,
Yu-Hua Guoc,
Lin Fand,
Ying-Bo Zhanga and
Gang Li*b
aTropical Crops Genetic Resources Institute, Chinese Academy of Tropical Agricultural Sciences CATAS, Haikou 571101, People's Republic of China
bDepartment of Natural Medicinal Chemistry and Pharmacognosy, School of Pharmacy, Qingdao University, Qingdao 266021, People's Republic of China. E-mail: gang.li@qdu.edu.cn; Tel: +86-532-8299-1172
cShandong Drug and Food Vocational College, Weihai, Shandong 264210, People's Republic of China
dWeihai Vocational College, Weihai 264210, People's Republic of China
First published on 2nd September 2019
Abstract
Four new α-pyrones, hypotiens A–D (1–4), were isolated from a fungal endophyte, Hypoxylon investiens J2, harbored in the medicinal plant Blumea balsamifera. Their structures were determined through detailed HRMS and NMR spectroscopic data. Compounds 1–4 are new α-pyrone derivatives containing an unusual dimethyl substitution in the highly unsaturated side chain. Their plausible biosynthetic pathway was discussed. Biological assay indicated that compounds 1–4 showed no antimicrobial, quorum sensing inhibitory, and cytotoxic activities. The specific side chain in α-pyrone derivatives 1–4 might be responsible for the weak pharmacological activities.
Introduction
Fungal endophytes asymptomatically colonize living tissues of healthy plants.1–3 They are now recognized as an invaluable source of structurally diverse and biologically active natural products.4 More than one hundred endophytic fungi-derived secondary metabolites with new carbon skeletons, rare ring systems, or unusual structural units have been reported.5 Exploration of these novel and bioactive secondary metabolites greatly facilitates the discovery of lead compounds.From the endophytic fungus Chaetomium sp. IFB-E015 living in the leaves of Adenophora axilliflora, an unprecedented alkaloid, chaetominine containing an unusual alanine-derived δ-lactam ring, was isolated and structurally elucidated.6 It exhibited more potent cytotoxicity to the human colon cancer SW1116 and leukemia K562 cell lines than the positive drug 5-fluorouracil, and has received considerable attention from chemists and biologists in the field of total synthesis and biological investigations.6–8 Papeo and co-workers reported a total synthesis of chaetominine based on a straightforward (nine steps) sequence and found that this compound exhibited negligible cytotoxic activities on several cancer cell lines.9 Rhizoctonia solani, an endophyte isolated from the medical plant Cyperus rotundus, was discovered to biosynthesize a degraded and rearranged steroid, solanioic acid with an unprecedented carbon skeleton.10,11 It showed significant antibacterial activities against Gram-positive bacteria, especially the problematic human pathogen methicillin-resistant Staphylococcus aureus with an MIC of 1 μg mL−1.10 The healthy plant Paris polyphylla contained an endophytic fungus Aspergillus versicolor.12 Its chemical investigation resulted in the isolation and purification of a highly oxygenated cyclopiazonic acid-derived alkaloid, aspergilline E.12 This compound has a new hexacyclic 6/5/6/5/5/5 scaffold and displayed significant biological activities, including anti-virus and cytotoxicity.12
As part of an ongoing program aimed at finding biologically active natural products from endophytic fungi,13,14 Hypoxylon investiens J2 as a fungal endophyte, was isolated from the medicinal plant Blumea balsamifera. Chemical investigation on its rice cultures led to the isolation of four new α-pyrone derivatives, hypotiens A–D (1–4). Compounds 1–4 possess a highly unsaturated side chain containing an unusual dimethyl substitution, which is similar to that of oxazolomycins with potent antibacterial, antiviral and cytotoxic activities.15 Details of the isolation, structure elucidation, and biological activity, together with a proposed biosynthesis of compounds 1–4 are reported here.
Results and discussion
Compound 1 (Fig. 1), yellow powder, has a molecular formula C18H22O4 as established by ESI-HRMS. The 1H NMR spectrum (Table 1, and Fig. S1†) showed the presence of five singlet methyls (δH 1.27, 1.27, 1.95, 2.04, and 2.14). In addition, six olefinic protons associated with three double bonds were also suggested in the 1H NMR spectrum (δH 6.56, d, J = 15.0 Hz; δH 7.07, dd, J = 15.0, 11.0 Hz; δH 6.47, dd, J = 15.0, 11.0 Hz; δH 6.55, dd, J = 15.0, 10.0 Hz; δH 6.31, dd, J = 15.5, 10.0 Hz; δH 5.98, d, J = 15.5 Hz). The 13C NMR spectrum (Table 1, and Fig. S2†) indicated the presence of 18 carbon signals, including a ketone group at δC 213.0 and five methyls at δC 9.2, 9.6, 24.4, 24.4, and 25.9. The 1D NMR data in combination with the HSQC spectrum (Fig. S3†) revealed five methyls, three trans-disubstituted double bonds, a ketone unit, and the remaining six quaternary carbons including a saturated carbon at δC 51.8.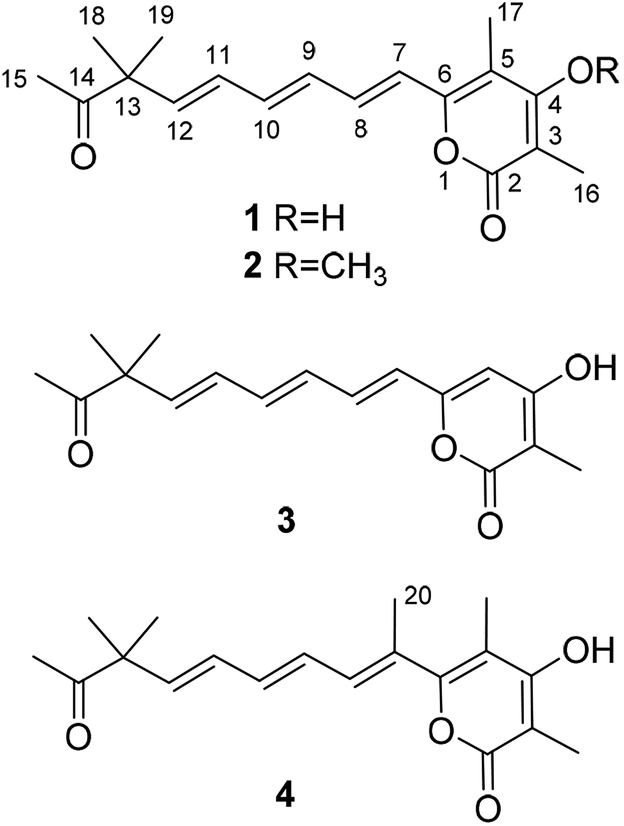 | ||
| Fig. 1 Chemical structures of compounds 1–4. | ||
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | |
| 2 | 167.4, C | 167.1, C | 168.0, C | 168.0, C | ||||
| 3 | 100.6, C | 111.7, C | 100.6, C | 99.6, C | ||||
| 4 | 167.5, C | 170.4, C | 168.2, C | 168.8, C | ||||
| 5 | 110.5, C | 112.6, C | 6.13, s | 102.4, C | 109.8, C | |||
| 6 | 153.7, C | 154.3, C | 157.9, C | 159.4, C | ||||
| 7 | 6.56, d (15.0) | 120.9, CH | 6.54, d (15.0) | 120.8, CH | 6.20, d (15.0) | 123.2, CH | 129.5, C | |
| 8 | 7.07, dd (15.0, 11.0) | 135.6, CH | 7.07, dd (15.0, 11.0) | 135.8, CH | 7.06, dd (14.5, 11.5) | 135.9, CH | 6.35, d (11.5) | 135.5, CH |
| 9 | 6.47, dd (15.0, 11.0) | 133.0, CH | 6.48, dd (15.0, 11.0) | 132.9, CH | 6.40, dd (13.0, 11.0) | 132.3, CH | 6.64, dd (14.5, 11.0) | 128.7, CH |
| 10 | 6.55, dd (15.0, 10.0) | 138.2, CH | 6.56, dd (15.0, 10.0) | 138.5, CH | 6.56, dd (14.5, 10.5) | 138.8, CH | 6.45, dd (14.5, 10.5) | 137.5, CH |
| 11 | 6.31, dd (15.5, 10.0) | 131.1, CH | 6.31, dd (15.5, 10.0) | 131.0, CH | 6.29, dd (15.5, 11.0) | 130.9, CH | 6.36, dd (14.5, 11.5) | 131.3, CH |
| 12 | 5.98, d (15.5) | 141.6, CH | 6.00, d (15.5) | 141.9, CH | 5.99, d (15.5) | 141.9, CH | 5.94, d (14.5) | 141.0, CH |
| 13 | 51.8, C | 51.8, C | 51.8, C | 51.8, C | ||||
| 14 | 213.0, C | 212.8, C | 213.8, C | 213.0, C | ||||
| 15 | 2.14, s | 25.9, CH3 | 2.14, s | 25.9, CH3 | 2.15, s | 26.0, CH3 | 2.13, s | 25.9, CH3 |
| 16 | 1.95, s | 9.2, CH3 | 2.02, s | 9.8, CH3 | 1.88, s | 8.7, CH3 | 1.93, s | 9.0, CH3 |
| 17 | 2.04, s | 9.6, CH3 | 2.04, s | 10.5, CH3 | 2.04, s | 12.1, CH3 | ||
| 18 | 1.27, s | 24.4, CH3 | 1.27, s | 24.3, CH3 | 1.26, s | 24.3, CH3 | 1.27, s | 24.4, CH3 |
| 19 | 1.27, s | 24.4, CH3 | 1.27, s | 24.3, CH3 | 1.26, s | 24.3, CH3 | 1.27, s | 24.4, CH3 |
| 20 | 2.05, d (1.0) | 15.2, CH3 | ||||||
| 4-OMe | 3.85, s | 61.2, CH3 | ||||||
The planar structure of compound 1 was further constructed through the detailed analysis of the HMBC spectrum (Fig. 2, and S4†). The key HMBC correlations (Fig. 2) from H3-15 to C-13 and C-14 coupled with the requirement of chemical shifts of C-14 (δC 213.0) and H3-15 (δH 2.14, s) confirmed the connection from C-13 to C-15. Two singlet methyls (C-18 and C-19) were further located at the C-13, which was confirmed by the HMBC correlations of H3-18 and H3-19 with C-13 and C-14. Based on the key HMBC correlations from H-12 to C-10, C-11, C-13, and C-14, from H-11 to C-9 and C-10, from H-9 to C-7 and C-8, and from H-7 and H-8 to C-6, a side chain from C-6 to C-15 was tentatively deduced. It contained three trans-disubstituted double bonds at C-7(8), C-9(10), and C-11(12), which was strongly supported by their chemical shifts and relatively large coupling constants.
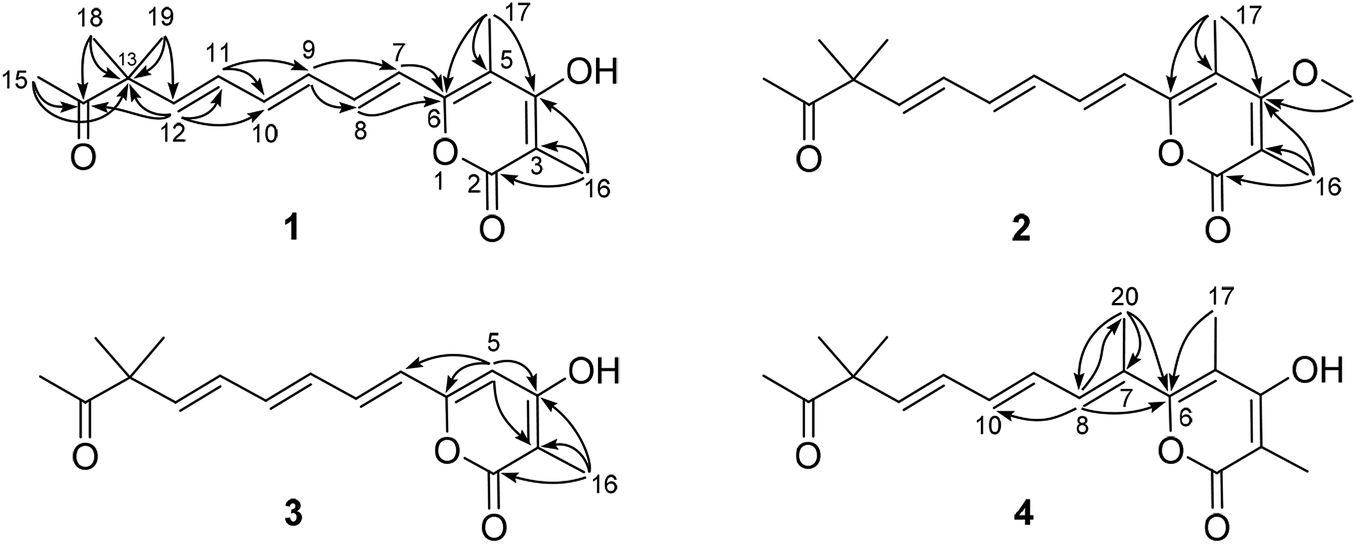 | ||
| Fig. 2 Key HMBC correlations of compounds 1–4. | ||
Further analysis of the HMBC cross-peaks of H3-16/C-2, H3-16/C-3, H3-16/C-4, H3-17/C-4, H3-17/C-5, and H3-17/C-6 verified the connections from C-2 to C-6 (Fig. 2). A hydroxyl group was placed at C-4 based on its chemical shift (δC 167.5). The remaining one degree of unsaturation and the chemical shifts of C-2 (δC 167.4) and C-6 (δC 153.7) suggested that C-2 and C-6 in compound 1 were both linked to the same oxygen atom to form a α-pyrone ring, which was consistent with its molecular formula. In the NOESY spectrum of compound 1, a key correlation between olefinic H-7 and aliphatic CH3-17 was observed (Fig. S5†), indicating these protons were close in space. Accordingly, the structure of compound 1 was established as depicted and it was named hypotien A.
Compound 2 (Fig. 1) was also obtained as a yellow powder and named as hypotien B. Based on the ESI-HRMS data, it was assigned the molecular formula C19H24O4, corresponding to one CH2 group more than 1. Analysis of its 1H, 13C, and HSQC NMR spectra (Table 1, and Fig. S7–S9†) indicated similar structural features to those of compound 1, except for the presence of a methoxy group (δH 3.85; δC 61.2) in 2 and the significant downfield shifts of C-3 and C-4. The above analysis revealed that a methoxy moiety in 2 instead of a hydroxyl group in 1 was linked to C-4, which was further supported by detailed analysis of the HMBC spectrum of compound 2 (Fig. 2).
For compound 3 (Fig. 1), its molecular formula C17H20O4 was determined by the same strategy as above and corresponded to one CH2 group less than compound 1. The 1H NMR spectrum of 3 (Table 1 and Fig. S12†) was also close to that of 1 except for the presence of an olefinic proton (H-5, δH 6.13) in 3 and the absence of a methyl signal at C-5 in 1. Key HMBC correlations (Fig. 2) from H-5 to C-3, C-4, C-6, and C-7 indicated the location of H-5 and assigned the structure of compound 3 as shown. Compound 3 was named hypotien C.
Hypotien D (4, Fig. 1) was a yellow powder. ESI-HRMS spectrum determined its molecular formula as C19H24O4. Detailed analysis of the 1H, 13C, and HSQC data of 4 (Table 1, and Fig. S17–S19†) suggested that compound 4 has similar structural characteristics to compound 1 and indicated a α-pyrone derivative. By comparing the 1D NMR data of 4 with that of 1, in addition to the absence of an olefinic proton signal in compound 4, one more methyl group (δH 2.05; δC 15.2) was observed in compound 4. The above methyl group was located at the olefinic C-7 based on the HMBC correlations of H3-20 with C-6, C-7, and C-8 (Fig. 2). Further analysis of key HMBC correlations confirmed the structure of compound 4, which was in accordance with the requirement of its molecular formula.
α-Pyrone, a six-membered lactone, is frequently discovered in microorganisms, plants, and animals, and is often substituted with a side chain.16 The diverse substitutions of the six-membered lactone, as well as the variations in length and substitutions of the side chain, greatly contribute to the structural diversity and complexity of α-pyrone derivatives.17–21 Compounds 1–4 are new α-pyrone derivatives containing an unusual dimethyl substitution in the highly unsaturated side chain (Fig. 1). Their plausible biosynthetic pathway was proposed through a polyketide synthase.16 A linear polyketide chain was first constructed from an acetyl coenzyme A (CoA) and six malonyl-CoA followed by reduction, dehydration, methylation, oxidation, or cyclization to generate the α-pyrone derivatives.
Natural products containing a α-pyrone have exhibited diverse biological activities, such as the mostly reported antimicrobial efficacy,17–19 quorum sensing (QS) inhibitory activity,22 and cytotoxicity.19,21 In this work, the antibacterial activities of new α-pyrones 1–4 were evaluated against four bacteria Staphylococcus aureus (ATCC 6538), Bacillus subtilis (ATCC 9372), Pseudomonas aeruginosa (ATCC 27853), and Escherichia coli (ATCC 25922), and their antifungal efficacies were tested against three agricultural pathogens Colletotrichum musae (ACCC 31244), Colletotrichum coccodes (ACCC 36067), and Colletotrichum orbiculare (ACCC 36095). Furthermore, the QS inhibitory activity against Chromobacterium violaceum and the cytotoxic assay against three human cancer cell lines A549, CT-26, and MCF-7 were also applied for compounds 1–4. Unfortunately, in contrast to the positive controls, none of them at the given concentrations (Experimental section) were effective against the tested microorganisms or cancer cells. The specific side chain in new α-pyrones 1–4 might be responsible for the weak pharmacological activities.
Experimental section
General experimental procedures
Mass spectra were measured on an LTQ-Orbitrap spectrometer equipped with an ESI source. 1D and 2D NMR spectra were recorded on a Bruker 500 MHz spectrometer. The semi-preparative HPLC was performed on an Agilent 1260 system (Agilent technologies, Germany) equipped with an RP-18 column (250 × 10 mm, YMC Park, 5 μM). Silica gel GF254 plates (Qingdao Haiyang Chemical Co., Ltd., China) was applied for thin-layer chromatography (TLC). Silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd., China) and Sephadex LH-20 (25–100 μm; Pharmacia, Uppsala, Sweden) were used for column chromatography and size exclusion chromatography, respectively.Fungal material
The fungal strain Hypoxylon investiens J2 was isolated from the medicinal plant Blumea balsamifera collected from Danzhou, Hainan Province, People's Republic of China. It was identified based on its internal transcribed spacer sequence (Genbank no. MK757895). The fungus was deposited at the Tropical Crops Genetic Resources Institute, Chinese Academy of Tropical Agricultural Sciences CATAS, Hainan, People's Republic of China, and was maintained at −80 °C. For the large-scale fermentation, the fungus H. investiens J2 was cultured in rice culture (20 flasks each containing 80 g rice and 120 mL water) in an incubator at 28 ± 2 °C for one month.Extraction and isolation
The fermented material was extracted by ethyl acetate for three times. The organic solvent was evaporated to give a crude extract (15 g), which was then fractionated into six fractions (Fr.1–Fr.6) by column chromatography on silica gel. Fr.1 was separated by a Sephadex LH-20 column eluting with MeOH, following by HPLC (MeOH/H2O, 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23, 2 mL min−1) to afford compound 2 (15.0 mg, tR = 29.6 min). Fr.2 was directly purified by HPLC (MeOH/H2O, 77:23, 2 mL min−1) to give compounds 1 (20.0 mg, tR = 26.8 min), 3 (15.0 mg, tR = 24.0 min), and 4 (10.0 mg, tR = 27.3 min).
:23, 2 mL min−1) to afford compound 2 (15.0 mg, tR = 29.6 min). Fr.2 was directly purified by HPLC (MeOH/H2O, 77:23, 2 mL min−1) to give compounds 1 (20.0 mg, tR = 26.8 min), 3 (15.0 mg, tR = 24.0 min), and 4 (10.0 mg, tR = 27.3 min).
Antimicrobial assay
The disk diffusion method was applied to evaluate the antibacterial and antifungal activities of compounds 1–4.23 For bacteria, 200 μL inoculum suspension was spread on the nutrient agar plates. For fungi, the mycelia were first macerated with mortar and pestle to generate a homogeneous inoculum. In antibacterial assay, sterile paper disks containing 40 μL of the compounds with different concentrations (10, 5, 2, 1, 0.1 μg mL−1 in MeOH) were air-dried and then placed on inoculated plates. In antifungal test, paper disks were impregnated with 50 μg of the samples. The plates were incubated at 37 °C for 24 h for bacteria or at 28 °C for 48 h for fungi. Streptomycin was used as the positive control for antibacterial evaluation, while actidione was employed as reference for antifungal efficacy.QS inhibitory activity
The strain Chromobacterium violaceum CV026 was inoculated in a 20 mL LB broth media overnight to afford seed culture.24 0.2 mL of seed broth was mixed with 15 mL of molten LB agar media. Kanamycin (0.72 mg) and N-hexanoyl-L-homoserine-lactone (C6-HSL, 1.5 μg) were further added to the culture. Then, the agar was poured into a sterile Petri dish and then punched with a sterile cork borer. Compound at 40 μg mL−1 in MeOH was pipetted into each well. The positive control is furanone C30 at 10 μg mL−1. Finally, the Petri dish was incubated overnight at 37 °C.Cytotoxicity assay
The in vitro cytotoxic activities of compounds 1–4 were evaluated using the MTT method.25 The cancer cells were properly seeded in 96-well culture plates and then treated with different concentrations of compounds (40, 20, 10, 5, 2, 1 μM) for 24 h. After treatment, cells were incubated with MTT for 4 h. The plates were recorded at 570 nm by a plate reader. Adriamycin was applied as the positive control in the cytotoxicity assay.Conclusions
In summary, we isolated and characterized four new α-pyrones, hypotiens A–D (1–4), from a fungal endophyte Hypoxylon investiens J2 living in the medicinal plant Blumea balsamifera. Their structures were determined by extensive spectroscopic analyses. Compounds 1–4, as α-pyrone derivatives, possess an unusual dimethyl substitution in the highly unsaturated side chain. All compounds were measured for their antimicrobial, quorum sensing inhibitory, and cytotoxic activities but proved to be inactive. These results indicated that the specific side chain in compounds 1–4 might be responsible for the weak pharmacological activities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Hainan Provincial Natural Science Foundation of China (No. 219MS079) and Central Public-interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (No. 1630032019045) are gratefully acknowledged.Notes and references
- A. H. Aly, A. Debbab and P. Proksch, Pharmazie, 2013, 68, 499–505 CAS.
- S. Kusari, C. Hertweck and M. Spiteller, Chem. Biol., 2012, 19, 792–798 CrossRef CAS PubMed.
- H. W. Zhang, Y. C. Song and R. X. Tan, Nat. Prod. Rep., 2006, 23, 753–771 RSC.
- G. Li and H.-X. Lou, Med. Res. Rev., 2018, 38, 1255–1294 CrossRef PubMed.
- H. Gao, G. Li and H.-X. Lou, Molecules, 2018, 23, 646 CrossRef PubMed.
- R. H. Jiao, S. Xu, J. Y. Liu, H. M. Ge, H. Ding, C. Xu, H. L. Zhu and R. X. Tan, Org. Lett., 2006, 8, 5709–5712 CrossRef CAS PubMed.
- H. Geng and P.-Q. Huang, Chem. Rec., 2019, 19, 523–533 CrossRef CAS PubMed.
- J. Yao, J. Xiao, X. Wei and Y. Lu, Oncol. Lett., 2018, 16, 4671–4678 Search PubMed.
- B. Malgesini, B. Forte, D. Borghi, F. Quartieri, C. Gennari and G. Papeo, Chem.–Eur. J., 2009, 15, 7922–7929 CrossRef CAS PubMed.
- P. B. Ratnaweera, D. E. Williams, B. O. Patrick, E. D. de Silva and R. J. Andersen, Org. Lett., 2015, 17, 2074–2077 CrossRef CAS PubMed.
- R. Mohamad-Ramshan, P. B. Ratnaweera, D. E. Williams, E. D. de Silva and R. J. Andersen, J. Antibiot., 2019, 72, 246–251 CrossRef CAS PubMed.
- M. Zhou, M.-M. Miao, G. Du, X.-N. Li, S.-Z. Shang, W. Zhao, Z.-H. Liu, G.-Y. Yang, C.-T. Che, Q.-F. Hu and X.-M. Gao, Org. Lett., 2014, 16, 5016–5019 CrossRef CAS PubMed.
- G. Li, K. Xu, W.-Q. Chen, Z.-H. Guo, Y.-T. Liu, Y.-N. Qiao, Y. Sun, G. Sun, X.-P. Peng and H.-X. Lou, RSC Adv., 2019, 9, 12913–12920 RSC.
- H.-H. Wang, G. Li, Y.-N. Qiao, Y. Sun, X.-P. Peng and H.-X. Lou, Org. Lett., 2019, 21, 3319–3322 CrossRef CAS PubMed.
- M. G. Moloney, P. C. Trippier, M. Yaqoob and Z. Wang, Curr. Drug Discov. Technol., 2004, 1, 181–199 CrossRef CAS PubMed.
- T. F. Schäberle, Beilstein J. Org. Chem., 2016, 12, 571–588 CrossRef PubMed.
- L. Ding, L. Ren, S. Li, J. Song, Z. Han, S. He and S. Xu, Mar. Drugs, 2019, 17, 344 CrossRef PubMed.
- Z.-Y. Guo, L.-W. Lu, S.-S. Bao, C.-X. Liu, Z.-S. Deng, F. Cao, S.-P. Liu, K. Zou and P. Proksch, Phytochem. Lett., 2018, 28, 98–103 CrossRef CAS.
- H. Zhu, D. Li, Q. Yan, Y. An, X. Huo, T. Zhang, M. Zhang, C. Wang, M. Xia, X. Ma and Y. Zhang, Bioorg. Chem., 2019, 83, 129–134 CrossRef CAS PubMed.
- J. Werner, W. Ebrahim, F. C. Özkaya, A. Mándi, T. Kurtán, M. El-Neketi, Z. Liu and P. Proksch, Fitoterapia, 2019, 133, 80–84 CrossRef CAS PubMed.
- Y. Fan, Y. Liu, Y.-X. You, L. Rao, Y. Su, Q. He, F. Hu, Y. Li, W. Wei, Y.-K. Xu, B. Lin and C.-R. Zhang, Fitoterapia, 2019, 136, 104167 CrossRef CAS PubMed.
- P. Fu, P. Liu, Q. Gong, Y. Wang, P. Wang and W. Zhu, RSC Adv., 2013, 3, 20726–20731 RSC.
- G. Li, S. Kusari, M. Lamshöft, A. Schüffler, H. Laatsch and M. Spiteller, J. Nat. Prod., 2014, 77, 2335–2341 CrossRef CAS PubMed.
- H.-M. Zhang, C.-X. Ju, G. Li, Y. Sun, Y. Peng, Y.-X. Li, X.-P. Peng and H.-X. Lou, Mar. Drugs, 2019, 17, 383 CrossRef PubMed.
- G. Li, H. Wang, R. Zhu, L. Sun, L. Wang, M. Li, Y. Li, Y. Liu, Z. Zhao and H. Lou, J. Nat. Prod., 2012, 75, 142–147 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of compounds 1–4. See DOI: 10.1039/c9ra05308e |
| This journal is © The Royal Society of Chemistry 2019 |