
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective Wacker type oxidation of a macrocyclic diene to the corresponding monounsaturated ketone used as fragrance†
Tom Brunzel *a,
Johannes Heppekausenb,
Johannes Pantenb and
Angela Köckritza
*a,
Johannes Heppekausenb,
Johannes Pantenb and
Angela Köckritza
aLeibniz Institute for Catalysis at the University of Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: angela.koeckritz@catalysis.de
bSymrise AG, Mühlenfeldstraße 1, 37603 Holzminden, Germany
First published on 4th September 2019
Abstract
A selective reaction method for the efficient conversion of an isomeric mixture of 1,9-cyclohexadecadiene (1,9-CHDD) to the corresponding monounsaturated cyclohexadec-8-en-1-one (8-CHD) is described. 8-CHD was synthesized via Wacker type oxidation at room temperature using a highly electrophilic in situ formed dicationic palladium species. Isomerisation of the diene and over-oxidation of the substrate could be nearly suppressed by suitable reaction control, which has a positive effect on selectivity. The utilization of molecular oxygen as a green oxidant and environmentally benign iron(III) salts as co-catalysts was successfully applied. This reaction strategy is promising to overcome the low overall reactivity of internal olefins in Wacker type oxidations. In addition, larger scale experiments showed further potential for industrial application.
Introduction
Since the 1950s when the Wacker process was described for the first time and industrially established a few years later, the Wacker oxidation has become a powerful tool for organic synthesis of fine and platform chemicals.1,2 The initial Wacker process refers to the aerobic palladium(II) chloride catalysed oxidation of ethylene to acetaldehyde in the presence of copper(II) chloride in aqueous acidic medium.3–6 Instead, the usage of an organic solvent as reaction medium, known as the Wacker–Tsuji oxidation, has opened new possibilities for the lab scale transformation of higher α-olefins to the corresponding methyl ketones.7 These oxyfunctionalised compounds are of great interest for the cosmetic, flavour and fragrance industries, among others.8–10 However, the Wacker–Tsuji oxidation is limited to the oxidation of terminal olefins while the oxidation of internal olefins remains challenging not least because of their lower selectivity and reactivity.11–13 This diminished selectivity mainly arises from the formation of undesired oxygenated side products by fast skeletal double bond isomerisation, also favoured by palladium. Multi-step synthesis and drastic reaction conditions have been used to overcome the lack of an efficient transformation of ketones from internal olefins.14 Regarding the increasing demand for greener and efficient processes, simplified methods are highly desirable. A lot of internal olefin transformation reactions have been investigated in the past and are summarized in Scheme 1. First successful attempts to overcome the barriers for internal and cyclic olefin transformation were done by Wayner et al.15 They found a copper- and chloride-free reaction system utilizing benzoquinone (BQ) as reoxidant in presence of inorganic acids bearing weakly coordinating anions (WCAs) in an acetonitrile/water solvent mixture. Interestingly, the reaction was strongly inhibited when hydrochloric acid was used, suggesting that the formation of a highly electrophilic dicationic palladium species strongly depends on the nature of the inorganic acid.16 Recently, Kaneda et al. disclosed a simple reaction technique for the Wacker type oxidation of internal olefins to tackle the problems of skeletal double bond isomerisation forcing the selectivity of the desired ketone to decrease.12 When using N,N-dimethylacetamide (DMA) as solvent, which is supposed to stabilise the reduced palladium species, the oxidation with oxygen as green and sole oxidant proceeds without isomerisation of the double bond.17 Further investigations showed broad substrate scope, functional group tolerance and high regioselectivity for this oxidation reaction, opening new strategies for the synthesis of biomolecules and other fine chemicals.18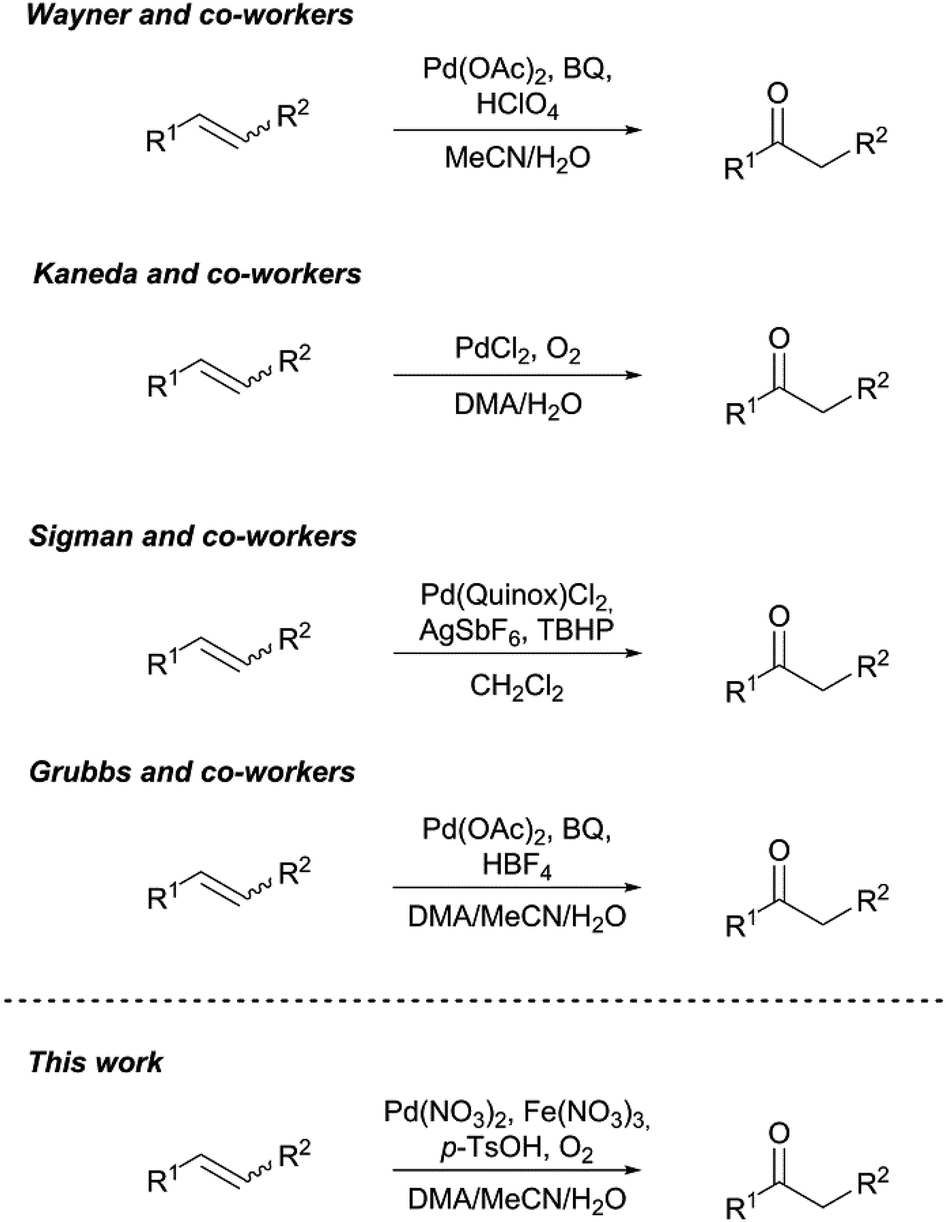 | ||
| Scheme 1 Reported Wacker type oxidation reactions of cyclic and internal olefins. | ||
More recently, Sigman and co-workers developed a special designed Pd-catalyst for the Wacker type oxidation of internal olefins.19 They used 2-(4,5-dihydro-2-oxazolyl)quinoline (quinox) as ligand and tert-butyl hydroperoxide (TBHP) as an easy to handle oxidant but they also required small amounts of silver salts with WCA. Using this method, ketones were obtained in comparably excellent yields and no double bond isomerisation was observed due to the elegantly designed catalyst. Nevertheless, the high price of this special catalyst, the use of an additional noble metal and the application of dichloromethane as solvent complicate the industrial implementation. At the same time, Grubbs and co-workers found a straightforward approach for the selective oxidation of internal olefins to the corresponding ketone at room temperature under ambient conditions.20 They used a reaction system related to the system already described by Wayner et al. (BQ as stoichiometric reoxidant and an acid with WCA) but they ingeniously added DMA (Kaneda) to the binary acetonitrile/water solvent mixture. With this improvement, the benefits of Kanedas reaction system (stabilizing reduced palladium) and Wayners method (dicationic palladium catalyst) were combined, where DMA stabilizes the reduced Pd(0) species, and acetonitrile participates in the formation of the dicationic palladium catalyst. Ketones were obtained in high yields and the reaction showed improved substrate scope, motivating us to further investigate this reaction procedure for a macrocyclic diene which is an industrially important raw material.21
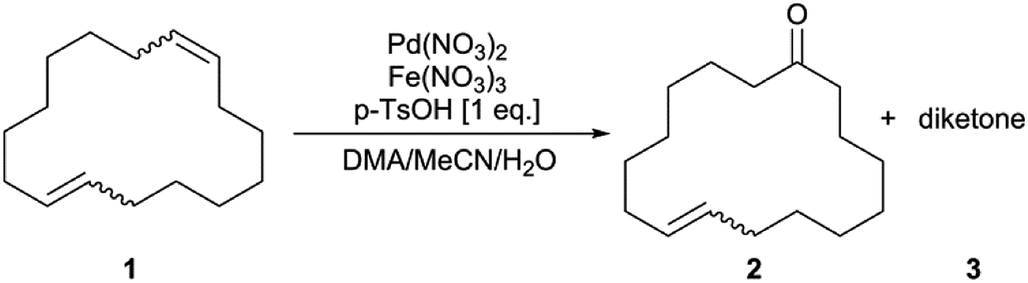
To the best of our knowledge, the selective oxidation of a macrocyclic diene to the corresponding monounsaturated ketone in one reaction step via Wacker type oxidation is not described in the literature. With this work, a simple chloride- and copper-free method for the palladium-catalysed oxidation of the macrocyclic diene 1,9-CHDD (1, isomeric mixture) to the musky-smelling 8-CHD (2, isomeric mixture) is reported, where only one double bond is selectively oxidised while the second double bond remains “untouched” (Scheme 2).22 The macrocyclic diene (1) was used as an isomeric mixture consisting of the cis/cis, cis/trans and trans/trans isomer, which complicates the reaction procedure owing to different kinetics and reactivity of the single alkene moieties. Chelation and subsequent deactivation of the palladium centre by dienes is reported in literature and should therefore be suppressed.23–27 The ratio of the used isomers (cis, cis/cis, trans/trans, trans) is about 1/2/1 and the product (2) is an isomeric mixture of two isomers of high value. Finally, the Wacker type oxidation of (1) can be coupled with molecular oxygen using an inexpensive, environmentally friendly and benign iron co-catalyst without affecting the catalyst activity. Different reaction parameters were extensively examined including reaction temperature, solvent system, catalyst concentration and oxidant. Interestingly, the oxidation proceeds better at room temperature while higher temperatures negatively affect the catalyst performance.
 | ||
| Scheme 2 Pathways in oxidation of 1,9-CHDD. | ||
Results and discussion
The industrial synthesis of (2) is accompanied by several disadvantages such as low overall yield, two-step process, subsequent oxidation of the monoepoxide (4) in the first stage to the unwanted diepoxide (5) and elaborate process management.28 Loss of expensive starting material should be therefore avoided in aspects of efficiency and economy. The different alkene moieties of the three diastereomers of (1) should be oxidised to the same extent, yielding the product (2) as an isomeric mixture of cis- and trans-cyclohexadec-8-en-1-one. The presence of two isomers in different ratios gives the product its olfactory properties, which is the reason why the starting materials were not used in their pure form. The favoured formation of either the cis- or the trans-isomer would negatively affect the product quality and olfactory properties, respectively, and should be prevented by the design of the catalyst. Initial experiments using either the Kaneda conditions or the Sigman conditions for the Wacker type oxidation of (1) resulted in low product selectivity caused by isomerisation of (1) and loss of starting material due to polymerisation, respectively. Encouraged by the state of the art, we performed the oxidation of (1) with Pd(OAc)2 and used a ternary solvent mixture consisting of DMA/MeCN/H2O. BQ was used as a stoichiometric oxidant for the reduced Pd-species at ambient air conditions and room temperature. At higher conversions of the isomeric diene mixture, it might be difficult to prevent over-oxidation of the substrate. For this reason, full conversion of (1) was not considered to be the main goal for this work. Hence, the focus was directed to obtain a high selectivity of the isomeric monoketone (2) at convenient conversion to minimise the formation of the unwanted and olfactorily interfering diketone (3). The conversion for this reaction is defined as the sum of the individual conversions of the single 1,9-CHDD isomers (1) while the selectivity is defined as the total amount of isomeric monoketones (2) and diketones (3) respectively, formed from the converted diene mixture (1). The main side product obtained in all reactions was the diketone which also appeared as an isomeric mixture of cyclohexadecane-1,8-dione and cyclohexadecane-1,9-dione (3), respectively. Isomerisation of the double bond and subsequent oxidation leading to oxidised side products took place only to a minor extent and was therefore not discussed any further in this work.The role of the noncoordinating acid is crucial for the formation of the active catalyst species, which is formed in situ. It is likely that the abstraction of the palladium ligand is accelerated in presence of an inorganic non-coordinating acid, forming the catalytically active dicationic palladium species (Table 1, entry 1 vs. entry 2).29–31 The requirement for the acid is that its anion interacts only weakly with the metal centre; otherwise a less electrophilic neutral palladium species is formed. Therefore, the ligand itself should not be strongly coordinating, too. When a binary solvent system consisting of either MeCN/H2O (Table 1, entry 2) or DMA/H2O (Table 1, entry 3) is used, the oxidation rate is quite low, especially in the absence of MeCN. After the third solvent component was added, the selectivity of (2) was higher than 70%, but the conversion of (1) was quite low (Table 1, entry 4). The performance of this oxidation reaction strongly depends on the DMA/MeCN ratio. Unlike reported, we observed decreasing conversions with increasing the concentration of MeCN.20 In turn, increased conversions were observed with increasing concentration of DMA (Table 1, entry 4 vs. entries 5, 6). The best ratio for the organic solvents was found to be DMA/MeCN = 4/3 (Table 1, entry 5). With the optimised solvent ratio, different palladium precursors were tested (Table 1, entries 5, 7–10). As soon as chloride-containing palladium salts were used, barely noticeable reaction took place (Table 1, entries 7, 8), indicating the lack of active catalyst formation. Chlorides are known to possess strong donor properties, which make them unsuitable and disruptive ligands for the active catalyst formation. Therefore, chloride-free reaction conditions were urgently needed. The best results regarding conversion and selectivity were obtained when Pd(NO3)2 was used as catalyst precursor (Table 1, entry 10). It was reported in the literature that nitrates can be involved in two steps of the catalytic cycle, recovery of the catalyst and oxygen transfer from the NOx− groups to the substrate.6,10,32 While small amounts of the catalyst led to lower activity, no further oxidation was observed at higher catalyst concentrations due to the accelerated irreversible formation of inactive palladium black (Table 1, entry 11 vs. entry 12). At the same time, the selectivity of (2) decreased at the expense of the diketone (3) when higher amounts of Pd(NO3)2 were used. Next, the influence of the water concentration used in the ternary solvent mixture was studied. By halving the amount of water, a selectivity of (2) higher than 80% was achieved, while the conversion was only slightly affected (Table 1, entry 10 vs. entry 15). Surprisingly, when the commercial dicationic palladium complex [Pd(MeCN)4](BF4)2 was used without additional WCA-containing acid, only low conversion of (1) was observed (Table 1, entry 16). The initial oxidation rate was reached again after addition of the inorganic acid HBF4 (Table 1, entry 16). This observation led to the assumption that the formation of the active catalyst is equilibrium-controlled and is accelerated in presence of sufficient amounts of the inorganic acid. The influence of the WCA by the use of different inorganic and organic acids was also studied (Table 1, entries 15, 18–20). All of the used acids were well applicable and conversions up to 54% were obtained without remarkably affecting the selectivity for (2). The use of para-toluenesulfonic acid (p-TsOH) resulted in a good compromise with regard to a sufficient conversion of the isomeric (1) and a high selectivity of (2) (Table 1, entry 20). The selectivity of (2) can be further increased by reducing the amount of p-TsOH hat was used (Table 1, entry 21). However, the increase in selectivity of (2) is accompanied by a slight loss in conversion of (1). Last, the scalability for the Wacker type oxidation of (1) was investigated in a small batch experiment for two different solvent ratios (Table 1, entries 13, 22, 23). When both, the concentration of the reactants and the amount of the solvent were increased proportionally, no noteworthy changes in observations were made (Table 1, entry 10 vs. entry 13; entry 21 vs. entry 22). Higher temperatures such as 80 °C led to a decrease in reactivity and a dark reaction solution formed, indicated the lack of sufficient reoxidation of the reduced palladium(0) at elevated temperatures (Table 1, entry 13 vs. entry 14). Further increase of the reactant concentration at constant solvent volume led to a decrease of reactivity due to the reduced solubility of (1) in the reduced solvent volume (Table 1, entry 22 vs. entry 23).
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | [H]+ | DMA/MeCN/H2O | X(1)b [%] | S(2)b [%] |
| a Reaction conditions: 1,9-CHDD (1, 0.2 mmol), [Pd] (5.0 mol%), benzoquinone (BQ, 1.0 eq.), [H]+ (415 eq.), solvent mixture DMA/MeCN/H2O (3.0 mL), room temperature, 20 h.b Conversion and selectivity determined by GC using n-hexadecane as internal standard.c 2.5 mol% of [Pd].d 10.0 mol% of [Pd].e 1.0 mmol of (1) in 15.0 mL solvent mixture.f Reaction at 80 °C.g Argon atmosphere.h 1.25 eq. of p-TsOH.i 5.0 mmol of (1) in 15.0 mL solvent mixture. | |||||
| 1 | Pd(OAc)2 | — | —/7/1 | 2 | 24 |
| 2 | Pd(OAc)2 | HBF4 | —/7/1 | 29 | 54 |
| 3 | Pd(OAc)2 | HBF4 | 7/—/1 | 11 | 19 |
| 4 | Pd(OAc)2 | HBF4 | 1/6/1 | 26 | 73 |
| 5 | Pd(OAc)2 | HBF4 | 4/3/1 | 43 | 72 |
| 6 | Pd(OAc)2 | HBF4 | 6/1/1 | 44 | 63 |
| 7 | PdCl2 | HBF4 | 4/3/1 | 5 | 64 |
| 8 | Pd(MeCN)2Cl2 | HBF4 | 4/3/1 | 3 | 87 |
| 9 | PdSO4 | HBF4 | 4/3/1 | 41 | 76 |
| 10 | Pd(NO3)2 | HBF4 | 4/3/1 | 45 | 79 |
| 11c | Pd(NO3)2 | HBF4 | 4/3/1 | 21 | 81 |
| 12d | Pd(NO3)2 | HBF4 | 4/3/1 | 42 | 64 |
| 13e | Pd(NO3)2 | HBF4 | 4/3/1 | 39 | 79 |
| 14e,f | Pd(NO3)2 | HBF4 | 4/3/1 | 31 | 67 |
| 15 | Pd(NO3)2 | HBF4 | 8/6/1 | 42 | 81 |
| 16g | [Pd(MeCN)4](BF4)2 | — | 8/6/1 | 10 | 49 |
| 17g | [Pd(MeCN)4](BF4)2 | HBF4 | 8/6/1 | 46 | 77 |
| 18 | Pd(NO3)2 | HClO4 | 8/6/1 | 49 | 67 |
| 19 | Pd(NO3)2 | MeSO3H | 8/6/1 | 51 | 75 |
| 20 | Pd(NO3)2 | p-TsOH | 8/6/1 | 54 | 76 |
| 21h | Pd(NO3)2 | p-TsOH | 8/6/1 | 47 | 79 |
| 22e,h | Pd(NO3)2 | p-TsOH | 8/6/1 | 48 | 81 |
| 23h,i | Pd(NO3)2 | p-TsOH | 8/6/1 | 37 | 77 |

For all experiments with sufficient product formation, the trans-8-CHD was observed as the main product, although the conversion of the trans/trans-1,9-CHDD was comparably low, indicating that the trans-8-CHD mainly arises from the cis/trans-isomer, where the cis-moiety is preferably oxidised. Therefore, the conversion of the single isomers was determined by evaluating the integrals of the chromatogram, before and after reaction with different catalyst concentrations (see Fig. S1†). Preliminary separation experiments for the three isomers of (1) by using several separation techniques did not give an isomer in its pure form, why further investigations were carried out with the provided isomeric mixture. With the optimised reaction conditions obtained from Table 1, the palladium concentration was varied between 0.1, 2.5, 5.0 and 10.0 mol%, respectively (see Table 2). It should be noted, that for these experiments, the formation of palladium black was not observed at higher palladium concentrations contrary to previous observations (Table 1, entry 12 vs. Table 2, entry 4). These observations lead to the assumption, that the amount of water that was used in the ternary solvent mixture plays an essential role in stability for the reduced catalyst. Higher volumes of water in the solvent mixture do not only reduce the solubility of the substrate but also cause the irreversible formation of palladium black (Table 1, entry 12), while the formation of palladium black was not observed at lower volumes of water (Table 2, entry 4). As already mentioned above, the isomer with the mixed cis/trans-configuration was preferably oxidised to the trans-product. Interestingly, the cis/cis isomer was less reactive than the cis/trans isomer although it is assumed that the cis-moiety is the most sterically accessible structure for this oxidation reaction. The trans/trans isomer was the most elusive structure for the conversion into the corresponding ketone (2). Coordination and subsequent chelation of the pure isomers cis/cis and trans/trans, respectively might be responsible for this diminished reactivity. To gain further insights into the reaction behaviour of the single isomers, several NMR experiments including 2D NMR experiments were done and the distinctive signals were assigned to the three configurational isomers. The range of the olefinic signals for the 13C-NMR spectrum is shown in Fig. 1. Surprisingly, the trans/trans signal disappeared after adding palladium nitrate to the isomeric mixture of 1,9-CHDD. The signals for the cis/trans isomer also decreased while the signals for the cis/cis isomer were least affected. In turn, new signals appeared in the range of the olefinic signals at 130–132 ppm and at higher fields around 40 and 80 ppm (see Fig. S3†) which might arise from an η2,η2-C16H32 palladium complex which is formed by coordination of the olefinic double bonds to the palladium centre. Here, the trans/trans-isomer seems to irreversibly coordinate at the palladium centre, which would explain its low oxidation rate and the missing signals in the 13C-NMR spectrum after addition of palladium to the isomeric mixture. With these results in our hands, the Wacker-type oxidation of 1,9-CHDD using molecular oxygen as final oxidant was investigated. The focus of this approach was directed on reducing and/or replacing stoichiometric amounts of harmful benzoquinone with more environmentally benign co-oxidants in catalytic quantities and molecular oxygen as final oxidant for potential large-scale synthesis. Oxygen-coupled reoxidation for the reduced palladium catalyst was described in literature, but direct reoxidation of the noble metal by molecular oxygen often fails due to insufficient electron transfer owing to a high activation barrier.33–39 The usage of potential co-catalysts serving as electron transfer mediators (ETMs) for the oxygen coupled reoxidation of the reduced palladium species according to Scheme 3 was therefore examined and is summarised in Table 3. The reactions were performed in a stainless steel autoclave pressurised with 3 bar of oxygen at room temperature. In order to obtain information on the activity of the catalytic system, the reaction time was set at four hours. The formation of the isomeric diketone (3) was observed in all experiments as well as slight isomerisation of the double bond(s) of the substrate.
|
|||||
|---|---|---|---|---|---|
| Entry | Pd(NO3)2 [mol%] | X(total)b [%] | X(trans/trans)c [%] | X(cis/trans)c [%] | X(cis/cis)c [%] |
| a Reaction conditions: 1,9-CHDD (1, 0.2 mmol), Pd(NO3)2 (0.1–10.0 mol%), benzoquinone (BQ, 1.0 eq.), p-TsOH (1.25 eq.), solvent mixture DMA/MeCN/H2O = 8/6/1 (3.0 mL), room temperature, 20 h.b Conversion and selectivity determined by GC using n-hexadecane as internal standard.c Conversion determined by the reduction of the GC integrals. | |||||
| 1 | 0.1 | 2 | 0 | 2 | 0 |
| 2 | 2.5 | 26 | 3 | 17 | 6 |
| 3 | 5.0 | 49 | 8 | 31 | 10 |
| 4 | 10.0 | 67 | 11 | 42 | 14 |

 | ||
| Fig. 1 13C-NMR spectrum in the range of the characteristic olefinic signals before and after addition of Pd(NO3)2 to the isomeric mixture of 1,9-CHDD: – before addition of Pd, – after addition of Pd, ◆ cis/cis-isomer, ▲ cis-moiety of the cis/trans-isomer, ● trans/trans-isomer, ▼ trans-moiety of the cis/trans-isomer. | ||
 | ||
| Scheme 3 Redox cycles for the oxygen-coupled Wacker oxidation. | ||
|
||||
|---|---|---|---|---|
| Entry | Co-cat. | X(1)b [%] | S(2)b [%] | S(3)b [%] |
| a Reaction conditions: 1,9-CHDD (1, 0.5 mmol), Pd(NO3)2 (5.0 mol%), co-catalyst (10 mol%), p-TsOH (1 eq.), solvent mixture DMA/MeCN/H2O = 10/4/1 (3.0 mL), room temperature, 3 bar O2, 4 h.b Conversion and selectivity determined by GC using n-hexadecane as internal standard. | ||||
| 1 | — | 28 | 64 | 12 |
| 2 | BQ | 29 | 73 | 12 |
| 3 | Cu(NO3)2 | 29 | 70 | 16 |
| 4 | Fe(NO3)3 | 35 | 77 | 9 |
| 5 | MnO2 | 19 | 78 | 10 |
| 6 | KMnO4 | 17 | 71 | 11 |
For all screening experiments, the product, although the conversion of the trans/trans-1,9-CHDD was comparably low indicating that the trans-8-CHD mainly arose from the cis/trans-isomer, where the cis-moiety was preferably oxidised. Isomerisation of (1) and subsequent oxidation to undesired oxidised side products was virtually suppressed. Surprisingly, in the absence of a co-catalyst, conversion of (1) was observed to some extent (Table 3, entry 1), indicating that direct reoxidation of the reduced palladium species occurred. Compared to the blank experiment, catalytic amounts of BQ or copper salts did not further improve catalyst turnover (Table 3, entries 2, 3). To our delight, the conversion of (1) could be accelerated and increased by using 10 mol% of iron nitrate (Table 3, entry 4). The selectivity of (2) that was previously achieved in Table 1 could almost be obtained by using iron(III) nitrate. The utilization of oxidic manganese salts for the reoxidation of the reduced palladium catalyst was described in literature.40 For the oxidation of 1,9-CHDD to 8-CHD, these oxides were rather unsuitable, leading to lower palladium turnovers than the blank experiment (Table 3, entry 1 vs. 5, 6). With these encouraging results in our hands, the palladium–iron redox system for the oxidation of (1) to the musky smelling (2) was further investigated. First, the influence of the reaction atmosphere was examined by using an inert atmosphere, ambient air and oxygen at different pressures, respectively. The results are given in Fig. 2. In absence of oxygen, the catalytic system was depleted after two palladium turnovers suggesting the lack of sufficient reoxidation and the fact that oxygen acts as final oxidant. Furthermore, isomerisation was observed to a certain extent. When the reaction was performed at ambient air, further palladium turnovers took place and the isomerisation of (1) dropped from 18% to 6%. The dependency on the atmosphere became evident when pure oxygen was used. The conversion was more than doubled and increased isomeric diketone formation occurred. The selectivity to (2) remained high and was not affected by the reaction atmosphere and pressure, respectively. Next, the influence of the ternary solvent composition was examined and is summarised in Table 4. Again, the cis-moiety of the cis/trans-isomer was preferably oxidised leading to an excess of the trans-product. We renounced the usage of a binary solvent system due to the reasons already described in Table 1. As shown in Table 4, the performance for this reaction strongly depends on the DMA/MeCN ratio. The best results in terms of conversion and selectivity to (2) were obtained with a solvent ratio of DMA/MeCN/H2O = 10/4/1 (Table 4, entry 4). With a selectivity of 82% to (2) and a conversion of (1) of 45%, these results were the best so far described for the selective and direct Wacker oxidation of a macrocyclic diene in just one reaction step. When the concentrations of DMA or MeCN were too high or too low, respectively, the conversion of (1) and selectivity of (2) dropped (Table 4, entry 4 vs. entries 1, 6). Successive increase of the water concentration led to a reduction in selectivity (Table 4, entry 4 vs. entries 2, 3). Furthermore, different iron salts were tested as potential co-catalysts for this oxidation reaction, but only Fe(NO3)3 had a positive effect on the performance for this reaction (Fig. 3).
 | ||
| Fig. 2 Influence of the reaction atmosphere for the Wacker type oxidation of (1). | ||
|
||||
|---|---|---|---|---|
| Entry | DMA/MeCN/H2O | X(1)b [%] | S(2)b [%] | S(3)b [%] |
| a Reaction conditions: 1,9-CHDD (1, 0.2 mmol), Pd(NO3)2 (5.0 mol%), Fe(NO3)3 (10 mol%), p-TsOH (1 eq.), solvent mixture DMA/MeCN/H2O (3.0 mL), room temperature, 18–20 h, 3–5 bar O2.b Conversion and selectivity determined by GC using n-hexadecane as internal standard. | ||||
| 1 | 6/1/1 | 38 | 69 | 8 |
| 2 | 5/2/1 | 46 | 75 | 11 |
| 3 | 5/2/1.5 | 49 | 57 | 12 |
| 4 | 10/4/1 | 45 | 82 | 13 |
| 5 | 3.5/3.5/1 | 44 | 70 | 8 |
| 6 | 2/5/1 | 35 | 69 | 7 |
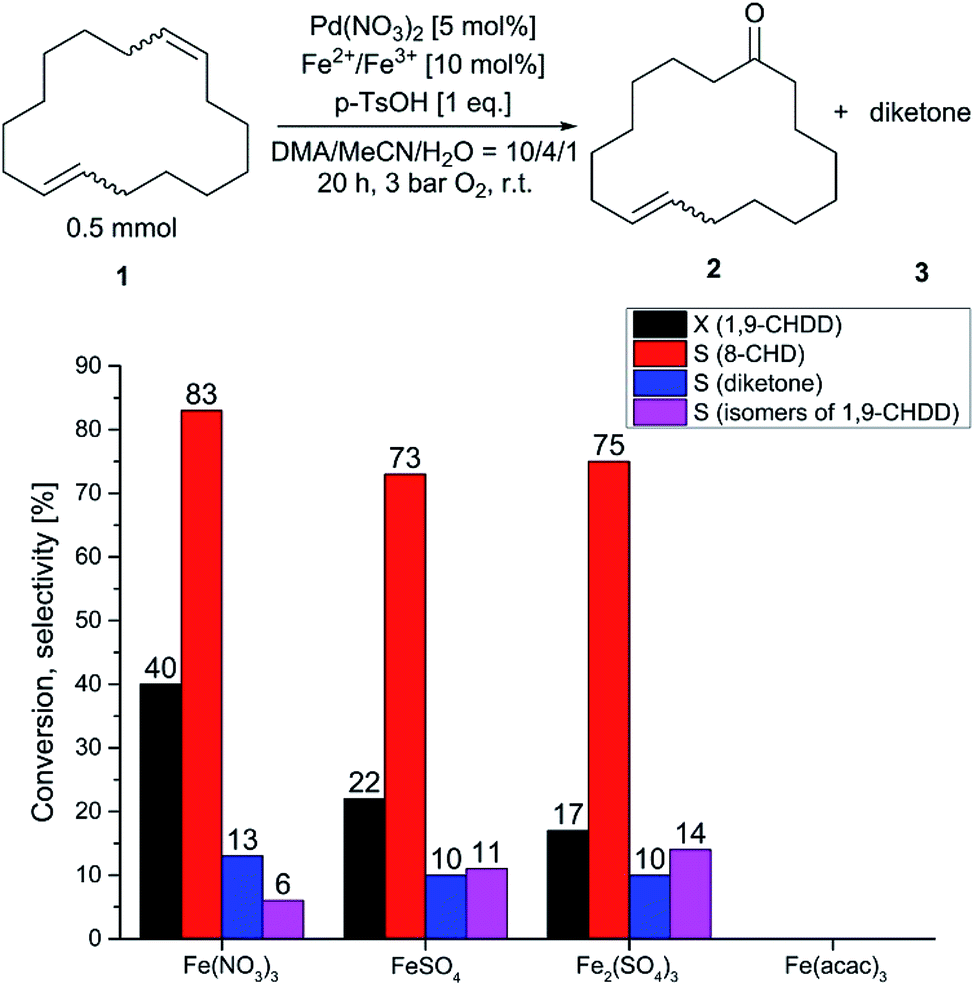 | ||
| Fig. 3 Wacker oxidation of an isomeric mixture of 1,9-CHDD to the corresponding ketone 8-CHD with different iron co-catalysts. | ||
Different sulphate salts of iron(II) and iron(III) tended to slow down the reaction rate. In addition, no oxidation was observed using iron(III) acetylacetonate (acac). This observation is in accordance with the previous statement, that strongly coordinating ligands restrict active catalyst formation. However, the importance of the nitrate salt has already been emphasised and is decisive for the oxidation rate. As already mentioned above, the oxidation of (1) proceeds preferably with the nitrate salts of palladium and iron, respectively. Therefore, the influence of the nitrate concentration was investigated and is represented in Fig. 4. Initial experiments using different amounts of concentrated nitric acid as nitrate source resulted in low selectivity of the desired monoketone (2) and diminished the recovery rate. Therefore, sodium nitrate was used as nitrate source and the reaction was carried out in an autoclave for 20 h and 3 bar of oxygen. For these experiments, the batch size was 0.5 mmol of (1) and the initial concentration of nitrate is marked with the red box and was 0.033 M without further addition of sodium nitrate due to the nitrate salts that were used for the reaction. When the reaction was run with small amounts of sodium nitrate, the conversion started to decrease. The selectivity of (2) and recovery rate was hardly affected but a downward trend can be anticipated. Regarding selectivity and recovery rate, the trend was confirmed when the nitrate concentration was increased. Surprisingly, the conversion started to increase again with higher amounts of the nitrate source. The drop in conversion after the first addition of sodium nitrate might be attributed to the sodium cation, which is supposed to impair the Wacker type oxidation of 1,9-CHDD. If the concentration of the nitrate anion passed 0.05 M, the selectivity and recovery rate dropped noticeably indicating the formation of higher molecular side products that could not be detected with standard GC/MS techniques. The increase in activity with elevated nitrate concentrations can be explained by the ability of the nitrate to act as an oxidising agent. Different 2D 1H–15N-NMR experiments confirmed the formation of reduced ammonium species that were formed over time and might be attributed to nitrate reduction (see Fig. S11 and S12 in the ESI†). Wacker type oxidation reactions where palladium nitro complexes are involved in the direct oxidation process are described in literature and have to be considered for this reaction as well.41–43
 | ||
| Fig. 4 Conversion, selectivity and recovery rate of the Wacker type oxidation of 1,9-CHDD with sodium nitrate used as nitrate source and dependency of the nitrate concentration. | ||
Finally, the scalability and metal concentration was evaluated in Table 5. Higher concentrations of the iron nitrate negatively affected the selectivity for the unsaturated monoketone (2) similar to the observations that was made in Fig. 4 (Table 5, entry 1 vs. entries 2, 4). As expected, the conversion increased with higher palladium concentration. Besides, over-oxidation leading to the diketone (3) was frequently observed when the palladium concentration was 10.0 mol% (Table 5, entries 3, 4). Again, the trans-8-CHD was observed as the main product while the trans/trans-1,9-CHDD was the least oxidised isomer. Furthermore, the applicability was investigated on a larger scale synthesis where the desired monounsaturated ketone was obtained in acceptable good yield at good selectivity (Table 5, entry 5).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd(NO3)2 [mol%] | Fe(NO3)3 [mol%] | X(1)b [%] | S(2)b [%] | S(3)b [%] | Y(2)b [%] |
| a Reaction conditions: 1,9-CHDD (1, 0.5 mmol), Pd(NO3)2, Fe(NO3)3, p-TsOH (1.0 eq.), solvent mixture DMA/MeCN/H2O = 10/4/1 (3.0 mL), room temperature, 3 bar O2.b Conversion and selectivity determined by GC using n-hexadecane as standard.c 5.0 mmol of (1) in 29.0 mL solvent mixture. | ||||||
| 1 | 5 | 10 | 45 | 82 | 13 | 37 |
| 2 | 5 | 20 | 37 | 65 | 8 | 24 |
| 3 | 10 | 10 | 62 | 71 | 17 | 44 |
| 4 | 10 | 20 | 63 | 63 | 14 | 39 |
| 5c | 5 | 10 | 36 | 75 | 9 | 27 |
Conclusions
In summary, a novel, simple and selective reaction strategy for the direct oxidation of an isomeric mixture of the macrocyclic 1,9-cyclohexadecadiene to the corresponding mono unsaturated cyclohexadec-8-en-1-one was developed, fulfilling several aspects of green chemistry. It is the first time reported, that a macrocyclic diene has been directly and selectively oxidised by Wacker type oxidation to the monounsaturated carbonyl compound in a single reaction step. The reaction proceeds at room temperature and the usage of molecular oxygen as final oxidant and iron nitrate as a benign and less ecologically hazardous co-catalyst was possible, without negatively affecting the conversion and selectivity. Interestingly, the presence and concentration of the nitrate anion plays a crucial role for the reoxidation of the reduced palladium species. Additionally, heteronuclear 2D-NMR experiments showed the formation of ammonium signals that might be attributed to the reduction of the nitrate. Side reactions such as isomerisation and subsequent oxidation were nearly suppressed, thus selectivities up to 83% were achieved at sufficient conversion of 41%. Moreover, the three isomers of the 1,9-CHDD showed different reactivity regarding the formation of the desired monounsaturated isomeric ketone mixture of 8-CHD. The trans-8-CHD was observed as the main product, although the conversion of the trans/trans-1,9-CHDD was comparably low indicating that the trans-8-CHD mainly arose from the cis/trans-isomer, where the cis-moiety was preferably oxidised. However, NMR experiments showed that especially the trans/trans isomer interacts preferentially with the palladium catalyst. Larger-scale experiments were successfully done for both reaction systems, reaction with equimolar amounts of BQ at ambient air conditions and reaction with iron nitrate as co-catalyst under oxygen atmosphere, respectively.Experimental
General information
Unless otherwise noted, all solvents, reagents and catalysts were purchased from commercial suppliers and used without further purification. 1,9-Cyclohexadecadiene and cyclohexadec-8-en-1-one were obtained from Symrise AG. NMR spectra were recorded on a Bruker AV 400 (400 MHz) and calibrated to the corresponding solvent signal. High-resolution mass spectra analysis was conducted on an Agilent 1200/6210 time-of-flight LC-MS. Infrared spectra were recorded on a Bruker Alpha FT-IR-spectrometer. Elemental analysis was measured on a Leco TruSpec Micro instrument. Conversion, selectivity, yield and recovery rate were determined by GC-MS using a SHIMADZU GCMS-QP2010 SE instrument equipped with a HP-5MS capillary column (30 m × 0.25 μm × 0.25 mm) from Agilent. For GC-MS calibration, the diketone (3) was prepared by oxidation of (2) and isolated as an isomeric mixture consisting of cyclohexadecane-1,8-dione and cyclohexadecane-1,9-dione by flash column chromatography (heptane/ethyl acetate = 8/1). Response factors were collected for 1,9-cyclohexadecadiene, cyclohexadec-8-en-1-one and the isomeric mixture of the diketone consisting of cyclohexadecane-1,8-dione and cyclohexadecane-1,9-dione using n-hexadecane as internal standard.General procedure for ambient air Wacker oxidation with BQ
The corresponding palladium salt (0.01 mmol, 5 mol%), the acid (0.83 mmol, 4.15 eq.) and benzoquinone (0.2 mmol, 1.0 eq.) were placed in a 4 mL glass vial with screw cap under air. Then, 1,9-cyclohexadecadiene (44.1 mg, 0.2 mmol) was added and the components were dissolved in 3 mL of a ternary DMA/MeCN/H2O solvent mixture. Under ambient conditions, the homogeneous mixture was vigorously stirred overnight for 20 h. The crude reaction mixture was transferred into a 5 mL volumetric flask and subsequently diluted with THF. Conversion, selectivity, yield and recovery rate were determined by GC-MS with internal standard technique using n-hexadecane as standard.General procedure for aerobic Wacker oxidation
Palladium nitrate dihydrate (6.66 mg, 0.025 mmol, 5 mol%), p-toluensulfonic acid monohydrate (96.56 mg, 0.5 mmol, 1.0 eq.) and the corresponding co-catalyst (0.05 mmol, 10 mol%) were weighed in a 4 mL glass vial. Then, 1,9-cyclohexadecadiene (110.21 mg, 0.5 mmol) was added and the components were dissolved in 3 mL of a ternary DMA/MeCN/H2O (10/4/1) solvent mixture. Next, the vial was sealed with a septum and a screw cap with a hole. The septum was punctured with a needle and placed in a stainless steel autoclave. The autoclave was then purged three times with oxygen and finally pressurised to 3 bar with oxygen. The homogeneous reaction mixture was stirred vigorously at room temperature for the indicated reaction time. After reaction, the crude reaction mixture was transferred into a 5 mL volumetric flask and subsequently diluted with THF. Conversion, selectivity, yield and recovery rate were determined by GC-MS with internal standard technique using n-hexadecane as standard.Synthesis of the isomeric mixture of cyclohexadecane-1,8-dione and cyclohexadecane-1,9-dione (3) for GC calibration
Palladium nitrate dihydrate (666.15 mg, 2.5 mmol, 10 mol%), p-toluensulfonic acid monohydrate (4.828 g, 25 mmol, 1.0 eq.) and iron nitrate nonahydrate (1.010 g, 2.5 mmol, 10 mol%) were weighed in a glass inlet for a 300 mL autoclave. Then, cyclohexadec-8-en-1-one (5.910 g, 25 mmol) was added and the components were dissolved in 150 mL of a ternary DMA/MeCN/H2O (10/4/1) solvent mixture. The glass inlet was placed in a 300 mL stainless steel autoclave. The autoclave was then purged three times with oxygen and finally pressurised to 5 bar with oxygen. The homogeneous reaction mixture was stirred vigorously at room temperature for 20 h. After reaction, the crude reaction mixture was transferred into a separation funnel and extracted with heptane (8 × 150 mL). After washing with distilled water and brine (each 100 mL), the combined organic layers were dried over anhydrous sodium sulphate. The sodium sulphate was filtered off and the solvent was evaporated in vacuo. Finally, the isomeric mixture consisting of cyclohexadecane-1,8-dione and cyclohexadecane-1,9-dione was isolated by flash column chromatography (heptane/ethyl acetate = 8/1). The product was isolated as a white crystalline product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge our analytical department for providing analytical data.Notes and references
- J. M. Escola, J. A. Botas, J. Aguado, D. P. Serrano, C. Vargas and M. Bravo, Appl. Catal., A, 2008, 335, 137–144 CrossRef CAS.
- B. Weiner, A. Baeza, T. Jerphagnon and B. L. Feringa, J. Am. Chem. Soc., 2009, 131, 9473–9474 CrossRef CAS PubMed.
- J. M. Takacs and X. T. Jiang, Curr. Org. Chem., 2003, 7, 369–396 CrossRef CAS.
- T. V. Baiju, E. Gravel, E. Doris and I. N. N. Namboothiri, Tetrahedron Lett., 2016, 57, 3993–4000 CrossRef CAS.
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger and H. Kojer, Angew. Chem., 1959, 71, 176–182 CrossRef CAS.
- P. M. Henry, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. i. Negishi, John Wiley & Sons, New York, 1st edn, 2003, ch. 5, vol. 2, pp. 2119–2139 Search PubMed.
- Q. Cao, D. S. Bailie, R. Z. Fu and M. J. Muldoon, Green Chem., 2015, 17, 2750–2757 RSC.
- J. A. Goncalves and E. V. Gusevskaya, Appl. Catal., A, 2004, 258, 93–98 CrossRef CAS.
- L. A. Parreira, A. M. da Cunha, L. Menini and E. V. Gusevskaya, RSC Adv., 2015, 5, 56987–56992 RSC.
- M. J. da Silva, R. R. Teixeira and D. M. Carari, J. Organomet. Chem., 2009, 694, 3254–3261 CrossRef CAS.
- A. S. Carlson, C. Calcanas, R. M. Brunner and J. J. Topczewski, Org. Lett., 2018, 20, 1604–1607 CrossRef CAS PubMed.
- T. Mitsudome, K. Mizumoto, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 1238–1240 CrossRef CAS PubMed.
- J. Tsuji, Synthesis, 1984, 5, 369–384 CrossRef.
- M. Winkler and M. A. R. Meier, Green Chem., 2014, 16, 1784–1788 RSC.
- D. G. Miller and D. D. M. Wayner, J. Org. Chem., 1990, 55, 2924–2927 CrossRef CAS.
- A. Sen, Accounts Chem. Res., 1988, 21, 421–428 CrossRef CAS.
- M. von Czapiewski, M. Rhein and M. A. R. Meier, ACS Sustain. Chem. Eng., 2018, 6, 15170–15179 CrossRef CAS.
- T. Mitsudome, S. Yoshida, Y. Tsubomoto, T. Mizugaki, K. Jitsukawa and K. Kaneda, Tetrahedron Lett., 2013, 54, 1596–1598 CrossRef CAS.
- R. J. DeLuca, J. L. Edwards, L. D. Steffens, B. W. Michel, X. X. Qiao, C. Y. Zhu, S. P. Cook and M. S. Sigman, J. Org. Chem., 2013, 78, 1682–1686 CrossRef CAS PubMed.
- B. Morandi, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 2944–2948 CrossRef CAS PubMed.
- B. Morandi, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 9751–9754 CrossRef CAS PubMed.
- H. Surburg and J. Panten, Individual Fragrance and Flavor Materials, Wiley-VCH, Weinheim, 2006 Search PubMed.
- J. Chatt, L. M. Vallarino and L. M. Venanzi, J. Chem. Soc., 1957, 3413–3416 RSC.
- C. Eaborn, N. Farrell and A. Pidcock, J. Chem. Soc., Dalton Trans., 1976, 289–292 RSC.
- J. K. Stille and R. A. Morgan, J. Am. Chem. Soc., 1966, 88, 5135–5141 CrossRef CAS.
- W. T. Wipke and G. L. Goeke, J. Am. Chem. Soc., 1974, 96, 4244–4249 CrossRef CAS.
- R. A. Widenhoefer, Accounts Chem. Res., 2002, 35, 905–913 CrossRef CAS PubMed.
- B. D. Mookherjee, R. W. Trenkle and R. R. Patel, J. Org. Chem., 1972, 37, 3846–3848 CrossRef CAS.
- K. Mitsudo, T. Kaide, E. Nakamoto, K. Yoshida and H. Tanaka, J. Am. Chem. Soc., 2007, 129, 2246–2247 CrossRef CAS PubMed.
- T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, Beilstein J. Org. Chem., 2016, 12, 1040–1064 CrossRef CAS PubMed.
- F. Yang, F. Song, W. Li, J. Lan and J. You, RSC Adv., 2013, 3, 9649–9652 RSC.
- M. A. Andrews and K. P. Kelly, J. Am. Chem. Soc., 1981, 103, 2894–2896 CrossRef CAS.
- E. N. Johnston, E. A. Karlsson, S. A. Lindberg, B. Akermark and J. E. Backvall, Chem. Eur J., 2009, 15, 6799–6801 CrossRef CAS PubMed.
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed.
- L. Jin and A. Lei, Sci. China Chem., 2012, 55, 2027–2035 CrossRef CAS.
- B. V. Popp and S. S. Stahl, in Topics in Organometallic Chemistry, ed. F. Meyer and C. Limberg, Springer Berlin Heidelberg, Berlin, Heidelberg, 1st edn, 2007, ch. 7, vol. 22, pp. 149–189 Search PubMed.
- B. V. Popp and S. S. Stahl, Chem. Eur J., 2009, 15, 2915–2922 CrossRef CAS PubMed.
- B. W. Purse, L. H. Tran, J. Piera, B. Akermark and J. E. Backvall, Chem. Eur J., 2008, 14, 7500–7503 CrossRef CAS PubMed.
- J. Piera and J. E. Backvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- A. Vavasori and L. Toniolo, J. Mol. Catal. A: Chem., 1999, 139, 109–119 CrossRef CAS.
- I. E. Bekk, E. V. Gusevskaya, V. A. Likholobov and Y. I. Yermakov, React. Kinet. Catal. Lett., 1987, 33, 209–214 CrossRef CAS.
- N. H. Kiers, B. L. Feringa and P. W. M. N. Vanleeuwen, Tetrahedron Lett., 1992, 33, 2403–2406 CrossRef CAS.
- M. J. da Silva and E. V. Gusevskaya, J. Mol. Catal. A: Chem., 2001, 176, 23–27 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04971a |
| This journal is © The Royal Society of Chemistry 2019 |