
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceX-ray powder diffraction to analyse bimetallic core–shell nanoparticles (gold and palladium; 7–8 nm)
A. Rosteka,
K. Lozaa,
M. Heggenb and
M. Epple *a
*a
aInorganic Chemistry and Center for Nanointegration Duisburg-Essen (CeNIDE), University of Duisburg-Essen, Universitätsstr. 5-7, 45117 Essen, Germany. E-mail: matthias.epple@uni-due.de
bErnst Ruska-Center for Microscopy and Spectroscopy with Electrons, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
First published on 27th August 2019
Abstract
A comparative X-ray powder diffraction study on poly(N-vinyl pyrrolidone) (PVP)-stabilized palladium and gold nanoparticles and bimetallic Pd–Au nanoparticles (both types of core–shell nanostructures) was performed. The average diameter of Au and Pd nanoparticles was 5 to 6 nm. The two types of core–shell particles had a core diameter of 5 to 6 nm and an overall diameter of 7 to 8 nm, i.e. a shell thickness of 1 to 2 nm. X-ray powder diffraction on a laboratory instrument was able to distinguish between a physical mixture of gold and palladium nanoparticles and bimetallic core–shell nanoparticles. It was also possible to separate the core from the shell in both kinds of bimetallic core–shell nanoparticles due to the different domain size and because it was known which metal was in the core and which was in the shell. The spherical particles were synthesized by reduction with glucose in aqueous media. After purification by multiple centrifugation steps, the particles were characterized with respect to their structural, colloid-chemical, and spectroscopic properties, i.e. particle size, morphology, and internal elemental distribution. Dynamic light scattering (DLS), differential centrifugal sedimentation (DCS), atomic absorption spectroscopy (AAS), ultraviolet-visible spectroscopy (UV-vis), high-angle annular dark field imaging (HAADF), and energy-dispersed X-ray spectroscopy (EDX) were applied for particle characterization.
Introduction
Nanoparticles of noble metals constitute an important area of research due to their many applications in imaging, biomedicine, and heterogeneous catalysis.1–9 Their application spectrum is enhanced in bimetallic nanoparticles where two or more metals are present in the same nanoparticle. For instance, the catalytic activity of nanoparticles depends on the nature of the particle surface, both chemically and electronically, which can be varied by alloying two metals.10–12 The optical properties of nanoparticles (e.g. surface plasmon resonance bands) are adjustable by the presence of two metals.6,13–15 In the biomedical area, the release of antibacterial silver from nanoparticles can be fine-tuned by alloying with other metals.16–18 For their characterization, a range of techniques is usually applied, with dynamic light scattering and electron microscopy being the most prominent.19–24 If bimetallic nanoparticles are considered, the question of the internal distribution of the metals inside each nanoparticle arises.10,25–28 This is typically addressed by EDX and similar techniques where the elemental distribution inside a number of nanoparticles is measured. However, this analysis can only be performed for a small number of nanoparticles. Another question of interest is the inner domain structure, i.e. the crystallographic nature of a particle. A particle could be single-crystalline, but more often it consists of several domains, a phenomenon known as twinning. Integral methods for particle size determination (like dynamic light scattering or centrifugation techniques) do not provide information on the internal structure of a particle.X-ray powder diffraction is a well-established method to analyse crystalline solids. Moderate amounts of material are needed (a few mg), and it gives averaged information about millions or billions of particles, depending on the particles size. The application to nanoparticles is often difficult due to broad diffraction peaks, caused by small crystalline domains and strong peak broadening, making the data interpretation sometimes complicated.29–36 Of course, X-ray powder diffraction does not distinguish between two domains present in the same particle and two different single-domain particles that lie adjacent in a sample.
In the following, we demonstrate how core–shell systems can be quantitatively distinguished and analysed by powder diffraction on a laboratory instrument. As example, we chose the catalytically relevant system gold/palladium. Bimetallic palladium–gold nanoparticles (Pd–Au) are of considerable interest in heterogeneous catalysis, e.g. for various redox reactions. This is mainly ascribed to the modulation of the properties of palladium by alloying it with gold, both electronically and structurally (see Zhang et al. for a comprehensive review about synthesis and properties of Pd/Au bimetallic nanoparticles).10 Only rarely, X-ray powder diffraction has been applied to such systems in closer detail.37–39 Here, well-defined samples with high monodispersity were prepared to exclude the effects of impurities and larger crystals. We will show that X-ray powder diffraction is well suited to distinguish between different systems, making it suitable for a rapid analysis of many nanoparticles in the same experiment.
Results and discussion
First, we prepared gold and palladium nanoparticles with a diameter of about 5 nm40 that were then used as seeds (i.e. cores) for the following synthesis of bimetallic core–shell nanoparticles as described earlier.39 The core–shell nanoparticles Pd–Au and Au–Pd were then prepared by reduction of the second metal, using a modified seeded growth approach.10,39,41 The presence of the reducing agent glucose prevented the galvanic replacement of the palladium seeds by the gold ions that would have led to a hollow gold nanoshell.42 The colloidal stabilization was accomplished by an external layer of PVP in all cases. The dispersed particles were analysed by dynamic light scattering (DLS), differential centrifugal sedimentation (DCS), and transmission electron microscopy (TEM) (Fig. 1–3). The diameter of the seeds was 5 to 6 nm, and the diameter of the core–shell particles was 7 to 8 nm, indicating a thickness of the outer metal shell of about 1 to 2 nm. The fact that the hydrodynamic diameter from DCS and from DLS was comparable with the size of the metallic core as determined by TEM confirms the well-dispersed state of the particles in water without relevant agglomeration.19 The only exception were Au–Pd nanoparticles that showed some degree of agglomeration as shown in DLS (hydrodynamic diameter 18 nm). | ||
| Fig. 1 Dynamic light scattering (DLS) data of palladium seeds (A), gold seeds (B), Pd–Au core–shell nanoparticles (C), and Au–Pd core–shell nanoparticles (D). The particle size distributions were analysed by a log-normal fit. | ||
 | ||
| Fig. 2 Differential centrifugal sedimentation (DCS) data of palladium seeds (A), gold seeds (B), Pd–Au core–shell nanoparticles (C), and Au–Pd core–shell nanoparticles (D). The particle size distributions were analyzed by a log-normal fit. | ||
 | ||
| Fig. 3 HAADF-STEM images and elemental mapping by EDX analysis of Au–Pd (top) and Pd–Au core–shell nanoparticles (bottom). | ||
Note that the particle diameter from DCS is systematically smaller than that determined by dynamic light scattering. This is due to the polymer shell around the particles that decreases the effective density of the particles. Consequently, the sedimentation time is higher, leading to an apparently smaller particle size.43 Elemental analysis of the bimetallic nanoparticles was performed by atomic absorption spectroscopy (AAS) after dissolution in aqua regia. All characterization data are comprised in Table 1.
| Analytical method | Pd seeds | Au seeds | Pd seeds + Au seeds (physical mixture 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 = n:n) :1 = n:n) |
Au–Pd (core–shell) | Pd–Au (core–shell) | |
|---|---|---|---|---|---|---|
| a Particle size distribution by number. | ||||||
| Diameter/nm | DLSa | 6.7(12) | 5.3(20) | — | 18.3(55) | 8.2(19) |
| PDI (DLS) | 0.16 | 0.48 | — | 0.43 | 0.21 | |
| DCSa | 5.1(15) | 4.7(15) | — | 6.9(19) | 6.7(23) | |
| TEM | 5.4(12) | 5.8(13) | — | 8.7(12) | 8.9(11) | |
| Pd:Au ratio/at% |
AAS | — | — | 49:51 |
57:43 |
68:32 |
| XRD | — | — | 44:56 |
63:37 |
26:74 |
|
The gold and palladium seeds were further analysed by high-resolution electron microscopy (HAADF-STEM), showing spherical polycrystalline nanoparticles (see ref. 40 for corresponding images). Both types of core–shell nanoparticles had an almost spherical shape with a diameter in good agreement with the DLS and DCS results. Energy-dispersive X-ray spectroscopy (EDX) showed the expected core–shell structure of the nanoparticles in both cases (Fig. 3). The poorly pronounced boundary between the core and the shell indicates some alloying at the interface which had probably occurred during the synthesis by the interaction of metal seeds and ions of the shell metal. The core had a diameter of 5–6 nm as in the seeds, the outer metal layer had a thickness of 1–2 nm, again in good agreement with the data from DLS and DCS.
Pure gold seeds showed the typical surface plasmon resonance (SPR) absorption with an absorption maximum around 520 nm, also in a physical mixture with palladium seeds.44 As expected for the outer layer of palladium metal, palladium seeds and Au–Pd core–shell nanoparticles showed no distinct absorption in the visible range.40 Clearly, palladium prevented the SPR of gold, confirming a dense layer of palladium metal around the gold core. After the addition of a gold layer to the surface of the palladium seeds to form Pd–Au core–shell nanoparticles, the SPR absorption band of gold was observed. Thus, the TEM-EDX results of the bimetallic core–shell nanoparticles are well supported by their optical properties (Fig. 4).
 | ||
| Fig. 4 UV-vis spectra of Pd seeds (5–6 nm), Au seeds (5–6 nm), a physical mixture of Au and Pd seeds (1:1 = n:n), and bimetallic Au–Pd and Pd–Au (core–shell) nanoparticles. The surface plasmon resonance of gold is visible for all systems where gold is at the particle surface (maximum at about 520 nm). | ||
After the comprehensive structural characterization of the gold and palladium seeds and the two kinds of bimetallic core–shell nanoparticles, the applicability of X-ray powder diffraction to analyse the structure of such systems was investigated. Fig. 5 shows the X-ray powder diffractograms of the gold and palladium seeds. Both metals crystallize in the fcc lattice (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) but with different lattice constants: a(Pd) = 3.89019 Å and a(Au) = 4.0786 Å.45 The diffraction peaks were very broad due to the small particle size.46 Table 2 gives the lattice parameter and the crystallite sizes as computed by Rietveld refinement.
m) but with different lattice constants: a(Pd) = 3.89019 Å and a(Au) = 4.0786 Å.45 The diffraction peaks were very broad due to the small particle size.46 Table 2 gives the lattice parameter and the crystallite sizes as computed by Rietveld refinement.
 | ||
| Fig. 5 X-ray powder diffraction pattern and Rietveld refinement of Au seeds (5–6 nm), showing the baseline-corrected fit of nanocrystalline fcc gold metal (top), and of Pd seeds (5–6 nm), showing the baseline-corrected fit of nanocrystalline fcc palladium metal (bottom). Note the broad diffraction peaks due to the small crystalline domains. | ||
| Sample | Measured lattice parameter a/Å | Measured crystal size/nm | ||
|---|---|---|---|---|
| Au | Pd | Au | Pd | |
| Au seeds | 4.063(1) | — | 3.0(1) (core) | — |
| Pd seeds | — | 3.900(2) | — | 2.6(1) (core) |
| Au + Pd physical mixture | 4.073(3) | 3.906(2) | 3.3(1) (core) | 5.8(3) (core) |
| Au–Pd core–shell | 4.07(1) | 3.99(8) | 5.9(2) (core) | 1.9(8) (shell) |
| Pd–Au core–shell | 4.05(2) | 3.97(1) | 3.7(4) (shell) | 6.7(6) (core) |
Next, a heterogeneous mixture of gold and palladium seeds (1:1) was analysed by X-ray powder diffraction (Fig. 6). Despite the strong peak broadening, the major (111) peak was clearly split, indicating the presence of both metals as separate phases. Rietveld refinement permitted a quantitative phase analysis and a clear separation of both metal phases, including the determination of the lattice parameter a and the individual crystallite size for both metals (Table 2).
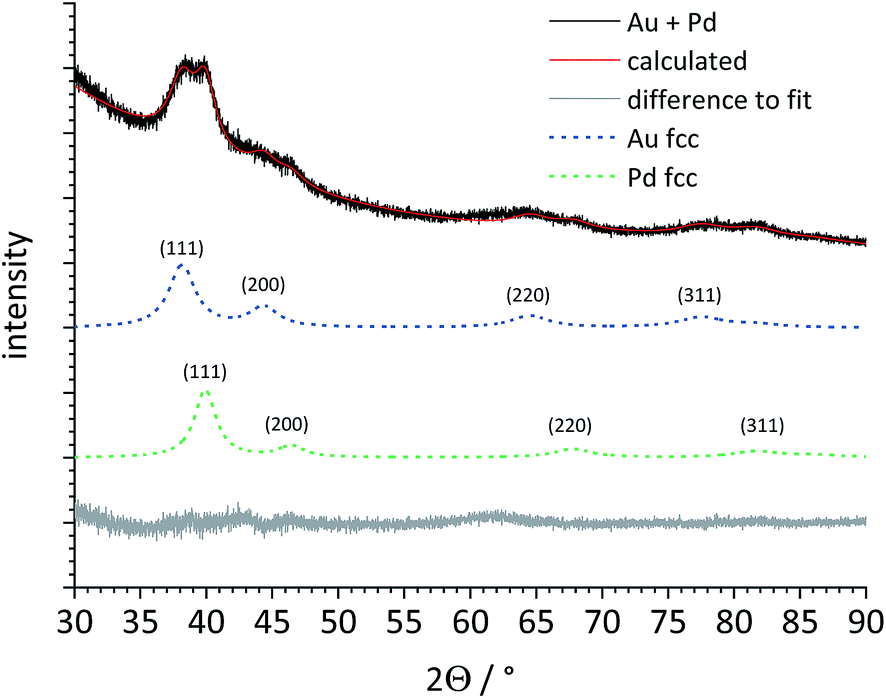 | ||
| Fig. 6 X-ray powder diffraction pattern and Rietveld refinement of a physical mixture of Pd and Au seeds (both 5–6 nm). The dashed lines for Au fcc and Pd fcc represent the baseline-corrected fits of these two metals. Au and Pd are clearly distinguishable, despite the broad diffraction peaks. | ||
Then, both types of core–shell nanoparticles were studied by X-ray powder diffraction (Fig. 7). Gold and palladium form an unlimited row of mixed crystals,47,48 i.e. an averaged lattice parameter a would be expected for fully alloyed nanoparticles. In accordance with the TEM images, Rietveld refinement showed that both metals were present as separate phases. The core metal showed more narrow diffraction peaks than the shell metal, in good agreement with a core diameter of 5–6 nm and a shell thickness of 1–2 nm. All quantitative results from Rietveld refinement are summarized in Table 2.
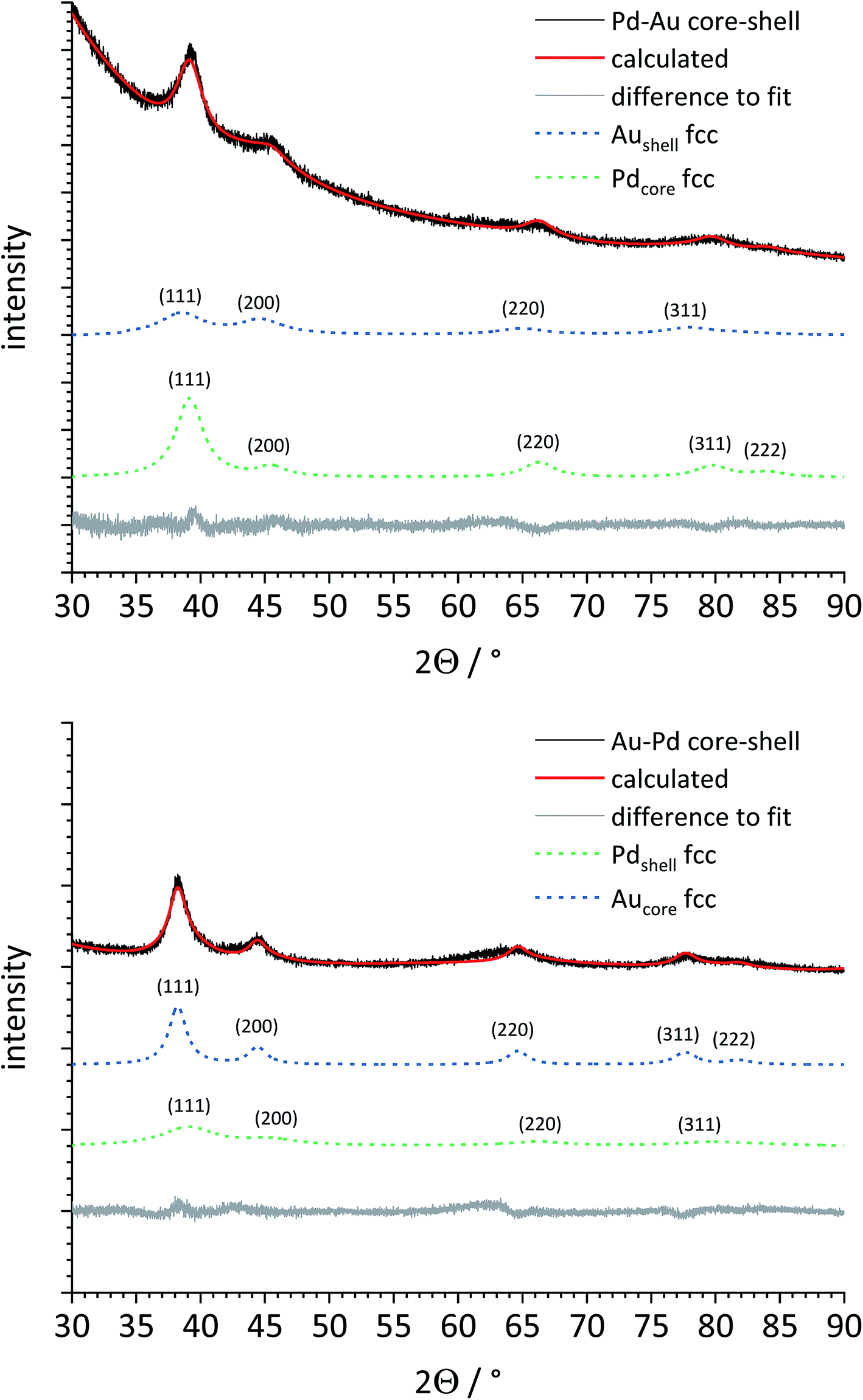 | ||
| Fig. 7 X-ray powder diffraction patterns and Rietveld refinement of Pd–Au (top) and Au–Pd core–shell nanoparticles (bottom). The dashed lines for Au fcc and Pd fcc represent the baseline-corrected fits of these two metals. The core metal (Au or Pd) and the shell metal (Pd or Au) are well distinguishable in both cases. The bigger core size (5–6 nm) leads to narrower peaks for the core metal compared to the shell metal (shell thickness 1–2 nm). | ||
The lattice parameter of gold was not significantly decreased in comparison to the pure gold seeds. In contrast, the palladium lattice was expanded compared to the pure metal, indicating a partial alloying with gold in both cases. This was not the case for the physical mixture of gold and palladium nanoparticles. The crystallite size as computed from the peak width showed the same trend: the core consists of bigger domains (but is probably still twinned), and the shell consists of smaller domains. The fact that the determined crystallite size does not fully match the core and shell diameters from TEM can be safely ascribed to the limitations of X-ray powder diffraction for such small particles due to twinning and local alloying in the core–shell interface.
Conclusions
X-ray powder diffraction is able to identify the inner and outer part of bimetallic core–shell nanoparticles of gold and palladium even at a size of a few nanometres. It can be used to discriminate between core and shell metals, provided that the inherent structure of the nanoparticle is known. Usually, it is known or at least suspected from the synthetic procedure which metal is in the core and which is in the shell. Given that knowledge, both lattice parameter and crystallite size can be used to elucidate the internal structure. In contrast, a physical mixture of gold and palladium nanoparticles shows the superposition of the diffraction patterns of both metals. We conclude that X-ray powder diffraction with a laboratory instrument is applicable to analyze such nanoparticulate samples in a quantitative way, looking at many particles at the same time. However, care must be given that the samples are indeed homogeneous and highly monodisperse because the presence of larger crystals would cause dominant narrow diffraction signals and lead to wrong conclusions. Furthermore, the analysis of more than one kind of bimetallic nanoparticles will only be possible if the diffraction peaks are well separated, i.e. if the lattice parameters and crystal systems are sufficiently different.Materials and methods
We used D-glucose (Baker, p.a.), poly(N-vinyl pyrrolidone) (PVP K30, Povidone 30, Fluka, M = 40000 g mol−1), Pd(NO3)2 and Na2PdCl4 (both Sigma-Aldrich, p.a.). HAuCl4 was prepared by dissolution of gold in hot aqua regia, followed by removal of the solvent to dryness and multiple dissolution and precipitation in hydrochloric acid. Ultrapure water (Purelab ultra instrument from ELGA, 18.2 MΩ cm−1) was used for all syntheses.
Pd nanoparticles (seeds), Au nanoparticles (seeds), and Pd–Au core–shell nanoparticles were synthesized as described earlier.39,40 Au–Pd core–shell nanoparticles were synthesized in analogy to Pd–Au core–shell nanoparticles as follows: In a two-neck round bottom flask, 600 mg (3 mmol) D-glucose and 50 mg (0.125 μmol) PVP were dissolved in 50 mL water. The mixture was vigorously stirred (700 rpm) and heated to 100 °C under reflux. Then, 5 mL of an aqueous solution containing Pd2+ (either as Pd(NO3)2 or as Na2PdCl4 with no notable difference between the two precursors; containing 5 mg or 0.05 mmol Pd) were added and the reaction mixture was boiled at 100 °C for 1 h under reflux. This led to dispersed palladium seeds. For the gold shell, 5 mL of a HAuCl4 solution (containing 9.85 mg or 0.05 mmol Au) were quickly added. The mixture was kept boiling for another 5 min at 100 °C. Then, the reaction mixture was rapidly quenched with an ice bath. The nanoparticle dispersion was centrifuged with a Millipore spin filter (molecular weight cut-off 3 kDa, remaining volume 500 μL) at 4000 rpm for 45 min. The remaining concentrated dispersion was centrifuged twice at 66000g (30000 rpm) for 30 min and redispersed in 5 mL water.
For X-ray powder diffraction, the nanoparticle dispersion was shock-frozen with liquid nitrogen and lyophilized at 0.31 mbar and −10 °C in a Christ Alpha 2-4 LSC instrument. The 1:1 mixture (n:n) was prepared by mixing the two nanoparticle dispersions, followed by shock-freezing and lyophilization. X-ray powder diffraction was performed with a D8 Advance instrument (Bruker) in Bragg–Brentano geometry with Cu Kα radiation (λ = 1.54 Å; 40 kV; 40 mA; step size 0.01° 2Θ, counting time 1.7 s) on a silicon single-crystal sample holder with a crystallographic (911) plane to minimize scattering. The instrumental peak broadening was determined with lanthanum hexaboride (LaB6) from NIST (National Institute of Standards and Technology) as internal standard. Rietveld refinement was performed with the program package TOPAS 4.2 (Bruker). The standard diffraction data of palladium and gold were taken from the International Centre of Diffraction Data (ICDD), Powder Diffraction File (PDF) cards 00-004-0784 (Au) and 00-046-1043 (Pd).
Dynamic light scattering for particle size analysis and zeta-potential determination was carried out with a Malvern Zetasizer Nano ZS ZEN 3600 instrument (Malvern Panalytical Ltd.; 25 °C, laser wavelength 633 nm). The scattering was monitored at a fixed angle of 173° in backward scattering mode.
Analytical disc centrifugation was performed with a DC 24000 disc centrifuge (CPS Instruments, Inc.). The rotor speed was set to 24000 rpm (28978g). Two aqueous sucrose solutions (8 wt% and 24 wt%) formed a density gradient which was capped with 0.5 mL dodecane as a stabilizing agent. The calibration standard was a poly(vinyl chloride) (PVC) latex in water with a particle size of 483 nm provided by CPS Instruments. The calibration was carried out prior to each run. Calibration and sample volumes were both 100 μL.
Ultraviolet-visible spectroscopy (UV/vis) was performed with a Varian Cary 300 instrument (Agilent Technologies, Inc.). Suprasil® micro quartz cuvettes with a sample volume of 750 μL were used after background correction.
Atomic absorption spectrometry was carried out with a Thermo Electron M-Series instrument (ThermoFisher Scientific) according to DIN EN lSO/lEC 17025:2005. Before the measurement, the nanoparticles were dissolved in aqua regia.
Scanning transmission electron microscopy (STEM) was performed with a FEI Titan microscope, equipped with a Cs-probe corrector (CEOS Company) and a high-angle annular dark field (HAADF) detector, operated at 200 kV. Z-contrast conditions were achieved with a probe semi-angle of 25 mrad and an inner collection angle of the detector of 70 mrad. Elemental mapping using energy-dispersive X-ray spectroscopy (EDX) was conducted on a probe-corrected FEI Titan 80-300 ChemiSTEM electron microscope, equipped with four symmetrical SDD detectors.49,50
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support of this work by the Deutsche Forschungsgemeinschaft (DFG) in the projects EP 22/44-1 and HE 7192/2-1. We thank Kerstin Brauner and Robin Meya for AAS measurements.References
- R. A. Sperling, P. Rivera, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- D. Pedone, M. Moglianetti, E. De Luca, G. Bardi and P. P. Pompa, Chem. Soc. Rev., 2017, 46, 4951–4975 RSC.
- L. A. Dykman and N. G. Khlebtsov, Biomaterials, 2016, 108, 13–34 CrossRef CAS PubMed.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- C. M. Cobley, J. Y. Chen, E. C. Cho, L. V. Wang and Y. N. Xia, Chem. Soc. Rev., 2011, 40, 44–56 RSC.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- M. Homberger and U. Simon, Philos. Trans. R. Soc., A, 2010, 368, 1405–1453 CrossRef CAS PubMed.
- L. Zhang, Z. Xie and J. Gong, Chem. Soc. Rev., 2016, 45, 3916–3934 RSC.
- V. Beermann, M. Gocyla, E. Willinger, S. Rudi, M. Heggen, R. E. Dunin-Borkowski, M. G. Willinger and P. Strasser, Nano Lett., 2016, 16, 1719–1725 CrossRef CAS PubMed.
- L. Masliuk, M. Heggen, J. Noack, F. Girgsdies, A. Trunschke, K. E. Hermann, M. G. Willinger, R. Schlögl and T. Lunkenbein, J. Phys. Chem. C, 2017, 121, 24093–24103 CrossRef CAS.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- J. J. Giner-Casares, M. Henriksen-Lacey, M. Coronado-Puchau and L. M. Liz-Marzán, Mater. Today, 2016, 19, 19–28 CrossRef CAS.
- J. Langer, S. M. Novikov and L. M. Liz-Marzán, Nanotechnology, 2015, 26, 322001 CrossRef PubMed.
- C. Rehbock, J. Jakobi, L. Gamrad, S. van der Meer, D. Tiedemann, U. Taylor, W. Kues, D. Rath and S. Barcikowski, Beilstein J. Nanotechnol., 2014, 5, 1523–1541 CrossRef PubMed.
- V. Grasmik, C. Rurainsky, K. Loza, M. V. Evers, O. Prymak, M. Heggen, K. Tschulik and M. Epple, Chem.–Eur. J., 2018, 24, 9051–9060 CrossRef CAS PubMed.
- V. Grasmik, M. Breisch, K. Loza, M. Heggen, M. Köller, C. Sengstock and M. Epple, RSC Adv., 2018, 8, 38582–38590 RSC.
- D. Mahl, J. Diendorf, W. Meyer-Zaika and M. Epple, Colloids Surf., A, 2011, 377, 386–392 CrossRef CAS.
- Y. Dieckmann, H. Colfen, H. Hofmann and A. Petri-Fink, Anal. Chem., 2009, 81, 3889–3895 CrossRef CAS PubMed.
- M. T. Zhu, G. J. Nie, H. Meng, T. Xia, A. Nel and Y. L. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- I. Montes-Burgos, D. Walczyk, P. Hole, J. Smith, I. Lynch and K. Dawson, J. Nanopart. Res., 2010, 12, 47 CrossRef.
- D. G. Thomas, J. N. Smith, B. D. Thrall, D. R. Baer, H. Jolley, P. Munusamy, V. Kodali, P. Demokritou, J. Cohen and J. G. Teeguarden, Part. Fibre Toxicol., 2018, 15, 22 CrossRef PubMed.
- N. Feliu, X. Sun, R. A. Alvarez Puebla and W. J. Parak, Langmuir, 2017, 33, 6639–6646 CrossRef CAS PubMed.
- G. Guisbiers, R. Mendoza-Cruz, L. Bazan-Diaz, J. J. Velazquez-Salazar, R. Mendoza-Perez, J. A. Robledo-Torres, J. L. Rodriguez-Lopez, J. M. Montejano-Carrizales, R. L. Whetten and M. Jose-Yacaman, ACS Nano, 2016, 10, 188–198 CrossRef CAS PubMed.
- H. L. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- F. Calvo, Phys. Chem. Chem. Phys., 2015, 17, 27922–27939 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- F. Bertolotti, D. Moscheni, A. Guagliardi and N. Masciocchi, Eur. J. Inorg. Chem., 2018, 3789–3803 CrossRef CAS.
- B. Palosz, E. Grzanka, S. Gierlotka and S. Stelmakh, Z. Kristallogr. - Cryst. Mater., 2010, 225, 588–598 CrossRef CAS.
- S. Xiong, H. Ozturk, S. Y. Lee, P. M. Mooney and I. C. Noyan, J. Appl. Crystallogr., 2018, 51, 1102–1115 CrossRef CAS.
- S. Banerjee, C. H. Liu, J. D. Lee, A. Kovyakh, V. Grasmik, O. Prymak, C. Koenigsmann, H. Liu, L. Wang, A. M. M. Abeykoon, S. S. Wong, M. Epple, C. B. Murray and S. J. L. Billinge, J. Phys. Chem. C, 2018, 122, 29498–29506 CrossRef CAS.
- S. J. L. Billinge and I. Levin, Science, 2007, 316, 561–565 CrossRef CAS PubMed.
- O. Prymak, J. Jakobi, C. Rehbock, M. Epple and S. Barcikowski, Mater. Chem. Phys., 2018, 207, 442–450 CrossRef CAS.
- S. Ristig, O. Prymak, K. Loza, M. Gocyla, W. Meyer-Zaika, M. Heggen, D. Raabe and M. Epple, J. Mater. Chem. B, 2015, 3, 4654–4662 RSC.
- C. Weidenthaler, Nanoscale, 2011, 3, 792–810 RSC.
- D. Jose and B. R. Jagirdar, J. Phys. Chem. C, 2008, 112, 10089–10094 CrossRef CAS.
- O. V. Belousov, N. V. Belousova, A. V. Sirotina, L. A. Solovyov, A. M. Zhyzhaev, S. M. Zharkov and Y. L. Mikhlin, Langmuir, 2011, 27, 11697–11703 CrossRef CAS PubMed.
- A. Rostek, M. Breisch, K. Loza, P. R. A. F. Garcia, C. L. P. Oliveira, O. Prymak, M. Heggen, M. Köller, C. Sengstock and M. Epple, ChemistrySelect, 2018, 3, 4994–5001 CrossRef CAS.
- A. Rostek, M. Breisch, K. Pappert, K. Loza, M. Heggen, M. Köller, C. Sengstock and M. Epple, Beilstein J. Nanotechnol., 2018, 9, 2763–2774 CrossRef CAS PubMed.
- P. Hou, H. Liu, J. Li and J. Yang, CrystEngComm, 2015, 17, 1826–1832 RSC.
- A. Papaderakis, I. Mintsouli, J. Georgieva and S. Sotiropoulos, Catalysts, 2017, 7, 34 CrossRef.
- H. Fissan, S. Ristig, H. Kaminski, C. Asbach and M. Epple, Anal. Methods, 2014, 6, 7324–7334 RSC.
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881–3891 RSC.
- H. E. Swanson and E. Tatge, J. Res. Natl. Bur. Stand., 1951, 46, 318–327 CrossRef CAS.
- H. P. Klug and L. E. Alexander, X-ray diffraction procedures for polycrystalline and amorphous materials, Wiley-Interscience, New York, 1974 Search PubMed.
- A. Maeland and T. B. Flanagan, Can. J. Phys., 1964, 42, 2364 CrossRef CAS.
- H. Okamoto and T. B. Massalski, Bull. Alloy Phase Diagrams, 1985, 6, 229–235 CrossRef CAS.
- A. Thust, J. Barthel and K. Tillmann, Journal of Large-scale Research Facilities, 2016, 2, A41 CrossRef.
- M. Heggen, M. Luysberg and K. Tillmann, Journal of Large-scale Research Facilities, 2016, 2, A42 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |