
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA theoretical study on the formation and oxidation mechanism of hydroxyalkylsulfonate in the atmospheric aqueous phase
Danna Zhang a,
Guochun Lva,
Xiaomin Sun*a,
Chenxi Zhangb and
Zhiqiang Li*c
a,
Guochun Lva,
Xiaomin Sun*a,
Chenxi Zhangb and
Zhiqiang Li*c
aEnvironment Research Institute, Shandong University, Jinan 250100, China. E-mail: sxmwch@sdu.edu.cn
bCollege of Biological and Environmental Engineering, Binzhou University, Binzhou 256600, China
cCenter for Optics Research and Engineering (CORE), Shandong University, Qingdao 266237, China. E-mail: lzq@sdu.edu.cn
First published on 30th August 2019
Abstract
Hydroxymethanesulfonate (HMS) is an important organosulfur compound in the atmosphere. In this work, we studied the formation mechanism of HMS via the reaction of formaldehyde with dissolved SO2 using the quantum chemistry calculations. The results show that the barrier (9.7 kcal mol−1) of the HCHO + HSO3− reaction is higher than that (1.6 kcal mol−1) of the HCHO + SO32− reaction, indicating that the HCHO + SO32− reaction is easier to occur. For comparison, the reaction of acetaldehyde with dissolved SO2 also was discussed. The barriers for the CH3CHO + HSO3− reaction and CH3CHO + SO32− reaction are 16.6 kcal mol−1, 2.5 kcal mol−1, respectively. This result suggests that the reactivity of HCHO with dissolved SO2 is higher than that of CH3CHO. The further oxidation of CH2(OH)SO3− and CH3CH(OH)SO3− by an OH radical and O2 shows that the SO5˙− radical can be produced.
Introduction
Organosulfur compounds (OS), including organosulfates (ROSO3−), sulfones (RSO2R′), and sulfonates (RSO3−),1,2 have been identified to widely exist in fog, rainwater and in ambient atmosphere aerosols.3–6 These OS can be produced from marine sources including dimethylsulfide (DMS) emissions and oxidation of primary marine biomass,7,8 and their formation can be affected by aerosol acidity, relative humidity and concentration of nitrogen oxides (NOx).3,9,10 Hydroxymethanesulfonate (HMS) is one of the important organosulfur compounds and is a significant contributor to secondary aerosol formation.6 The other hydroxyalkylsulfonate species are considered to be less important than HMS.11 HMS has been misidentified as inorganic sulfate (SO42−) for a long time, which results in discrepancies between sulfate observation and model results. As a matter of fact, HMS is an important OS compound, and it may account for about 1/3 of the missing sulfate in Beijing winter haze aerosols.12The formation of HMS has been studied by many researchers. The reaction between SO2 and HCHO contributes to its formation. Wagner et al. proposed that the reaction rate was determined by both HSO3− and SO32− (HCHO + HSO3− → CH2(OH)SO3−; H2O + HCHO + SO32− → CH2(OH)SO3− + OH−).13 But in subsequent study, Peter et al. disagreed with Wagner's conclusion and thought that termolecular process (H2O + HCHO + SO32− → CH2(OH)SO3− + OH−) was impossible, and the kinetics should be explained by such reactions: HCHO + HSO3− → CH2(OH)SO3−; HSO3− ↔ SO32− + H+. They believed that only HSO3− could react with HCHO and determine the reaction rate.14 However, the experiment using spectrophotometer showed that the SO32− reacts rapidly with HCHO.15 In the spectrophotometric study on the reaction between dissolved SO2 and HCHO, it can be concluded that HSO3− and SO32− can react with HCHO, and the rate for SO32− is higher than HSO3− obviously.16 In a recent research, the important organosulfur compound (HMS) was been investigated for its important role in haze aerosols.12 And this paper proposed a potentially HMS chemical mechanism. According to the analysis above, the chemical formation of HMS can be illustrated as:16,17
| SO2·H2O ↔ HSO3− + H+ | (1) |
| HSO3− ↔ SO32− + H+ | (2) |
| HCHO + HSO3− ↔ CH2(OH)SO3− | (3) |
| HCHO + SO32− ↔ CH2(O−)SO3− | (4) |
| CH2(OH)SO3− ↔ CH2(O−)SO3− + H+ | (5) |
| CH2(OH)SO3H ↔ CH2(OH)SO3− + H+ | (6) |
Although the HMS formation mechanism has been proposed in many researches, there is no theoretical calculation on it. Thus, in order to confirm whether the mechanism is reliable, theoretical calculation is necessary to be done.
In this paper, we investigated the reaction HCHO with HSO3− and SO32− using quantum chemical calculations. In order to understand the influence of different aldehydes for the reactions, we also discussed the reaction between CH3CHO and HSO3− or SO32−. Besides, the total rate constant (ktotal, M−1 s−1) for the individual reaction pathway within the range of 200 to 298 K were calculated. The further oxidation of the reaction products (CH2(OH)SO3− and CH3CH2(OH)SO3−) by OH radical and O2 also be talked about.
Computational methods
The Gaussian-09 suite of programs was used to perform all the quantum chemistry calculations described in this paper. The density functional theory (DFT) was used for calculations.18 All the geometrical structures (including reactants, pre-reactive complexes, transition states and products) calculated in this paper were optimized using the M06-2X functional at the 6-311++G(d,p) basis set.19,20 Vibrational frequencies were calculated at the same level of theory to ascertain the local minimum points and the transition states, which supposed to have zero and one imaginary frequency. We also performed intrinsic reaction coordinate (IRC)21 calculations in order to prove whether the transition state we found were the correct. Single-point energies were refined using the CCSD(T) method22 with the aug-cc-pVTZ basis set.23–25 Very recently, many high level quantum chemical methods have been used for the atmosphere reactions in order to obtain more reliable and excellent intrinsic accuracy.26–28 In this article, under the consider of computational speed and accuracy, we think the dual-level strategy (CCSD(T)/aug-cc-pVTZ//M06-2X/6-311++G(d, p) level of theory) is appropriate. M06-2X functional has been widely used in theoretical calculation and can be better for ionic hydrogen-bonding interactions and identifying the global minimum conformer.29–36 Been considered as the “gold standard” of quantum chemistry, CCSD(T) method was widely been used for reactions of organic matters and has high level accuracy. Thus we choose M06-2X functional at the 6-311++G(d,p) basis set for geometrical structures optimization and CCSD(T) method with the aug-cc-pVTZ basis set for single point energy calculation in this paper.22,37–40 The Gibbs free energies were calculated by the following equation: G = E (single-point energy) + Gcorr (thermal correction to Gibbs free energy). The geometries were drawn using the CYLview software package.41For the kinetics analysis, the conventional transition-state theory (TST)42 with Wigner tunneling correction was used to calculate the rate constants. All rate constants were calculated by using the KiSThelP program.43
Results and discussion
Reaction of HCHO with dissolved SO2
It aqueous phase, dissolved SO2 can be dissociated to form HSO3− and SO32−. Thus, we focus on the reactions of HSO3− + HCHO and SO32− + HCHO.As shown in Fig. 1(a), when HCHO approaches HSO3−, the reaction occurs via a five-membered cyclic prereactive complex HCHO⋯HSO3− (C1). C1 is regarded as the initial step of the HCHO and HSO3− reaction because C1 is connected with the transition state TS1. The C1 is held together through a hydrogen bond and an electron donor–acceptor type of interaction between the two molecules. The change of the distances of Cb⋯Od and Sc⋯Ce are all shorten from 2.11 Å (C1) to 1.70 Å (TS1) and 3.24 Å (C1) to 2.11 Å (TS1), respectively. The reaction proceeds via a transition state TS1 with a barrier of 9.7 kcal mol−1 to produce CH2(OH)SO3− (HMS).
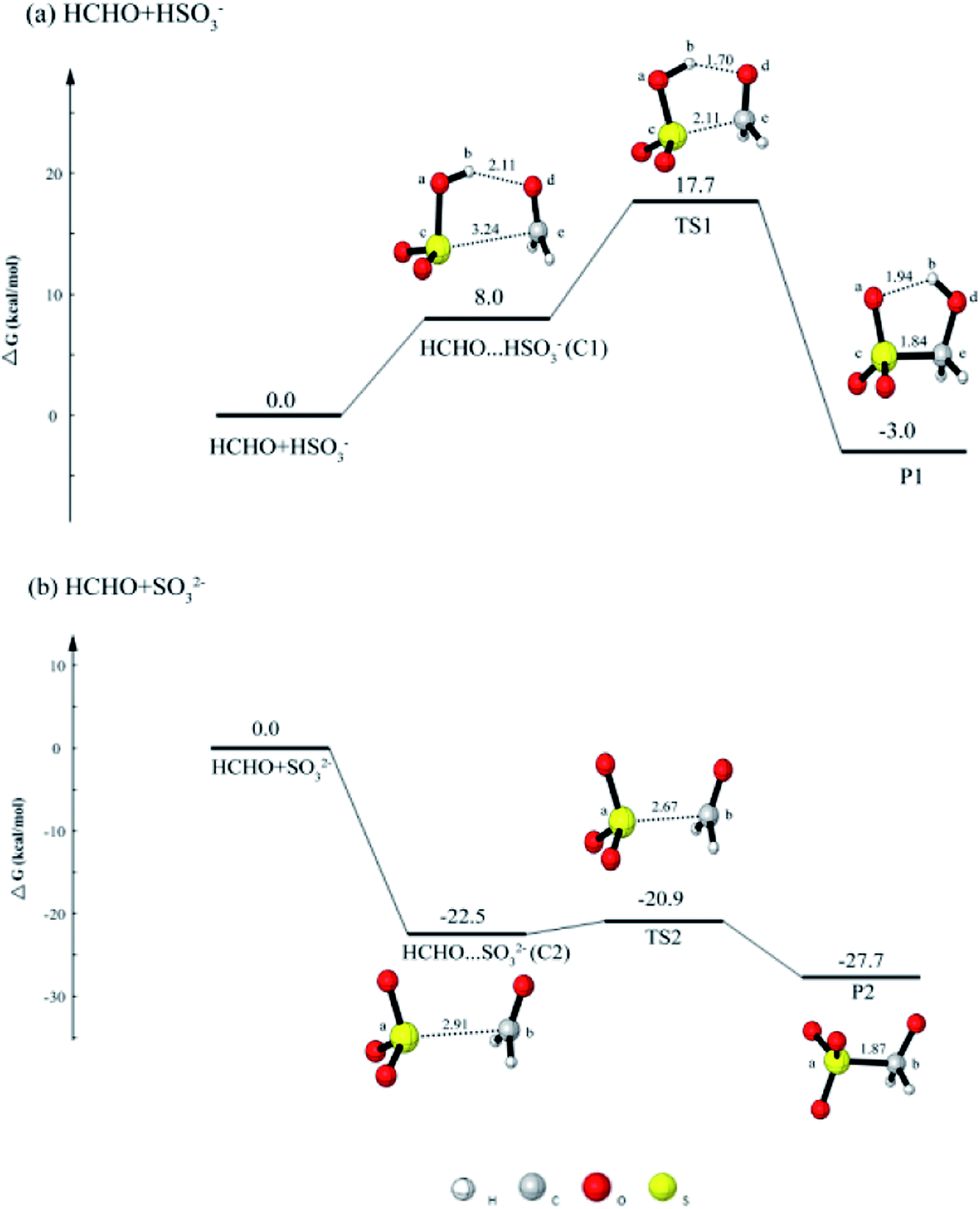 | ||
| Fig. 1 The calculated free energy diagram for (a) the HCHO + HSO3− reaction, and for (b) the HCHO + SO32− reaction calculated at the CCSD(T)/aug-cc-pVTZ//M06-2X/6-311++G(d, p) level of theory. | ||
When SO32− reacts with HCHO, the process is similar with the HCHO + HSO3− reaction. From Fig. 1(b), it can be found that the free energy of the complex C2 is 22.5 kcal mol−1 lower than reactants. Similar with C1, C2 is regarded as the initial step of the HCHO and SO32− reaction. The complex C2 is held together by one van der Waals interaction. The change of distance between Sa⋯Cb is from 2.91 Å to 2.67 Å. The result is contrary to that of the HCHO + HSO3−, in which the higher free energy of the C1 can be observed. Once the C2 is formed, it can easily transform to product CH2(O−)SO3− because the free energy of transition state TS2 is only 1.6 kcal mol−1 higher than the complex C2.
According to the analysis above, it is clear that the SO32− is more likely to react with HCHO. However, the dissolved SO2 exist in the form of HSO3− in the pH range of 2–7,44 and the acidic condition of aerosol particles can be found in Beijing winter haze.45 Thus, the HSO3− predominates in aerosol particles of Beijing winter haze so as to that the main reaction is HCHO + HSO3− in these aerosol particles. And only the further oxidation of HMS will be discussed in the latter part.
Reaction of CH3CHO with dissolved SO2
In order to study the effect of different aldehydes on the reaction, we also discussed the reaction between CH3CHO with dissolved SO2. As shown in Fig. 2(a), CH3CHO + HSO3− reaction is firstly considered. The reaction is initiated with the formation of a five-ringlike structure complex CH3CHO⋯HSO3− (C3), followed by the formation of a transition state TS3 to produce CH3CH(OH)SO3− (HES). The complex C3 is held together by one hydrogen bond and one van der Waals interaction. As shown in Fig. 2(a), the hydrogen atom (Hb) of HSO3− interacts with the oxygen atom (Od) of CH3CHO to form one hydrogen bond, and the sulfur atom (Sc) of HSO3− reacts with the carbon atom (Ce) of CH3CHO. It is obviously that the distances between Sc⋯Ce and Hb⋯Od are all shorten. The barrier in this reaction is 16.6 kcal mol−1, which is larger than that of HCHO + HSO3− reaction.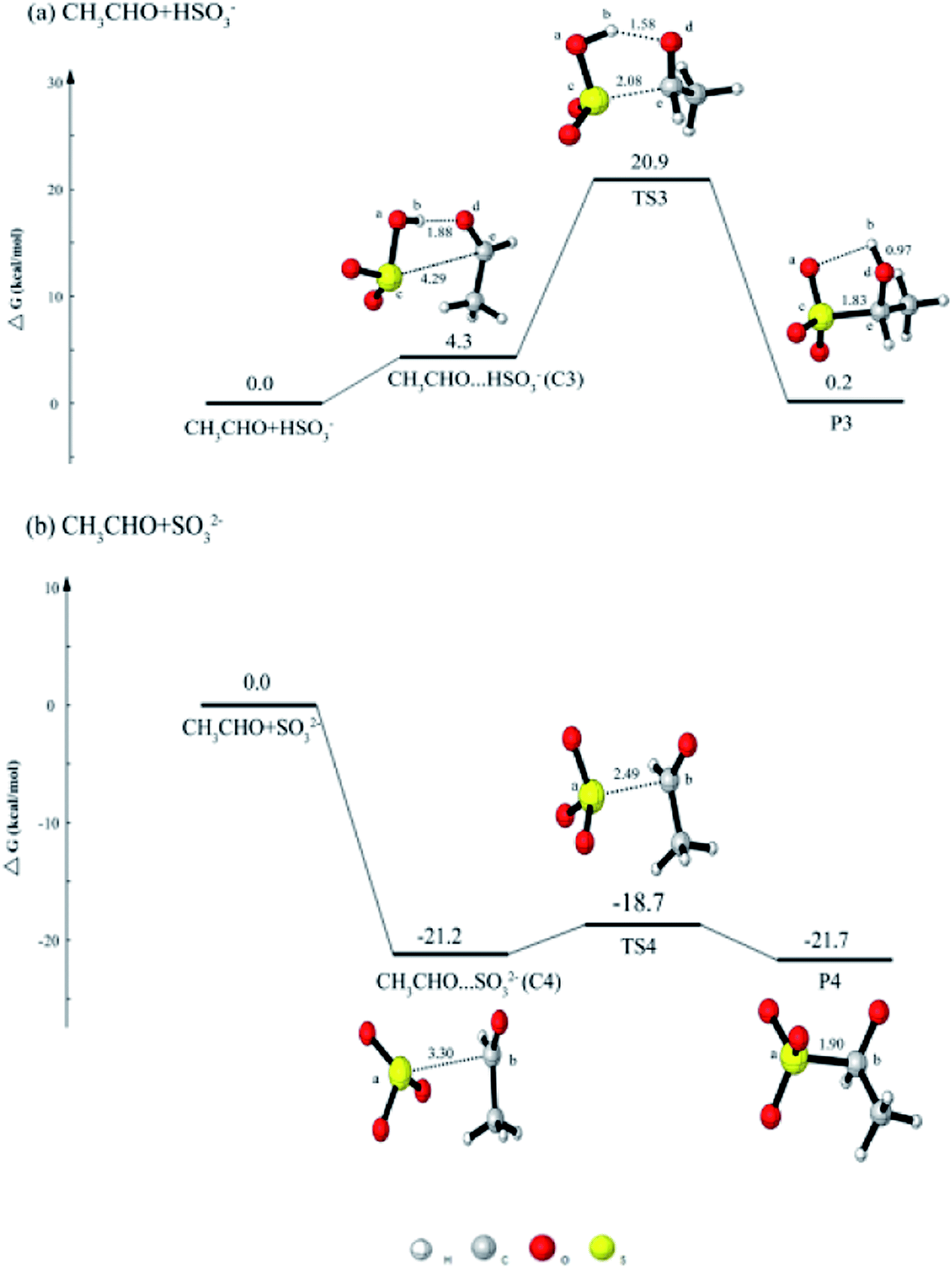 | ||
| Fig. 2 The calculated free energy diagram for (a) the CH3CHO + HSO3− reaction, and for (b) the CH3CHO + SO32− reaction calculated at the CCSD(T)/aug-cc-pVTZ//M06-2X/6-311++G(d, p) level of theory. | ||
For CH3CHO + SO32− reaction in Fig. 2(b), the formed complex C4 is also lower in energy than the reactants, which is similar with the HCHO + SO32− reaction. The product CH3CH(O−)SO3− can be produced from the C4. The complex C4 is held together by one van der Waals interaction. As shown in Fig. 2(b), and sulfur atom (Sa) is involved in the formation of van der Waals interaction with a carbon atom (Cb) of HSO3−. The distance between Sa and Cb changes from 3.30 Å (C4) to 2.49 Å (TS4). The process needs to cross a transition state TS4 and to overcome the barrier of 2.5 kcal mol−1, which is higher than that in HCHO + SO32− reaction.
On the basis of the results, it can be concluded that the reactivity of HCHO with dissolved SO2 is higher than that of another aldehydes with dissolved SO2.
Rate calculation
In terms of the theoretical results discussed above, the reactions occur through a two-step mechanism, involving firstly a fast pre-equilibrium between the reactants and a pre-reactive complex, and the irreversible formation of the products, which can be characterized by eqn (7) and (8).
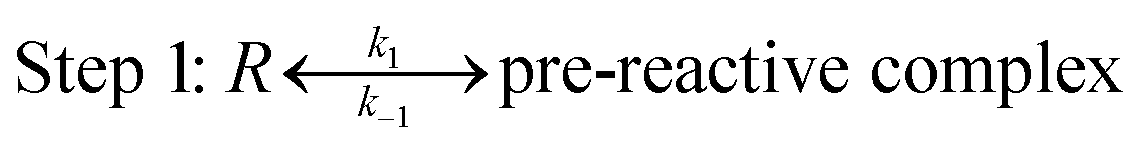 | (7) |
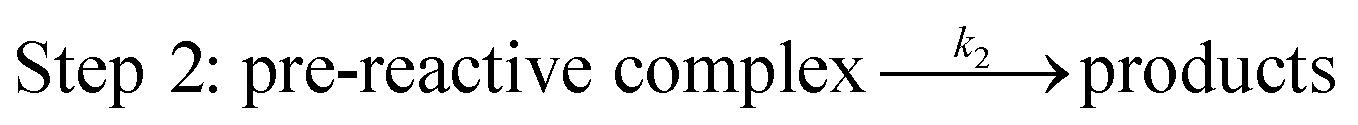 | (8) |
In the above reactions, k1 is the kinetic rate constant characterizing the forward bimolecular reaction step (in cm3 per molecule per s), whereas k−1 and k2 represent the backward and forward unimolecular reaction rate constants (in s−1). A steady-state analysis of the total reaction pathway's rate constant is formulated as:
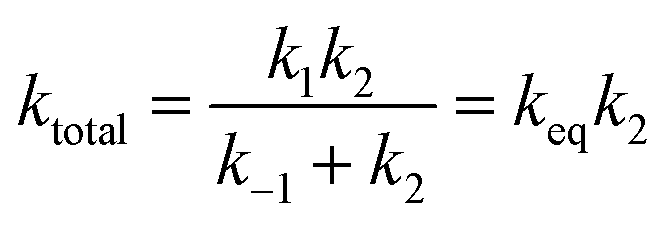 | (9) |
| 200 K | 220 K | 240 K | 260 K | 280 K | 298 K | |
|---|---|---|---|---|---|---|
| HCHO + HSO3− → CH2(OH)SO3− | 4.00 × 10−3 | 1.70 × 10−2 | 6.06 × 10−2 | 1.73 × 10−1 | 4.27 × 10−1 | 8.60 × 10−1 |
| HCHO + SO32− → CH2(O−)SO3− | 3.21 × 106 | 2.74 × 106 | 2.42 × 106 | 2.20 × 106 | 2.04 × 106 | 1.93 × 106 |
| CH3CHO + HSO3 → CH3CH(OH)SO3− | 3.52 × 10−6 | 2.41 × 10−5 | 1.20 × 10−4 | 4.69 × 10−4 | 1.52 × 10−3 | 3.80 × 10−3 |
| CH3CHO + SO32− → CH3CH(O−)SO3− | 3.96 × 102 | 5.85 × 102 | 8.23 × 102 | 1.10 × 103 | 1.43 × 103 | 1.76 × 103 |
The rate constants of HCHO + SO32− are ∼109 to 107 times than that of HCHO + HSO3−, whereas rate constants of CH3CHO + SO32− is 8–6 orders of magnitude larger than that of CH3CHO + HSO3− within range of 200–298 K. These results show that the reaction between aldehydes and SO32− is faster, which is consistent with the analysis above. For HCHO and CH3CHO, it is clear that the rate constants between HCHO and HSO3− or SO32− are larger than that between CH3CHO and HSO3− or SO32−, which is coincident with the discussion above.
Although the reaction between aldehydes and SO32− is faster, HSO3− is the main form of dissolved SO2 in the aerosol particles, leading to that the reaction of aldehydes with HSO3− predominates in the aqueous phase of aerosol particles. Thus, in the next part, only CH2(OH)SO3− and CH3CH(OH)SO3− will be talked about.
Oxidation of HMS and HES
As the oxidation of HMS is of great importance, it is meaningful and necessary to investigate the oxidation mechanism. Previous research46 has investigated the oxidation of HMS with H2O2 and O3, there was no calculation on the oxidation by OH radical. Thus, we will calculate the HMS + OH reaction so as to confirm whether peroxysulfate radicals (SO5˙−) can be produced.Fig. 3(a) shows the potential energy profile for CH2(OH)SO3− + OH reaction. In the reaction process, HMS firstly reacts with OH radical to form the complex CH2(OH)SO3−···OH (C5) with the free energy release of 35.2 kcal mol−1. The C5 can evolve via TS5 with a barrier of 29.9 kcal mol−1 into CH2(OH)2 and SO3˙−. The SO3˙− can continue to react with O2 to produce SO5˙−. The addition reaction between SO3˙− and O2 can occur via a transition state TS6 with the barrier of 12.2 kcal mol−1.
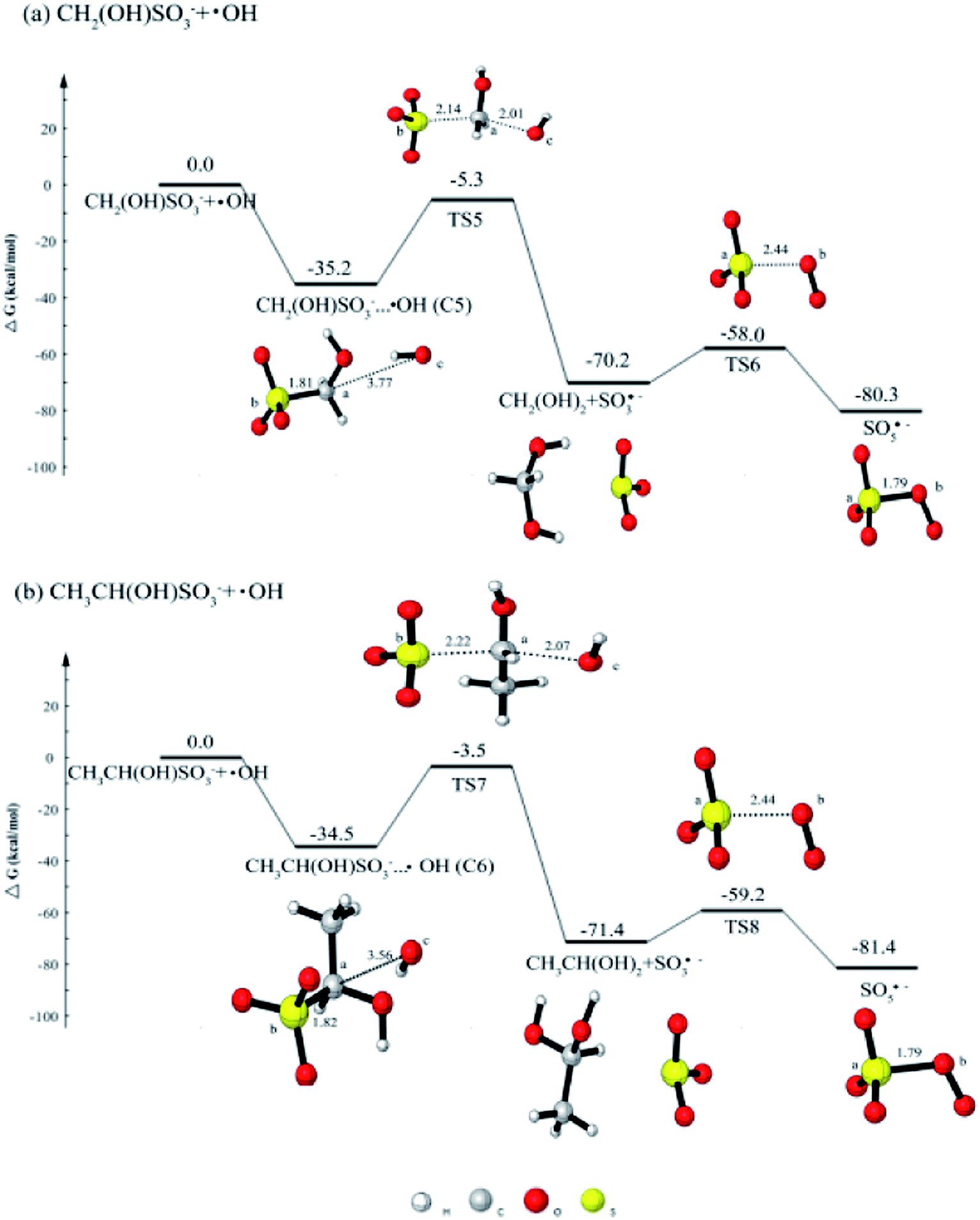 | ||
| Fig. 3 The calculated free energy diagram for (a) the CH2(OH)SO3− + ·OH reaction, and for (b) the CH3CH(OH)SO3− + ·OH reaction calculated at the CCSD(T)/aug-cc-pVTZ//M06-2X/6-311++G(d, p) level of theory. | ||
The further oxidation mechanism of CH3CH(OH)SO3− (HES) by OH radical is similar with that of HMS by OH radical. As can be seen from Fig. 3(b), OH radical is added to HES to produce the complex CH3CH(OH)SO3−···OH (C6). The reaction proceeds via a transition state TS7 with a barrier of 31.0 kcal mol−1. The formed SO3˙− in this reaction also react with O2 to form SO5˙−, and the process has the same barrier of 12.2 kcal mol−1.
Conclusions
HMS is the major OS species. In this paper, we investigated the formation of HMS using the quantum chemical calculations. Besides, other aldehydes like acetaldehyde also exist in the atmosphere. The similar structure between formaldehyde and acetaldehyde makes us to think the effect of different aldehydes on their reaction with dissolved SO2. Thus, the reaction between acetaldehyde and dissolved SO2 also was discussed.The result shows that the energy barrier for CH3CHO + HSO3− reaction is 16.6 kcal mol−1, which is a little higher than 9.7 kcal mol−1 for reaction of HCHO with HSO3−. The barrier (2.5 kcal mol−1) of CH3CHO + SO32− reaction is larger than that (1.6 kcal mol−1) of HCHO and SO32− reaction. These results indicate that the reaction of aldehydes with SO32− is easier than that with HSO3−. However, the HSO3− is the main form of dissolved SO2 in the aerosol particles, leading to that the aldehydes + HSO3− reaction dominates. Thus, the main products are CH2(OH)SO3− and CH3CH(OH)SO3−. Their further oxidation by OH radical and O2 shows that the SO3˙− and SO5˙− radical can be formed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Natural Science Foundation of China (21607011), Natural Science Foundation of Shandong Province (ZR2018MB043), The Fundamental Research Funds of Shandong University (2018JC027) and Focus on Research and Development Plan in Shandong Province (2019GSF109037).Notes and references
- D. J. Eatough and L. D. Hansen, Sci. Total Environ., 1984, 36, 319–328 CrossRef CAS.
- R. W. Dixon and H. Aasen, Atmos. Environ., 1999, 33, 2023–2029 CrossRef CAS.
- J. D. Surratt, J. H. Kroll, T. E. Kleindienst, E. O. Edney, M. Claeys, A. Sorooshian, N. L. Ng, J. H. Offenberg, M. Lewandowski, M. Jaoui, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2007, 41, 517–527 CrossRef CAS PubMed.
- Y. Iinuma, C. Müller, T. Berndt, O. Böge, M. Claeys and H. Herrmann, Environ. Sci. Technol., 2007, 41, 6678–6683 CrossRef CAS.
- J. D. Blando, R. J. Porcja, T.-H. Li, D. Bowman, P. J. Lioy and B. J. Turpin, Environ. Sci. Technol., 1998, 32, 604–613 CrossRef CAS.
- M. P. Tolocka and B. Turpin, Environ. Sci. Technol., 2012, 46, 7978–7983 CrossRef CAS PubMed.
- R. J. Charlson, J. E. Lovelock, M. O. Andreae and S. G. Warren, Nature, 1987, 326, 655–661 CrossRef CAS.
- M. Claeys, W. Wang, R. Vermeylen, I. Kourtchev, X. Chi, Y. Farhat, J. D. Surratt, Y. Gómez-González, J. Sciare and W. Maenhaut, J. Aerosol Sci., 2010, 41, 13–22 CrossRef CAS.
- J. Liao, K. D. Froyd, D. M. Murphy, F. N. Keutsch, G. Yu, P. O. Wennberg, J. M. St. Clair, J. D. Crounse, A. Wisthaler, T. Mikoviny, J. L. Jimenez, P. Campuzano-Jost, D. A. Day, W. Hu, T. B. Ryerson, I. B. Pollack, J. Peischl, B. E. Anderson, L. D. Ziemba, D. R. Blake, S. Meinardi and G. Diskin, J. Geophys. Res.: Atmos., 2015, 120, 2990–3005 CAS.
- D. D. Huang, Y. J. Li, B. P. Lee and C. K. Chan, Environ. Sci. Technol., 2015, 49, 3672–3679 CrossRef CAS PubMed.
- T. M. Olson and M. R. Hoffmann, Atmos. Environ., 1989, 23, 985–997 CrossRef CAS.
- S. Song, M. Gao, W. Xu, Y. Sun, D. R. Worsnop, J. T. Jayne, Y. Zhang, L. Zhu, M. Li, Z. Zhou, C. Cheng, Y. Lv, Y. Wang, W. Peng, X. Xu, N. Lin, Y. Wang, S. Wang, J. W. Munger, D. J. Jacob and M. B. McElroy, Atmos. Chem. Phys., 2019, 19, 1357–1371 CrossRef CAS.
- C. Wagner, Ber. Dtsch. Chem. Ges., 1929, 62, 2873–2877 CrossRef.
- P. Jones and K. Oldham, The theory of the formaldehyde clock reaction, 1963 Search PubMed.
- P. Bell Ronald and P. G. Evans, Proc. R. Soc. London, Ser. A, 1966, 291, 297–323 CrossRef.
- S. D. Boyce and M. R. Hoffmann, J. Phys. Chem., 1984, 88, 4740–4746 CrossRef CAS.
- J. W. Munger, D. J. Jacob, J. M. Waldman and M. R. Hoffmann, J. Geophys. Res., 1983, 88, 5109–5121 CrossRef CAS.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- K. Fukui, in Frontier Orbitals and Reaction Paths, World Scientific, 1997, vol. 7, pp. 471–476 Search PubMed.
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef CAS.
- A. K. Wilson, T. van Mourik and T. H. Dunning, J. Mol. Struct.: THEOCHEM, 1996, 388, 339–349 CrossRef CAS.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 CrossRef CAS.
- A. Godfrey-Kittle and M. Cafiero, Int. J. Quantum Chem., 2006, 106, 2035–2043 CrossRef CAS.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6135–6140 CrossRef CAS PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2019, 141, 611–617 CrossRef CAS PubMed.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS PubMed.
- D. S. Huh and S. J. Choe, J. Porphyrins Phthalocyanines, 2010, 14, 592–604 CrossRef CAS.
- H. A. Rypkema, A. Sinha and J. S. Francisco, J. Phys. Chem. A, 2015, 119, 4581–4588 CrossRef CAS PubMed.
- B. Long, X.-F. Tan, C.-R. Chang, W.-X. Zhao, Z.-W. Long, D.-S. Ren and W.-J. Zhang, J. Phys. Chem. A, 2013, 117, 5106–5116 CrossRef CAS PubMed.
- S. Pari, I. A. Wang, H. Liu and B. M. Wong, Environ. Sci.: Processes Impacts, 2017, 19, 395–404 RSC.
- X. Shi, R. Zhang, Y. Sun, F. Xu, Q. Zhang and W. Wang, Phys. Chem. Chem. Phys., 2018, 20, 1005–1011 RSC.
- N. Rashidian, E. Zahedi and A. Shiroudi, Sci. Total Environ., 2019, 679, 106–114 CrossRef CAS PubMed.
- A. S. M. Gad El-Hak, A. A. K. Mohammed, A. F. Abdel Hakiem and R. M. Mahfouz, Spectrochim. Acta, Part A, 2019, 222, 117200 CrossRef CAS PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- P. R. Nagy, G. Samu and M. Kállay, J. Chem. Theory Comput., 2018, 14, 4193–4215 CrossRef CAS PubMed.
- M. B. Oviedo, N. V. Ilawe and B. M. Wong, J. Chem. Theory Comput., 2016, 12, 3593–3602 CrossRef CAS PubMed.
- J. Řezáč and P. Hobza, Chem. Rev., 2016, 116, 5038–5071 CrossRef PubMed.
- C. Y. Legault, CYLview 1.0 b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- S. Canneaux, F. Bohr and E. Henon, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- R. Zhang, G. Wang, S. Guo, M. L. Zamora, Q. Ying, Y. Lin, W. Wang, M. Hu and Y. Wang, Chem. Rev., 2015, 115, 3803–3855 CrossRef CAS PubMed.
- S. Song, M. Gao, W. Xu, J. Shao, G. Shi, S. Wang, Y. Wang, Y. Sun and M. B. McElroy, Atmos. Chem. Phys., 2018, 18, 7423–7438 CrossRef CAS.
- J. W. Munger, C. Tiller and M. R. Hoffmann, Science, 1986, 231, 247 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |