
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt/Fe co-loaded mesoporous zeolite beta for CO oxidation with high catalytic activity and water resistance
Qinru Liab,
Xiaoxia Zhou *b,
Wanpeng Zhaob,
Chunlei Pengb,
Huixia Wu*a and
Hangrong Chenb
*b,
Wanpeng Zhaob,
Chunlei Pengb,
Huixia Wu*a and
Hangrong Chenb
aThe Key Laboratory of Resource Chemistry of Ministry of Education, Shanghai Key Laboratory of the Rare Earth Functional Materials, Shanghai Municipal Education Committee Key Laboratory of Molecular Imaging Probes and Sensors, Shanghai Normal University, Shanghai 200234, China
bState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai, 200050, P. R. China. E-mail: zhouxiaoxia@mail.sic.ac.cn
First published on 6th September 2019
Abstract
A series of Pt/Fe co-loaded mesoporous zeolite beta (Pt/Fe–mBeta) catalysts with different Fe contents have been successfully synthesized by an ion exchange and subsequent ethylene glycol reduction method. The catalysts were characterized by XRD, N2 adsorption–desorption, TEM, SEM, XPS and H2-TPR. The optimized sample Pt/Fe(3)–mBeta shows high catalytic activity for CO oxidation under dry conditions, and the complete conversion temperature of CO is as low as 90 °C. More importantly, the sample Pt/Fe(3)–mBeta also shows excellent water resistance and good durability, which could meet the practical needs of exhaust purification of diesel vehicles. It is believed that the synergistic effect between varied-valence Pt/Fe species and the mesoporous zeolite support with high surface area and good water resistance jointly contribute to the excellent catalytic performance.
Introduction
Carbon monoxide (CO) is one of the main pollutants in the exhaust of diesel vehicles, which is very harmful to human health.1 The catalytic oxidation of CO is generally considered as the most efficient way to eliminate carbon monoxide from the environment. Recently, large numbers of catalysts have been developed for CO oxidation, such as metal oxides MnO2,2–4 CuO5,6 and Fe2O3,7 perovskite oxide La0.5Sr0.5MnO3,8 and precious metal-based catalysts. Generally, non-noble metal-based catalysts can promote the oxidation of CO by the synergetic effect of the different active components, but the complete conversion temperature of CO is generally higher than 150 °C. Especially, the catalytic activity of non-noble metal-based catalysts drops substantially under harsh conditions. e.g., higher humidity. In contrast, the precious metal-based catalysts show relatively high catalytic activity and stability for CO oxidation.9–11 For example, Pd/Al2O3 shows a great enhancement in the CO and C3H6 oxidation due to the highly dispersed Pd species.12 Nevertheless, Pd-based catalysts still face the challenge of water resistance.13 Therefore, the design and preparation of a highly-efficient and water-resistant catalyst for CO oxidation is of essential importance.Irons oxides, as a highly active component, are usually used in the catalytic oxidation of CO.14,15 However, the pure iron oxides may lead to aggregation at higher temperatures and affect the catalytic activity. Therefore, the support with high surface area is needed for dispersing Fe species. Recently, mesoporous zeolites as the catalyst support have received much attention owing to the high surface area, high adsorption capacity, and excellent hydrothermal stability.16 Moreover, the doping of heteroatoms Al+ in the [SiO4] makes zeolite have rich acidic sites and a larger number of oxygen vacancies, which is helpful to the adsorption of oxygen.17 Therefore, it is promising to construct a kind of noble metal and Fe species co-loaded mesoporous zeolite as a highly-efficient catalyst for the CO oxidation by reasonably modulating the pore structure and active species.
Herein, a novel mesoporous zeolite beta as the support has been successfully synthesized by a one-pot hydrothermal approach. Then, both Fe and Pt active species could be highly dispersed onto/into the support by an ion exchange and following ethylene glycol reduction method. The catalytic activity and water-resistance of the prepared samples towards CO oxidation is investigated under different reaction conditions. Based on the characterization analyses and related catalytic results, a possible catalytic mechanism of CO oxidation on the prepared catalyst Pt/Fe–mBeta is proposed.
Results and discussion
Structural and morphology characteristics
The powder XRD patterns of pure mBeta, and the samples Pt/Fe co-loaded mBeta are shown in Fig. 1. It is clear that all prepared samples keep the typical diffraction peaks of mBeta structure, and no diffraction peaks assigned to FeOx and Pt species can be observed, suggesting that the Fe and Pt species could be highly dispersed onto/into the zeolite carrier. Notably, the intensity of the diffraction peaks of the samples gradually decreases with the increase of Fe species content, indicating that part of Fe ions could enter into the zeolite framework by replacing cations of zeolite. Besides, compared to the support mBeta, the Si/Al ratio of the samples Pt/Fe–mBeta gradually increases with the increase of Fe content, as shown in Table 1, further confirming that the part of Al in zeolite beta can be replaced by Fe by an ion-exchange process.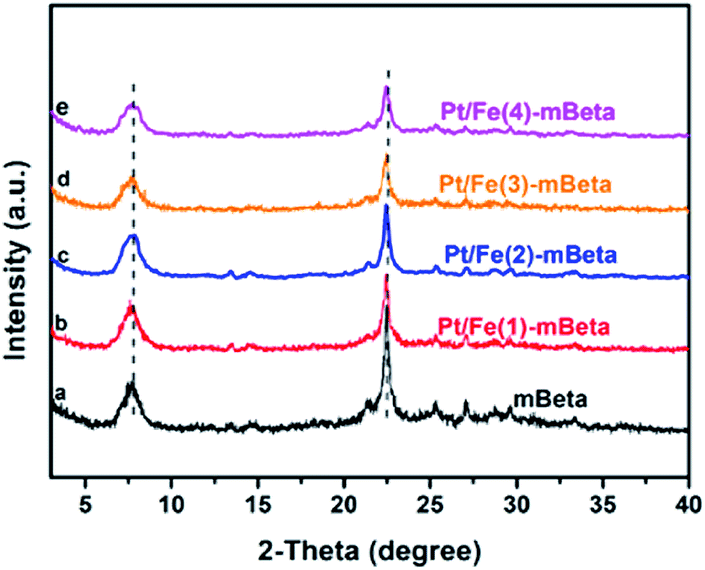 | ||
| Fig. 1 The powder XRD patterns of a series of prepared samples, (a) mBeta, (b) Pt/Fe(1)-mBeta, (c ) Pt/Fe(2)-mBeta, (d) Pt/Fe(3)-mBeta and (e) Pt/Fe(4)-mBeta. | ||
| Sample | Stotal (m2 g−1) | Smesoa (m2 g−1) | d (nm) | Vtotal (cm3 g−1) | Feb (%) | Si/Alb |
|---|---|---|---|---|---|---|
| a Determined by the difference between Stotal and Smicro.b Determined by the ICP-AES. | ||||||
| mBeta | 595 | 238 | 3.4 | 0.52 | — | 26.0 |
| Pt/Fe(1)–mBeta | 570 | 278 | 3.1 | 0.44 | 1.2 | 26.2 |
| Pt/Fe(2)–mBeta | 568 | 239 | 3.0 | 0.47 | 4.6 | 27.5 |
| Pt/Fe(3)–mBeta | 590 | 251 | 3.1 | 0.48 | 9.3 | 28.3 |
| Pt/Fe(4)–mBeta | 528 | 214 | 3.2 | 0.43 | 13.0 | 28.5 |
The typical SEM images of as-prepared Pt/Fe(3)–mBeta are shown in Fig. 2a. It is found that the mBeta possesses a rough surface morphology with diameter about 200–400 nm, suggesting the generation of the mesoporous structure.16 After loading with Fe and Pt species, no obvious aggregates could be found (Fig. 2b and c), further indicating that Fe and Pt species are highly dispersed onto/into the zeolite support, consistent with the XRD results. From the DFI, TEM and HRTEM images of the sample Pt/Fe(3)–mBeta, it is clearly found that the crystalline Pt nanoparticles with the size of 2–5 nm are highly dispersed on the support, as shown in Fig. 2b–d.
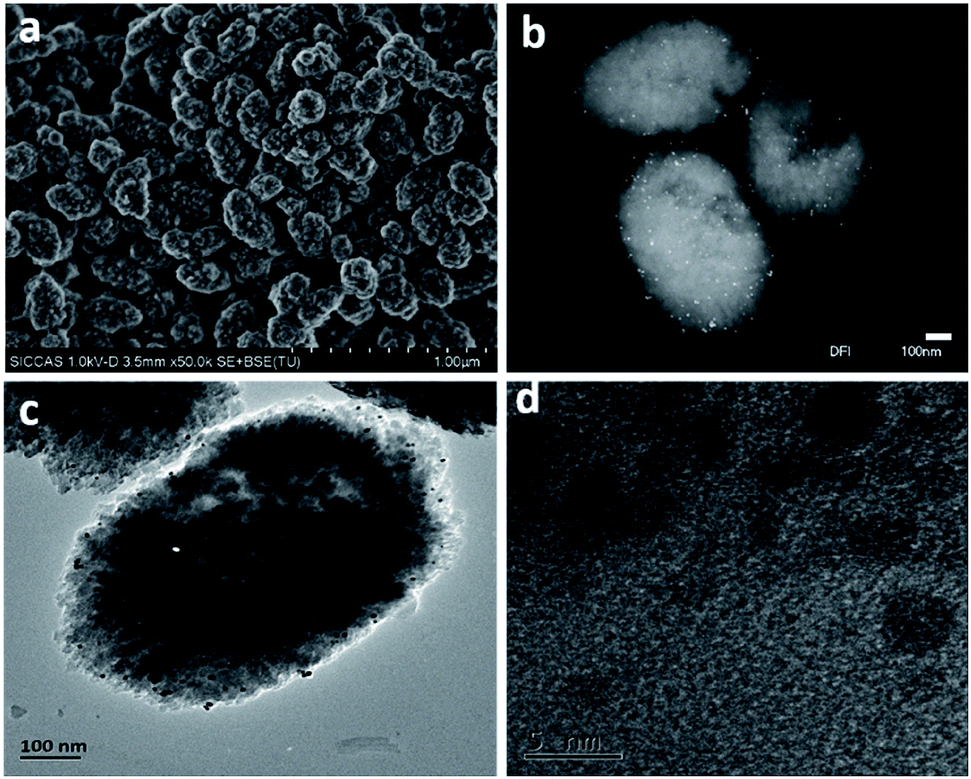 | ||
| Fig. 2 (a) Typical SEM images, (b) the DFI image and (c and d) TEM images of Fe(3)–mBeta. | ||
The N2 adsorption–desorption isotherms and the corresponding pore size distribution curves of the different samples are shown in Fig. 3. The corresponding pore structure parameters are shown in Table 1. All samples possess typical Langmuir IV isotherms, confirming the presence of mesopore structure. It is noting that the support mesopore zeolite beta shows the highest BET surface area (Stotal = 595 m2 g−1) and the largest mesopore volume (Vtotal = 0.52 cm3 g−1) with the average pore size of 3.4 nm. After co-loading Fe and Pt species, the sample Pt/Fe(3)–mBeta still keeps large BET surface area and high total pore volume (590 m2 g−1, 0.48 cm3 g−1), suggesting that the active species could be highly dispersed into the sample without blocking the mesopores, which is beneficial to provide more active sites for CO adsorption and oxidation.
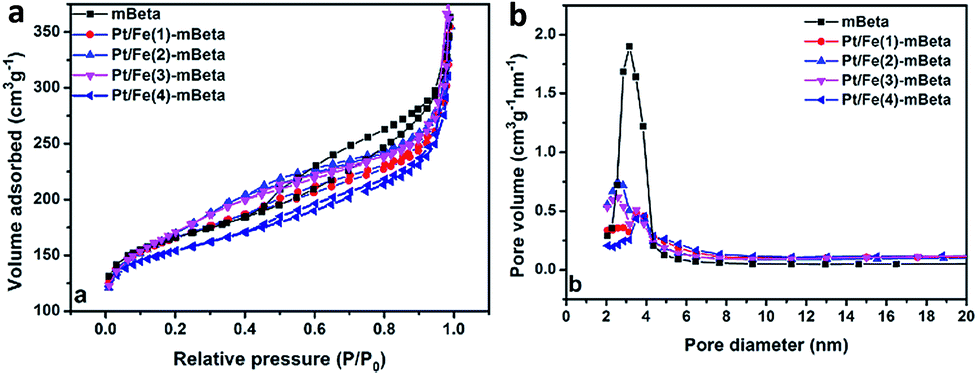 | ||
| Fig. 3 (a) N2 adsorption/desorption and (b) the corresponding pore size distribution curves of the samples. | ||
Surface chemical environment analysis
Fig. 4 shows the XPS results of the catalyst Pt/Fe(3)–mBeta. The Pt 4f peaks could be deconvoluted to two peaks at 71.3 eV and 72.4 eV, which can be ascribed to Pt0 and Pt2+,20,21 respectively. The Pt2+ is calculated to be 33.0% in the catalyst Pt/Fe(3)–mBeta (Table 2). It is believed that the presence of Pt with low oxidation states can reduce the adsorption strength of CO on the Pt sites, which is beneficial for improving the reaction rate.22 For the Fe 2p3/2, the peak at 714.4 eV can be assigned to the Fe3+ species,22,23 and the peak at 711.2 eV is ascribed to the Fe2+,24 as shown in Fig. 5b, which accounts for 51.8% and 48.2% of the total Fe content, respectively (Table 2). In Fig. 5c, the O 1s spectrum can be fitted with two Gaussian peaks. Thereinto, the peaks at 530.5 eV and 532.8 eV could be assigned to the lattice oxygen (Olatt) and the chemisorbed oxygen species (Oads),25 respectively. The presence of Oads is related to the oxygen vacancies derived from the doping of heteroatoms Fe and/or Al in the zeolite [SiO4]. By further calculating peak area, the content of Oads of Pt/Fe(3)–mBeta is as high as 69.0%, as shown in Table 2, implying that the adsorbed oxygen is much beneficial to promote the catalytic oxidation of CO, similarly to the previous report.26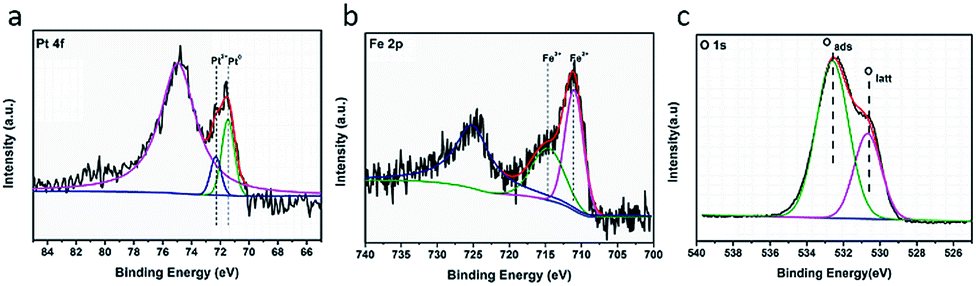 | ||
| Fig. 4 XPS spectra of (a) Pt 4f, (b) Fe 2p and (c) O 1s of the sample Pt/Fe(3)–mBeta. | ||
| Elements | Pt | Fe | O | |||
|---|---|---|---|---|---|---|
| Pt0 | Pt2+ | Fe2+ | Fe3+ | Oads | Olatt | |
| Binding energy/eV | 71.3 | 72.4 | 711.2 | 714.4 | 532.8 | 530.5 |
| Percentage of total area/% | 67.0 | 33.0 | 48.2 | 51.8 | 69.0 | 31.0 |
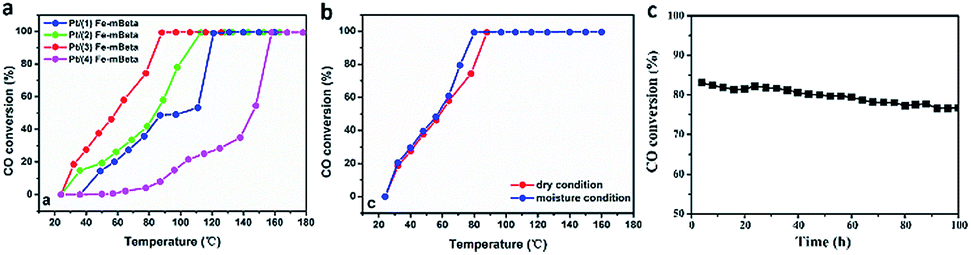 | ||
| Fig. 5 (a) The catalytic performances of Pt/Fe(x)–mBeta catalysts with different Fe contents; (b) the catalytic performance of Pt/Fe(3)–mBeta under different conditions and (c) the stability of Pt/Fe(3)–mBeta at 75 °C under moisture condition. | ||
The catalytic oxidation of CO
The CO catalytic oxidation for different samples under dry condition is shown in Fig. 5a. It is found that the complete conversion temperature of CO on the sample Pt/Fe–mBeta decreases with the increase of Fe content, indicating that the addition of Fe species is beneficial for the catalytic oxidation of CO. However, excess Fe content would seriously affect the catalytic, which could be associated with the possible aggregation of Fe species on the surface of mBeta. As result, the optimal sample Pt/Fe(3)–mBeta shows the highest catalytic activity to CO oxidation among a series of Pt, Fe co-loaded mBeta, and the complete oxidation temperature of CO is as low as 90 °C. Furthermore, the water resistance of the catalyst Pt/Fe(3)–mBeta was also investigated, as shown in Fig. 5b. When 4.0 vol% moisture is introduced into the reaction atmosphere, the activity of Pt/Fe(3)–mBeta has a further improvement, and the CO complete conversion temperature decreases to 79 °C. This shows that OH groups from the adsorbed water on the surface of catalyst, as active species, can also interact with the CO molecule, similarly as previous reports.18,19 The CO catalytic stability on the sample Pt/Fe(3)–mBeta is also investigated under moisture condition, as shown in Fig. 5c. It is found the CO conversion decreases by only 6% on the sample Pt/Fe(3)–mBeta within 100 h, suggestive of excellent catalytic durability.Furthermore, the morphology and structure characterizations of the spent catalyst Pt/Fe(3)–mBeta after stability test are shown in Fig. 6. It is interesting to find that though the BET surface area (365 m2 g−1) of the spent sample obviously decreases, the spent catalyst well keeps typical diffraction peak assigned to zeolite BEA with narrow and larger pore size distribution (ca., 5.3 nm), as shown in Fig. 6d–f. The slight decrease of catalytic activity during the stability test could be ascribed to the damage of pore structure to some extent. Nevertheless, the spent catalyst Pt/Fe(3)–mBeta still shows high catalytic in 100 h at 75 °C under moisture condition (Fig. 5c), which can be assigned to the highly dispersed Pt nanoparticles and the stable zeolite carrier. As shown in Fig. 6a–c, highly dispersed Pt nanoparticles on the catalyst are still remained after catalytic stability test.
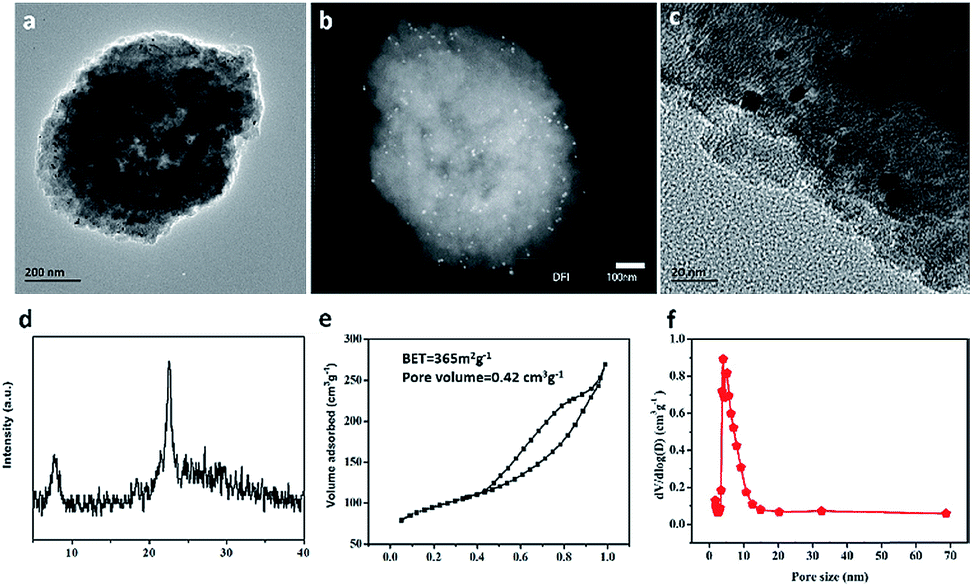 | ||
| Fig. 6 (a) TEM image, (b) DFI image, (c) HRTEM image, (d) XRD, (e and f) N2 adsorption/desorption and the corresponding pore size distribution curves of the spent catalyst Pt/Fe(3)–mBeta after stability test. | ||
The synergistic effect between active Pt and Fe species
The H2 temperature-programmed reduction (H2-TPR) profiles of the optimized Pt/Fe(3)–mBeta and reference samples were used to investigate the redox behaviour, as shown in Fig. 7a. It is found that the mBeta carrier is nearly inactive, and the Pt–mBeta has only one weak reduction peak at around 150 °C, which can be ascribed to the reduction of Pt2+. For reference sample Fe(3)–mBeta, two obvious redox peaks can be found. More details, the peak at around 360 °C is ascribed to the reduction of Fe2O3 to Fe3O4, and the broad peak at around 680 °C can be corresponding to the further reduction of Fe3O4 to metallic iron.27–29 Interestingly, after co-loading with Pt and Fe species, the obtained sample Pt/Fe(3)–mBeta shows four obvious reduction peaks. Among, the peak at around 150 °C is ascribed to the reduction of Pt2+, and the strong peak at 400–500 °C and the weak peak at 600 °C can be related to the reduction of Fe2O3 to Fe3O4 to Fe, which shifts to lower temperature range compared to that of Fe(3)–mBeta, indicating that there are the synergetic effect between Pt species and Fe(3)–mBeta. Besides, the new redox peak at 250 °C can be ascribed to the synergistic effect between the active species Pt and Fe, which can greatly promote the redox potential of the catalyst.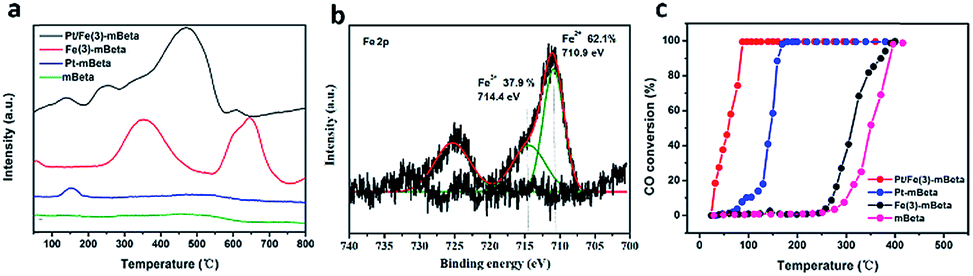 | ||
| Fig. 7 (a) H2-TPR profiles, (b) XPS spectrum of Fe 2p of the sample Fe(3)–mBeta and (c) CO catalytic oxidations of different catalysts. | ||
XPS spectrum of Fe 2p of the reference Fe(3)–mBeta without Pt loading is offered to further explore the synergetic effect between Pt and Fe species, as shown in Fig. 7b. For the optimized sample Pt/Fe(3)–mBeta (711.2 eV, Fig. 4b), the peak assigned to Fe2+ shifts toward higher binding energy than Fe(3)–mBeta (710.9 eV), which can be ascribed to the electron transfer from Fe species to Pt nanoparticles. Besides, the content of Fe3+ in the sample Pt/Fe(3)–mBeta (51.8%) is much higher than that of the reference sample Fe(3)–mBeta (37.9%), further indicating the presence of electron transfer from Fe2+ species to Pt nanoparticles.
Furthermore, the catalytic activity of CO oxidation over different samples is also shown in Fig. 7C. Though the mBeta can promote the oxidation of CO oxidation as an acidic catalyst, the CO complete conversion temperature is as high as 400 °C. The reference Fe(3)–mBeta shows an enhanced activity to CO oxidation due to the adsorption performance of Fe species to CO. Additionally, the Pt–mBeta shows an excellent catalytic activity, and the complete oxidation temperature is as low as 160 °C. Notably, the optimized sample Pt/Fe(3)–mBeta shows the best catalytic activity among these catalysts, and the complete oxidation temperature can reduce to 90 °C due to the strong synergistic effect between Fe and Pt species, which can be ascribed to the electron transport between varied-valence Pt and Fe. Besides, the mBeta as support with large surface area is also favor for the dispersion of active species Fe and Pt, and the larger pore volume of the mBeta is much helpful to the adsorption and diffusion of reaction gas, which jointly promote the CO catalytic oxidation.
According to the above results and discussion, a possible catalytic reaction mechanism is proposed for the catalytic oxidation of CO on the optimized sample Pt/Fe(3)–mBeta, as shown in Scheme 1. Firstly, in the presence of CO and O2, Fe3+ species gain electrons from Pt0 to produce Pt2+ and Fe2+. Then, CO molecule can be adsorbed on Pt2+ and activated into CO+ species by capturing one electron from Pt2+, at the same time, the Pt2+ becomes to Pt0. Meanwhile, Fe2+ adsorbs O2 molecule and donates an electron to the O atom while Fe2+ turns back to Fe3+. The O2 is activated into O− species on the Fe2+ species. (I) Finally, the reaction between the activated CO+ and O− species can quickly produce CO2, as shown in Step I. Besides, there are varied-valence Fe and Pt in the catalyst Pt/Fe(3)–mBeta, and the reaction gas CO molecules can be also adsorbed on the FeOx, therefore, another reaction route is also possible, as shown in Step I. More details, Pt2+ gains two electrons from neighbouring Fe2+, and then becomes to Fe3+ and Pt0, respectively. Afterwards, similar CO and O2 activation process can happen on Fe3+ and Pt0, respectively, leading to the CO oxidation. Notably, when a certain amount of water vapor is introduced, both O2 and H2O can be adsorbed on the varied-valence Fe species and activated into O− and/or OH groups, respectively. Meanwhile, the CO can be adsorbed and activated into CO− on the Pt species. Finally, the reaction between CO− and O−/OH can produce CO2, as shown in Step II in Scheme 1.
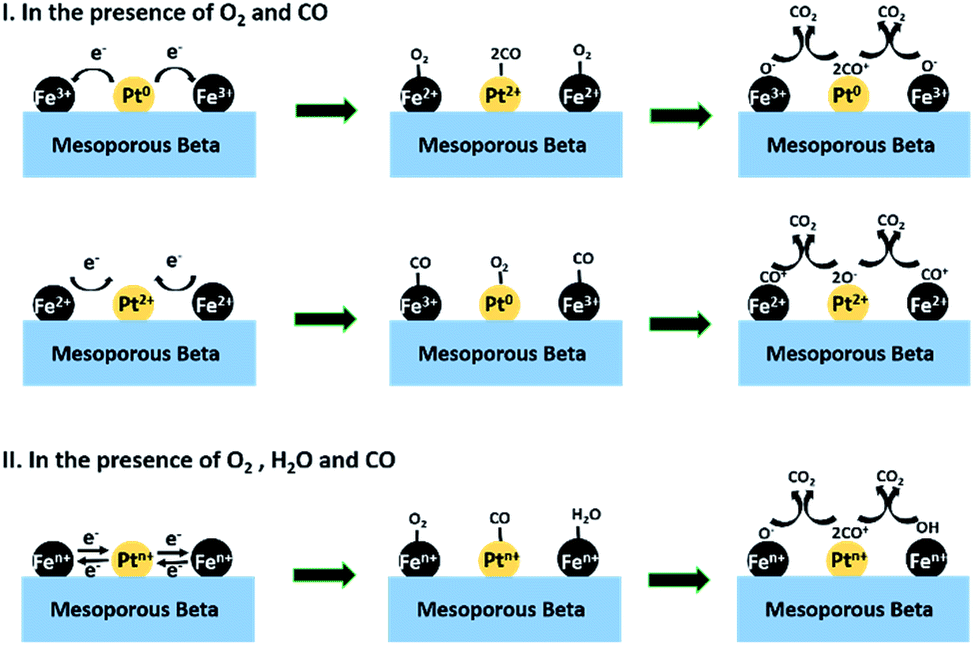 | ||
| Scheme 1 The possible catalytic pathways of CO oxidation over the catalyst Pt/Fe(3)–mBeta under different conditions. | ||
Experimental
Chemicals and reagents
Tetraethylammonium hydroxide (TEAOH, 25 wt%) and cetyltrimethylammonium chloride (CTAB, 99%) were obtained from Shanghai J&K, China. Silicic acid hydrate (H2SiO3, AR, Sinopharm Chemical Reagent Co., Ltd) and other regents were purchased from Sinopharm Chemical Reagent Co., Ltd.Synthesis of the mesoporous zeolite beta (mBeta)
The synthesis process of mesoporous zeolite beta (mBeta) was conducted according to our previous report.16 In detail, 0.09 g of Na2CO3 and 3.9 g of H2SiO3 were added to 6 mL distilled water solution including 10 g TEAOH with stirring 8 h at 40 °C. Next, the solution containing 0.016 g of Na2CO3, 0.16 g of NaAlO2 and 3 mL distilled water were slowly added to the resultant solution and further stirred at 40 °C for 8 h. Under magnetic stirring, 3 mL distilled water including 0.5 g CTAB was added to the obtained solution. Subsequently, the obtained mixed solution was hydrothermally treated for 24 h at 150 °C. Finally, the solids were washed with distilled water and dried at 100 °C for 12 h. The final product mesoporous zeolite was calcined at 550 °C for 8 h to remove any organics.Synthesis of the Fe–mBeta
A series of Fe–mBeta catalysts with varied Fe contents were prepared by an ion exchange method. In a typical synthesis, 0.5 g of mBeta was added to 20 mL distilled water. Then, the Fe(NO3)3 with different concentrations (0.01 M, 0.025 M, 0.1 M, 0.2 M) was slowly added into the above solution and further stirred at 80 °C for 4 h. Next, the obtained solution was washed and then dried at 100 °C for 24 h. Finally, the sample Fe–mBeta was collected after calcination at 550 °C for 8 h. The obtained samples are labelled as Fe(1)–mBeta, Fe(2)–mBeta, Fe(3)–mBeta and Fe(4)–mBeta, respectively.Synthesis of the Pt/Fe(x)–mBeta
Typically, 500 mg of Fe–mBeta was dispersed in 50 mL distilled water containing 10 mL ethylene glycol. Next, 5 mg of Pt(NO3)2 dissolved in water solution was added into the above solution, and the resulting mixture solution was further stirred for 6 h at 40 °C. Next, the obtained solution was washed and then dried at 100 °C for 24 h. Finally, the sample Pt/Fe(x)–mBeta was collected after calcined at 450 °C for 6 h.Catalyst characterization
The crystalline structure of the catalysts were tested by a Rigaku D/Max-2200 PC X-ray diffractometer using Cu reaction at 40 kV and 40 mA. Micromeritics Tristar 3000 was used to measure the specific surface area and pore structure (pore volume and pore size) of the samples by N2 adsorption–desorption. The specific surface area and the pore size distribution were calculated using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods at 77 K. Field emission scanning electron microscopy (SEM) analysis was obtained using JEOL JSM6700F electron microscope. Field emission transmission electron microscopy (TEM) analysis was conducted with a JEOL200CX electron microscope operated at 200 keV. X-ray photoelectron spectroscopy (XPS) signals were collected on a Thermo Scientific ESCALAB 250 instrument. The temperature-programmed reduction with hydrogen (H2-TPR) were performed on Micromeritics Chemisorb 2750 instrument attached with ChemiSoft TPx software. TPR was carried out from room temperature to 800 °C under 5% H2 in Ar at a flow rate of 50 mL min−1. The contents of Fe and Pt were measured by using Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) analyzer on a Vista AX.The oxidation of CO
The catalytic activity for CO oxidation was carried out in a fixed-bed quartz reactor using 0.1 g catalyst without any pretreatment. The feed gas contained 1.0 vol% CO, 20 vol% O2 and a high-purity N2 (99.99%). The total flow rate was 50 mL min−1, corresponding to a space velocity of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL h−1 gcat−1. Under steady-state condition, the activity date of CO was collected by online gas chromatograph (GC). In the presence of moisture, the catalytic activity was tested by passing through a water vapor saturator.
000 mL h−1 gcat−1. Under steady-state condition, the activity date of CO was collected by online gas chromatograph (GC). In the presence of moisture, the catalytic activity was tested by passing through a water vapor saturator.
The CO conversion was calculated by
| X (%) = (COinlet − COoutlet)/COinlet × 100% |
Conclusions
In conclusion, a series of Pt/Fe co-loaded mBeta have been successfully prepared by an ion exchange and subsequent ethylene glycol reduction method. Among, the optimal sample Pt/Fe(3)–mBeta has high BET surface area (Stotal = 590 m2 g−1) and large volume (V = 0.48 cm3 g−1) with average mesopore size of 3.1 nm, which shows the excellent catalytic activity and high water resistance. The CO complete conversion temperature on the sample Pt/Fe(3)–mBeta is as low as 78 °C, and the catalytic activity still keeps unchanged in 100 h in the presence of H2O. Such good catalytic performance can be ascribed to the following aspects: (1) the support beta with large surface area favors the dispersion of active species Fe and Pt, and the high pore volume of the sample is helpful to the adsorption and diffusion of reaction gas; (2) the synergetic effect between Pt and Fe from the electron transport between varied-valence Pt and Fe greatly accelerates CO oxidation. This work demonstrates a broad application prospect in the practical application of catalytic purification of diesel exhaust gas.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was sponsored by National Natural Science Foundation of China (51502330, 51961165107), the Shanghai Natural Science Foundation (19ZR1464500), and Science Foundation for Youth Scholar of State Key Laboratory of High Performance Ceramics and Superfine Microstructures (SKL201604).Notes and references
- P. Kast, M. Friedrich, D. Teschner, F. Girgsdies, T. Lunkenbein, R. Naumann d'Alnoncourt, M. Behrens and R. Schlögl, Appl. Catal., A, 2015, 502, 8 CrossRef CAS.
- S. Liang, F. Teng, G. Bulgan, R. Zong and Y. Zhu, J. Phys. Chem. C, 2008, 112, 5307 CrossRef CAS.
- J. H. Park, D. C. Kang, S. J. Park and C. H. Shin, J. Ind. Eng. Chem., 2015, 25, 250 CrossRef CAS.
- Y. Xi and J. Ren, J. Phys. Chem. C, 2016, 120, 24302 CrossRef CAS.
- F. Balıkçı Derekaya, C. Kutar and Ç. Güldür, Mater. Chem. Phys., 2009, 115, 496 CrossRef.
- C. Wang, Q. Cheng, X. Wang, K. Ma, X. Bai, S. Tan, Y. Tian, T. Ding, L. Zheng, J. Zhang and X. Li, Appl. Surf. Sci., 2017, 422, 932 CrossRef CAS.
- T. Yan, D. W. Redman, W. Y. Yu, D. W. Flaherty, J. A. Rodriguez and C. B. Mullins, J. Catal., 2012, 294, 216 CrossRef CAS.
- Y. F. Y. Yao, J. Catal., 1975, 36, 266 CrossRef CAS.
- G. Li, L. Li, Y. Yuan, J. Shi, Y. Yuan, Y. Li, W. Zhao and J. Shi, Appl. Catal., B, 2014, 158–159, 341 CrossRef CAS.
- J. Luo, M. Meng, J. Yao, X. Li, Y. Zha, X. Wang and T. Zhang, Appl. Catal., B, 2009, 87, 92 CrossRef CAS.
- E. Kolobova, A. Pestryakov, G. Mamontov, Y. Kotolevich, N. Bogdanchikova, M. Farias, A. Vosmerikov, L. Vosmerikova and V. Cortes Corberan, Fuel, 2017, 188, 121 CrossRef CAS.
- M. Haneda, M. Todo, Y. Nakamura and M. Hattori, Catal. Today, 2017, 281, 447 CrossRef CAS.
- F. Wang, K. Zhao, H. Zhang, Y. Dong, T. Wang and D. He, Chem. Eng. J., 2014, 242, 10 CrossRef CAS.
- L. Liu, F. Zhou, L. Wang, X. Qi, F. Shi and Y. Deng, J. Catal., 2010, 274, 1 CrossRef CAS.
- B. Zheng, G. Liu, L. Geng, J. Cui, S. Wu, P. Wu, M. Jia, W. Yan and W. Zhang, Catal. Sci. Technol., 2016, 6, 1546 RSC.
- X. Zhou, H. Chen, G. Zhang, J. Wang, Z. Xie, Z. Hua, L. Zhang and J. Shi, J. Mater. Chem. A, 2015, 3, 9745 RSC.
- J. Lin, B. Qiao, L. Li, H. Guan, C. Ruan, A. Wang, W. Zhang, X. Wang and T. Zhang, J. Catal., 2014, 319, 142 CrossRef CAS.
- S. Ivanova, V. Pitchon, C. Petit and V. Caps, ChemCatChem, 2010, 2, 556 CrossRef CAS.
- J. Wang, J. Zhu, X. Zhou, Y. Du, W. Huang, J. Liu, W. Zhang, J. Shi and H. Chen, J. Mater. Chem. A, 2015, 3, 7631–7638 RSC.
- T. Zhang, Z. Cheng, C. Yi and Z. Xu, Chem. Commun., 2018, 54, 9143 RSC.
- N. An, W. Zhang, X. Yuan, B. Pan, G. Liu, M. Jia, W. Yan and W. Zhang, Chem. Eng. J., 2013, 215–216, 1 CrossRef CAS.
- X. Zhou, L. Chen, G. Wan, Y. Chen, Q. Kong, H. Chen and J. Shi, ChemSusChem, 2016, 9, 2337 CrossRef CAS.
- V. M. Shinde and G. Madras, Int. J. Hydrogen Energy, 2012, 37, 18798 CrossRef CAS.
- P. Szarek and W. Grochala, J. Phys. Chem. A, 2015, 119, 2483 CrossRef CAS.
- C. L. Corkhill, P. L. Wincott, J. R. Lloyd and D. J. Vaughan, Geochim. Cosmochim. Acta, 2008, 72, 5616 CrossRef CAS.
- J. Xu, X. Xu, L. Ouyang, X. Yang, W. Mao, J. Su and Y. Han, J. Catal., 2012, 287, 114 CrossRef CAS.
- F. Gao, S. M. McClure, Y. Cai, K. K. Gath, Y. Wang, M. S. Chen, Q. L. Guo and D. W. Goodman, Surf. Sci., 2009, 603, 65 CrossRef CAS.
- A. Biabani-Ravandi and M. Rezaei, Chem. Eng. J., 2012, 184, 141 CrossRef CAS.
- H. Einaga, N. Urahama, A. Tou and Y. Teraoka, Catal. Lett., 2014, 144, 1653 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |