
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of miR-16 enhances the sensitivity of fibroblast-like synovial cells to methotrexate by restraining MDR1/P-gp expression via NF-κB pathway†
Jing Wangab,
Ni Maob,
Yiming Liub,
Xi Xieb,
Jing Tianb,
Fen Lib and
Jinwei Chen *b
*b
aDepartment of Rheumatology and Immunology, The First People's Hospital of Yunnan Province, Kunming 650034, Yunnan Province, China
bDepartment of Rheumatology and Immunology, The Second Xiangya Hospital of Central South University, No. 139 Middle Renmin Road, Changsha 410011, Hunan Province, China. E-mail: xieiv31@126.com; Fax: +86-731-85533525; Tel: +86-731-85295888
First published on 27th August 2019
Abstract
MicroRNAs (miRNAs) are demonstrated to contribute to the regulation of drug resistance in a number of diseases. Nevertheless, little is known about the role and the underlying mechanism of miR-16 in rheumatoid arthritis (RA) methotrexate resistance. In this study, we firstly examined the miR-16 expression in the serum and synovial fluid from RA patients who were unresponsive to methotrexate monotherapy (UR-MTX patients) and responsive RA patients (R-MTX patients). Secondly, the miR-16 expression was measured in both fibroblast-like synovial cells (FLS) and methotrexate resistance RA-FLS cells (FLS-MTX). FLS cells used in this study were isolated from synovial tissue specimens obtained from patients with RA who underwent total joint replacement. FLS-MTX cells were conducted by gradually increasing the concentration of methotrexate in the medium. The construction of FLS-MTX cells was confirmed by 3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide (MTT) assay. Thirdly, in order to further investigate the role of miR-16 in FLS-MTX cells, we introduced miR-16 inhibitor into FLS-MTX cells to knockdown the expression of miR-16, used fluorescence quantitative PCR to detect the inhibition efficiency. The effects of miR-16 inhibition on cell viability, cell cycle arrest and apoptosis in FLS-MTX cells were monitored with MTT and flow cytometry analysis, respectively. And the regulation of miR-16 on P-glycoprotein (P-gp) was performed using qRT-PCR, western blotting, and immunofluorescence staining. Fourthly, ammonium pyrrolidinedithiocarbamate (PDTC), a NF-κB pathway inhibitor, was applied to verify the mechanism by which miR-16 involved in to regulate the P-gp expression, and thus contributing to the methotrexate resistance in FLS-MTX cells. MiR-16 was upregulated in the in serum and synovial fluid from UR-MTX patients as well as in FLS-MTX cells. Inhibition of miR-16 re-sensitized the FLS-MTX cells to methotrexate by suppressing the cell viability, cell promoting cycle arrest at G0/G1 phase and enhancing apoptosis. Knockdown of miR-16 significantly reduced MDR1 mRNA expression and P-gp protein expression in FLS-MTX cells. Furthermore, inhibition of NF-κB pathway by PDTC reinforced the effect of miR-16 knockdown on P-gp expression, cell viability, cell cycle arrest and apoptosis. In conclusion, our study illustrated that inhibition of miR-16 in FLS-MTX cells alleviated methotrexate resistance by inhibiting MDR1/P-gp expression through inactivation of the NF-κB pathway.
1. Introduction
Rheumatoid arthritis (RA) is a serious autoimmune disease characterized by chronic multi-joint inflammation with pain and swelling. The persistent development of rheumatoid arthritis can lead to cartilage and bone damage, which can finally result in irreversible deformity and disability.1 Though the incidence of RA has decreased significantly in recent decades, the prevalence of RA worldwide remains at 0.5% to 1.1%.2 The key to effectively control the development of RA is early diagnosis and timely establishment of effective and active treatment. Methotrexate is a competitive inhibitor of dihydrofolate reductase, and low-dose methotrexate is recommended as the first-line pharmacotherapy for patients newly diagnosed with RA.3 However, there are significant differences in the efficacy of methotrexate among individual RA patients. Evidence suggests that 30% to 50% of RA patients are unresponsive/resistant to methotrexate treatment.4,5 So far, the mechanisms for unresponsiveness to methotrexate treatment are complex and unclear. It has been proved that TGF-β signaling defect induced low expression of CD39 on Tregs could be a noninvasive biomarker for identifying MTX-resistant RA patients.6,7 Hence, understanding the methotrexate resistance would be of great importance and valuable to develop more effective therapies for RA patients.MicroRNAs (miRNAs) are endogenous non-coding single-stranded small RNAs that have been widely found in eukaryotic cells in recent years.8 MiRNAs regulate post-transcriptional gene expression by binding to the 3′ untranslated region (3′ UTR) of target gene, thereby inhibiting the specific gene expression.9 Mounting evidences prove that dysregulation of miRNAs is involved in chemotherapeutic resistance in various diseases. miR-204 increases the sensitivity to arsenic trioxide in acute myeloid leukemia cells through enhancing cell apoptosis mediated by BIRC6/p53.10 Restoration of miR-431 in vitro reduces the half maximal inhibitory concentrations of doxorubicin and mitotane, and notably promotes the cell apoptosis in adrenocortical carcinoma.11 MicroRNA-383 restrains doxorubicin resistance in hepatocellular carcinoma by targeting eukaryotic translation initiation factor 5A2.12 Previous studies illustrated that miR-16 were increased in the sera of established RA patients in comparison with early RA.13,14 MiR-16 were shown to be overexpressed at the systemic level: in both the periphery and RA joints.15 Upregulation of miR-16 is demonstrated to be involved in Th17/Treg cell imbalance in patients with RA.16 miR-16 was suggested as a biomarker of RA.17 Therefore, we could conclude that miR-16 plays an important role in the occurrence and development of RA. Nevertheless, little work has been conducted to explore the relationship between miR-16 and methotrexate resistance in RA.
NF-κB pathway was reported to be involved in RA progression. Carnosic acid could inhibit NF-κB phosphorylation, thus cause the down-regulation of pro-inflammatory cytokines in RA.18 CD147 was capable of preventing TNF-induced apoptosis of FLS in RA by promoting the IKK/IκB/NF-κB signaling pathway.19 Insulin-like growth factor binding protein 3 reduced the inflammatory response and bone destruction by blocking NF-κB activation and inducing apoptosis in RA FLS.20 But whether NF-κB contributed to resistance in RA remains unknow.
Here, we observed that the expression of miR-16 was elevated in serum and synovial fluid of RA patients who were unresponsive to methotrexate monotherapy (UR-MTX patients) and constructed methotrexate-resistant fibroblast-like synovial cells (FLS-MTX). Inhibition of miR-16 re-sensitized the FLS-MTX cells to methotrexate treatment. Moreover, knockdown of miR-16 significantly reduced P-gp expression at mRNA and protein levels in FLS-MTX cells. Additionally, inactivation of NF-κB pathway by PDTC reinforced the effect of miR-16 knockdown on re-sensitizing the FLS-MTX cells to methotrexate treatment.
2. Materials and methods
2.1 Patient samples collection
Altogether 64 patients diagnosed with RA according to the criteria of the American College of Rheumatology were recruited between July 2016 and December 2017 from the Department of Rheumatology and Immunology, The Second Xiangya Hospital of Central South University. All patients were newly diagnosed with RA, and before sample collection, all patients were treated with methotrexate monotherapy (15–20 mg per week) for at least four weeks. DAS28 (Disease Activity Score, including a 28-joint count) was applied to assess the RA activity. When DAS28 > 4.0, patients were stratified to be unresponsive RA patients (UR-MTX, n = 32); and when DAS28 < 3.0, patients were classified into responsive RA patients (R-MTX, n = 32). No other drugs were used before or during sample collection. All related experiments were performed in accordance with the Guidelines of Declaration of Helsinki, and Experiments were approved by the ethics committee of The Second Xiangya Hospital of Central South University. Informed consents were obtained from human participants of this study.2.2 Cell culture and treatment
FLS were isolated from synovial tissue specimens obtained from an unresponsive RA patient who underwent total joint replacement. Fresh synovial tissue was cut into small pieces and digested with tissue enzyme digestion solution, following incubation with RPMI 1640 medium at 37 °C for six hours. Then, cells were maintained in complete RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). Cell culture medium was replaced every five days, and unadhered cells nonadherent cells were discarded. RA-FLS cells were grown, and cells at four to eight passages were used for the following experiments.RA-FLS cells obtained above were used to construct methotrexate resistance cells. RA-FLS cells were cultured in complete RPMI 1640 medium supplemented with 10% FBS overnight, then the culture medium was replaced with complete medium added with 0.01 μg mL−1 methotrexate to induce FLS cells to MDR following a gradual increase in the drug concentrations to 1.5 μg mL−1. After eight months, the cells were able to steadily grow in the culture medium containing 1.5 μg mL−1 methotrexate, and were named as FLS-MTX cells.
2.3 RNAs and cell transfection
MiR-16 inhibitor and its corresponding negative controls (NC) were derived from Gene-Pharma (Shanghai, China). MiR-16 inhibitor or NC was transfected into FLS-MTX cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.2.4 3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide (MTT) assay
RA-FLS and FLS-MTX cells were seeded in quintuplicate at the density of 1 × 104 per well in 96-well plates and incubated overnight. And then cells were treated to with indicated concentrations of methotrexate. After incubating for 24 hours, 20 μL of MTT (Sigma, St. Louis, USA) solution (5 mg mL−1) was added into each well, and incubated for another 4 hours. Cell supernatants were carefully removed after centrifugation, and cells in each well were dissolved in 200 μL of dimethyl sulfoxide (DMSO). Optical density (OD) was measured at 570 nm with a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). Cell viability was calculated according to the following formulas: (OD value of the experimental group/OD value of the control group) × 100%.2.5 Cell cycle attest
Cells were centrifuged and collected 48 hours after transfection, and then cells were washed thrice with pre-cooled phosphate buffer saline (PBS) and fixed with 70% ethanol at 4 °C for 12 hours. Cells were washed with PBS again. Straight after, cells were incubated in 0.5 mL RNase A (50 μg mL−1) for 30 minutes at 37 °C, and propidium iodide (PI) (50 μg mL−1) was added and incubated for 30 minutes in the dark. Finally, cells were analyzed with flow cytometry analysis (Coulter Epics XL).2.6 Cell apoptosis analysis
Cells were digested with trypsin (without EDTA), washed twice with PBS and centrifuged. Cells (3 × 105 cells per mL) were then resuspended in 500 μL binding buffer (7SeaPharmTech, Shanghai, China), 5 μL Annexin V-EGFP (7SeaPharmTech) and 5 μL PI (7SeaPharmTech) was added into cells for incubation for fifteen minutes at room temperature in the dark. And then the cells were analyzed by flow cytometry (guava easyCyte HT; EMD Millipore, Billerica, MA, USA).2.7 Immunofluorescence staining
Cells were planted on cover slips and incubated in 24-well plates overnight. Before fixing with pre-cold 4% paraformaldehyde for twenty minutes, cells were washed twice with PBS softly. And then cells were permeabilized with 0.2% Triton X-100 in PBS for 10 minutes. After blocking with BSA for 30 minutes, cells were incubated with P-Glycoprotein Monoclonal Antibody (F4), (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100; Thermo Fisher scientific) for overnight at 4 °C, followed by incubation with secondary antibody conjugated with FITC for 2 hours at room temperature in the dark. DAPI was employed to stain the nuclei. Fluorescence images were pictured with a CKX41 microscope (Olympus, Tokyo, Japan).
:100; Thermo Fisher scientific) for overnight at 4 °C, followed by incubation with secondary antibody conjugated with FITC for 2 hours at room temperature in the dark. DAPI was employed to stain the nuclei. Fluorescence images were pictured with a CKX41 microscope (Olympus, Tokyo, Japan).
2.8 Reverse transcription and quantitation of miRNAs and mRNA by real-time PCR
Total RNA was isolated from serum/synovial fluid or cells using TRIzol Reagent according to the manufacturer's protocol (Invitrogen, CA, USA). 1 μg of total RNA was used to synthesize first strand cDNA with PrimeScript RT reagent kit (TaKaRa, Shiga, Japan). qPCR was conducted with the SYBR Premix Ex Taq (TaKaRa), and PCR-specific amplification was performed with ViiA™ 7 system (Thermo Fisher Scientific, Waltham, MA, USA). For quantification of miRNA expression, stem-loop RT-PCR was employed, and U6 was employed as an endogenous control. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as endogenous control to mRNA.2.9 Western blot analysis
Total protein of the cells was extracted with total protein extraction (Millipore, Bedford, MA, USA). Protein concentration was determined using the BCA protein assay kit. Cell lysates containing an equivalent amount of protein (10 μg) were electrophoresed through 12% SDS polyacrylamide gels, and subsequently transferred onto a nitrocellulose membrane (Sigma-Aldrich, Shanghai, China). Western blot analysis was performed with P-Glycoprotein Monoclonal Antibody (F4), (1:100; Thermo Fisher scientific), anti-IKB alpha (ab32518) (1:5000; Abcam), anti-IKB alpha (phospho S36) (ab133462), (1:50000; Abcam), NFkB p50 Monoclonal Antibody (5D10D11) (1:1000; Thermo Fisher scientific), anti-NF-κB p65 (ab16502) (1:5000; Abcam), GAPDH was used as an internal control. Afterwards, membrane was washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. Protein bands were photoed with ChemiDoc system (Bio-Rad). The expression levels of these proteins were quantified with Image J software.
2.10 Statistical analysis
All data are presented as the mean ± standard deviation (SD) from 3 independent experiments. The difference between means of different groups was analyzed with Student's t test. SPSS 12.0 software (SPSS Inc., Chicago, IL, USA) was employed to conduct the statistical analysis. P value less than 0.05 was considered significant.3. Results
3.1 miR-16 expression was evaluated in methotrexate resistant RA patients
Previous studies showed that levels of miR-16 increased (p = 0.008) after 3 months of therapy and was associated with the decrease in DAS28.13 Hence, we planted to figure out whether miR-16 is associated with methotrexate resistant in RA patients. We detected the miR-16 expression in serum and synovial fluid in R-MTX patients and UR-MTX patients by q-PCR. The data showed that miR-16 expression was significantly evaluated in the in serum and synovial fluid from UR-MTX patients than R-MTX patients (Fig. 1A and B). | ||
| Fig. 1 miR-16 expression was evaluated in methotrexate resistant RA patients. miR-16 expression in serum (A) and synovial fluid (B) in R-MTX patients and UR-MTX patients by q-PCR. *p < 0.5 compared with R-MTX patients. | ||
3.2 miR-16 expression was up-regulated in methotrexate resistant RA-FLS cells
In order to investigate the role of miR-16 in methotrexate resistant RA-FLSs, we performed in vitro studies. Firstly, we constructed the methotrexate resistance RA-FLSs and named as FLS-MTX, and the RA-FLSs that have not undergone any additional processing were named as FLS. FLS and FLS-MTX cells were treated with different concentrations of methotrexate (0.001, 0.01, 0.1, 1.0 μg mL−1) for 48 hours and MTT assay was used to detect the cell viability. As shown in Fig. 2A, compared with FLS, FLS-MTX cells exhibited stronger cell viability after the addition of methotrexate, and the significant difference between the two was more obvious with the increase of concentration. Besides, the IC50 value of methotrexate was 0.178 μg mL−1 in the FLS cells, and 1.152 μg mL−1 in FLS-MTX cells. Therefore, the resistance index between FLS and FLS-MTX cells was calculated to be 6.472, indicating the successful construction of methotrexate-resistant strains. Secondly, we explored the miR-16 mRNA expression in FLS and FLS-MTX cells by qRT-PCR, and observed that the expression level of miR-16 in the FLS-MTX cells were observably boosted in comparison with FLS cells (Fig. 2B). These data manifested that miR-16 may be involved in chemoresistance in RA disease. | ||
| Fig. 2 miR-16 expression was up-regulated in methotrexate resistant RA-FLS cells. (A) The cell viability of FLS and FLS-MTX cells were determined by MTT assay after cells were treated with indicated concentrations of methotrexate for 48 h. (B) miR-16 expression in FLS and FLS-MTX cells were detected by qRT-PCR. *p < 0.5 compared with FLS cells. All the experiments were repeated three times independently. | ||
3.3 Suppression of miR-16 enhanced methotrexate sensitivity in methotrexate resistant RA-FLS cells
To explore the potential functions of miR-16 played on chemoresistance in RA, the miR-16 inhibitor or inhibitor NC were transfected into FLS-MTX cells respectively. qRT-PCR analysis showed that the expression level of miR-16 in the FLS-MTX cells was significantly restrained in miR-16 inhibitor group than NC group (Fig. 3A), confirming the transfection efficiency. Subsequently, the cell viability of FLS-MTX cells transfected with miR-16 inhibitor or inhibitor NC were monitored by MTT assay after treating with different concentrations of methotrexate (0.001, 0.01, 0.1, 1.0 μg mL−1) for 48 hours. The results showed that compared to inhibitor NC group, inhibition of miR-16 results in a significantly decreased cell viability as the concentration of methotrexate goes up (Fig. 3B), suggested that miR-16 suppression restored the sensitivity of FLS-MTX cells to methotrexate treatment. Then, cell cycle of FLS-MTX cells transfected with miR-16 inhibitor or inhibitor NC were evaluated with FCM analyses under 0.01 μg mL−1 methotrexate treatment. MiR-16 inhibition notably increased the cell proportion of G0/G1 phase of FLS-MTX cells (Fig. 3C), indicating that knockdown of miR-16 restrained the cell growth of FLS-MTX cells. Finally, apoptosis of FLS-MTX cells was analyzed via FCM analyses under 0.01 μg mL−1 methotrexate treatment. The data exhibited that the apoptotic rate in miR-16 inhibitor group was prominently enhanced compared to inhibitor NC groups (Fig. 3D), leading to the decreased number of FLS-MTX cells.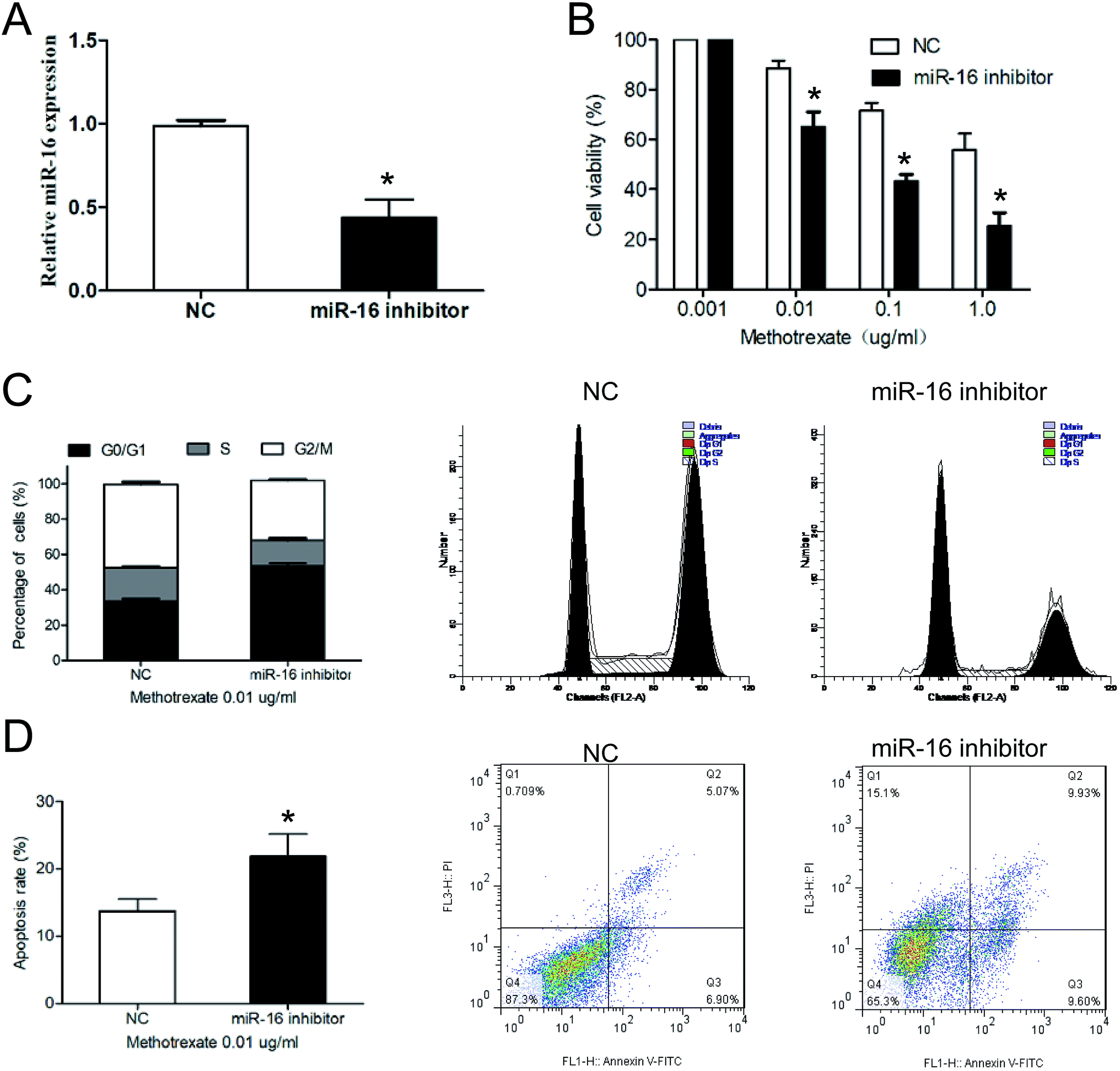 | ||
| Fig. 3 Suppression of miR-16 enhanced methotrexate sensitivity in methotrexate resistant RA-FLS cells. (A) miR-16 expression in FLS-MTX cells transfected with miR-16 inhibitor or inhibitor NC were measured by qRT-PCR. (B) The cell viability of FLS-MTX cells transfected with miR-16 inhibitor or inhibitor NC were determined by MTT assay after cells were treated with indicated concentrations of methotrexate for 48 h. (C) Cell cycle of FLS-MTX cells were evaluated with FCM analyses under 0.01 μg mL−1 methotrexate treatment. (D) Apoptosis of FLS-MTX cells were analyzed via FCM analyses under 0.01 μg mL−1 methotrexate treatment. *p < 0.5 compared with NC group. All the experiments were repeated three times independently. | ||
3.4 Suppression of miR-16 enhanced the sensitivity of FLS-MTX cells to methotrexate by inhibiting P-gp expression
To elucidate the molecular mechanisms of miR-16 is involved in the regulation of methotrexate resistant in RA-FLS cells, the well-known drug resistance related gene, MDR1, and its gene relative protein, P-gp were examined. The results of qRT-PCR and western blot analysis showed that suppression of miR-16 in FLS-MTX cells decreased both MDR1/P-gp expression compared with inhibitor NC group (Fig. 4A and B). Meanwhile, the results of P-gp immunofluorescent staining revealed that the expression of P-gp was restrained by miR-16 knockdown (Fig. 4C). | ||
| Fig. 4 Suppression of miR-16 enhanced the sensitivity of FLS-MTX cells to methotrexate by inhibiting P-gp expression. (A) The MDR1 mRNA repression level in FLS-MTX cells transfected with miR-16 inhibitor or inhibitor NC were measured by qRT-PCR. (B) The P-gp protein expression level in FLS-MTX cells transfected with miR-16 inhibitor or inhibitor NC were measured by western blotting. (C) Expression of P-gp was measured by immunofluorescence staining (100×). *p < 0.5 compared with NC group. All the experiments were repeated three times independently. | ||
3.5 Suppression of miR-16 restrained MDR1/P-gp expression by the NF-κB pathway in FLS-MTX cells
Studies have proven that the NF-κB pathway plays a regulatory role on MDR1 expression in cancer cells drug resistance.21 Hence, we hypothesized that miR-16 may play a fundamental role in promoting P-gp expression through regulating NF-κB pathway. PDTC, a NF-κB pathway inhibitor, was applied to verify our hypothesis. The results of western blot showed that miR-16 inhibition decreased the expression of p-IκBα, p50 and p65 in FLS-MTX cells, verifying that knockdown of miR-16 restrained the phosphorylation of NF-κB pathway (Fig. 5A). The addition of PDTC in miR-16-knockdown-FLS-MTX cells further suppressed the P-gp expression (Fig. 5A), indicating that the P-gp expression was modulated by NF-κB pathway in FLS-MTX cells. In addition, addition of PDTC prominently retarded the cell viability of miR-16-knockdown-FLS-MTX cells (Fig. 5B). Furthermore, the increase of cell proportion of G0/G1 phase of FLS-MTX cells induced by miR-16 inhibition was strengthened by additional treatment of PDTC (Fig. 5C), similar phenomenon was noted in apoptosis assay (Fig. 5D). Results above demonstrated that inhibition of miR-16 suppressed the P-gp expression through inhibiting the NF-κB pathway in FLS-MTX cells to restore the sensitivity of RA-FLS cells to methotrexate. | ||
| Fig. 5 Suppression of miR-16 restrained P-gp expression by the NF-κB pathway in FLS-MTX cells. (A) The protein expression of P-gp, p-IκBα, IκBα, p50 and p65 in FLS-MTX cells were measured by western blotting. (B) The cell viability of FLS-MTX cells were determined by MTT assay after cells were treated with indicated concentrations of methotrexate for 48 h. (C) Cell cycle of FLS-MTX cells were evaluated with FCM analyses under 0.01 μg mL−1 methotrexate treatment. (D) Apoptosis of FLS-MTX cells were analyzed via FCM analyses under 0.01 μg mL−1 methotrexate treatment. *p < 0.5 compared with NC group, #p < 0.5 compared with miR-16 inhibitor group. All the experiments were repeated three times independently. | ||
4. Discussion
Until now, it has been generally accepted that RA are primary caused by synovitis, which is characterized not only by the increased activity of lymphocytes and macrophages, but also by the number of synovial tissue-specific cells called fibroblast-like synovial cells (FLS).22 Stimulation and proliferation of FLS is thought to be the major underlying event in the joint inflammation and destruction in the process of RA.23 Therefore, RA-FLS and the FLS-MTX was employed to investigate the drug resistance mechanism of RA.miR-16 was reported to participate in a series of pathophysiological processes. Combination of vitamin B12 active forms facilitated fetal growth by enhancing placental miR-16 and miR-21 expression levels in Wistar rats.24 In a rat model of depression, reflecting raphe miR-16 level, cerebrospinal fluid miR-16 was involved in the pathogenesis of depression and regulated raphe serotonin transporter expression.25 MiR-16 regulated myoblast proliferation and apoptosis by targeting SESN1, and thus contributed to myoblast differentiation.26 Some reports have described that miR-16 was also involved in the regulation of drug resistance in diseases. MiR-16 was verified to down-regulated in astrocytic gliomas, and miR-16-5p overexpression increased sensitivity to irradiation and TMZ in A172 glioma cells.27 Levels of miR miR-16 were the lowest in serum of Multi Drug Resistant (MDR) tuberculosis patients and associate with responses to drug therapy.28 Forced expression of miR-16 sensitized the ErbB-2-positive breast and gastric cancer to trastuzumab and lapatinib therapy via targeting CCNJ and FUBP1.29 Previous studies illustrated that miR-16 were increased in the sera of established RA patients in comparison with early RA.13,14 However, whether miR-16 is involved in chemoresistance in RA remains uncertain. In this study, we observed that miR-16 expression was notably upregulated in the serum and synovial fluid of methotrexate-resistant RA patient than methotrexate-sensitive RA patient. Meanwhile, the miR-16 was shown upregulated in FLS-MTX cells in comparison with normal RA-FLS cells. These results suggested that the expression of miR-16 is most likely related to the methotrexate resistance in RA. In addition, the CCK-8 assay suggested that miR-16 suppression restored the sensitivity of FLS-MTX cells to methotrexate treatment. Flow cytometry illustrated that miR-16 inhibition increased the cell proportion of G0/G1 phase and enhanced apoptosis of FLS-MTX cells. There data implied that knockdown of miR-16 may weaken the methotrexate resistance of FLS-MTX cells.
P-Glycoprotein (P-gp), encoded by MDR1, is the most classical and most studied transmembrane transporter associated with MDR. Targeting P-gp to improve the sensitivity of chemotherapy has been widely used in the treatment of various drug-resistant diseases.30,31 Studies showed that RA patients with disease-modifying antirheumatic drugs (DMARD) failure had higher serum P-gp levels than patients with a therapeutic response, and high P-gp levels increased the risk of DMARD failure.32 These studies suggested that P-gp may be a therapeutic target to reverse drug resistance, restore and enhance drug sensitivity of anti-rheumatic drugs. Here, we illustrated that inhibition of miR-16 effectively reduced MDR1/P-gp expression, resulting in moderation of the methotrexate resistant in FLS-MTX cells. These results suggested that inhibition of miR-16 enhances methotrexate-resistance by down-regulating the expression of MDR1/P-gp in RA.
MDR1 is the downstream gene of NF-κB pathway, and the transcription of MDR1 is activated under the regulation of NF-κB, leading to the excessive expression of P-gp, which in turn leads to MDR.21 NF-κB has been demonstrated to regulate the expression of MDR1 gene in many cell lines. RY10-4 application suppressed the MDR in MCF-7/ADR cells by P-gp downregulation through the PI3K/Akt/NF-κB pathway.33 Chronic inflammation increased P-gp expression in peripheral mononuclear blood cells via the STAT3/Nf-κb pathway in colitis mice.34 Here, we showed that miR-16 inhibition decreased the expression of p-IκBα, p50 and p65 in FLS-MTX cells. NF-κB inactivation by PDTC in miR-16-knockdown-FLS-MTX cells further suppressed the P-gp expression, confirming that the P-gp expression was modulated by NF-κB pathway in FLS-MTX cells. Furthermore, addition of PDTC prominently retarded the cell viability, further increase cell proportion of G0/G1 phase and apoptosis in miR-16-knockdown-FLS-MTX cells, indicating that inhibition of miR-16 suppressed the P-gp expression through inhibiting the NF-κB pathway in FLS-MTX cells to restore the sensitivity of RA-FLS cells.
5. Conclusion
In general, we verified that the miR-16 expression was significantly increased in the serum and synovial fluid of methotrexate-resistant RA patient and FLS-MTX cells. Most importantly, inhibition of miR-16 enhances methotrexate sensitivity by restraining P-gp protein via NF-κB pathway in FLS-MTX cells. These results suggested that a combination of miR-16 inhibitor and methotrexate might be an effective strategy for the RA treatment.Conflicts of interest
The authors declare no conflicts of interest associated with this manuscript.Acknowledgements
This study was supported by Yunnan Applied Basic Research Projects (Grant No. 2016FB131), Associated Project of Yunnan Province Science & Technology Department and Kunming Medical University Basic Research for Application (Grant No. 2017FE467(-207)), and National Natural Science Foundation of China (Grant No. 81760407).References
- H. S. Smith, A. R. Smith and P. Seidner, Pain Physician, 2011, 14, E427–E458 Search PubMed.
- D. Aletaha, T. Neogi, A. J. Silman, J. Funovits, D. T. Felson, C. O. Bingham, N. S. Birnbaum, G. R. Burmester, V. P. Bykerk, M. D. Cohen, B. Combe, K. H. Costenbader, M. Dougados, P. Emery, G. Ferraccioli, J. M. Hazes, K. Hobbs, T. W. Huizinga, A. Kavanaugh, J. Kay, T. K. Kvien, T. Laing, P. Mease, H. A. Ménard, L. W. Moreland, R. L. Naden, T. Pincus, J. S. Smolen, E. Stanislawska-Biernat, D. Symmons, P. P. Tak, K. S. Upchurch, J. Vencovský, F. Wolfe and G. Hawker, Arthritis Rheum., 2010, 62, 2569–2581 CrossRef PubMed.
- B. N. Cronstein, Pharmacol. Rev., 2005, 57, 163–172 CrossRef CAS PubMed.
- A. Lima, M. Bernardes, R. Azevedo, R. Medeiros and V. Seabra, Int. J. Mol. Sci., 2015, 16, 13760–13780 CrossRef CAS PubMed.
- J. Braun, P. Kästner, P. Flaxenberg, J. Währisch, P. Hanke, W. Demary, U. von Hinüber, K. Rockwitz, W. Heitz, U. Pichlmeier, C. Guimbal-Schmolck and A. Brandt, Arthritis Rheum., 2008, 58, 73–81 CrossRef CAS PubMed.
- R. S. Peres, P. B. Donate, J. Talbot, N. T. Cecilio, P. R. Lobo, C. C. Machado, K. W. A. Lima, R. D. Oliveira, V. Carregaro, H. I. Nakaya, T. M. Cunha, J. C. Alves-Filho, F. Y. Liew, P. Louzada-Junior and F. Q. Cunha, J. Autoimmun., 2018, 90, 49–58 CrossRef CAS PubMed.
- R. S. Peres, F. Y. Liew, J. Talbot, V. Carregaro, R. D. Oliveira, S. L. Almeida, R. F. Franca, P. B. Donate, L. G. Pinto, F. I. Ferreira, D. L. Costa, D. P. Demarque, D. R. Gouvea, N. P. Lopes, R. H. Queiroz, J. S. Silva, F. Figueiredo, J. C. Alves-Filho, T. M. Cunha, S. H. Ferreira, P. Louzada-Junior and F. Q. Cunha, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2509–2514 CrossRef CAS PubMed.
- J. Krützfeldt, M. N. Poy and M. Stoffel, Nat. Genet., 2006, 38, S14–S19 CrossRef PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed.
- Z. Wang, Z. Fang, R. Lu, H. Zhao, T. Gong, D. Liu, L. Hong, J. Ma and M. Zhang, Oncol. Res., 2019 DOI:10.3727/096504019X15528367532612 [Epub ahead of print].
- G. T. Y. Kwok, J. T. Zhao, A. R. Glover, A. J. Gill, R. Clifton-Bligh, B. G. Robinson, J. C. Y. Ip and S. B. Sidhu, Oncologist, 2019, 24, e241–e250 CrossRef CAS PubMed.
- C. Tu, W. Chen, S. Wang, W. Tan, J. Guo, C. Shao and W. Wang, J. Cell. Mol. Med., 2019 DOI:10.1111/jcmm.14197 [Epub ahead of print].
- M. Filková, B. Aradi, L. Senolt, C. Ospelt, S. Vettori, H. Mann, A. Filer, K. Raza, C. D. Buckley, M. Snow, J. Vencovský, K. Pavelka, B. A. Michel, R. E. Gay, S. Gay and A. Jüngel, Ann. Rheum. Dis., 2014, 73, 1898–1904 CrossRef PubMed.
- M. Dunaeva, J. Blom, R. Thurlings and G. J. M. Pruijn, Clin. Exp. Immunol., 2018, 193, 376–385 CrossRef CAS PubMed.
- A. V. Churov, E. K. Oleinik and M. Knip, Autoimmun. Rev., 2015, 14, 1029–1037 CrossRef CAS PubMed.
- Y. H. Wu, W. Liu, B. Xue, L. Zhang, X. Y. Liu, B. Liu, Y. Wang, Y. Cai and R. Duan, DNA Cell Biol., 2016, 35, 853–860 CrossRef CAS PubMed.
- M. Dunaeva, J. Blom, R. Thurlings and G. J. M. Pruijn, Clin. Exp. Immunol., 2018, 193, 376–385 CrossRef CAS PubMed.
- G. Xia, X. Wang, H. Sun, Y. Qin and M. Fu, Free Radical Biol. Med., 2017, 108, 418–432 CrossRef CAS PubMed.
- Y. Zhai, B. Wu, J. Li, X. Y. Yao, P. Zhu and Z. N. Chen, J. Mol. Med., 2016, 94, 71–82 CrossRef CAS PubMed.
- H. S. Lee, S. J. Woo, H. W. Koh, S. O. Ka, L. Zhou, K. Y. Jang, H. S. Lim, H. O. Kim, S. I. Lee and B. H. Park, Arthritis Rheumatol., 2014, 66, 863–873 CrossRef CAS PubMed.
- M. Bentires-Alj, V. Barbu, M. Fillet, A. Chariot, B. Relic, N. Jacobs, J. Gielen, M. P. Merville and V. Bours, Oncogene, 2003, 22, 90–97 CrossRef CAS PubMed.
- M. T. Leech and E. F. Morand, Curr. Opin. Pharmacol., 2013, 13, 565–569 CrossRef CAS PubMed.
- B. Bartok and G. S. Firestein, Immunol. Rev., 2010, 233, 233–255 CrossRef CAS PubMed.
- T. Shah, S. Mishra, A. More, S. Otiv, K. Apte and K. Joshi, Life Sci., 2017, 191, 97–103 CrossRef CAS PubMed.
- Q. Y. Shao, F. You, Y. H. Zhang, L. L. Hu, W. J. Liu, Y. Liu, J. Li, S. D. Wang and M. F. Song, J. Affective Disord., 2018, 238, 609–614 CrossRef CAS PubMed.
- B. Cai, M. Ma, B. Chen, Z. Li, B. A. Abdalla, Q. Nie and X. Zhang, Cell Death Dis., 2018, 9, 367 CrossRef PubMed.
- A. Krell, M. Wolter, N. Stojcheva, C. Hertler, F. Liesenberg, M. Zapatka, M. Weller, B. Malzkorn and G. Reifenberger, Neuropathol. Appl. Neurobiol., 2018, 45, 441–458 Search PubMed.
- V. Wagh, A. Urhekar and D. Modi, Tuberculosis, 2017, 102, 24–30 CrossRef CAS PubMed.
- L. Venturutti, R. I. Cordo Russo, M. A. Rivas, M. F. Mercogliano, F. Izzo, R. H. Oakley, M. G. Pereyra, M. De Martino, C. J. Proietti, P. Yankilevich, J. C. Roa, P. Guzmán, E. Cortese, D. H. Allemand, T. H. Huang, E. H. Charreau, J. A. Cidlowski, R. Schillaci and P. V. Elizalde, Oncogene, 2016, 35, 6189–6202 CrossRef CAS PubMed.
- R. G. Tuguntaev, S. Chen, A. S. Eltahan, A. Mozhi, S. Jin, J. Zhang, C. Li, P. C. Wang and X. J. Liang, ACS Appl. Mater. Interfaces, 2017, 9, 16900–16912 CrossRef CAS PubMed.
- A. A. De Vera, P. Gupta, Z. Lei, D. Liao, S. Narayanan, Q. Teng, S. E. Reznik and Z. S. Chen, Cancer Lett., 2019, 442, 91–103 CrossRef CAS PubMed.
- E. E. Perez-Guerrero, L. Gonzalez-Lopez, J. F. Muñoz-Valle, J. C. Vasquez-Jimenez, M. Ramirez-Villafaña, E. N. Sanchez-Rodriguez, S. R. Gutierrez-Ureña, S. Cerpa-Cruz, E. A. Aguilar-Chavez, E. G. Cardona-Muñoz, M. L. Vazquez-Villegas, A. M. Saldaña-Cruz, N. A. Rodriguez-Jimenez, N. S. Fajardo-Robledo and J. I. Gamez-Nava, Inflammopharmacology, 2018 DOI:10.1007/s10787-018-0529-2 [Epub ahead of print].
- X. Yang, Y. Ding, M. Xiao, X. Liu, J. Ruan and P. Xue, Chem.-Biol. Interact., 2017, 278, 22–31 CrossRef CAS PubMed.
- J. Liu, F. Zhou, Q. Chen, A. Kang, M. Lu, W. Liu, X. Zang, G. Wang and J. Zhang, Sci. Rep., 2015, 5, 13558 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04991f |
| This journal is © The Royal Society of Chemistry 2019 |