
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: LncRNA NEXN-AS1 attenuates proliferation and migration of vascular smooth muscle cells through sponging miR-33a/b
Leiming Wu†
,
Yapeng Li†,
Dianhong Zhang,
Zhen Huang,
Binbin Du,
Zheng Wang,
Lulu Yang and
Yanzhou Zhang *
*
Department of Cardiology, The First Affiliated Hospital of Zhengzhou University, No. 1 Jianshe East Road, Zhengzhou 450052, Henan, China. E-mail: chizi965014mb@126.com; Tel: +86-37167967661
First published on 4th September 2019
Abstract
Non-protein-coding RNAs (lncRNAs) are emerging as important regulators in disease pathogenesis, including atherosclerosis (AS). Here, we investigated the role and underlying mechanisms of nexilin F-actin binding protein antisense RNA 1 (NEXN-AS1) on the proliferation and migration of vascular smooth muscle cells (VSMCs). Our data revealed that ox-LDL treatment resulted in decreased NEXN-AS1 expression and increased miR-33a/b levels in human aorta VSMCs (HA-VSMCs) in dose- and time-dependent manners. Overexpression of NEXN-AS1 mitigated the proliferation and migration of HA-VSMCs under ox-LDL stimulation using CCK-8 and wound-healing assays. Moreover, dual-luciferase reporter and RNA immunoprecipitation assays verified that NEXN-AS1 acted as molecular sponges of miR-33a and miR-33b in HA-VSMCs. MiR-33a or miR-33b silencing attenuated the proliferation and migration of ox-LDL-treated HA-VSMCs. Furthermore, miR-33a or miR-33b mediated the inhibitory effects of NEXN-AS1 overexpression on the proliferation and migration of ox-LDL-treated HA-VSMCs. Our study suggested that high level of NEXN-AS1 mitigated VSMC proliferation and migration under ox-LDL stimulation at least partly through sponging miR-33a and miR-33b, illuminating NEXN-AS1 as a novel therapeutic approach for AS treatment.
Introduction
Atherosclerosis (AS) is a progressive, chronic immunoinflammatory disease, being a leading cause of death and giving rise to a loss of quality of life around the world. It underlies the majority of cardiovascular diseases, such as coronary heart disease, cerebral infarction, and stroke.1,2 The pathogenesis of AS is complicated and still largely unknown. Vascular smooth muscle cells (VSMCs) have recently been postulated to be involved in the progression of AS. Upon atheroma formation, VSMCs migrate from the media into the intima, and produce hyperproliferation, thereby contributing to AS progression.3,4 Additionally, oxidized low-density lipoprotein (ox-LDL) has been postulated to contribute to AS development and is widely used for AS model construction in vitro.5,6 Therefore, identifying novel biomarkers to interfere with the excessive migration and proliferation of VSMCs is very relevant for AS management.In recent years, non-protein-coding RNAs (ncRNAs) have been gaining more and more attention for their crucial functions from normal physiology to disease.7 Among these ncRNAs, long ncRNAs (lncRNAs), a loosely classified group of RNA molecules defined as being >200 nucleotides long, are the most prevalent and functionally diverse class.8 MicroRNAs (miRNAs) are approximately 18–25 nucleotides long, evolutionarily conserved, small non-coding RNAs that direct posttranscriptional suppression of target mRNAs.9 Dysregulation of lncRNAs and/or miRNAs has been widely accepted to play a key role in pathological processes with potential effects on the initiation and/or progression of human diseases, including AS.10–12 The competing endogenous RNA (ceRNA) concept proposes that lncRNA modulates gene expression through acting as a molecular sponge of miRNA, illuminating the importance of such interaction in multiple physiological and pathological processes.13 Emerging evidence has suggested that several lncRNAs regulate the occurrence and progression of AS via sponging special miRNAs.14,15
Nexilin F-actin binding protein antisense RNA 1 (NEXN-AS1), a previously uncharacterized lncRNA, has been reported to associate with risk of lung cancer and overall survival of cholangiocarcinoma.16,17 A recent document manifested that NEXN-AS1 reduced inflammatory cytokines production and adhesion molecules expression, and repressed monocyte adhesion via modulation of the actin-binding protein NEXN, eliciting its role as an anti-atherosclerotic agent for AS treatment.18 Herein, we further explored the function and molecular mechanisms of NEXN-AS1 on the migration and proliferation of VSMCs.
MiR-33 family, which consists of miR-33a and miR-33b, has been identified to play a pro-atherogenic role via various regulatory mechanisms.19,20 However, whether miR-33a/b influence VSMC proliferation and migration has not been reported. In the present study, our data firstly demonstrated that ox-LDL reduced NEXN-AS1 expression and NEXN-AS1 overexpression mitigated the proliferation and migration of ox-LDL-induced HA-VSMCs. Consequently, the underlying mechanisms of NEXN-AS1 on HA-VSMC proliferation and migration under ox-LDL exposure were further investigated.
Experimental section
Cell culture and treatment
Human aorta VSMCs (HA-VSMCs, ATCC® CRL-1999) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were grown in MCDB 131 medium (Gibco Laboratories, North Andover, MA, USA) containing 10% fetal bovine serum (FBS, Bovogen, Heidelberg West, Victoria, Australia), 1.5 mg mL−1 bFGF (Gibco Laboratories), 0.5 mg mL−1 EGF (Gibco Laboratories), 2 mM L-glutamine (Gibco Laboratories) and 1% antibiotics (penicillin/streptomycin, Gibco Laboratories) at 37 °C in an air atmosphere of 5% CO2.Ox-LDL was obtained from PeproTech (Tebu-Bio, Le Perray en Yvelines, France) and used following the instructions of manufacturers. To assess its impact on the expression of NEXN-AS1 and miR-33a/b, HA-VSMCs were exposed to various concentrations (0, 25, 50, 100 and 200 μg mL−1) of ox-LDL for 24 h or different time point (0, 6, 12, 24 and 48 h) at a final concentration of 100 μg mL−1 ox-LDL.
Cell transfection
For NEXN-AS1 overexpression, HA-VSMCs were transfected with NEXN-AS1 overexpression plasmid (pcDNA-NEXN-AS1), and non-targeting plasmid (pcDNA-NC) was used as a negative control. For miR-33a and miR-33b upregulation, HA-VSMCs were introduced with miR-33a mimic, miR-33b mimic, or a scrambled oligonucleotide sequence (miR-NC mimic) as the negative control. MiR-33a and miR-33b silencing were carried out using miR-33a inhibitor (anti-miR-33a), miR-33b inhibitor (anti-miR-33b), or inhibitor negative control (anti-miR-NC). The Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, Life Technologies, Ghent, Belgium) was used for each transfection, in accordance with the protocols of manufacturers. All plasmids and oligonucleotides were synthesized by GenePharma (Shanghai, China).RNA extraction and quantitative real-time PCR
The extraction of total cellular RNA was performed using RNAqueous™-Micro Total RNA Isolation Kit (Invitrogen) referring to the producer's instructions. Quantitative real-time PCR (qRT-PCR) was used to determine the expression of NEXN-AS1, miR-33a and miR-33b. For the detection of NEXN-AS1, cDNA was synthesized from 10 ng of total RNA using Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Bremen, Germany). For miR-33a/b quantification, a total of 10 ng RNA extracts was reverse-transcribed using TaqMan™ Advanced miRNA cDNA Synthesis Kit (Applied Biosystems, Darmstadt, Germany). qRT-PCR was carried out using QuantiTect SYBR Green PCR Kit (Qiagen GmbH, Hilden, Germany) on an Applied Biosystems 7500 RT PCR System following the manufacturer's guidance. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 snRNA were used as housekeeping genes for normalization. Relative expression levels of NEXN-AS1, miR-33a and miR-33b were calculated using the 2−ΔΔCt method.Cell proliferation assay
The Cell Counting Kit-8 (CCK-8, Dojindo Lab, Kumamoto, Japan) was applied to evaluate the proliferation ability of treated HA-VSMCs, in accordance with the manufacturer's direction. To be brief, transfected or untransfected cells were plated in 96-well plates, and then exposed to 100 μg mL−1 of ox-LDL. After 0, 24, 48 and 72 h treatment, cells were incubated with CCK-8 solution (10 μL per well) at 37 °C for 2 h. Cell proliferation ability was monitored at the optical density of 450 nm using a SUNRISE microplate reader (Tecan Japan Co., Ltd., Tokyo, Japan).Wound-healing assay
Cell migration capacity was determined using a wound-healing assay. HA-VSMCs (5 × 104) were seeded in 6-well plates and incubated under an air atmosphere of 5% CO2 and temperature of 37 °C. After reaching approximately 80% confluence, a scratch was created in the cell monolayer using a 200 μL pipette tip. Then, cells were transfected with or without the indicated plasmid or/and oligonucleotide for 24 h before treatment with 100 μg mL−1 of ox-LDL for 24 h (transfected group) or 48 h (untransfected group). Wound area was photographed using an inverted light microscope (Leica, Wetzlar, Germany) and analyzed by Image-Pro Plus v.6.0 software (Media Cybernetics, Dutch Vision Components, Breda, The Netherlands).Western blot
Treated HA-VSMCs were homogenized in RIPA lysis buffer (Sigma-Aldrich, Buchs, Switzerland) containing protease and phosphatase inhibitors (Thermo Fisher Scientific). Equal amounts of total protein (100 μg) were fractionated on a 10% sodium dodecyl sulfate–polyacrylamide gel and electrotransferred onto polyvinylidene difluoride membranes (PVDF, GE Healthcare, Piscataway, NJ. USA). Primary antibodies and dilutions were as follows: anti-matrix metalloproteinase-2 (anti-MMP-2, ab97779, Abcam, Cambridge, UK) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution, anti-MMP9 (ab219372, Abcam) at a 1:3000 dilution and anti-β-actin (ab8227, Abcam) at a 1:2000 dilution. Protein bands were determined using ECL detection reagents (Clarity Western ECL Substrate, Bio-Rad, Marnes-la-Coquette, France) and analyzed by Image Lab software (Bio-Rad).
:1000 dilution, anti-MMP9 (ab219372, Abcam) at a 1:3000 dilution and anti-β-actin (ab8227, Abcam) at a 1:2000 dilution. Protein bands were determined using ECL detection reagents (Clarity Western ECL Substrate, Bio-Rad, Marnes-la-Coquette, France) and analyzed by Image Lab software (Bio-Rad).
Bioinformatics
The targeted miRNAs of NEXN-AS1 were predicted using LncBase v.2 software available at http://diana.imis.athena-innovation.gr.Dual-luciferase reporter assay
NEXN-AS1 luciferase reporter plasmid (NEXN-AS1-WT) harboring miR-33a/b-binding site and its mutant in seeded sequence (NEXN-AS1-MUT) were obtained from GenePharma. To confirm the targeted correlation between NEXN-AS1 and miR-33a, HA-VSMCs were transfected with NEXN-AS1-WT or NEXN-AS1-MUT, together with miR-33a mimic or miR-NC mimic. To verify whether NEXN-AS1 directly bonded to miR-33b, HA-VSMCs were cotransfected with NEXN-AS1-WT or NEXN-AS1-MUT, and miR-33a mimic or miR-NC mimic. 48 h later, cells were lysed and assayed for both Firefly and Renilla luciferase activities, using the Dual-Luciferase reporter assay system (Promega, Fitchburg, WI, USA).RNA immunoprecipitation assay
The endogenous interactions between NEXN-AS1 and miR-33a/b were evaluated using RNA immunoprecipitation (RIP) assay. Briefly, HA-VSMCs were transfected with miR-33a mimic, miR-33b mimic or miR-NC mimic for 48 h. After that, cell lysates were prepared using RIPA lysis buffer and then incubated with anti-Argonaute2 (anti-Ago2, Abcam) or negative control IgG (Abcam) prior to adding protein A/G plus agarose beads (Santa Cruz Biotechnology, Dallas, TX, USA) for 4 h. Beads were harvested by centrifugation at 3000g for 10 min and washed three times with PBS. Finally, total RNA was extracted, and NEXN-AS1 enrichment was assessed by qRT-PCR.Statistical analysis
Data were analyzed by GraphPad Prism v.7 software (GraphPad, La Jolla, CA, USA) and presented as mean ± SD. Differences between groups were compared using a two-tailed Student's t-test or one-way analysis of variance (ANOVA). All experiments were performed as 3 biological replicates × 3 technical replicates, unless otherwise indicated. P values at 0.05 or smaller were considered significant.Results and discussion
NEXN-AS1 expression was reduced by ox-LDL in HA-VSMCs
In order to preliminarily observe the involvement of NEXN-AS1 in AS progression, HA-VSMCs were exposed to various concentrations (0, 25, 50, 100 and 200 μg mL−1) of ox-LDL for 24 h or different time point (0, 6, 12, 24 and 48 h) at a final concentration of 100 μg mL−1 ox-LDL. As shown by qRT-PCR, ox-LDL exposure significantly hindered the expression of NEXN-AS1 in dose- and time-dependent manners in HA-VSMCs (Fig. 1A and B). | ||
| Fig. 1 The expression of NEXN-AS1 was repressed by ox-LDL in HA-VSMCs. (A) HA-VSMCs were treated with various concentrations (0, 25, 50, 100 and 200 μg mL−1) of ox-LDL for 24 h, and then NEXN-AS1 expression was detected by qRT-PCR. (B) HA-VSMCs were exposed to 100 μg mL−1 of ox-LDL for different time point (0, 6, 12, 24 and 48 h), followed by the measurement of NEXN-AS1 expression by qRT-PCR. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
Overexpression of NEXN-AS1 mitigated the proliferation and migration in HA-VSMCs under ox-LDL stimulation
Given the data that ox-LDL decreased NEXN-AS1 expression in HA-VSMCs (Fig. 2A), our results also revealed that ox-LDL stimulation triggered the significant enhancement of cell proliferation and migration capacities by CCK-8 assay and wound-healing assay (Fig. 2B and C). MMP-9 and MMP-2 have been proposed to contribute to AS pathogenesis via accelerating VSMCs migration.21,22 As a result, ox-LDL treatment resulted in increased MMP-9 and MMP-2 levels compared with vehicle control in HA-VSMCs (Fig. 2D), supporting the promotional impact of ox-LDL on cell migration.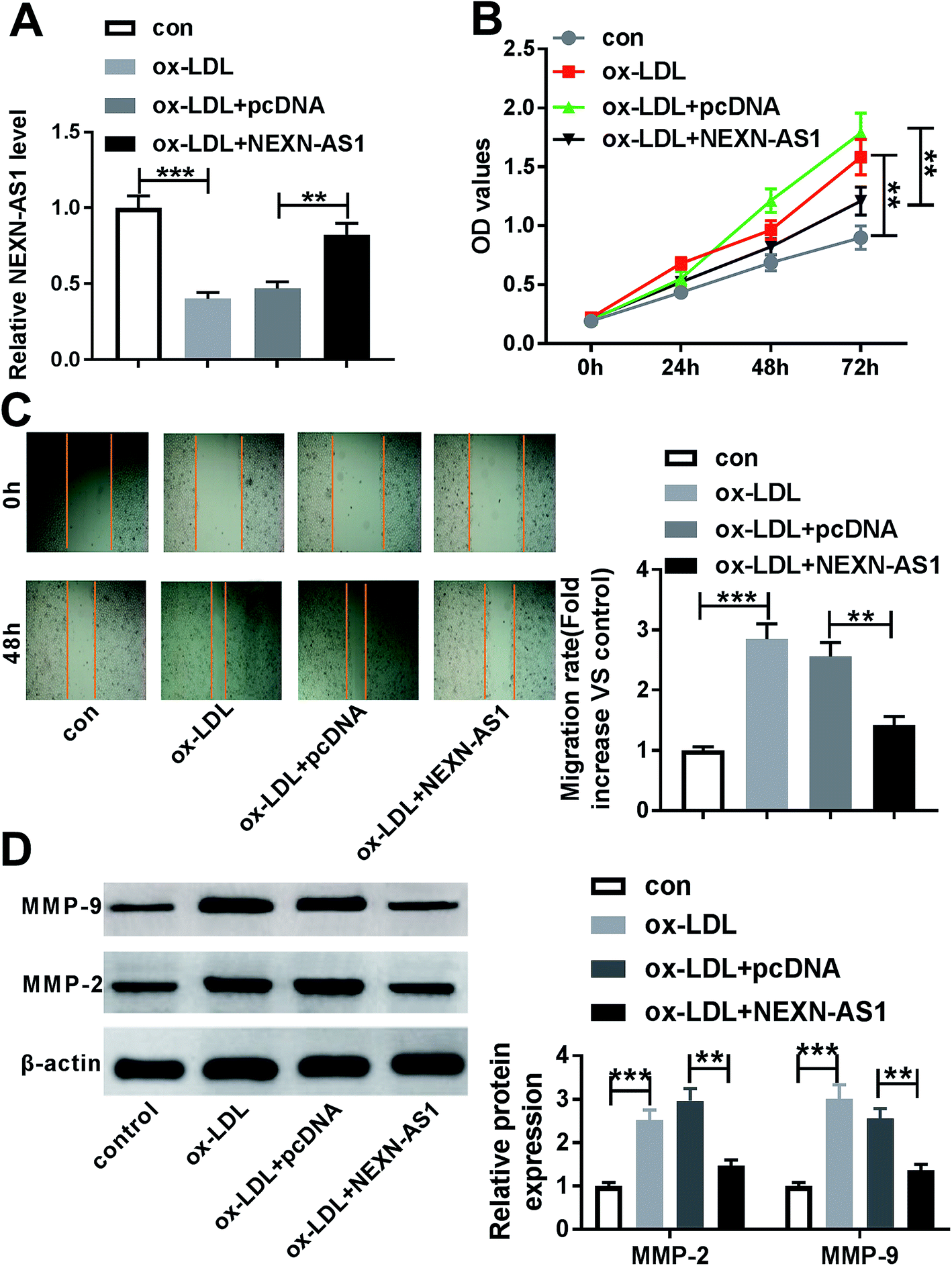 | ||
| Fig. 2 Inhibitory effect of NEXN-AS1 overexpression on HA-VSMC proliferation and migration under ox-LDL stimulation. HA-VSMCs were transfected with or without pcDNA-NC or pcDNA-NEXN-AS1 for 24 h, and then were exposed to 100 μg mL−1 of ox-LDL for the indicated time. (A) 24 h after ox-LDL treatment, NEXN-AS1 expression was assessed by qRT-PCR. (B) After 0, 24, 48 and 72 h ox-LDL exposure, cell proliferation capacity was detected by CCK-8 assay. (C) 48 h (untransfected group) or 24 h (transfected group) after ox-LDL stimulation, cell migration ability was determined using wound-healing assay. (D) After 24 h ox-LDL treatment, the expression levels of MMP-9 and MMP-2 were evaluated by western blot. **P < 0.01 or ***P < 0.001. | ||
To explore the effect of NEXN-AS1 on AS progression in vitro, we manipulated NEXN-AS1 expression by its overexpression plasmid (pcDNA-NEXN-AS1). Transient introduction of pcDNA-NEXN-AS1, but not a negative plasmid, remarkably elevated the expression of NEXN-AS1 in ox-LDL-induced HA-VSMCs (Fig. 2A). CCK-8 assay demonstrated that in contrast to negative control, NEXN-AS1 upregulation strikingly hampered HA-VSMC proliferation under ox-LDL exposure (Fig. 2B). Moreover, NEXN-AS1 overexpression prominently mitigated cell migration in ox-LDL-treated HA-VSMCs, as evidenced by the reduction of migrated distance, and MMP-9 and MMP-2 levels (Fig. 2C and D). These data together established that NEXN-AS1 overexpression hindered the proliferation and migration in ox-LDL-treated HA-VSMCs.
NEXN-AS1 acted as molecular sponges for miR-33a and miR-33b
Our above results demonstrated that high level of NEXN-AS1 weakened cell proliferation and migration. Next, we further explored how NEXN-AS1 achieved it. Using LncBase v.2 software, the predicted data revealed that NEXN-AS1 harbored a putative target sequence (AAUGCAC) for miR-33a and miR-33b (Fig. 3A and B). To validate this, we carried out dual-luciferase reporter assays using NEXN-AS1 luciferase reporter (NEXN-AS1-WT). Cotransfection of NEXN-AS1-WT and miR-33a mimic into cells produced lower luciferase activity than in cells cotransfected with miR-NC mimic (Fig. 3C). Likewise, with the wide-type reporter and miR-33b upregulation caused a significant reduction in luciferase activity (Fig. 3D). However, site-directed mutant in seeded sequence no longer elicited such effects (Fig. 3C and D). MiRNAs are known to be present in the form of miRNA ribonucleoprotein complexes that also contain Ago2, the core component of the RNA-induced silencing complex (RISC).9 Thus, RIP experiments were performed to confirm the endogenous interactions between NEXN-AS1 and miR-33a/b in the RISC of transfected HA-VSMCs. In comparison to their counterparts, NEXN-AS1 enrichment was substantially elevated by miR-33a or miR-33b overexpression (Fig. 3E and F). All these data strongly pointed the role of NEXN-AS1 as molecular sponges for miR-33a and miR-33b. | ||
| Fig. 3 MiR-33a and miR-33b were directly targeted by NEXN-AS1. (A and B) Schematic of NEXN-AS1 illustrating the putative binding sequence for miR-33a and miR-33b identified by LncBase v.2 software, and the mutant of target sequence. HA-VSMCs were transfected with NEXN-AS1-WT or its mutant in seeded region (NEXN-AS1-MUT) and miR-33a mimic (C), miR-33b mimic (D) or miR-NC mimic, and then the luciferase activity was determined. The enrichment of NEXN-AS1 by qRT-PCR in HA-VSMCs transfected with miR-33a mimic (E), miR-33b mimic (F) or miR-NC mimic. ***P < 0.001. | ||
MiR-33a expression was increased by ox-LDL and miR-33a silencing attenuated the proliferation and migration in ox-LDL-treated HA-VSMCs
To observe the role of miR-33a in AS progression in vitro, firstly, we determined whether ox-LDL influenced miR-33a expression in HA-VSMCs. In contrast to negative control, ox-LDL treatment resulted in increased miR-33a expression in dose- and time-dependent manners (Fig. 4A and B). Then, we implemented “phenocopy” silencing by inhibitor of miR-33a (anti-miR-33a). Transfection of anti-miR-33a, but not a scrambled control sequence, dramatically weakened the expression of miR-33a in ox-LDL-induced HA-VSMCs (Fig. 4C). Subsequent experiments results showed that miR-33a silencing led to a distinct repression of cell proliferation (Fig. 4D) and an obvious inhibition of cell migration (Fig. 4E), as well as a significant reduction of MMP-9 and MMP-2 levels (Fig. 4F) in ox-LDL-treated HA-VSMCs. Together, these data suggested that miR-33a silencing retarded HA-VSMC proliferation and migration under ox-LDL stimulation. | ||
| Fig. 4 Suppressive role of miR-33a silencing on the proliferation and migration of ox-LDL-treated HA-VSMCs. (A) HA-VSMCs were treated with various concentrations (0, 25, 50, 100 and 200 μg mL−1) of ox-LDL for 24 h, and then miR-33a expression was assessed by qRT-PCR. (B) HA-VSMCs were exposed to 100 μg mL−1 of ox-LDL for different time point (0, 6, 12, 24 and 48 h), and then miR-33a level was detected. HA-VSMCs were transfected with or without anti-miR-33a or anti-miR-NC, and then exposed to 100 μg mL−1 of ox-LDL for the indicated time, followed by the determination of miR-33a expression by qRT-PCR 24 h after ox-LDL treatment (C), cell proliferation ability by CCK-8 assay after 0, 24, 48 and 72 h ox-LDL exposure (D), cell migration capacity by wound-healing assay after 48 h (untransfected group) or 24 h (transfected group) ox-LDL stimulation (E), the levels of MMP-9 and MMP-2 by western blot 24 h after ox-LDL treatment (F). *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
MiR-33b level was elevated by ox-LDL and miR-33b knockdown retarded the proliferation and migration in ox-LDL-induced HA-VSMCs
Consistent with the above data that ox-LDL increased miR-33a expression, qRT-PCR results also demonstrated that ox-LDL dose- and time-dependently promoted the expression of miR-33b in HA-VSMCs (Fig. 5A and B). Next, we further investigated the impact of miR-33b in AS progression in vitro by anti-miR-33b. In comparison to a scrambled control sequence, miR-33b expression was remarkably reduced by transfection of anti-miR-33b (Fig. 5C). Subsequent experiments data showed that miR-33b knockdown significantly mitigated cell proliferation and migration of ox-LDL-treated HA-VSMCs (Fig. 5D–F). | ||
| Fig. 5 Repressive effect of miR-33b depletion on the proliferation and migration of ox-LDL-treated HA-VSMCs. HA-VSMCs were treated with various concentrations (0, 25, 50, 100 and 200 μg mL−1) of ox-LDL for 24 h (A), or exposed to 100 μg mL−1 of ox-LDL for different time point (0, 6, 12, 24 and 48 h) (B), followed by the detection of miR-33b expression by qRT-PCR. HA-VSMCs were transfected with or without anti-miR-33b or anti-miR-NC prior to treatment with 100 μg mL−1 of ox-LDL for the indicated time, followed by the measurement of miR-33b expression by qRT-PCR (C), cell proliferation capacity by CCK-8 assay (D), cell migration ability by wound-healing assay (E), MMP-9 and MMP-2 levels by western blot (F). *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
MiR-33a mediated the inhibitory effect of NEXN-AS1 overexpression on the proliferation and migration in ox-LDL-treated HA-VSMCs
In order to provide further mechanistic insight into the link between NEXN-AS1 and miR-33a on AS progression in vitro, HA-VSMCs were cotransfected with pcDNA-NEXN-AS1 and miR-33a mimic. As demonstrated by qRT-PCR, NEXN-AS1 overexpression triggered a significant decrease on miR-33a expression, while this effect was markedly reversed by cotransfection of miR-33a mimic (Fig. 6A). Moreover, in contrast to control group, NEXN-AS1 overexpression-mediated anti-proliferation, anti-migration and decreased levels of MMP-9 and MMP-2 were strikingly abated by miR-33a expression restoration (Fig. 6B–D). These data together strongly pointed a notion that NEXN-AS1 overexpression-mediated anti-proliferation and anti-migration effect was mediated by miR-33a. | ||
| Fig. 6 MiR-33a mediated the inhibitory effect of NEXN-AS1 overexpression on proliferation and migration in ox-LDL-treated HA-VSMCs. HA-VSMCs were transfected with pcDNA-NC, pcDNA-NEXN-AS1, pcDNA-NEXN-AS1 + miR-NC mimic or pcDNA-NEXN-AS1 + miR-33a mimic, and then exposed to 100 μg mL−1 of ox-LDL for the indicated time point. (A) After 24 h ox-LDL treatment, miR-33a expression was assessed by qRT-PCR. (B) At 0, 24, 48 and 72 h after treatment, cell proliferation ability was detected by CCK-8 assay. (C) 24 h after ox-LDL exposure, cell migration capacity was determined by wound-healing assay. (D) After 24 h ox-LDL stimulation, the levels of MMP-9 and MMP-2 were evaluated by western blot. *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
NEXN-AS1 overexpression-mediated anti-proliferation and anti-migration effect was abated by miR-33b expression restoration
Lastly, we further explored whether the inhibitory effect of NEXN-AS1 upregulation on cell proliferation and migration was mediated by miR-33b. qRT-PCR results revealed that compared with negative control, NEXN-AS1 upregulation also resulted in reduced miR-33b expression in ox-LDL-induced HA-VSMCs, while this effect was strongly reversed by cotransfection of miR-33b mimic (Fig. 7A). Notably, the suppressive effect of NEXN-AS1 overexpression on cell proliferation and migration was prominently abolished by the restoration of miR-33b expression (Fig. 7B–D). Taken together, these results strongly implied that miR-33b mediated the inhibitory effect of NEXN-AS1 upregulation on the proliferation and migration of ox-LDL-treated HA-VSMCs. | ||
| Fig. 7 The inhibitory effect of NEXN-AS1 overexpression on cell proliferation and migration was mediated by miR-33b. HA-VSMCs were transfected with pcDNA-NC, pcDNA-NEXN-AS1, pcDNA-NEXN-AS1 + miR-NC mimic or pcDNA-NEXN-AS1 + miR-33b mimic before treatment with 100 μg mL−1 of ox-LDL for the indicated time point, followed by the determination of miR-33b expression by qRT-PCR (A), cell proliferation ability by CCK-8 assay (B), cell migration capacity by wound-healing assay (C), the levels of MMP-9 and MMP-2 by western blot (D). *P < 0.05, **P < 0.01 or ***P < 0.001. | ||
Discussion
In recent years, dysregulation of lncRNAs is being found to have relevance to human diseases, including AS. For instance, Wu et al. reported that lncRNA-p21 hindered VMSC proliferation and accelerated cell apoptosis by promoting p53 transcriptional activity.23 Zhang et al. demonstrated that H19 was upregulated in AS and H19 silencing inhibited the proliferation and enhanced apoptosis of ox-LDL-treated HA-VSMCs through sponging miR-148b.24 Tian et al. manifested that urothelial carcinoma-associated (UCA1) acted as a sponge of miR-26a to regulate the proliferation and migration of VSMCs.25 These researches highlighted that lncRNAs served as novel promising therapeutic targets for AS management. In the present study, our results firstly suggested that NEXN-AS1 alleviated the proliferation and migration of HA-VSMCs under ox-LDL stimulation through sponging miR-33a/b.NEXN-AS1 has recently been found to be closely related to lung cancer susceptibility and prognosis of patients with cholangiocarcinoma.16,17 Moreover, NEXN-AS1 expression in human head and neck cancer cells was enhanced by fisetin, a novel anti-tumor agent, providing evidence for NEXN-AS1 as a potential molecular target of fisetin.26 A previous report proposed that downregulated NEXN-AS1 might act as a vital biomarker in ischemic stroke.27 Intriguingly, a recent document manifested that high level of NEXN-AS1 retarded AS progression through repression of adhesion molecules expression and inflammatory cytokines secretion by regulating NEXN.18 These findings from our study described above prompted us to explore NEXN-AS1 as a potential regulator on VSMC proliferation and migration in AS progression. To confirm this, we firstly established AS model in vitro using ox-LDL, and our data demonstrated that NEXN-AS1 expression was downregulated in ox-LDL-induced HA-VSMCs. Moreover, we firstly illuminated that NEXN-AS1 overexpression mitigated the proliferation and migration in HA-VSMCs under ox-LDL stimulation. In a word, NEXN-AS1 performed an anti-atherosclerotic effect through inhibition of VMSC proliferation and migration.
It is widely acknowledged that lncRNAs function as molecular sponges of special miRNAs, and thus play important roles in pathological processes. Therefore, we carried out a detailed analysis for its targeted miRNAs using LncBase v.2 software. Among these predicted candidates, miR-33a and miR-33b were of interest in our research owing to their important functions in AS etiology and pathophysiology.28–30 Antagonism of miR-33a/b was validated to lead to a prominent reduction of plaque size and lipid content in AS mice.29,31 Moreover, miR-33a/b silencing was reported to perform atheroprotective effects through regulating cholesterol efflux, lipid accumulation and macrophage inflammation.19,30,32 Subsequently, we firstly manifested that NEXN-AS1 acted as molecular sponges for miR-33a and miR-33b. Based on the above, our study continued from this hypothesis that NEXN-AS1 regulated the proliferation and migration of VSMCs via miR-33a and miR-33b. As expected, our data demonstrated that miR-33a/b were upregulated in ox-LDL-treated HA-VSMCs, and the knockdown of miR-33a or miR-33b alleviated VSMC proliferation and migration under ox-LDL treatment. In short, the silencing of miR-33a and miR-33b played atheroprotective roles, highlighting their inhibitors as therapeutic strategies for AS treatment. More importantly, our results substantiated that miR-33a or miR-33b mediated the inhibitory effect of NEXN-AS1 overexpression on the proliferation and migration of ox-LDL-treated HA-VSMCs. The present study was limited in vitro research, and more researches in vivo AS mice model will be carried out to validate this new regulatory mechanism in further work.
Conclusions
In conclusion, our study suggested that NEXN-AS1 overexpression alleviated VSMC proliferation and migration under ox-LDL stimulation at least partly through acting as molecular sponges of miR-33a and miR-33b. Our study provided a piece of evidence for NEXN-AS1 as a promising effective therapeutic approach for AS treatment.Conflicts of interest
The authors declare that they have no financial conflicts of interest.References
- P. Libby, P. M. Ridker and G. K. Hansson, Nature, 2011, 473, 317–325 CrossRef CAS PubMed.
- I. Tabas, G. Garcia-Cardena and G. K. Owens, J. Cell Biol., 2015, 209, 13–22 CrossRef CAS PubMed.
- A. C. Doran, N. Meller and C. A. McNamara, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 812–819 CrossRef CAS PubMed.
- M. R. Bennett, S. Sinha and G. K. Owens, Circ. Res., 2016, 118, 692–702 CrossRef CAS PubMed.
- J. Hulthe and B. Fagerberg, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 1162–1167 CrossRef CAS PubMed.
- D. Tian, Y. Sha, J. M. Lu and X. J. Du, J. Cell. Biochem., 2018, 119, 6231–6237 CrossRef CAS PubMed.
- M. Esteller, Nat. Rev. Genet., 2011, 12, 861–874 CrossRef CAS PubMed.
- J. J. Quinn and H. Y. Chang, Nat. Rev. Genet., 2016, 17, 47–62 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2018, 173, 20–51 CrossRef CAS PubMed.
- J. M. Lorenzen and T. Thum, Nat. Rev. Nephrol., 2016, 12, 1–14 Search PubMed.
- I. Fernandez-Ruiz, Nat. Rev. Cardiol., 2018, 15, 195 CrossRef PubMed.
- A. Schober and C. Weber, Annu. Rev. Phytopathol., 2016, 11, 583–616 CrossRef CAS PubMed.
- R. V. Kartha and S. Subramanian, Front. Genet., 2014, 5, 8 Search PubMed.
- X. Zhong, X. Ma, L. Zhang, Y. Li, Y. Li and R. He, Biomed. Pharmacother., 2018, 97, 1078–1085 CrossRef CAS PubMed.
- Y. Tang, X. Jin, Y. Xiang, Y. Chen, C. X. Shen, Y. C. Zhang and Y. G. Li, FEBS Lett., 2015, 589, 3189–3196 CrossRef CAS PubMed.
- H. Yuan, H. Liu, Z. Liu, K. Owzar, Y. Han, L. Su, Y. Wei, R. J. Hung, J. McLaughlin, Y. Brhane, P. Brennan, H. Bickeboeller, A. Rosenberger, R. S. Houlston, N. Caporaso, M. T. Landi, J. Heinrich, A. Risch, D. C. Christiani, Z. H. Gumus, R. J. Klein, C. I. Amos and Q. Wei, Sci. Rep., 2016, 6, 34234 CrossRef CAS PubMed.
- W. Song, D. L. Miao and L. Chen, Biochem. Biophys. Res. Commun., 2018, 506, 1004–1012 CrossRef CAS PubMed.
- Y. W. Hu, F. X. Guo, Y. J. Xu, P. Li, Z. F. Lu, D. G. McVey, L. Zheng, Q. Wang, J. H. Ye, C. M. Kang, S. G. Wu, J. J. Zhao, X. Ma, Z. Yang, F. C. Fang, Y. R. Qiu, B. M. Xu, L. Xiao, Q. Wu, L. M. Wu, L. Ding, T. R. Webb, N. J. Samani and S. Ye, J. Clin. Invest., 2019, 129, 1115–1128 CrossRef PubMed.
- N. L. Price, N. Rotllan, A. Canfran-Duque, X. Zhang, P. Pati, N. Arias, J. Moen, M. Mayr, D. A. Ford, A. Baldan, Y. Suarez and C. Fernandez-Hernando, Cell Rep., 2017, 21, 1317–1330 CrossRef CAS PubMed.
- T. Horie, O. Baba, Y. Kuwabara, Y. Chujo, S. Watanabe, M. Kinoshita, M. Horiguchi, T. Nakamura, K. Chonabayashi, M. Hishizawa, K. Hasegawa, N. Kume, M. Yokode, T. Kita, T. Kimura and K. Ono, J. Am. Heart Assoc., 2012, 1, e003376 Search PubMed.
- J. L. Johnson, A. Dwivedi, M. Somerville, S. J. George and A. C. Newby, Arterioscler., Thromb., Vasc. Biol., 2011, 31, e35–e44 CAS.
- H. J. Park, M. K. Kim, Y. Kim, S. S. Bae, H. J. Kim, S. K. Bae and M. K. Bae, BMB Rep., 2017, 50, 628–633 CrossRef CAS PubMed.
- G. Wu, J. Cai, Y. Han, J. Chen, Z. P. Huang, C. Chen, Y. Cai, H. Huang, Y. Yang, Y. Liu, Z. Xu, D. He, X. Zhang, X. Hu, L. Pinello, D. Zhong, F. He, G. C. Yuan, D. Z. Wang and C. Zeng, Circulation, 2014, 130, 1452–1465 CrossRef CAS PubMed.
- L. Zhang, H. Cheng, Y. Yue, S. Li, D. Zhang and R. He, J. Biomed. Sci., 2018, 25, 11 CrossRef PubMed.
- S. Tian, Y. Yuan, Z. Li, M. Gao, Y. Lu and H. Gao, Gene, 2018, 673, 159–166 CrossRef CAS PubMed.
- D. H. Won, S. H. Chung, J. A. Shin, K. O. Hong, I. H. Yang, J. W. Yun and S. D. Cho, J. Clin. Biochem. Nutr., 2019, 64, 97–105 CrossRef PubMed.
- Y. Zheng, S. Sun, M. Yu and X. Fu, J. Cell. Biochem., 2019, 120, 12832–12842 CrossRef CAS PubMed.
- W. J. Chen, M. Zhang, G. J. Zhao, Y. Fu, D. W. Zhang, H. B. Zhu and C. K. Tang, Atherosclerosis, 2013, 227, 201–208 CrossRef CAS PubMed.
- K. J. Rayner, F. J. Sheedy, C. C. Esau, F. N. Hussain, R. E. Temel, S. Parathath, J. M. van Gils, A. J. Rayner, A. N. Chang, Y. Suarez, C. Fernandez-Hernando, E. A. Fisher and K. J. Moore, J. Clin. Invest., 2011, 121, 2921–2931 CrossRef CAS PubMed.
- M. Ouimet, H. N. Ediriweera, U. M. Gundra, F. J. Sheedy, B. Ramkhelawon, S. B. Hutchison, K. Rinehold, C. van Solingen, M. D. Fullerton, K. Cecchini, K. J. Rayner, G. R. Steinberg, P. D. Zamore, E. A. Fisher, P. Loke and K. J. Moore, J. Clin. Invest., 2015, 125, 4334–4348 CrossRef PubMed.
- N. Rotllan, C. M. Ramirez, B. Aryal, C. C. Esau and C. Fernandez-Hernando, Arterioscler., Thromb., Vasc. Biol., 2013, 33, 1973–1977 CrossRef CAS PubMed.
- T. J. Marquart, R. M. Allen, D. S. Ory and A. Baldan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12228–12232 CrossRef CAS PubMed.
Footnote |
| † The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |