
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteractions between marmatite and bornite during the oxidative dissolution process in abiotic and biotic systems†
Yanjun Zhang ab,
Hongbo Zhao*ab,
Yisheng Zhangab,
Lu Qianab,
Luyuan Zhangab,
Xiaoyu Mengab,
Xin Lvab,
Abhilashc,
Hussnain Ahned Janjuad and
Guanzhou Qiuab
ab,
Hongbo Zhao*ab,
Yisheng Zhangab,
Lu Qianab,
Luyuan Zhangab,
Xiaoyu Mengab,
Xin Lvab,
Abhilashc,
Hussnain Ahned Janjuad and
Guanzhou Qiuab
aSchool of Minerals Processing & Bioengineering, Central South University, No. 932 Lushan South Road, Yuelu District, Changsha, Hunan, China 410083. E-mail: alexandercsu@126.com; zhbalexander@csu.edu.cn
bKey Lab of Biohydrometallurgy of Ministry of Education, Changsha, Hunan, China
cCSIR – National Metallurgical Laboratory, Jamshedpur 831007, India
dAtta-ur-Rahman School of Applied Biosciences, National University of Sciences and Technology (NUST), Sector H-12, Islamabad, Pakistan
First published on 27th August 2019
Abstract
Marmatite and bornite are commonly associated together in nature, and their interactions in an acidic environment are vital for both (bio)hydrometallurgy and acid mine drainage (AMD) production. In this work, dissolution experiments (marmatite![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bornite = 2:0, 3:1, 1:1, 1:3 and 0:2) accompanied by analytic techniques such as electrochemical methods, Raman spectroscopy and synchrotron radiation-XRD (SR-XRD) were utilized to interpret the interactions between marmatite and bornite in acidic abiotic and biotic systems. The dissolution experiments showed that marmatite can significantly accelerate the oxidative dissolution of bornite, especially in the abiotic system. On the contrary, bornite inhibited the oxidative dissolution of marmatite when the percentage of bornite was high. Electrochemical measurements proved that the galvanic interactions between marmatite and bornite were slight and should not be the main cause for the interactions. Combined with the dissolution experiments, analytic techniques and previous references, it could be speculated that marmatite accelerated bornite dissolution mainly by providing an iron source, which acted as the energy source for microorganisms and oxidants. Bornite affected the dissolution of marmatite mainly by Cu2+ ions dissolving from bornite. Bornite inhibited the oxidative dissolution of marmatite mainly because a high Cu2+ concentration could significantly hinder marmatite dissolution. In addition, the formation of elemental sulfur or jarosite was also one important cause. Bornite intensified marmatite dissolution when the percentage of bornite or the Cu2+ concentration was extremely low and then, a synergic dissolution process occurred.
:bornite = 2:0, 3:1, 1:1, 1:3 and 0:2) accompanied by analytic techniques such as electrochemical methods, Raman spectroscopy and synchrotron radiation-XRD (SR-XRD) were utilized to interpret the interactions between marmatite and bornite in acidic abiotic and biotic systems. The dissolution experiments showed that marmatite can significantly accelerate the oxidative dissolution of bornite, especially in the abiotic system. On the contrary, bornite inhibited the oxidative dissolution of marmatite when the percentage of bornite was high. Electrochemical measurements proved that the galvanic interactions between marmatite and bornite were slight and should not be the main cause for the interactions. Combined with the dissolution experiments, analytic techniques and previous references, it could be speculated that marmatite accelerated bornite dissolution mainly by providing an iron source, which acted as the energy source for microorganisms and oxidants. Bornite affected the dissolution of marmatite mainly by Cu2+ ions dissolving from bornite. Bornite inhibited the oxidative dissolution of marmatite mainly because a high Cu2+ concentration could significantly hinder marmatite dissolution. In addition, the formation of elemental sulfur or jarosite was also one important cause. Bornite intensified marmatite dissolution when the percentage of bornite or the Cu2+ concentration was extremely low and then, a synergic dissolution process occurred.
1. Introduction
Acid mine drainage (AMD) is primarily derived from mining tailings, rock pile-ups, and oxidation of pyrite and other sulfide minerals.1–3 It has caused serious environmental problems. AMD has extremely low pH values, which lead to the dissolution of some minerals and the release of metal ions such as cupric, ferric, zinc, and nickel ions into the solution. Heavy metal ions and the acidity of the solution contaminate the surrounding groundwater layer. Its production, release, movement and attenuation are conducted in accordance with complex physical, chemical and biological factors. Although the chemistry of AMD generation is straightforward, the final product is a function of the geology of the mining region, presence of microorganisms, temperature and also the availability of water and oxygen.4 Recent advances concentrate more on the development of treatment techniques involved in source control and migration control.5 Source control is an effective method that directly controls the formation of AMD by avoiding direct contact between oxygen or water and sulfide minerals.6,7Bio-dissolution or bacterial dissolution refers to the process of using some bacteria's oxidizing activities to dissolve metal sulfide minerals.8 It has been successfully implemented to extract secondary copper sulfide minerals.9,10 There are many bioleaching microorganisms such as A. ferrooxidans, L. ferrooxidans, Acidithiobacillus caldus (A. caldus), and Sulfobacillus thermosulfidooxidans (S. thermosulfidooxidans). A. ferrooxidans is one of the most commonly used species in bio-dissolution or biomineralization; it is frequently observed in nature and is present in sulfide minerals such as pyrite, coal mines, and also in acid mine drainage.11–13 It is a Gram-negative and chemoautotrophy bacterium, which fixes carbon dioxide in the air as a carbon source. Furthermore, it can oxidize ferrous ions and reduce sulfides as energy sources.14
Bornite (Cu5FeS4) is an extremely important copper sulfide mineral. It is commonly found in the earth with other sulfide minerals. The economic benefit of bornite ranks third among copper sulfide ores and second only to chalcopyrite and chalcocite.15 The chemical and biological oxidation of bornite is relatively fast and easy; also, residual bornite was not detected by X-ray diffraction in solids after 2 days of contact in an acid solution and virtually, 100% of copper solubilization was achieved in A. ferrooxidans cultures with or without ferrous ions.16 Theoretically, the sum of the number of iron and zinc atoms in marmatite is similar to the number of sulfur atoms. Therefore, the chemical formula of marmatite can be approximately regarded as (Zn1−xFex)S, and it is one of the most important zinc containing resources. Marmatite is formed by replacing some zinc sites in sphalerite (ZnS) crystals with iron elements, and the iron contents are about 6–25%.17 According to the percentage of iron elements, it can be categorized into low-iron marmatite, medium-iron marmatite and high-iron marmatite. Marmatite is analogous to bornite and often coexists with other sulfide minerals such as chalcopyrite, pyrite and bornite. On account of the relatively low zinc content in marmatite, it is difficult to effectively treat with conventional techniques.18 Nevertheless, as a result of the presence of iron, it has a faster oxidation dissolution rate than sphalerite.19 Therefore, it is more suitable for bio-dissolution.20–22
Some recent studies have shown the interactions of different sulfide minerals in sulfuric acid and also in the presence of bacteria. Zhao et al.23 reported that bornite decreased the oxidation reduction potential (ORP) and maintained it at an appropriate range (380–480 mV vs. Ag/AgCl) to promote chalcopyrite (A) dissolution; however, this caused the redox potential to be out of the optimum range and inhibited chalcopyrite (B) dissolution in the presence of L. ferriphilum. However, they suggested that a synergistic effect existed between chalcopyrite and bornite during bioleaching in the presence of A. ferrooxidans.24 R. G. Acres et al.25 proposed that in the presence of hydrochloric acid, bornite can decrease the oxidation rate of chalcopyrite, thus resulting in the slow dissolution of chalcopyrite. Saavedra et al.26 suggested that CuFeS2 and Fe3O4 behaved as an anode and a cathode, respectively, in a natural mixed mineral, thus stimulating and increasing the chalcopyrite reactivity and preventing the formation of a passive layer commonly formed during chalcopyrite oxidation in both the sterile and L. ferriphilum systems. Liao et al.27 investigated the effect of marmatite on the bioleaching of chalcopyrite and observed that marmatite hindered the dissolution of chalcopyrite. The reason for this finding may be that the redox potential of the mixed minerals was not in the optimum range.
In a natural environment, the contact of sulfide minerals cannot be avoided. Thus, it is essential to study the interaction of sulfide minerals in a geochemical environment. Both marmatite and bornite are iron-bearing sulfide minerals, but their interactions in an acidic environment have not been studied yet. However, the use of bacteria for the bio-dissolution of flotation marmatite concentrates and bornite has been proven to be feasible.28,29 Therefore, the dissolution processes of marmatite and bornite in the abiotic system and in the presence of A. ferrooxidans were conducted in this work. Analytic techniques such as electrochemical experiments, synchrotron radiation XRD (SR-XRD) and Raman spectroscopy were conducted to provide some insights into the interaction mechanisms between marmatite and bornite during the mixed dissolution process in the acidic abiotic and biotic systems. This would be useful for the research fields of both (bio)hydrometallurgy and AMD. On the one hand, pyrite is the main cause of AMD production, but other sulfide minerals such as zinc sulfide and copper sulfide can also release heavy metal ions and contribute to the production of AMD, resulting in serious environmental problems.30,31 Therefore, the interaction between them plays an important role in understanding the mechanism of AMD production and would also provide a reference for preventing the production of AMD from the source. On the other hand, bornite (Cu5FeS4) and chalcopyrite (CuFeS2) are the most abundant copper mineral resources in the world as both usually co-exist in raw ores.32 Moreover, studies have shown that chalcopyrite is reduced to bornite in the initial stage of bioleaching, which is also the rate-limiting step in the dissolution of chalcopyrite since the dissolution of bornite is very rapid.33,34 As stated in several recent reports, R. G. Acres et al.25 found that bornite can reduce the oxidation rate of chalcopyrite. However, Zhao et al.24 proposed that a synergistic effect existed between chalcopyrite and bornite bioleaching in the presence of A. ferrooxidans. Therefore, this work plays an important role in understanding the biological leaching behaviors of mixed ores and the dissolution mechanism of other copper sulfide ores, especially chalcopyrite, which represents approximately 70% of the known copper reserves;35 this also provides an important research example for hydrometallurgy.
2. Experimental section
2.1. Dissolution experiments
000 × the dilution ratio.:bornite = 2:0, 3:1, 1:1, 1:3 and 0:2) was conducted in 250 mL flasks containing 99 mL sterilized 9K basal medium and 1 mL bacteria culture. In order to investigate the effect of Cu2+ on the dissolution of marmatite, we conducted a second batch of dissolution experiments. Similarly, a series of concentration gradients of Cu2+ (0, 0.01, 0.05, 0.25, 0.5, 0.75, 1.0 and 2.5 g L−1) were conducted in 250 mL flasks containing 100 mL sterilized 9K basal medium and 2 g marmatite. Then, all dissolution experiments were conducted at 30 °C and 170 rpm in an orbital shaker for 14 days. Water evaporation was compensated for by adding sterilized deionized water periodically. During the testing period, the liquid sample loss was supplemented with the same amount of distilled water, and the values of pH were maintained at 1.7–1.80 by adding sulfuric acid. During the dissolution process, the variations in pH, ORP and bacterial concentrations, and cupric, iron and zinc ion concentration values were measured.2.2. Electrochemical experiments
The electrodes used in all electrochemical experiments were carbon paste electroactive electrodes (CPEEs). The composition was 0.7 g minerals, 0.2 g graphite, and 0.1 g solid paraffin. The mixtures were manually homogenized, and they were mechanically pressed at 20 tons to obtain a uniform sample surface.39 All the experiments were performed with a three-electrode system with the graphite rod as the counter electrode and the Ag/AgCl (using 3.0 mol L−1 KCl) electrode as the reference electrode. The open circuit potential (OCP), Tafel tests and cyclic voltammetry tests of bornite, marmatite and the mixed minerals with different ratios of marmatite and bornite were performed at the electrochemical work station. Meanwhile, the galvanic current effect between marmatite and bornite was measured. Prior to each test, the corresponding electrodes were smoothed by 1200 mesh sandpapers. The electrolyte was deionized water with pH of 1.70. During the test, nitrogen was introduced into the electrolyte to remove oxygen. OCP was conducted for 900 seconds. Tafel tests were performed at a sweep rate of 2 mV s−1. Cyclic voltammetry (CV) tests were conducted at a sweep rate of 20 mV s−1. The galvanic current between marmatite and bornite was analyzed by an electrochemical noise measurement program for 900 s.2.3. Analysis of liquid and solid samples
The analytic process for the liquid sample during the (bio)dissolution process is shown in Fig. 1a. The dissolved Fe2+ and Fe3+ were quantified through the o-phenanthroline spectrophotometry method40 with a microplate reader (BOX 998, Biotek Instruments, HIGHLAND PARK, USA). Iron, Cu2+, and Zn2+ concentrations were analyzed by inductively coupled plasma-atomic emission spectrometry (ICP-AES) (ICAP-7400 Radial, Thermo Fisher Scientific, MA, USA); meanwhile, the pH and oxidation–reduction potential (ORP) values were monitored with a pH meter (PHSJ-4A, Shanghai LEICI Co., Shanghai, China) and ORP meter (BPH-221, Dalian BELL Co., Dalian, China), respectively. The bacterial density was detected using a hemocytometer in a bright field microscope (CX31RTSF, Tokyo OLYMPUS Co., Tokyo, Japan). The mineralogical contents of bornite and marmatite were detected by XRF (Axios MAX, PANalytical B.V., Almelo, Holland). All electrochemical experiments were performed by electrochemical TAB (CHI700E, Shanghai CH Instruments, Shanghai, China). The mineralogical phases of bornite, marmatite and the residues were analyzed by SR-XRD at beam line of 4B9A of the Beijing Synchrotron Radiation Facility (BSRF). The mineralogical changes were detected by a confocal micro-Raman spectrometer (inVia Qontor, Renishaw, United Kingdom).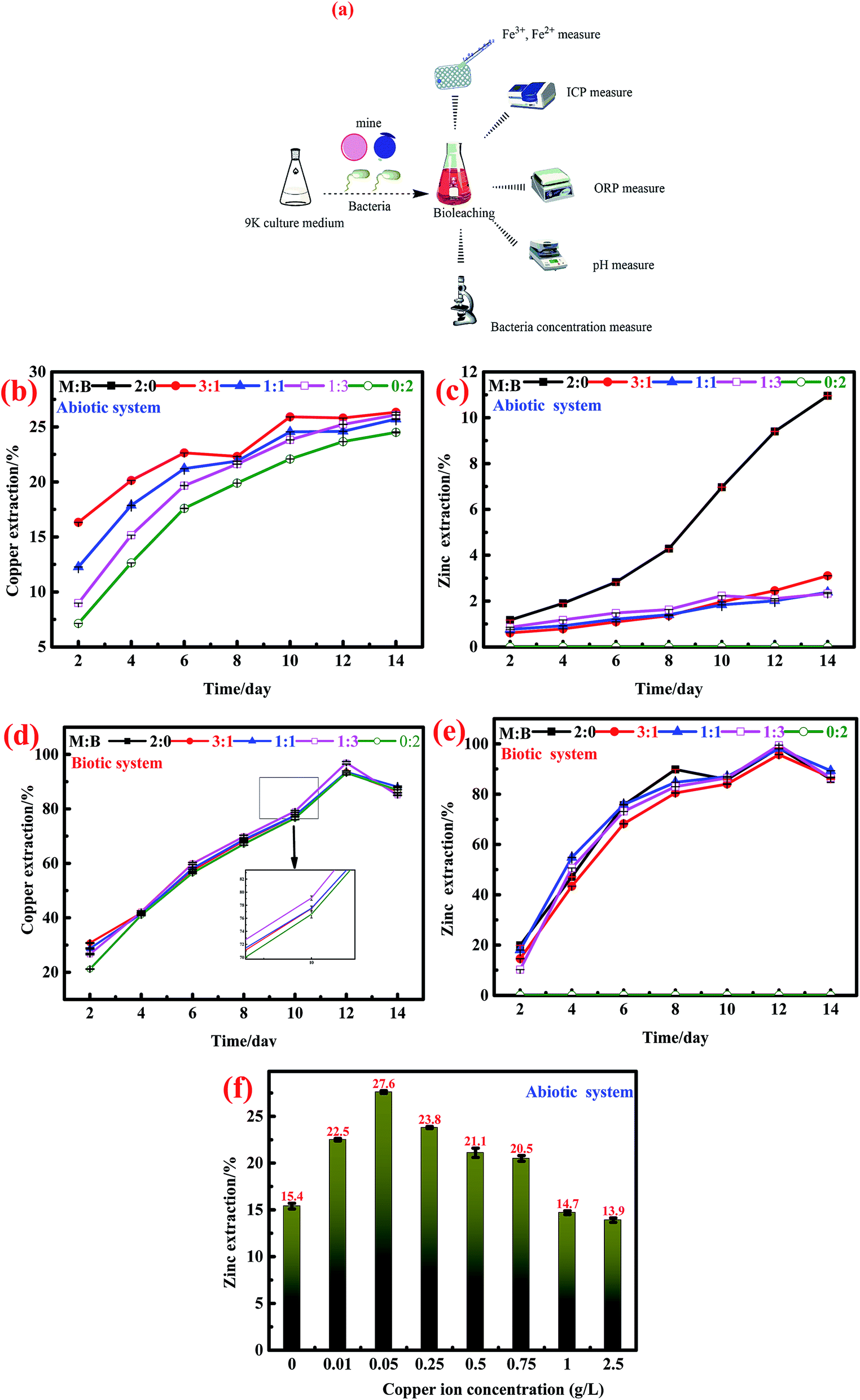 | ||
| Fig. 1 The dissolution behaviors of marmatite and bornite with different mass ratios in the absence and presence of bacteria. (a) Model diagram of the liquid sample detection during dissolution experiments. The ion extraction of copper and zinc in the absence of bacteria: (b) copper extraction and (c) zinc extraction. The ion extraction of copper and zinc in the presence of bacteria: (d) copper extraction and (e) zinc extraction. (f) The final zinc extractions of marmatite under different conditions of Cu2+ concentrations. M (marmatite) and B (bornite). | ||
3. Results and discussion
3.1. Results
:bornite = 3:1, 1:1 and 1:3).In the presence of A. ferrooxidans, the interactions between bornite and marmatite were similar to those in the abiotic system. The addition of marmatite accelerated the dissolution of bornite, but the final copper extractions of single bornite and mixed ores were not very different; all resulted in the final copper extraction of more than 90%. On the contrary, the addition of bornite inhibited the dissolution of marmatite but eventually, the zinc extraction of single marmatite and mixed ores reached higher than 90%.
The effect of cupric ions on the oxidative dissolution of marmatite is shown in Fig. 1e. The concentrations of cupric ions were 0, 0.01, 0.05, 0.25, 0.5, 0.75, 1.0 and 2.5 g L−1; the corresponding final zinc extraction values of marmatite were 15.4, 22.5, 27.6, 23.7, 21.1, 20.5, 14.6 and 13.9%. It could be found that the cupric ion concentration of 0–0.75 g L−1 accelerated the dissolution of marmatite. However, the cupric ions could obviously hinder the dissolution of marmatite when their concentration exceeded 1.0 g L−1, which was consistent with previous research.41
3.1.2.1. Open circuit potential and galvanic current effect test. OCP is an important reference for judging the presence of the galvanic current between different minerals. The OCP values for the minerals consisting different ratios of marmatite and bornite were 153 (2
:0), 213 (3:1), 216 (1:1), 226 (1:3) and 242 (0:2) mV (Fig. 2a). The OCP value for mixed sulfides was higher than that of single marmatite, and it increased as the proportion of bornite increased gradually.
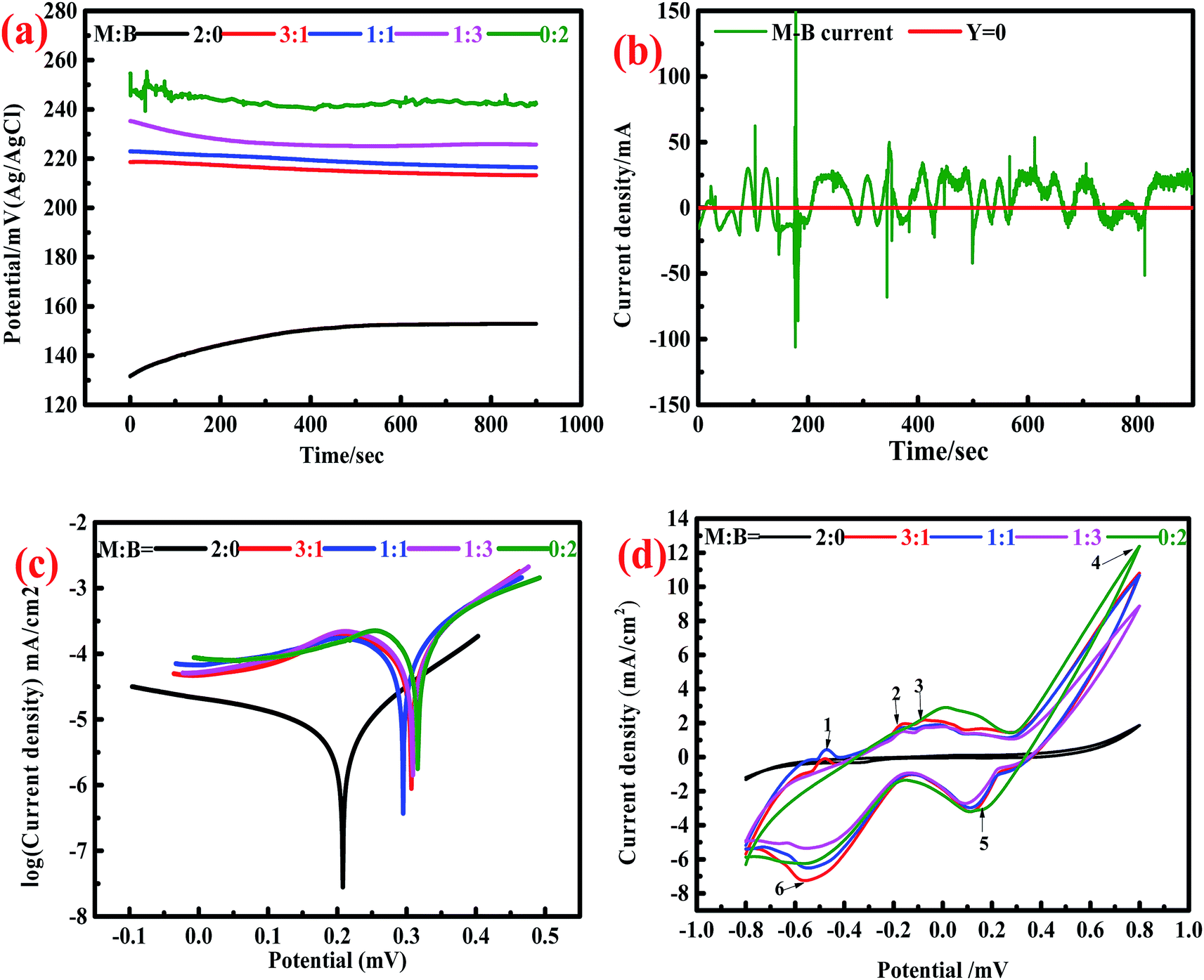 | ||
| Fig. 2 Comparison of the electrochemistry of electrodes with different mass ratios of marmatite and bornite: (a) open circuit potential, (b) galvanic current, (c) Tafel plots, (d) cyclic voltammograms; M (marmatite) and B (bornite). | ||
Theoretically, there is a galvanic current between marmatite and bornite. In the galvanic couple of marmatite-bornite, bornite would act as the cathode, marmatite would act as the anode, and the oxidation of marmatite would be accelerated. However, this conjecture was contrary to the results of the dissolution experiments. The electrochemical noise measurement results further proved that the galvanic current between marmatite and bornite was extremely weak (Fig. 2b). Hence, the galvanic effect between bornite and marmatite was extremely small and could be ignored.
3.1.2.2. Tafel studies. The corrosion current density indicates the kinetics for corrosion reactions. The higher the corrosion current, the faster the reaction rate. The corrosion potential indicates the thermodynamics for the degree of corrosion. A high corrosion potential means that it is difficult for the corrosion reaction to occur, and a low corrosion potential indicates that the reaction can take place easily. Fig. 2c shows the Tafel plots of the electrodes with different mass ratios of marmatite and bornite. The result calculated from the Tafel plot showed that the corrosion current densities of the samples containing different ratios of marmatite and bornite were 8.122 × 10−3 (2
:0), 1.785 × 10−1 (3:1), 2.103 × 10−1 (1:1), 2.018 × 10−1 (1:3), and 2.972 × 10−1 (0:2) mA cm−2; the corresponding corrosion potentials were 208.5, 307.1, 294.2, 305.2 and 316.9 mV.Fig. 2d indicates the oxidation and reduction reactions for the samples with different mass ratios of marmatite and bornite. The cyclic voltammetry curves of the mixed ores and single minerals were similar; they showed four anodic peaks (1–4) and two cathodic peaks 5 and 6). Therefore, the electrochemical interactions did not change the redox reactions of bornite and/or marmatite but did change the reaction kinetics. The dissolution reactions for marmatite and bornite can be summarized as follows in eqn (1)–(3):16,42
| Zn(1−x)FexS + 2H+ + 1/2O2 → (1 − x)Zn2+yFe2+ + (x − y)FeS + (1 − x + y)S + H2O | (1) |
| Zn(1−x)FexS + 2(1 − x + y)Fe3+ → (1 − x)Zn2+ + (2 − 2x + 3y)Fe2+ + (x − y)FeS + (1 − x + y)S | (2) |
| 4Cu5FeS4 + 37O2 + 20H+ → 20Cu2+ + 4Fe3+16SO2−4 + 10H2O | (3) |
Peak 1 was attributed to the oxidation reaction of metal copper to chalcocite (Cu2S); the reaction is summarized in eqn (4):34,43,44
| 2Cu0 + H2S → Cu2S + 2H+ + 2e− | (4) |
The reaction of peak 2 and may be the formation of the sulfur element as well as the reaction formula can be seen in eqn (5):45,46
| H2S → S0 + 2H+ + 2e− | (5) |
Peak 3 and 4 represent the stepwise oxidation of chalcocite. A large peak d was detected at about 600–800 mV. The reactions for the formation of chalcocite through a series of defect sulfides djurleite (Cu1.92S), digenite (Cu1.60S) and covellite (CuS) are shown in eqn (6)–(8):38,44,47
| Cu2S → Cu1.92S + 0.08Cu2+ + 0.16e− | (6) |
| Cu1.92S → Cu1.60S + 0.32Cu2+ + 0.64e− | (7) |
| Cu1.60S → CuS + 0.60Cu2+ + 1.2e− | (8) |
The cathodic peak 5 may represent the reduction reactions of elemental sulfur, chalcocite and bornite; the reactions are shown in eqn (9)–(11):43,44,47
| S0 + 2H+ + 2e− → H2S | (9) |
| Cu2S + 2H+ + 2e− → 2Cu0 + H2S | (10) |
| 3Cu5FeS4 + 6H+ + 2e− → 5Cu2S + 3H2S + 2Fe2+ | (11) |
The reactions for peak 6 involved the formation of chalcocite, which can be described as follows:34,47
| Cu2+ + CuS + 2e− → Cu2S | (12) |
| 2CuS + 2H+ + 2e− → Cu2S + H2S | (13) |
3.2. Discussions
Synchrotron X-ray powder diffraction analysis (Fig. 3a) showed that the peaks for bornite underwent massive changes, while those for marmatite remained unchanged in the abiotic system. The main intermediate of bornite dissolution was haycockite (Cu4Fe5S8); the peaks for marmatite were always observed in the residues of marmatite and mixed ores, indicating that bornite dissolved faster than marmatite. However, no metal mineralogical phases were obviously detected in the residues for the biotic system (Fig. 3b). Only an elemental sulfur phase was detected, which indicated that the minerals dissolved more completely.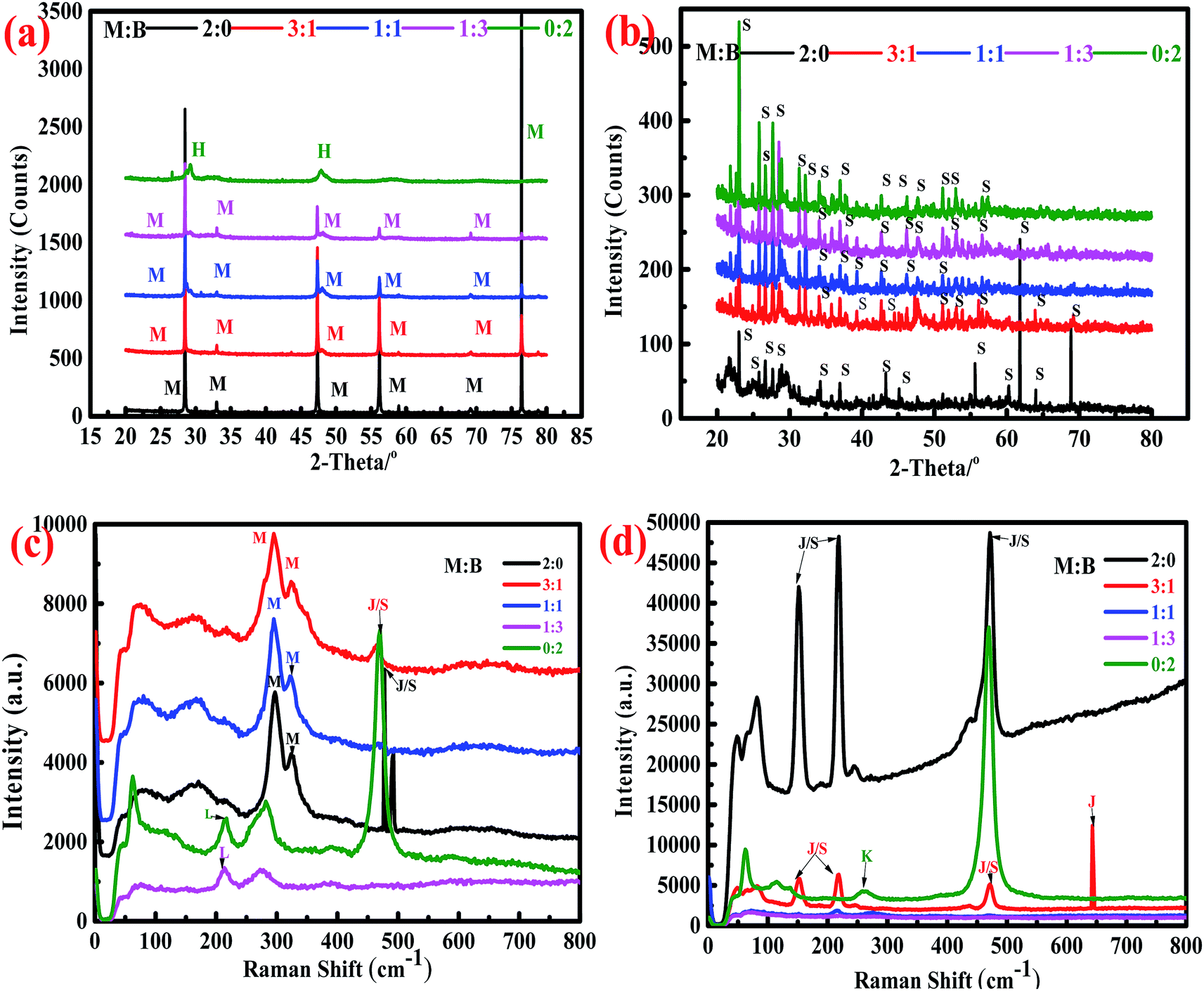 | ||
| Fig. 3 Synchrotron X-ray powder diffraction analysis and Raman analysis of residues in the abiotic and biotic systems. (a and b) Synchrotron X-ray powder diffraction analysis of dissolution residues for (bio)dissolution experiments: (a) dissolution residues in the abiotic system, (b) dissolution residues in the biotic system (H – haycockite, Cu4Fe5S8; L – chalcocite, Cu2S; J – jarosite, KFe3(SO4)2(OH)6; K – covellite, CuS; M – marmatite, (ZnxFey)S; S – sulfur, S8). (c and d) Raman analysis of dissolution residues for (bio)dissolution experiments: (c) dissolution residues in abiotic system, (d) dissolution residues in biotic system; M (marmatite) and B (bornite). | ||
The Raman database used for comparing the obtained spectra is the RRUFF database.48 The bands for sphalerite are mainly observed at 299 and 320 cm−1. Elemental sulfur has very strong bands at 154, 220 and 473 cm−1. The bands for jarosite are similar to those of elemental sulfur (154, 220 and 473 cm−1), but bands also exist at 138, 153, 220 and 639 cm−1. The band for chalcocite is mainly found at 213 cm−1, while the bands for covellite are mainly observed at 261 and 470 cm−1 (ESI, Fig. S1†).
The Raman spectra of the dissolution residues are shown in Fig. 3c–d. In the abiotic system, bands for sphalerite were detected, which were consistent with the analytic result of synchrotron radiation. There was a band for chalcocite (213 cm−1); it was speculated that chalcocite may exist in the dissolution residues of bornite and mixed ores (M:B = 1:3). The band was also detected at 477 cm−1, which was close to the band for elemental sulfur, indicating that elemental sulfur covered the surface of the residues. In the biotic system, the main bands for the residues were in accordance with those of elemental sulfur and jarosite. The band at 259 cm−1 could be attributed to covellite. This result was consistent with a previous research result.16
During the whole dissolution process, the values of pH in both the biotic and abiotic systems were always maintained at 1.70–1.80. Based on the dynamic monitoring of the daily pH values and according to eqn (14)–(16), we could calculate the acid consumption during the entire leaching process. The total acid consumption in both the biotic and abiotic systems is shown in Fig. 4a; it can be found that the whole acid consumption increased on increasing the bornite ratio.
| pH1 = −lg[H1+] | (14) |
| pH2 = −lg[H2+] | (15) |
| m = (10−pH1 − 10−pH2) × V | (16) |
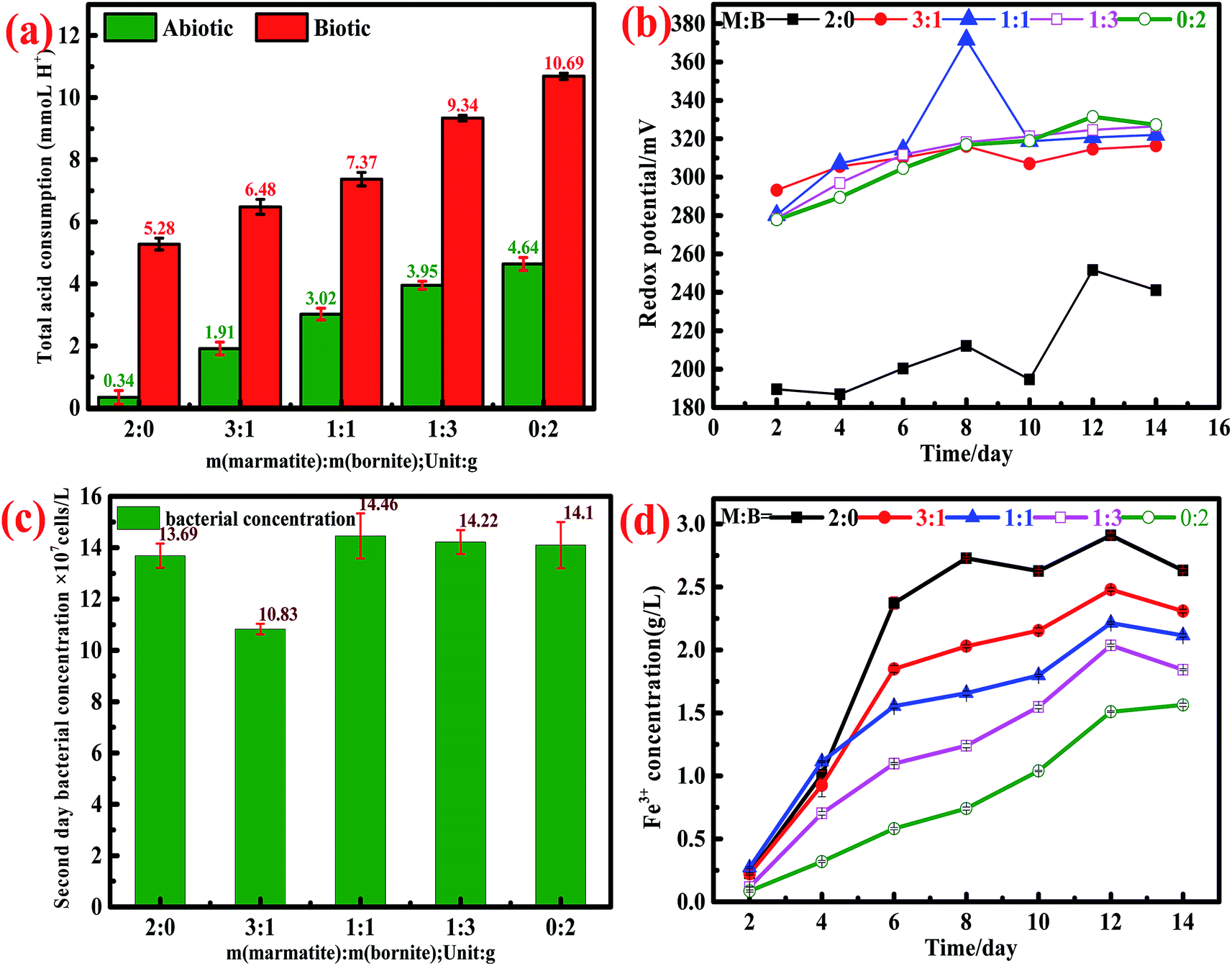 | ||
| Fig. 4 Characteristics of leachates: (a) total acid consumption in both biotic and abiotic environments. (b) Oxidation reduction potential in the abiotic system. (c) The concentration of bacteria in the biotic system on the 2nd day. (d) Fe3+ concentration in the biotic system; M (marmatite) and B (bornite). | ||
In the abiotic system, the interactions between bornite and marmatite were more obvious. As mentioned above, the addition of marmatite accelerated the dissolution of bornite, while the addition of bornite impeded the dissolution of marmatite. For the dissolution of bornite, ORP increased gradually on increasing the proportion of marmatite in the mixed ores because of the contributions of the dissolved iron from marmatite (Fig. 4b), thus accelerating the oxidative dissolution process. The dissolution of bornite in the mixed ores was accelerated, thus resulting in a higher cupric ion concentration. A high concentration of Cu2+ (>0.75 g L−1) was adsorbed on the unreacted marmatite surface to form a passivation layer mainly consisting of Cu–S, Cu–O and CuSn species, which reduced the electrochemical reactivity of marmatite and hindered the oxidative dissolution of marmatite.41 The lowest cupric ion concentration in the system was 0.8 g L−1 on the second day, which inhibited the further dissolution of marmatite according to the above result (Fig. 1f). Furthermore, elemental sulfur and jarosite were formed when the sulfide mineral was oxidized and adhered to the mineral surface, thus hindering the further dissolution of sulfide minerals.24,29 Since there were no bacteria in the system, the elemental sulfur hardly oxidized to sulfuric acid. Therefore, the inhibition effect was more obvious than that in the biotic system.
In the biotic system, since the strains used were domesticated, the growth and physiological activity were powerful. As shown in Fig. 4c, the bacterial concentration exceeds 108 mL−1 on the second day and remains above this concentration until the end of the dissolution experiments. The rapid increase in the bacterial concentration accelerated the redox reactions of marmatite and bornite. Therefore, the oxidative dissolution of minerals was accelerated. For the dissolution of bornite, the addition of marmatite increased the concentration of the ferric ions in the solution system (Fig. 4d). The high concentration of ferric ions not only acted as an oxidant for mineral dissolution but also increased the ORP of the system, thus accelerating bornite dissolution. For the dissolution of marmatite, on one hand, the rapid dissolution of bornite caused the concentration of cupric ions to rapidly exceed 1 g L−1 on the second day; on the other hand, the elemental sulfur and jarosite generated were attached to the mineral surface. It has been commonly reported that polysulfide is a possible passivating species in metal sulfide leaching.49,50 Therefore, the addition of bornite inhibited the dissolution of marmatite. The presence of bacteria attenuated the inhibition effect because the formed elemental sulfur was oxidized into sulfuric acid by the bacteria. However, the bio-oxidation of elemental sulfur lagged behind the generation process. Hence, the sulfur product layer was still present and became an obstacle for the dissolution of marmatite.42
A schematic is shown in Fig. 5 to explain the interaction mechanisms between bornite and marmatite in an acidic environment. Fe2+ dissolved from marmatite and bornite could provide an energy source for the bacterial growth and activity through oxidizing Fe2+ to Fe3+, which produced oxidants of Fe3+ and increased ORP. Hence, the oxidative dissolution process of bornite was accelerated. On the contrary, the dissolution of bornite could produce a high concentration of Cu2+ (>1.0 g L−1), thus hindering marmatite dissolution. Bornite intensified marmatite dissolution when the percentage of bornite or Cu2+ concentration (0–0.75 g L−1) was extremely low, resulting in the occurrence of a synergic dissolution process. In addition, the galvanic current between bornite and marmatite was negligible; thus, its role may be ignored.
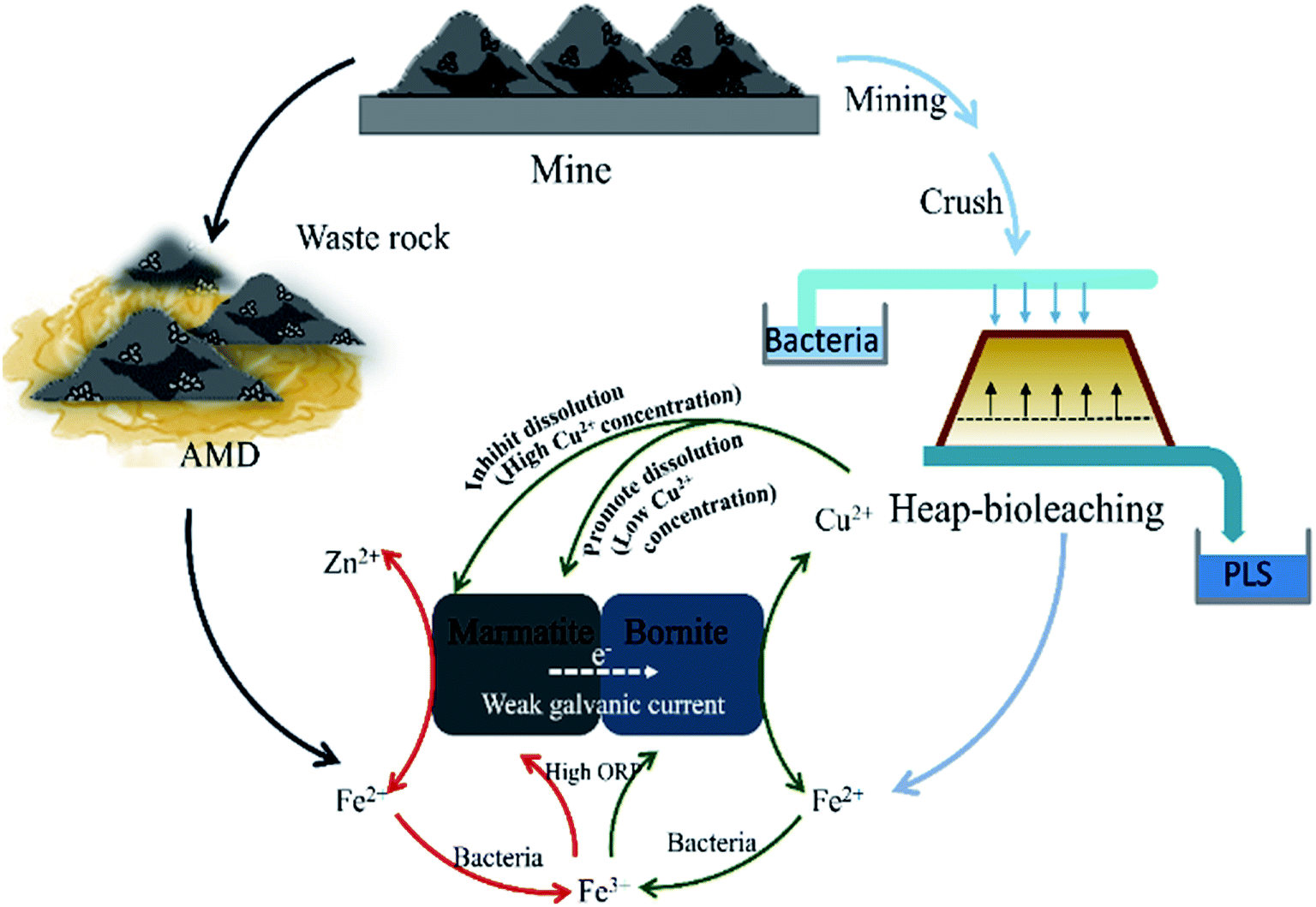 | ||
| Fig. 5 A model for interpreting the interaction mechanisms between bornite and marmatite in an acidic environment. | ||
4. Conclusion
The oxidative dissolution processes of marmatite and bornite were both acid-consuming processes, and bornite dissolution consumed more sulfuric acid. The dissolution of bornite could be accelerated by the addition of marmatite, which produced oxidants of Fe3+ and increased ORP. The acceleration effect in the biotic system was less obvious than that in the abiotic system. The dissolution of marmatite was inhibited by the addition of bornite because a high Cu2+ concentration (higher than 1 g L−1) could significantly hinder marmatite dissolution. Furthermore, the formation of elemental sulfur or jarosite was also one important cause for hindering marmatite dissolution. This was one reason for the different degrees of inhibition in the abiotic and biotic systems because the bacteria could oxidize the formed elemental sulfur. Theoretically, if the Cu2+ concentration caused by bornite dissolution would have been maintained at a relatively low concentration, the presence of bornite would have accelerated the dissolution of marmatite, thus resulting in a synergistic dissolution process. In addition, the galvanic effect between bornite and marmatite was negligible, and its role may be ignored. The research results can help understand the generation mechanisms of AMD and would also provide a reference for preventing the production of AMD from the source. This work can contribute to understanding the (bio)leaching process of complex ores such as selective and cooperative (bio)leaching. Furthermore, the obtained results would also be useful for studying the interactions of other sulfide minerals in abiotic and/or biotic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51704331), the Young Elite Scientists Sponsorship Program by CAST (2017QNRC001), the Innovation-Driven Project of Central South University (2018CX019), as well as the Postgraduate Independent Exploration and Innovation Project of Central South University (2019zzts705). The authors also thank Beamline 4B9A (Beijing Synchrotron Radiation Facility) for providing the experimental facilities.References
- V. P. Evangelou, J. Lab. Clin. Med., 1995, 82, 377–389 Search PubMed.
- D. B. Johnson and K. B. Hallberg, Sci. Total Environ., 2005, 338, 3–14 CrossRef CAS PubMed.
- A. Parbhakar-Fox and B. G. Lottermoser, Miner. Eng., 2015, 82, 107–124 CrossRef CAS.
- G. S. Simate and S. Ndlovu, J. Environ. Chem. Eng., 2014, 2, 1785–1803 CrossRef CAS.
- A. Akcil and S. Koldas, J. Cleaner Prod., 2006, 14, 1139–1145 CrossRef.
- N. Kuyucak, CIM Bull., 2002, 95, 96–102 CAS.
- N. O. Egiebor and O. Ben, Asia-Pac. J. Chem. Eng., 2010, 2, 47–62 CrossRef.
- E. R. Bobicki, Q. Liu, Z. Xu and H. Zeng, Prog. Energy Combust. Sci., 2012, 38, 302–320 CrossRef CAS.
- F. Anjum, M. Shahid and A. Akcil, Hydrometallurgy, 2012, 117–118, 1–12 CrossRef CAS.
- M. Chen, Y. Zhang, B. Zhong, Q. Liyou and B. Liang, Ind. Eng. Chem. Res., 2002, 41, 1329–1334 CrossRef CAS.
- R. A. Davis Jr, A. T. Welty, J. Borrego, J. A. Morales, J. G. Pendon and J. G. Ryan, Environ. Geol., 2000, 39, 1107–1116 CrossRef.
- D. E. Rawlings, Annu. Rev. Microbiol., 2002, 56, 65 CrossRef CAS PubMed.
- E. González-Toril, E. Llobet-Brossa, E. O. Casamayor, R. Amann and R. Amils, Ind. Eng. Chem. Res., 2003, 69, 4853 Search PubMed.
- F. S. Martín, W. Kracht and T. Vargas, Miner. Eng., 2018, 117, 127–131 CrossRef.
- Z. X. Liu, Z. L. Yin, S. F. Xiong, Y. G. Chen and Q. Y. Chen, Hydrometallurgy, 2014, 144–145, 86–90 CrossRef CAS.
- D. Bevilaqua, O. Garcia and O. H. Tuovinen, Process Biochem., 2010, 45, 101–106 CrossRef CAS.
- X. Tong, S. Song, J. He, F. Rao and A. Lopez-Valdivieso, Miner. Eng., 2007, 20, 259–263 CrossRef CAS.
- H. Zhao, X. Gan, J. Wang, L. Tao, W. Qin and G. Qiu, Hydrometallurgy, 2017, 171, 374–386 CrossRef CAS.
- F. K. Crundwell, Hydrometallurgy, 1988, 21, 155–190 CrossRef CAS.
- S. Y. Shi, Z. H. Fang and J. R. Ni, Bioelectrochemistry, 2006, 68, 113–118 CrossRef CAS PubMed.
- S. Y. Shi, Z. H. Fang and J. R. Ni, Process Biochem., 2006, 41, 438–446 CrossRef CAS.
- J. R. Ban, G. U. Guo-Hua and H. U. Ke-Ting, Trans. Nonferrous Met. Soc. China, 2013, 23, 494–500 CrossRef CAS.
- H. Zhao, J. Wang, X. Gan, X. Zheng, L. Tao, M. Hu, Y. Li, W. Qin and G. Qiu, Bioresour. Technol., 2015, 194, 28–35 CrossRef CAS PubMed.
- H. Zhao, J. Wang, M. Hu, W. Qin, Y. Zhang and G. Qiu, Bioresour. Technol., 2013, 149, 71–76 CrossRef CAS PubMed.
- R. G. Acres, S. L. Harmer and D. A. Beattie, Int. J. Miner. Process., 2010, 94, 43–51 CrossRef CAS.
- A. Saavedra, J. V. García-Meza, E. Cortón and I. González, Electrochim. Acta, 2018, 265, 569–576 CrossRef CAS.
- R. Liao, J. Wang, H. B. Zhao, X. X. Wang, X. T. Huang and M. X. Hong, Solid State Phenom., 2017, 262, 168–171 Search PubMed.
- D. Bevilaqua, H. A. Acciari, F. A. Arena, A. V. Benedetti, C. S. Fugivara, G. T. Filho and O. G. Júnior, Miner. Eng., 2009, 22, 254–262 CrossRef CAS.
- S.-Y. Shi and Z.-H. Fang, Hydrometallurgy, 2004, 75, 1–10 CrossRef CAS.
- J. Castillo, R. Pérez-López, M. A. Caraballo, J. M. Nieto, M. Martins, M. C. Costa, M. Olías, J. C. Cerón and R. Tucoulou, Sci. Total Environ., 2012, 423, 176–184 CrossRef CAS PubMed.
- A. Voegelin, O. Jacquat, S. Pfister, K. Barmettler, A. C. Scheinost and R. Kretzschmar, Environ. Sci. Technol., 2011, 45, 255–261 CrossRef CAS PubMed.
- H.-b. Zhao, M.-h. Hu, Y.-n. Li, S. Zhu, W.-q. Qin, G.-z. Qiu and J. Wang, Trans. Nonferrous Met. Soc. China, 2015, 25, 303–313 CrossRef CAS.
- A. N. Buckley, I. C. Hamilton and R. Woods, J. Appl. Electrochem., 1984, 14, 63–74 CrossRef CAS.
- G. Gu, K. Hu, X. Zhang, X. Xiong and H. Yang, Electrochim. Acta, 2013, 103, 50–57 CrossRef CAS.
- T. Rohwerder, T. Gehrke, K. Kinzler and W. Sand, Appl. Microbiol. Biotechnol., 2003, 63, 239 CrossRef CAS PubMed.
- T. J. Veasey and B. A. Wills, Miner. Eng., 1991, 4, 747–752 CrossRef.
- Y. Zhang, G. Zhao, X. Lv, Y. Tian, L. Yang, G. Zou, H. Hou, H. Zhao and X. Ji, ACS Appl. Mater. Interfaces, 2019, 11, 6154–6165 CrossRef CAS PubMed.
- X. Wang, R. Liao, H. Zhao, M. Hong, X. Huang, H. Peng, W. Wen, W. Qin, G. Qiu, C. Huang and J. Wang, Hydrometallurgy, 2018, 176, 9–16 CrossRef CAS.
- A. D. Bas, W. Zhang, E. Ghali and Y. Choi, Hydrometallurgy, 2015, 158, 1–9 CrossRef CAS.
- L. G. Saywell and B. B. Cunningham, Ind. Eng. Chem., Anal. Ed., 1937, 9, 67–69 CrossRef CAS.
- X. Meng, H. Zhao, M. Sun, Y. Zhang, Y. Zhang, X. Lv, H. Kim, M. Vainshtein, S. Wang and G. Qiu, Sci. Total Environ., 2019, 675, 213–223 CrossRef CAS PubMed.
- Z. Lan, Y. Hu and W. Qin, Miner. Eng., 2009, 22, 10–13 CrossRef CAS.
- L. I. Hong-Xu, G. Z. Qiu, H. U. Yue-Hua, D. Q. Cang and D. Z. Wang, Trans. Nonferrous Met. Soc. China, 2006, 16, 1240–1245 CrossRef.
- C.-L. Liang, J.-L. Xia, Y. Yang, Z.-Y. Nie, X.-J. Zhao, L. Zheng, C.-Y. Ma and Y.-D. Zhao, Hydrometallurgy, 2011, 107, 13–21 CrossRef CAS.
- M. Sauber and D. G. Dixon, Hydrometallurgy, 2011, 110, 1–12 CrossRef.
- A. E. Elsherief, Miner. Eng., 2002, 15, 215–223 CrossRef CAS.
- E. M. Arce and I. González, Int. J. Miner. Process., 2002, 67, 17–28 CrossRef CAS.
- D. T. U. o. Arizona, The RRUFF Project website containing an integrated database of Raman spectra for minerals, 2018 Search PubMed.
- H. Zhao, Y. Zhang, X. Zhang, L. Qian, M. Sun, Y. Yang, Y. Zhang, J. Wang, H. Kim and G. Qiu, Miner. Eng., 2019, 136, 140–154 CrossRef CAS.
- M. Khoshkhoo, M. Dopson, A. Shchukarev and Å. Sandström, Hydrometallurgy, 2014, 149, 220–227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03658j |
| This journal is © The Royal Society of Chemistry 2019 |