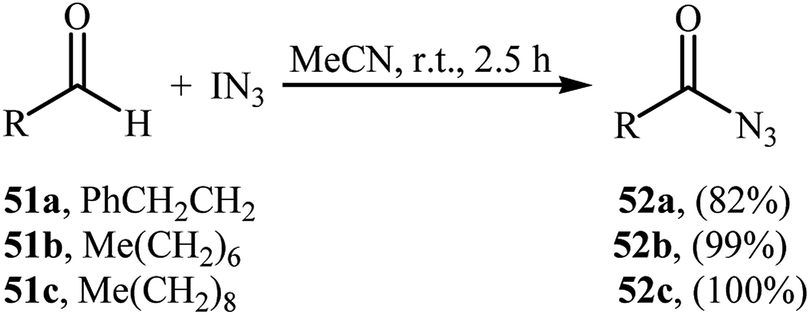DOI:
10.1039/C9RA04534A
(Review Article)
RSC Adv., 2019,
9, 25199-25215
Methods for direct C(sp2)–H bonds azidation
Received
17th June 2019
, Accepted 30th July 2019
First published on 13th August 2019
Abstract
Direct functionalization of C–H bonds has attracted great attention in recent years from the perspectives of atom and step economy. In this context, a variety of processes have been developed for the construction of synthetically and biologically important organic azides through the oxidative C–H bonds azidation. In this review, we have summarized recent progress in the direct azidation of C(sp2)–H bonds. The review is divided into three major sections: (i) direct azidation of aromatic C–H bonds; (ii) direct azidation of olefinic C–H bonds; and (iii) direct azidation of aldehydic C–H bonds. Mechanistic aspects of the reactions are considered and discussed in detail.
 Leila Youseftabar-Miri | Leila Youseftabar-Miri was born in Babol, Iran, in 1979. She received her B.S. degree in pure chemistry from University of Mazandaran, Iran, and her M.S. degree in organic chemistry from Zanjan University, Zanjan, Iran, in 2006 under the supervision of Prof. A. Ramazani. She completed her PhD degree in 2011 under the supervision of Prof. K. Rad-Moghadam. Now, she is working at Tehran Medical Sciences, Islamic Azad University as associate professor. Her research interests include organic synthesis, nano chemistry and nanotechnology. |
 Akbar Hassanpour | Akbar Hassanpour was born in Tabriz, Iran, in 1978. He received his B.S. degree in pure chemistry from Tabriz Islamic Azad University, Iran, and his M.S. degree in organic chemistry from Tabriz University, Tabriz, Iran, in 2004 under the supervision of Prof. Kazem D. Safa. He completed his PhD degree in 2008 under the supervision of Prof. Kazem D. Safa. He was a postdoctoral fellow in the lab of Prof. W. A. Stanczyk at the Centre of Molecular and Macromolecular Studies in Łódź, Poland. Now he is working at Islamic Azad University as assistant professor. His research interests include organometallic and organic synthesis, and new methodologies in nano material synthesis. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor of Organic Chemistry. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
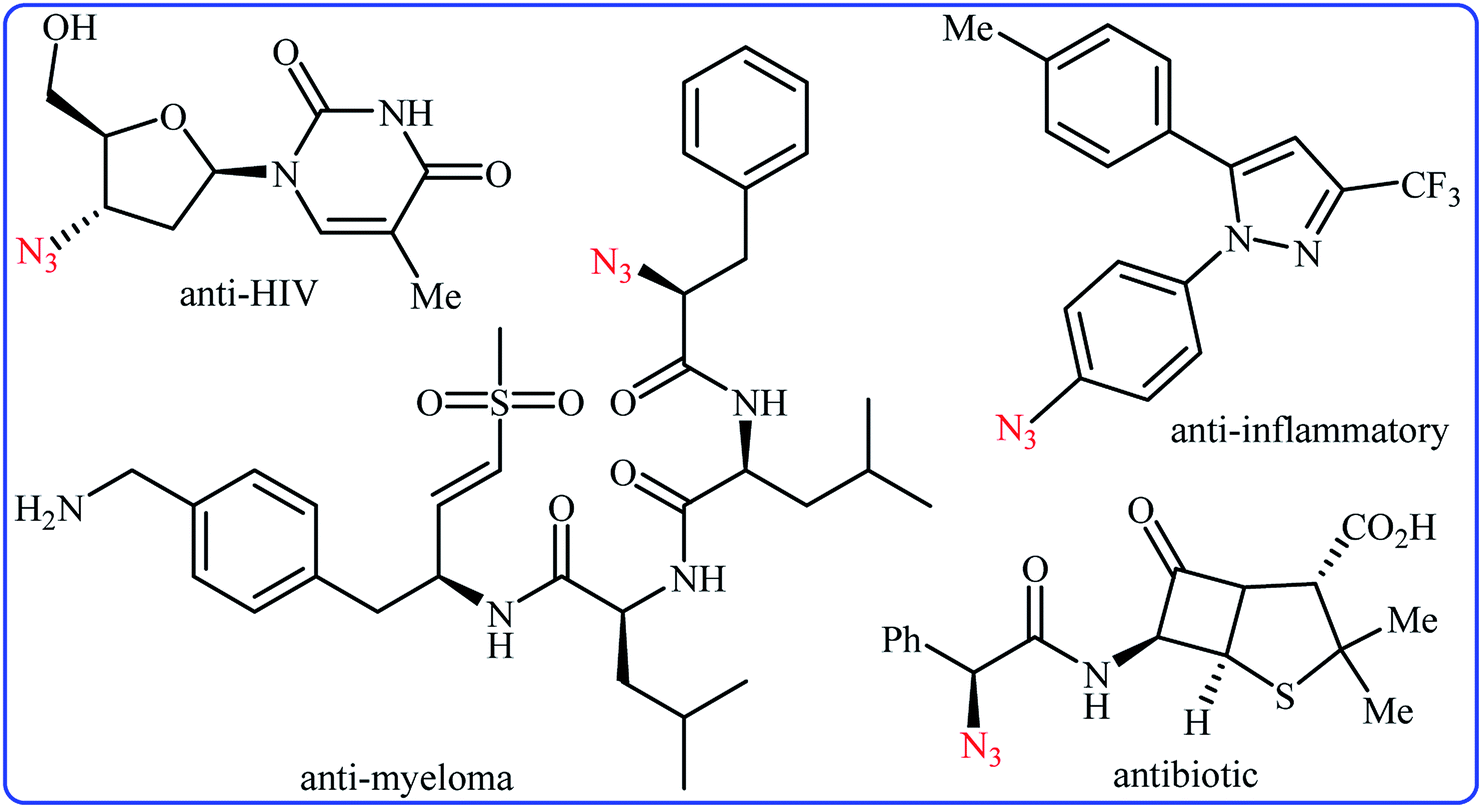
Needless to say, organic azides are among the most important and versatile intermediates in organic chemistry which are diversely utilized in the synthesis of various nitrogen-containing heterocycles,1 amines,2 amides,3 and imines4 owing to the unique chemical reactivity of the azido functionality. Moreover, compounds bearing this valuable functional group exhibit a variety of biological activities (Scheme 1), such as anti-HIV,5 anti-inflammatory,6 antibiotic,7 and anti-myeloma8 activities. Due to the wide importance of the title compounds in organic and medicinal chemistry, a number of methods for their preparation were developed. The most commonly used methods involve: (i) coupling of organometallic reagents with azido compounds;9 (ii) carbon-nitrogen coupling reaction between organic halides and azido compounds;10 and (iii) decarboxylative azidation of carboxylic acids.11 Despite their prevalence, these approaches not only require the pre-functionalized starting materials but also produce hazardous waste streams. Therefore, it is of considerable importance to develop straightforward and truly efficient procedures for the fabrication of organic azides without pre-activation of starting materials.
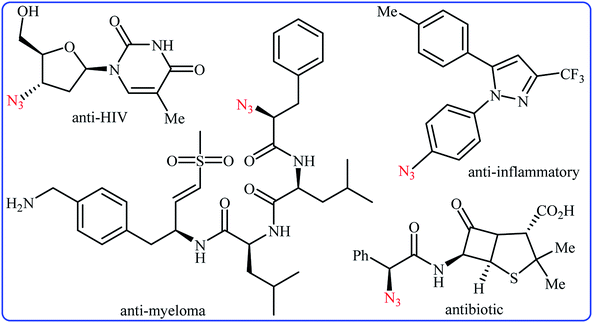 |
| | Scheme 1 Selected examples of biologically active molecules bearing azido group. | |
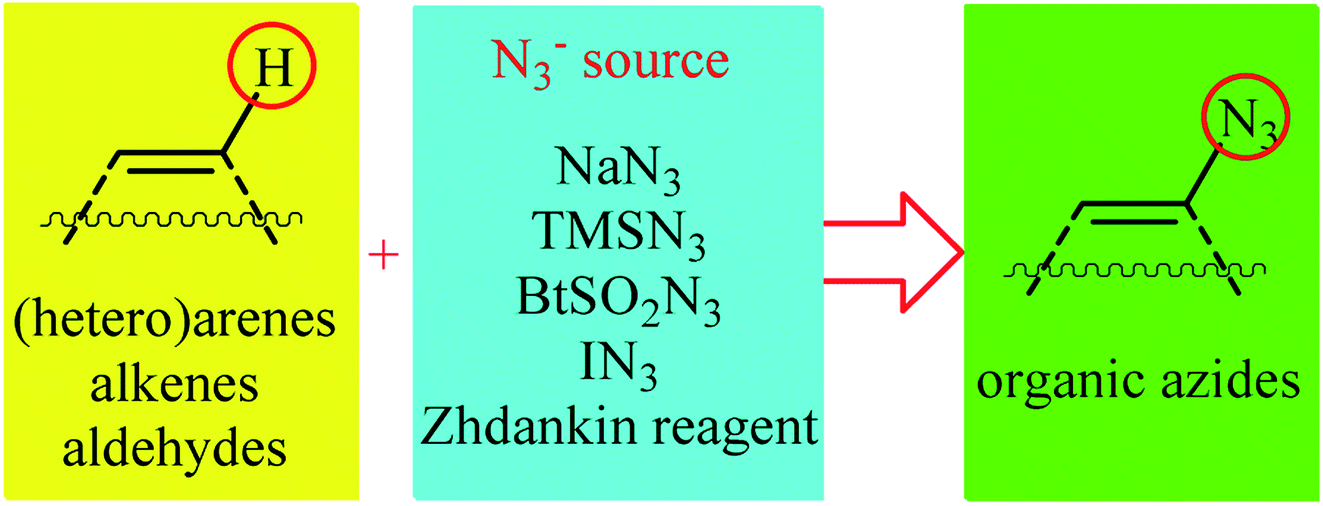
Direct functionalization of C–H bonds represents a powerful strategy for the rapid construction of diverse C–C and C–X (X= heteroatom) bonds due to its inherent efficiency.12 In contrast to traditional methods which rely on the use of pre-functionalized (often halogenated) starting materials, in this strategy the inactive C–H bonds act as a functional group, thereby the number of synthetic steps and chemical wastes are considerably reduced. Among various C–H bond functionalization reactions, direct C–N3 bond forming reactions have attracted much attention in recent years. In connection with our recent review papers on the direct functionalization of C–H bonds13 and cross-coupling reactions,14 we summarize here a variety of protocols for the direct oxidative C(sp2)–H bonds azidation reactions (Fig. 1). The review is divided into three major sections. The first section focuses exclusively on direct azidation of aromatic C–H bonds. The second will cover azidation of olefinic C–H bonds. The third section will discuss azidation of aldehydic C–H bonds. Of note, we have not discussed azidation of aliphatic C–H bonds, since it has recently been described in another publication.15
 |
| | Fig. 1 Direct C(sp2)–H bonds azidation. | |
2. Aromatic C–H bonds
In the present section, we'll describe the available literature on the direct azidation of (hetero)aromatic C–H bonds. For clarity, the reactions are classified by the type of azidation reagents (e.g., NaN3, TMSN3, Zhdankin reagent).
2.1. NaN3 as azide source
This subsection considers direct C–H azidation of (hetero)arene substrates utilizing inexpensive, easily available and stable sodium azide (NaN3) as the azide source.
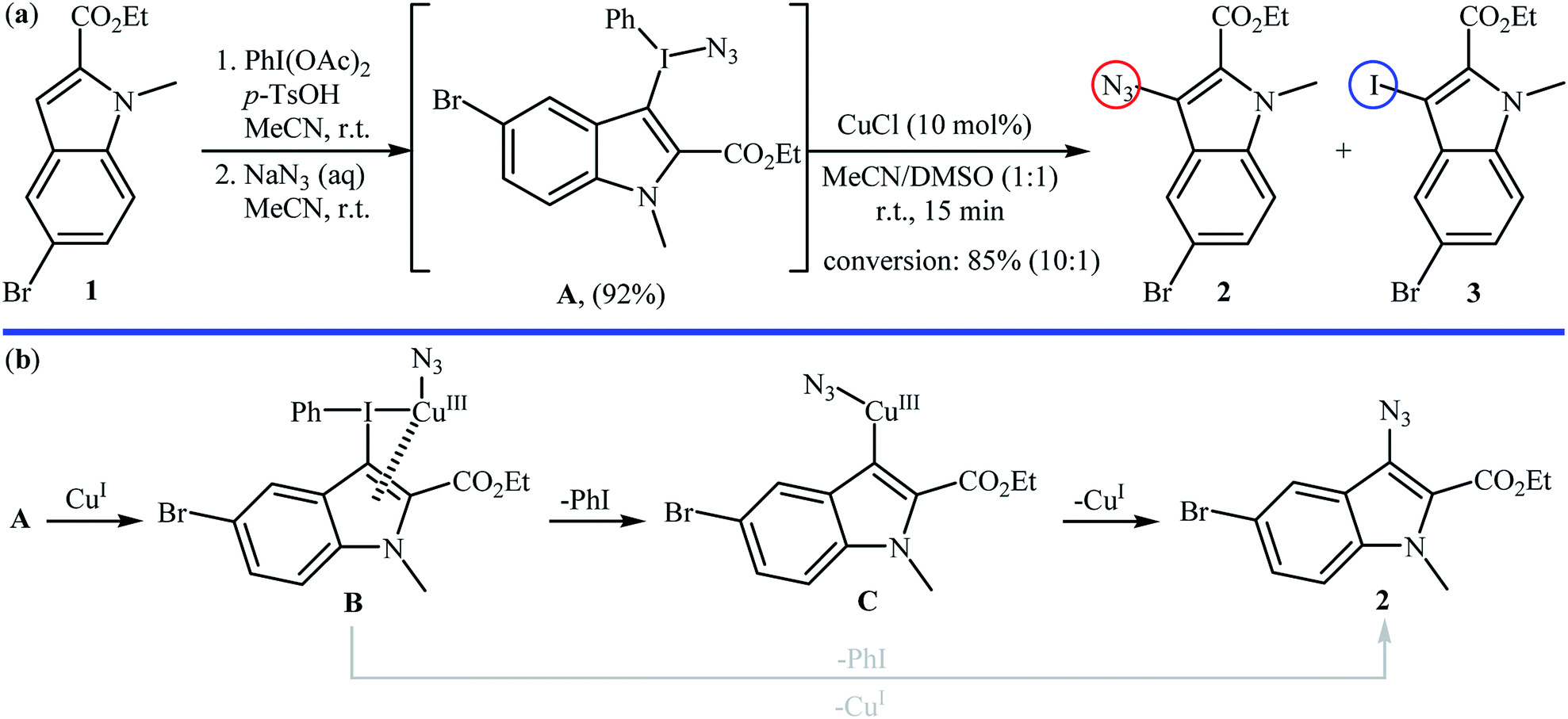
2.1.1. Metal-catalyzed reactions. Metal-catalyzed direct aromatic C–H bonds azidation using NaN3 as the azide source was first demonstrated by Suna and co-workers,16 where a functionalized indolyl azide 2 was achieved in good yield by the reaction of corresponding indole 1 with a mixture of PhI(OAc)2, TsOH, and NaN3 in MeCN followed by regioselective fragmentation of the resulted indolyl(phenyl)iodonium azide A via the treatment with a catalytic amount of CuCl in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeCN and DMSO at room temperature (Scheme 2a). The results proved that the choice of CuCl was critical to the success of this reaction and other metal salts (e.g., Pd(OAc)2, Zn(OTf)2, Sc(OTf)3, (Ph3P)AuCl) almost failed to give the target product. In the absence of catalyst, fragmentation of iodonium salt A is controlled by electronic factors, as evidenced by the formation of iodoindole 3 as the main product. The authors proposed the mechanistic pathway for the conversion of iodonium salt A into indolyl azide 2 shown in Scheme 2b that involves the initial formation of Cu(III) species B through the oxidative addition of this intermediate A to Cu(I) salts, which undergoes regioselective transformation to PhI and heteroarylcopper(III) species C. Finally, reductive elimination of intermediate C leads to the expected indolyl azide 2 and regenerate the Cu(I) species. In another possibility, complex B can directly collapse into azide product 2 via the highly regioselective coupling of the heterocycle with the azide.
:1 mixture of MeCN and DMSO at room temperature (Scheme 2a). The results proved that the choice of CuCl was critical to the success of this reaction and other metal salts (e.g., Pd(OAc)2, Zn(OTf)2, Sc(OTf)3, (Ph3P)AuCl) almost failed to give the target product. In the absence of catalyst, fragmentation of iodonium salt A is controlled by electronic factors, as evidenced by the formation of iodoindole 3 as the main product. The authors proposed the mechanistic pathway for the conversion of iodonium salt A into indolyl azide 2 shown in Scheme 2b that involves the initial formation of Cu(III) species B through the oxidative addition of this intermediate A to Cu(I) salts, which undergoes regioselective transformation to PhI and heteroarylcopper(III) species C. Finally, reductive elimination of intermediate C leads to the expected indolyl azide 2 and regenerate the Cu(I) species. In another possibility, complex B can directly collapse into azide product 2 via the highly regioselective coupling of the heterocycle with the azide.
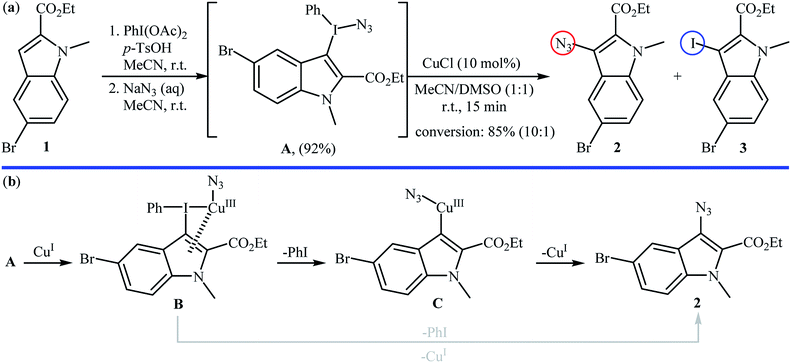 |
| | Scheme 2 (a) Suna's synthesis of indolyl azide 2; (b) mechanistic explanation of the synthesis of indolyl azide 2. | |
The procedure was nicely applied for the synthesis of a diverse range of 1,2,3-triazoles by 1,3-dipolar cycloaddition of in situ generated aizdes with acetylenes 5. As shown in Scheme 3, various electron-rich heterocycles 4 such as pyrroles, indoles, thienopyrroles, pyrrolopyridines, and pyrrolopyrimidines were applied in this transformation to give the corresponding 1,4-disubstituted 1,2,3-triazole products 6 in moderate to excellent yields. The authors also showed that in situ generated heteroaryl azides can be conveniently reduced to heteroarylamines by aqueous ammonium sulfide at room temperature.
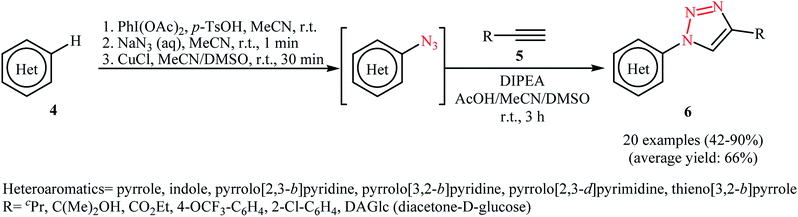 |
| | Scheme 3 . One-pot multistep synthesis of 1,4-disubstituted 1,2,3-triazoles 6. | |
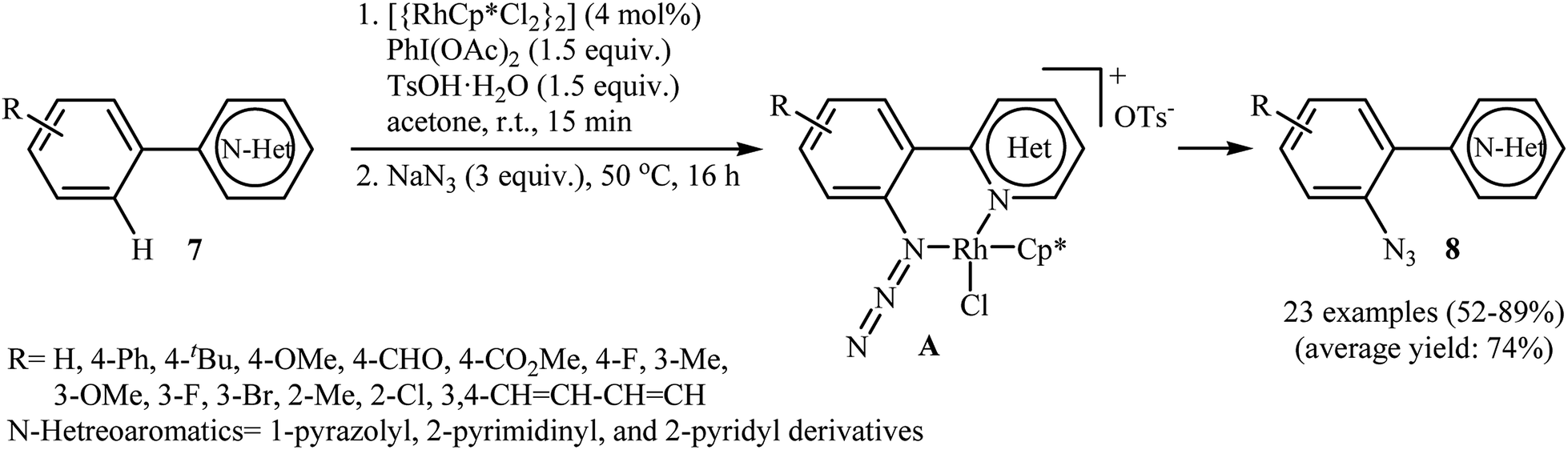
Inspired by this work, Li and colleagues developed an interesting direct ortho-selective C–H azidation of arene substrates bearing coordinating directing groups, such as 2-pyridyl, 2-pyrimidinyl, and 1-pyrazolyl using NaN3 as the azide source.17 Thus, in the presence of [{RhCp*Cl2}2] as a commercially available but expensive catalyst, PhI(OAc)2 as an oxidant and TsOH as an additive, various arene substrates 7 possessing electron-donating or electron-withdrawing functional groups underwent smooth o-azidation to afford the expected mono-azidated products 8 in 52–89% yield (Scheme 4). The authors suggested that the N,N chelation in intermediate A may account for the high selectivity of this reaction for mono-azidation. After evaluation of potential synthetic applicability of the prepared azido arenes, they found that simple heating (120 °C) of adducts in 1,4-dioxane for 10 h afforded the corresponding pyrido[1,2-b]indazole derivatives in almost quantitative yields. They also successfully expended their oxidative conditions to direct C–H nitration of a library of (hetero)arenes with NaNO2 as a readily available nitro source. Recently, density functional theory (DFT) calculations indicated that the active Rh(III) catalyst is oxidized to Rh(V) by trivalent iodine (PhIOAc+).18 Thus it was suggested that the reaction proceeds through an oxidation/electrophilic deprotonation/coordination/reductive elimination sequential process.
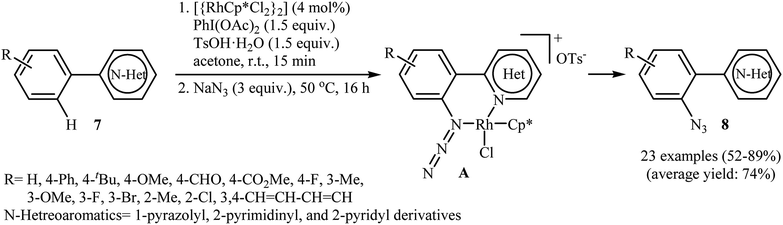 |
| | Scheme 4 Rh-catalyzed direct ortho-selective C–H azidation of arenes 7 with NaN3. | |
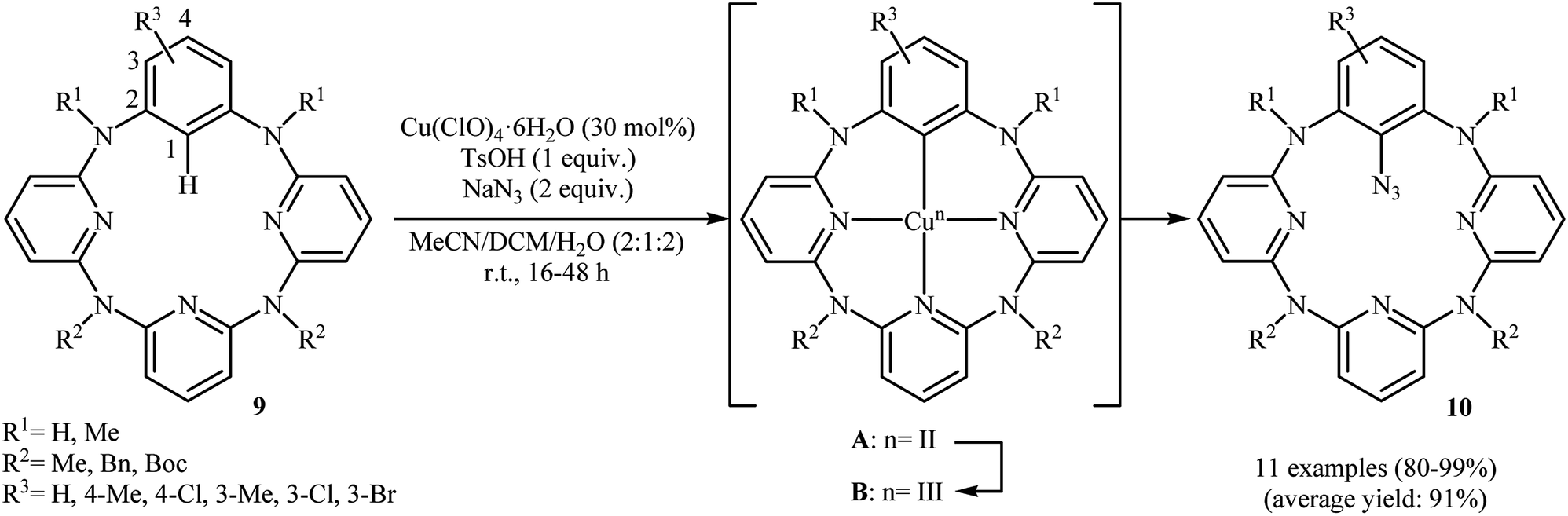
Shortly afterwards, an elegant Cu(II)-catalyzed direct azidation of azacalix[1]arene[3]pyridine derivatives with NaN3 was reported by Wang et al.19 The Cu(ClO)4·6H2O/TsOH/MeCN/DCM system was found to be optimal for this transformation, while addition of an small amount of water gave excellent results. The authors declared that water prevented precipitation of copper salts from the reaction mixture. These reaction were run under aerobic conditions at ambient temperature, tolerated various NH-free and N-protected azacalix[1]arene[3]pyridines 9, and generally provided mono-azidated products 10 in high to quantitative yields (Scheme 5). It should be mentioned that the reaction of NaN3 with isolated arylcopper(II) A and arylcopper(III) B compounds indicated clearly that the arylcopper(III) B was an active species in this azidation reaction. Thus, an unprecedented Cu(II)–ArCu(II)–ArCu(III)–Cu(I) catalytic cycle was proposed for this synthetic strategy. Just like previous works, the synthesized azido compounds were successfully utilized as starting materials in the fabrication of amino- and 1,2,3-triazole-bearing azacalix[1]-arene[3]pyridines.
 |
| | Scheme 5 Cu(II)-catalyzed direct azidation of azacalix[1]arene[3]pyridines 9 with NaN3. | |
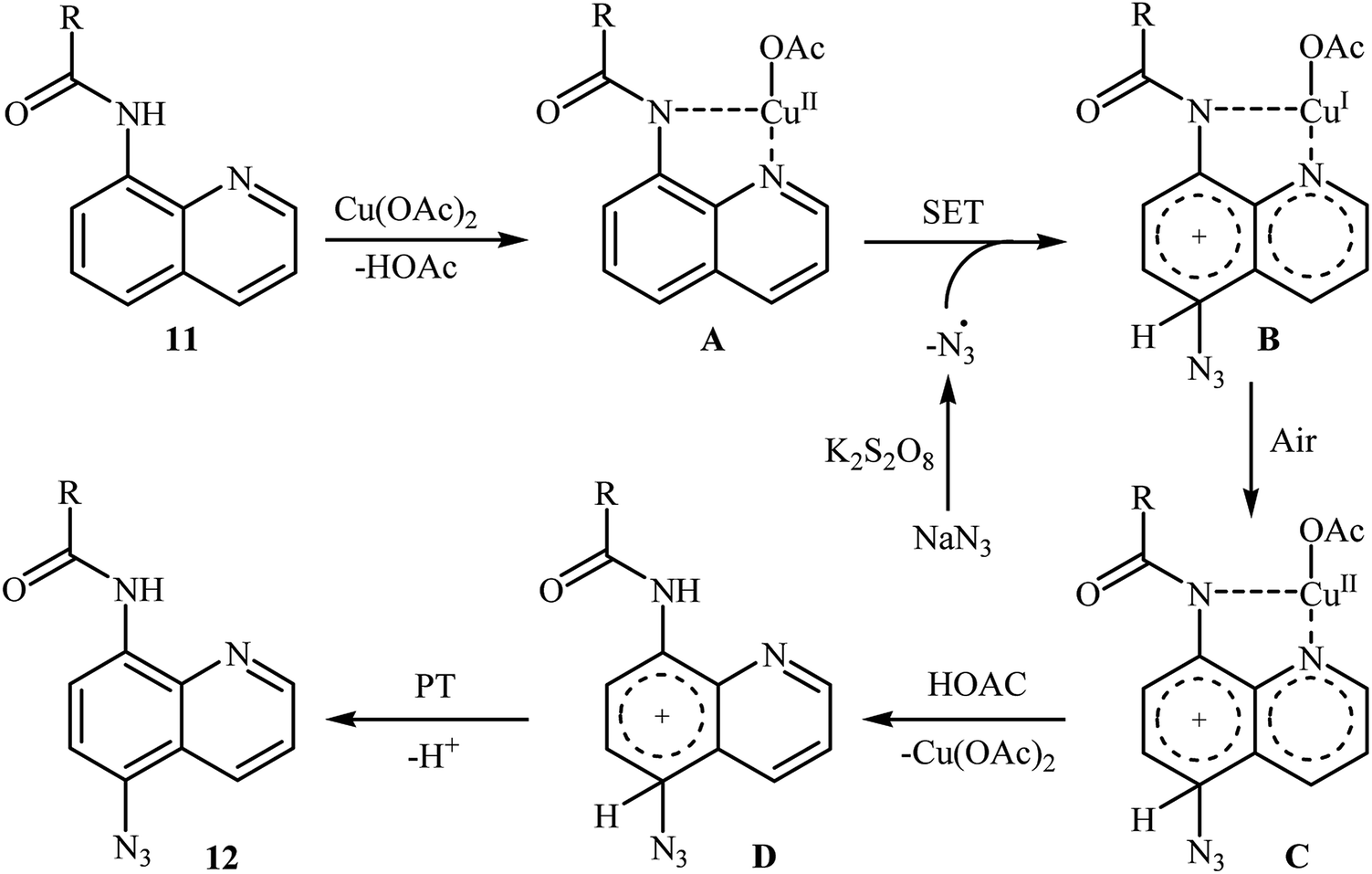
In 2016, the group of Zhu investigated Cu-catalyzed direct C–H azidation of N-acylated 8-aminoquinolines with NaN3.20 A careful screening of catalysts, oxidants, additives, and solvents resulted in the optimized reaction conditions shown in Scheme 6. Under these conditions, azidation took place selectively at the 5-position of various substituted 8-aminoquinolines 11 to form C5-azidated 8-aminoquinolines 12 in almost moderate to high yields. However, the reaction failed with the C-2 substituted quinolines due to the failure formation of Cu(II) complex coming from the steric hindrance of the ortho-substituted groups. The control experiments revealed that both amide N–H and quinoline nitrogen were indispensable for this reaction. Based on a series of other control experiments, the authors suggested that the reaction starts with the generation of an aryl-CuII complex A through the coordination of Cu(OAc)2 to the nitrogen atoms of 8-aminoquinoline 11, which is followed by reaction with the azide radical, which is generated in situ from the NaN3 and K2S2O8, to give the intermediate B via a single electron transfer (SET) process. Subsequently, oxidation of CuI complex B under air leading to the formation of CuII complex C that, after a metal dissociation process affords the intermediate D and regenerates the catalyst Cu(OAc)2. Finally, a proton transfer process from D delivers the target C5-azidated 8-aminoquinoline 12 (Scheme 7).
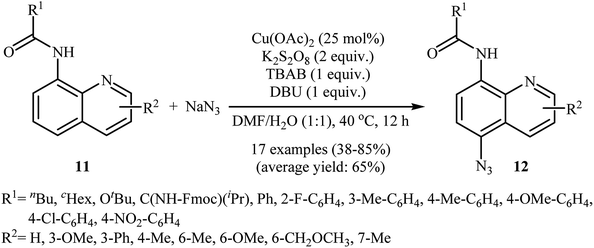 |
| | Scheme 6 Regioselective azidation of N-acylated 8-aminoquinolines 11 with NaN3. | |
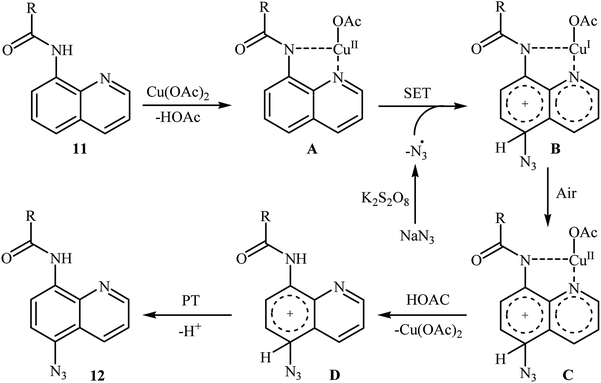 |
| | Scheme 7 The proposed mechanism for the formation of C5-azidated 8-aminoquinolines 12. | |
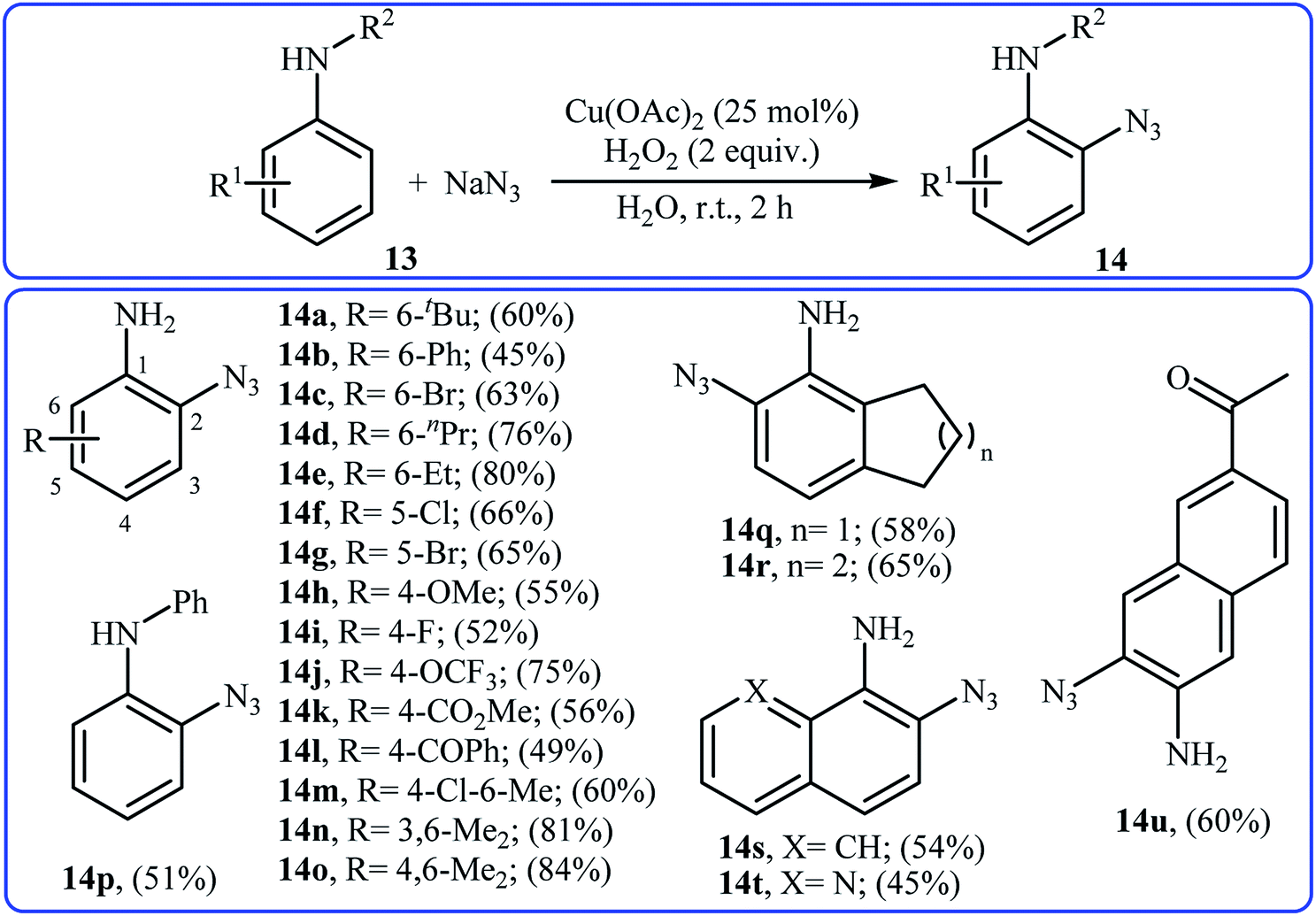
Recently, the same group developed an interesting Cu-catalyzed free amino group-directed C–H bond azidation of aromatic amines with NaN3.21 Thus, various electron-rich and electron-poor anilines 13 in the presence of 25 mol% of Cu(OAc)2 and 2 equiv. of H2O2 in neat water underwent ortho-selective azidation and afforded the target aryl azides 14 in moderate to high yields (Scheme 8). The reaction is noteworthy in that both primary and secondary anilines are tolerated. This protocol was also successfully applied to late-stage functionalization of aniline-based drugs, such as procaine, benzocaine, and aminoglutethimide. Mechanistically, a single electron transfer process was likely involved in this transformation, which could be completely inhibited by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), a radical scavenger.
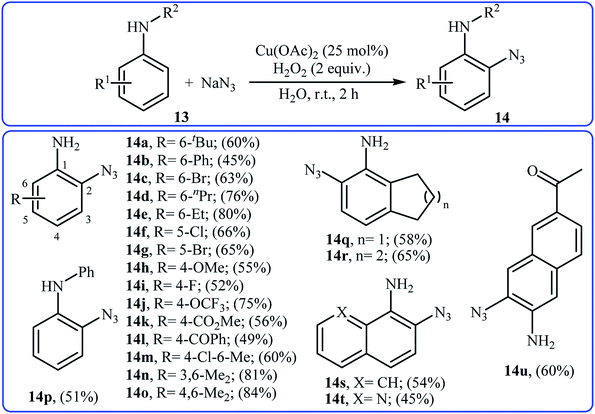 |
| | Scheme 8 ortho-Selective azidation of anilines 11 with NaN3. | |
2.1.2. Metal-free reactions. In 2016, Sudalai and colleagues reported an efficient I2-mediated regioselective C3 azidation of indole derivatives 15 with NaN3 under metal-free conditions at room temperature.22 1.0 equiv. molecular iodine and 1.0 equiv. Et3N in DMSO delivered the 3-azido indole products 16 in moderate to excellent yield (Scheme 9). The protocol was highly tolerable to the substrates bearing both electron-donating and -withdrawing substituents. The results showed that NH-free substrates furnished better yields compared to N-substituted indoles. The authors claimed that their methodology was the first example of the direct azidation of indoles at C-3 position. Under similar conditions, the azidation of a 1H-indazole substrate was also proceeded effectively to afford the corresponding 3-azido-1H-indazole in moderate yield with excellent regioselectivity. Mechanistic study revealed that the reaction starts with the formation of an iodonium ion intermediate A by reaction between starting indole 15 and I2. Next, its ring opening with azide anion (from the preferred benzylic position) leads to iodo-azide intermediate B that after HI elimination in the presence of base, affords the target 3-azido indoles 16 (Scheme 10).
 |
| | Scheme 9 I2-mediated C3–H azidation of indoles 15. | |
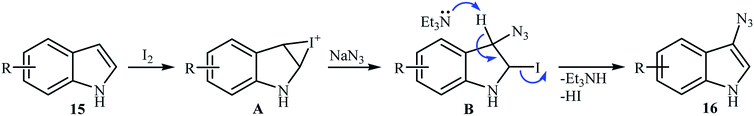 |
| | Scheme 10 Mechanistic pathway for the formation of 3-azido indoles 16. | |
2.2. TMSN3 as azide source
Significant progress has been made in the azidation of organic molecules using trimethylsilyl azide (TMSN3) as a versatile, commercially available azidating reagent in the past decades.23 In this sub-section, we wish to summarize the recent developments in the direct azidation of (hetero)aromatic C–H bonds utilizing the titled reagent.
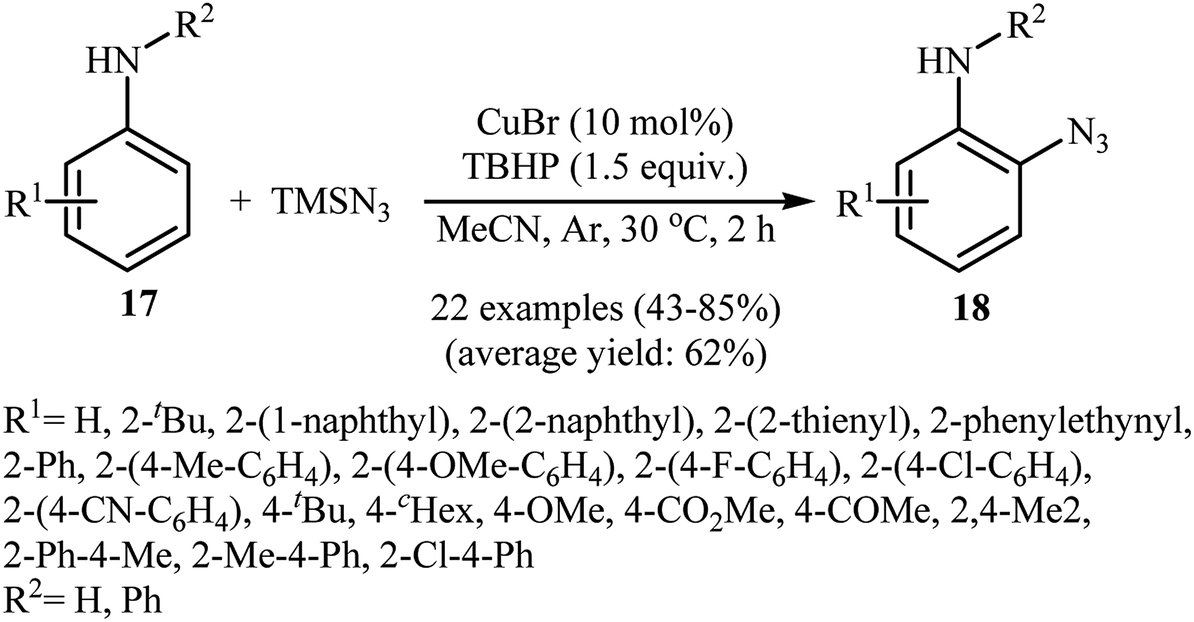
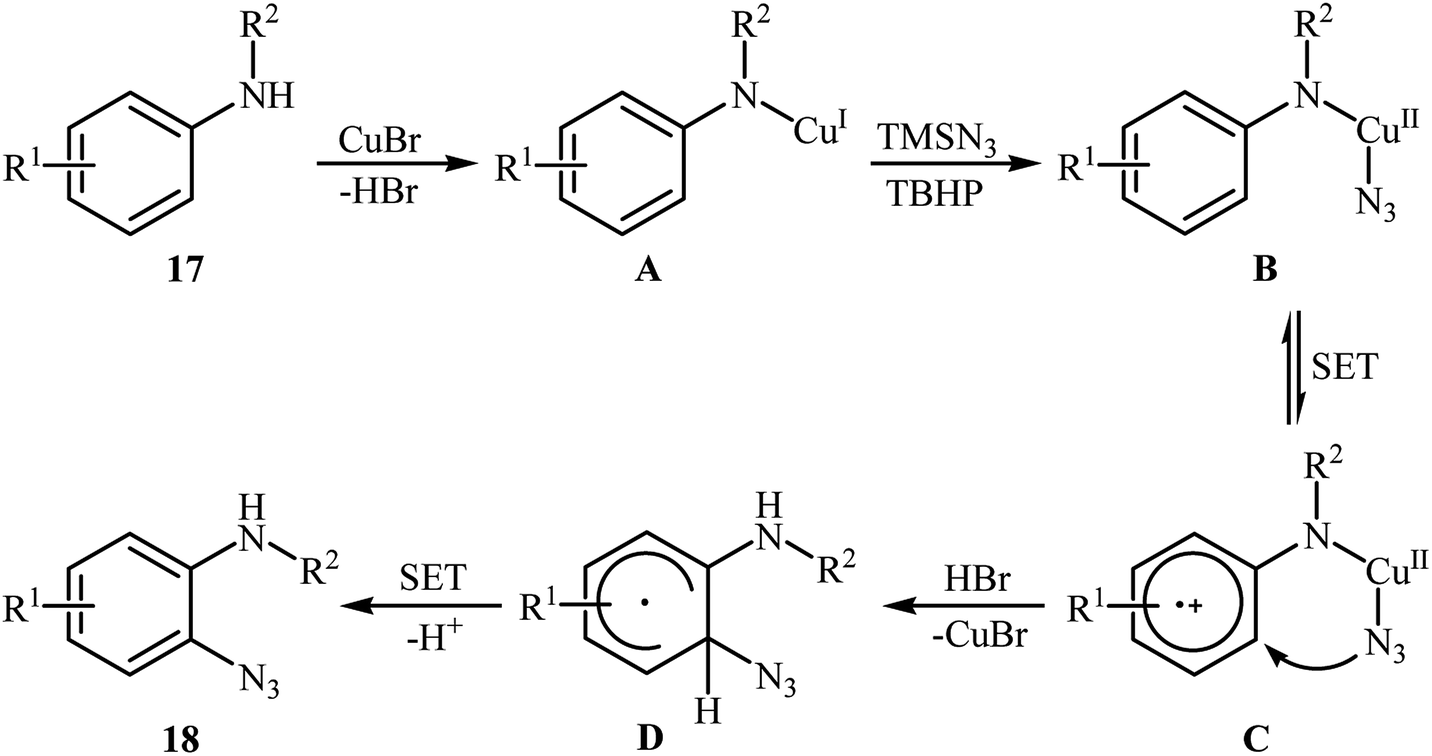
2.2.1. Metal-catalyzed reactions. In 2012, Tang and Jiao developed a copper-catalyzed ortho-directed azidation of anilines 17 with TMSN3 under rather mild conditions to provide 2-azidobenzenamines 18 in moderate to good yields (Scheme 11).24 The optimal conditions of the reaction involve the use of CuBr (10 mol%) as the catalyst and tert-butyl hydroperoxide (TBHP) as the oxidant. Among the various solvents like toluene, benzene, ethyl acetate, DMF, DCE, MeCN, MeNO2; MeCN proved to be the most efficient for this transformation. The established conditions exhibited very good functional group tolerance and regioselectivity. However, in the cases of para-substituted anilines, both mono- and di-azidated products were obtained. This protocol was compatible with primary and secondary amines, but not tertiary amines. According to the authors, the amino group was responsible for ortho C–H bond activation by coordinating with the metal center. A plausible mechanism for this C–N bond forming reaction involves the following steps (Scheme 12): (i) coordination of copper to the amino group of the substrate 17 to form intermediate A; (ii) combination of intermediate A with an azide radical, generated in situ from TMSN3 and TBHP, to yield intermediate B; (iii) single electron transfer from the aryl ring of the intermediate B to the metal center to afford intermediate C; (iv) migration of the azido group into the aryl ring to give intermediate D; and (v) deprotonation of intermediate D via SET process to afford the final product 18. Three years later, Lhoták and co-workers applied the similar protocol for the regioselective meta C–H azidation of calix[4]arene.25 Utilizing the same catalytic system in toluene (r.t., 15 min), the desired meta-monoazidated calix[4]arene was obtained in 52% yield.
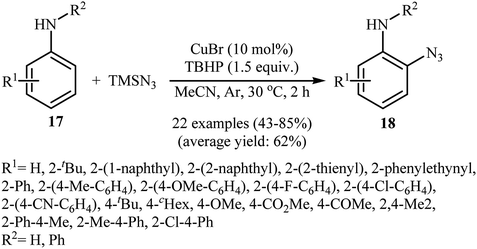 |
| | Scheme 11 Cu-catalyzed ortho-directed azidation of anilines 17 with TMSN3. | |
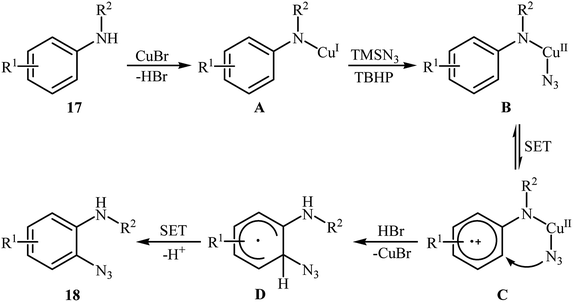 |
| | Scheme 12 Plausible mechanism for Cu(I)-catalyzed azidation of anilines 17 with TMSN3. | |
Very recently, the group of Prabhu realized a similar process which extended the scope to indoles.26 Thus, a variety of C2-azidated indoles 20 were prepared by treatment of indoles 19 bearing an ester or ketone moiety at the C3-position with TMSN3 in the presence of CuBr/TBHP combination as a catalytic system at room temperature (Scheme 13). Interestingly, when the substitutions on the C3-position of indoles were less radical stabilizing groups, such as an alkyl or amide groups, the reaction preferred to furnish the 3-azidooxindole products. Noteworthy, the replacement of CuBr by molecular iodine afforded better yields of 3-azidooxindole products 21. In this investigation, the authors found some limitation in their protocol when they attempted to react N-methylindole and 3-methylindole with TMSN3 under the standard reaction conditions (either CuBr or I2 catalysis conditions). In these cases, no desired product was observed. The application of both procedures to gram-scale synthesis was reported showing the scalability of these transformations.
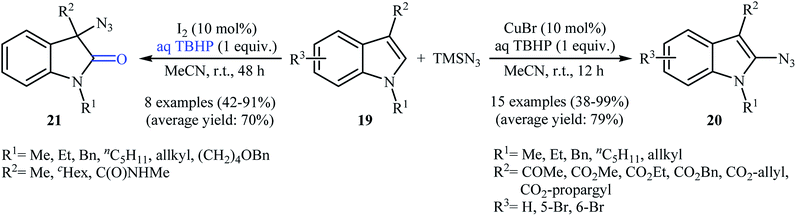 |
| | Scheme 13 Substituent-directed regioselective azidation of indoles 19 with TMSN3. | |
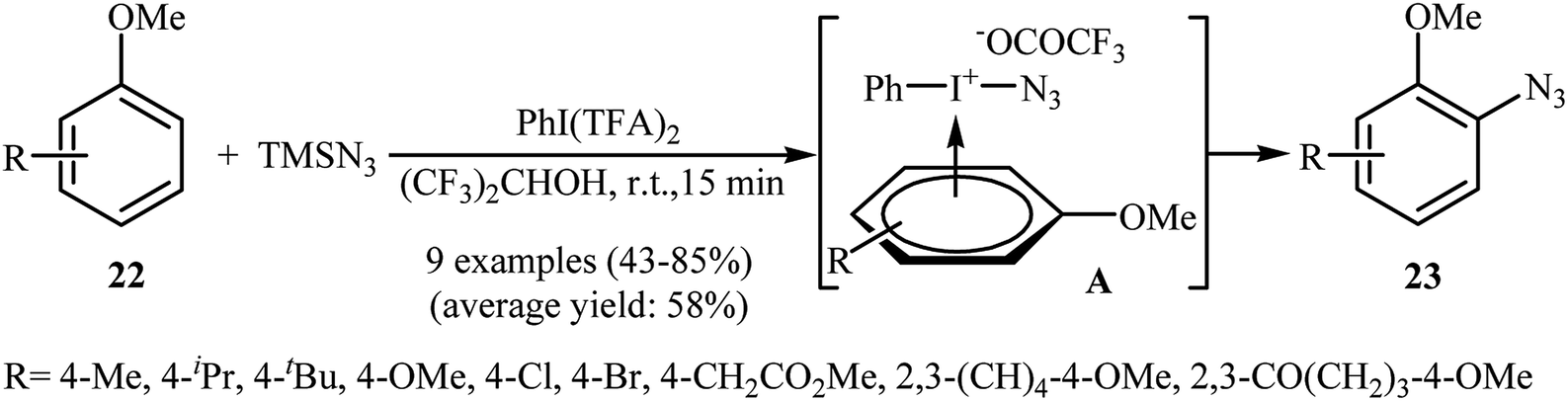
2.2.2. Metal-free reactions. In 1991, Kita and co-workers successfully accomplished a hypervalent iodine reagent, phenyliodine(III) bis(trifluoroacetate) PhI(TFA)2, mediated direct azidation of anisole derivatives 22 with TMSN3, presenting the first example of metal-free aromatic C–H bonds azidation utilizing TMSN3.27,28 They found that the choice of solvent was critical to the success of this reaction and polar protic solvents, such as 1,1,1,3,3,3-hexafluoro-2-propanol proved to be the best choice, giving the desired mono-azidated products 23 in moderate to high yields (Scheme 14). It was suggested that the π-complex A was the key intermediate in this transformation. Eighteen year later, Tsarevsky applied this strategy to azidation of polystyrene by using PhI(OAc)2 as a hypervalent iodine reagent.29 The amount of azide groups in the products was estimated to be about 1 in every 11 styrene units.
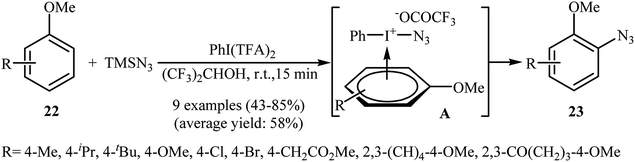 |
| | Scheme 14 PhI(TFA)2-mediated azidation of anisoles 22 with TMSN3. | |
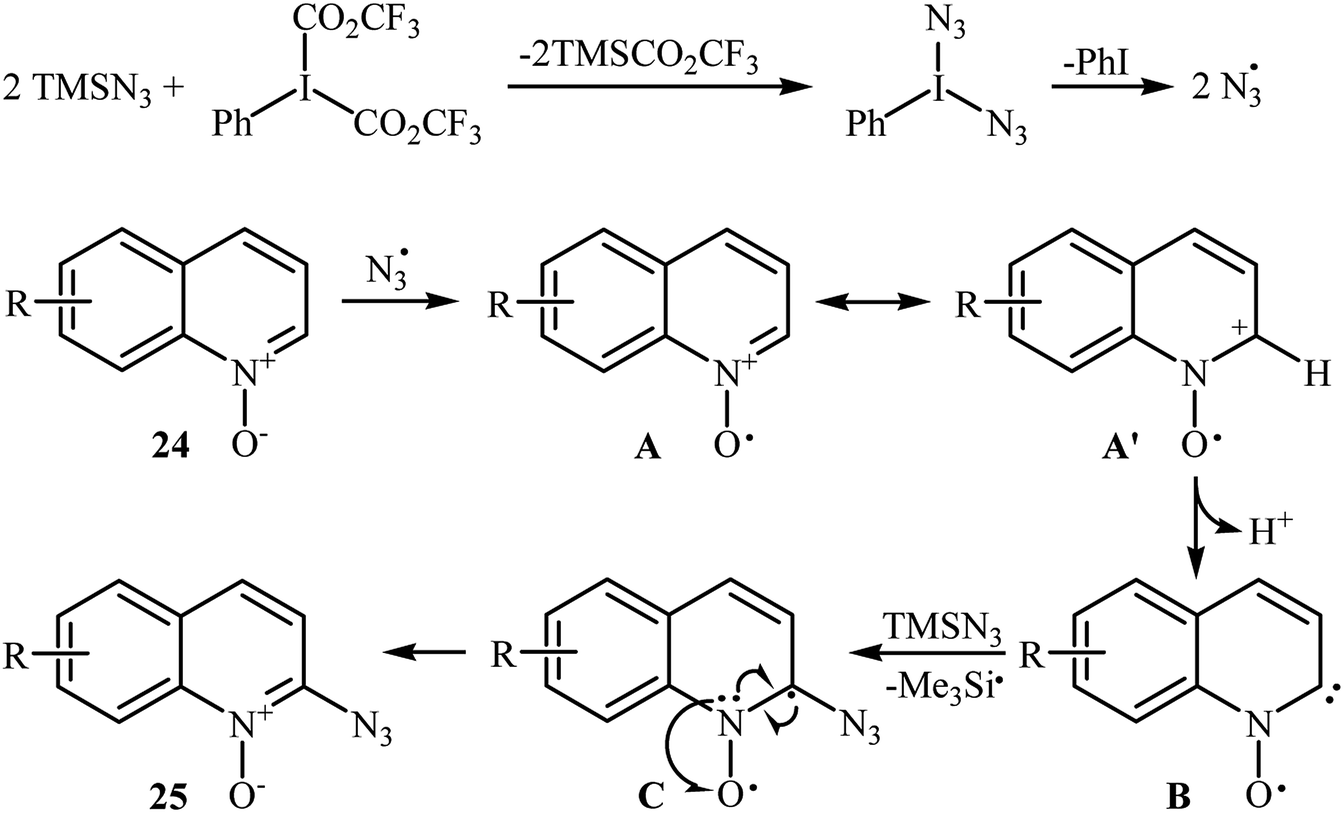
This azidation reaction was nicely extended in 2015 to quinoline N-oxides by Li et al.30 They showed that functionalized quinoline N-oxides 24 can be preferentially azidated at the C2-position with TMSN3 in EtOAc at room temperature (Scheme 15). This very fast reaction gave moderate to almost quantitate yields with a broad substrate scope. However, 2-substituted quinoline N-oxides failed to give the expected products under this reaction condition. Furthermore, isoquinoline N-oxide and pyridine N-oxide were incompatible in this reaction, indicating the importance of radical stabilization from the π-neighboring Ph group besides the other side of carbene. The mechanism of this azidation was proposed based on the control experiments and electron paramagnetic resonance (EPR) studies, determining that it proceeds through the following key steps (Scheme 16): (i) initial formation of two azide radicals via thermal hemolytic cleavage of in situ generated PhI(N3)2; (ii) single electron oxidation of quinoline N-oxide 24 with the help of the azide radical to give the radical cation A or A′; (iii) deprotonation of intermediate A (or A′) to afford the carbene stabilized N–O radical B; and (iv) single electron transfer from the radical B to TMSN3 to give the target product 25 through the intermediate C.
 |
| | Scheme 15 Metal-free regioselective C–H azidation of quinoline N-oxides 24 with TMSN3. | |
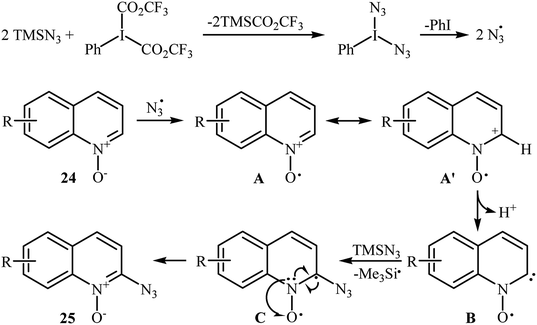 |
| | Scheme 16 Proposed mechanism for azidation of quinoline N-oxides 24 with TMSN3. | |
2.3. Azido-benziodoxolone as azide source
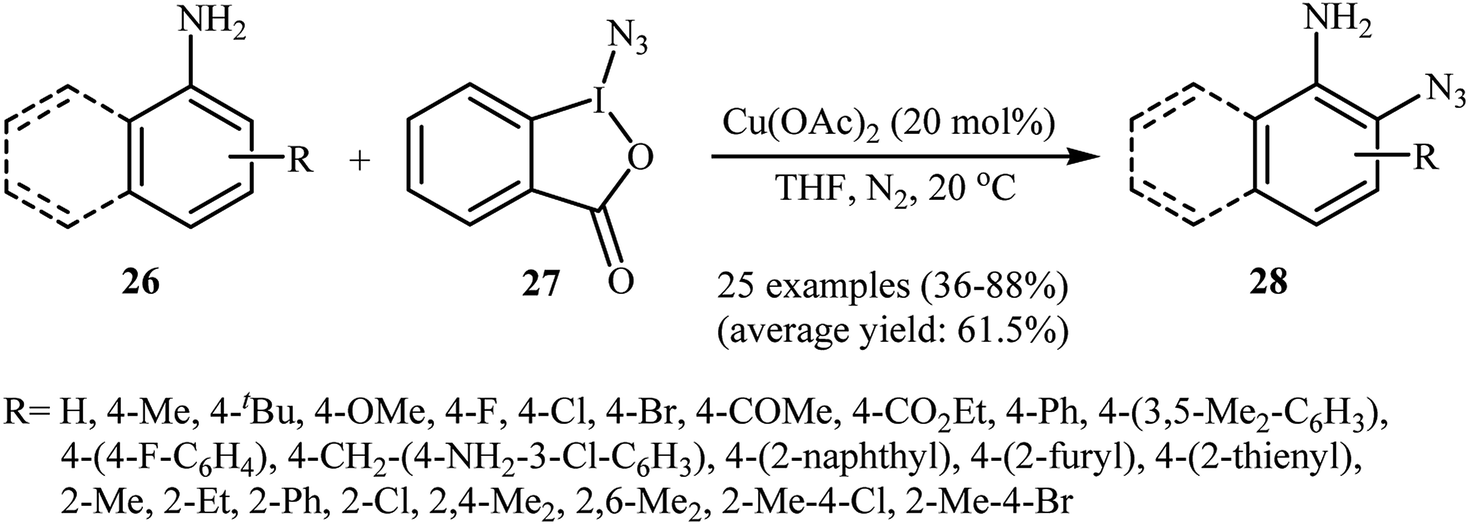
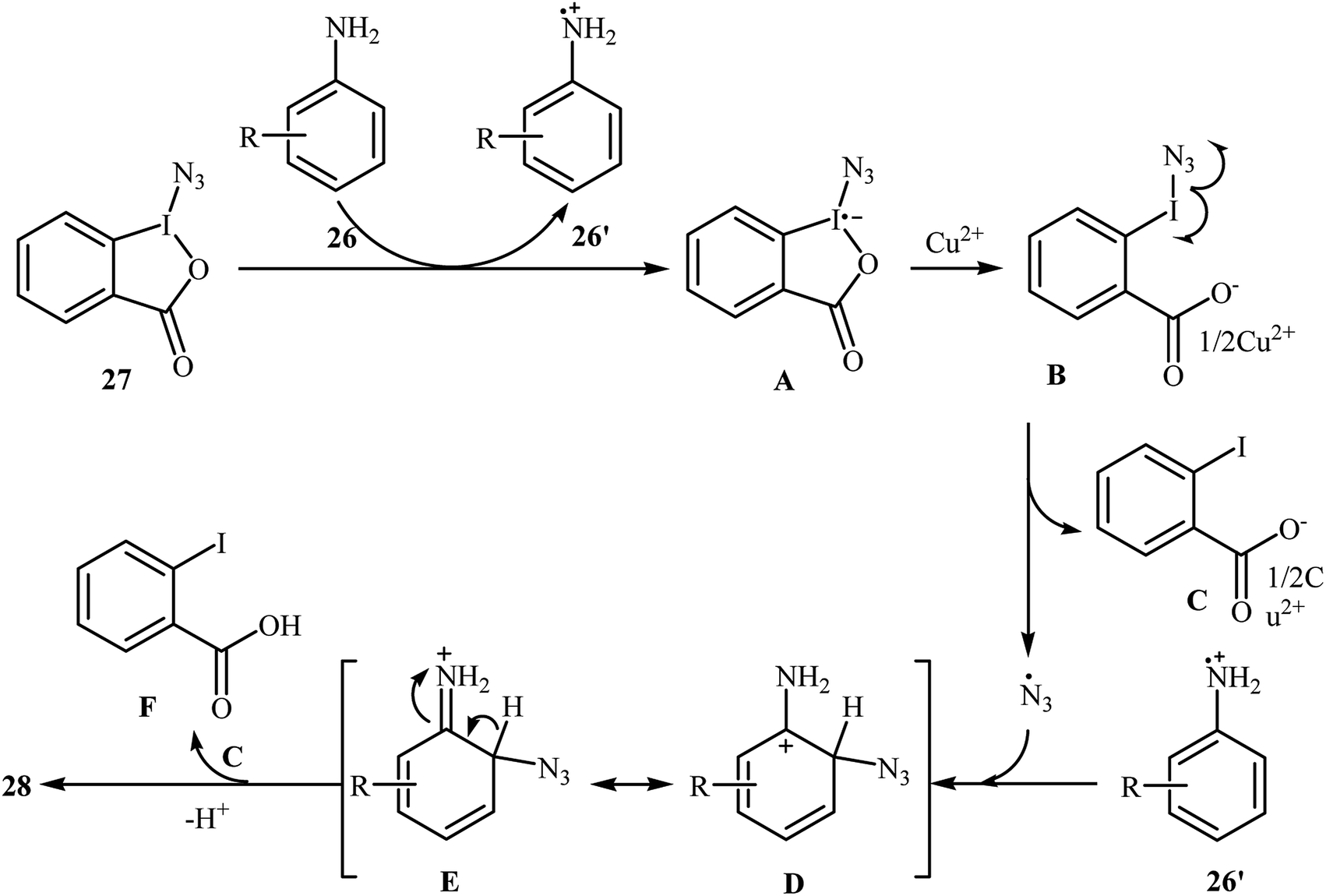
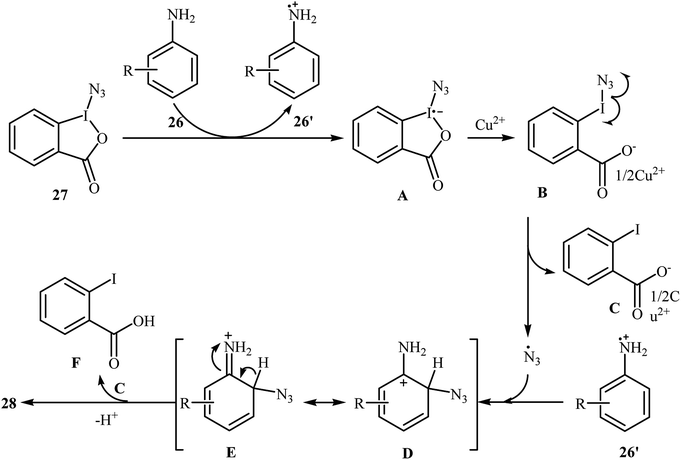
Azido-benziodoxolone (Zhdankin reagent) is a novel and valuable azidating reagent which is increasingly being used in the direct azidation of aliphatic, benzylic, olefinic, and aldehydic C–H bonds.31 However, recently it was found that this reagent has hazardous nature.32 The first protocol for the direct aromatic C–H bonds azidation utilizing Zhdankin reagent was reported by Hao et al. in 2014.33 Catalyzed by Cu(OAc)2, the reaction of aromatic amines 26 with azido-benziodoxolone 27 in THF furnished ortho-azido anilines 28 in almost moderate to high yields (Scheme 17). To compare the efficiency of this procedure to the Jiao's procedure (TMSN3, CuBr, TBHP, MeCN, 30 °C) it can be concluded that the former methodology is superior due to it was afforded higher yields of mono-azidated products with excellent regioselectivity even for para-substituted anilines. Scheme 18 shows the authors proposed mechanism for this transformation. Initially, one-electron oxidation of aniline 26 by oxidative hypervalent iodine(III) reagent 27 generates the aniline radical cation and radical anion A. Its N–I bond cleavage by the aim of Lewis acid leads to N3-containing 2-iodo-benzoate B that undergoes decomposition under the reaction conditions to form a copper(II) salt C and an azide radical. Subsequently, the N3 radical preferentially attacks the aromatic ring ortho to the primary amino group of the aniline radical cation to give the cyclohexadienyl cation species D which is in equilibrium with cation species E. Finally, deprotonation of E by 2-iodobenzoate C affords the target azidation product 28.
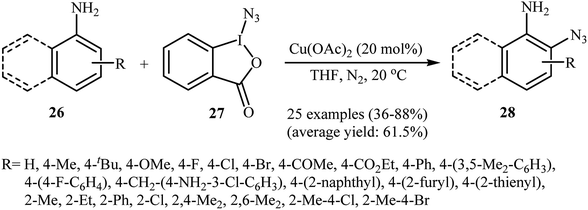 |
| | Scheme 17 Cu-catalyzed ortho-azidation of aromatic amines 26 with azido-benziodoxolone 27. | |
 |
| | Scheme 18 Proposed mechanism of the direct azidation of aromatic amines 26 with azido-benziodoxolone 27. | |
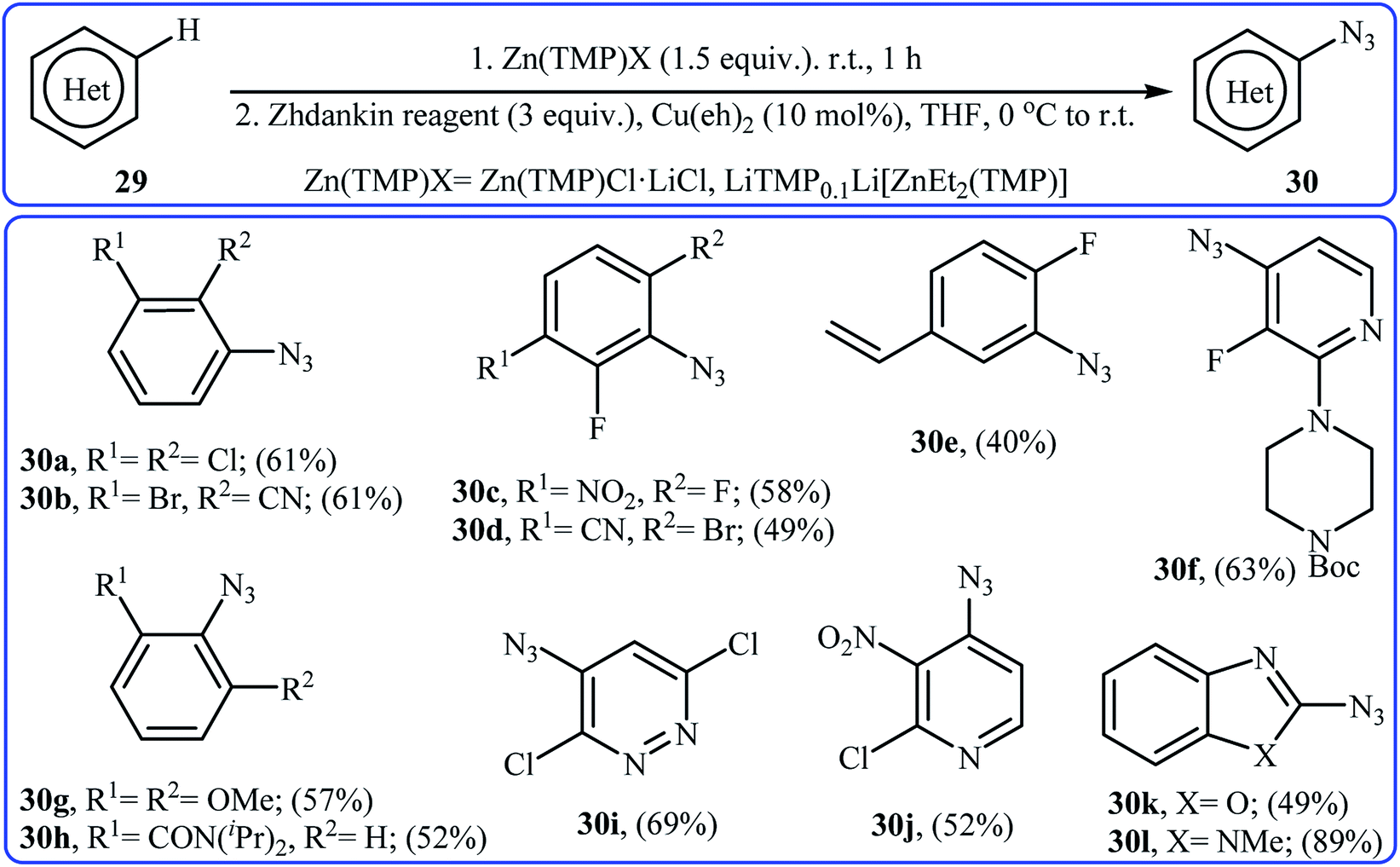
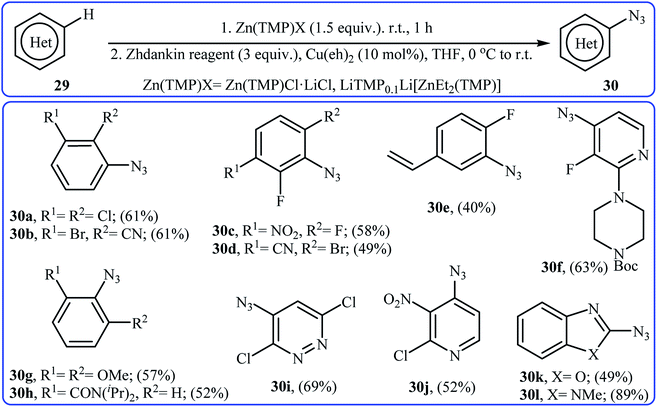
In 2017, Wang and co-workers synthesized a library of azido-(hetero)arenes 30 in moderate to good yields through the site-selective deprotonative zincation followed by copper-catalyzed azidation of the corresponding (hetero)arenes 29 with Zhdankin reagent.34 Among the various zincation reagents, Zn(TMP)Cl·LiCl was the most efficient for the strongly electron-deficient (hetero)arenes and LiTMP0.1Li[ZnEt2(TMP)] for the non-activated and electron-rich ones. As shown in Scheme 19, the azidation took place exclusively at the most acidic position.
 |
| | Scheme 19 Wang's synthesis of azido-(hetero)arenes 30. | |
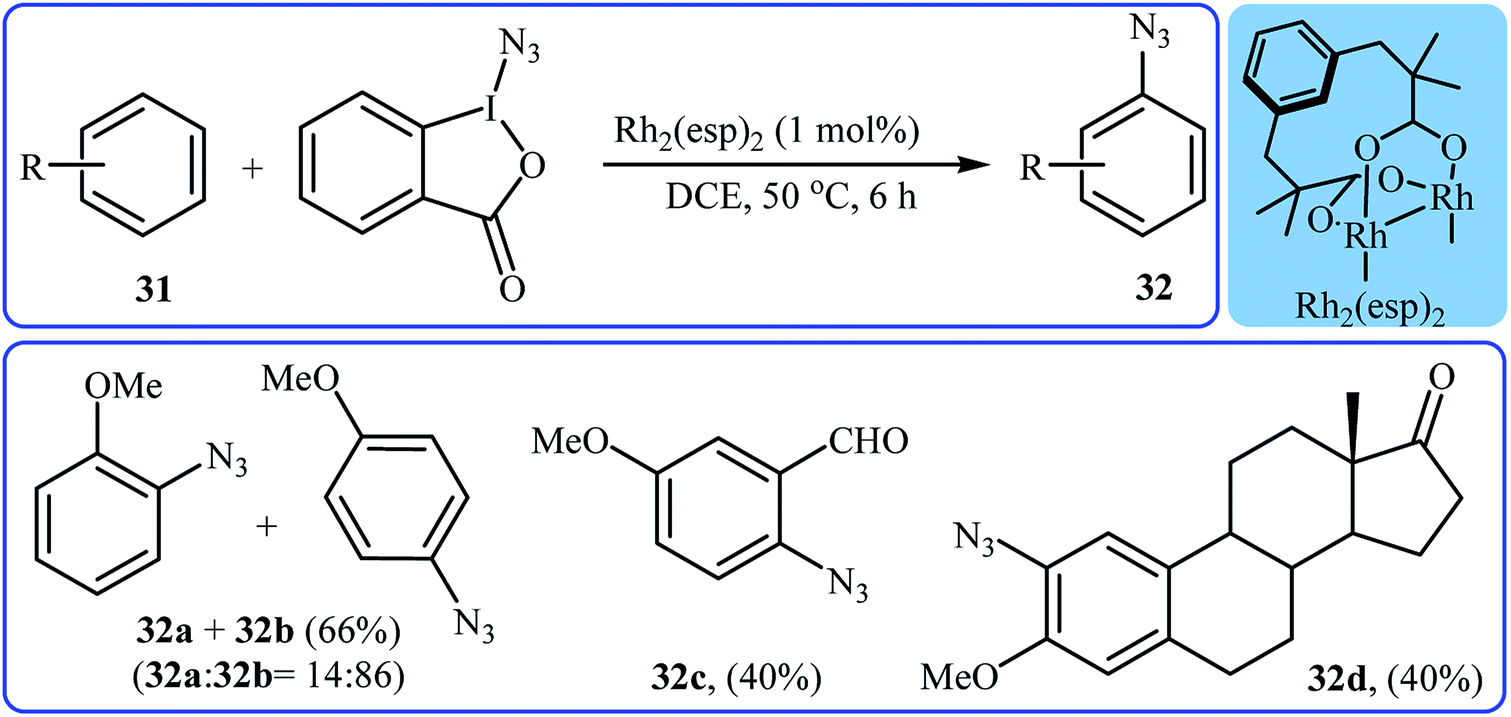
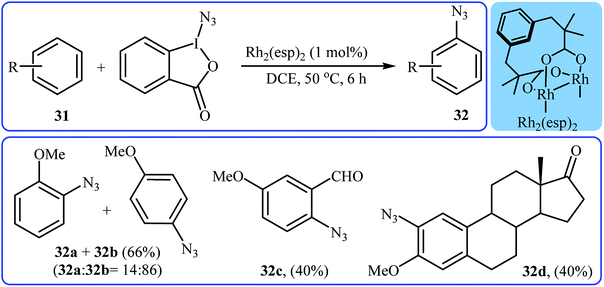
Recently, Chen and Wang along with their co-workers demonstrated that, in the presence of only 1 mol% of Rh2(esp)2 as a commercially available catalyst in DCE, various electron-rich aromatic substrates 31 undergo chemo- and regioselective azidation with Zhdankin's reagent to afford mono-azidated arenes 32 in moderate yields.35 Interestingly, under the standard conditions, the azidation of arenes bearing alkene moieties displayed different chemoselectivities, favoring aromatic C(sp2)–H azidation over alkeneic C(sp2)–H azidation. The mild reaction conditions allowed the tolerance of various functional groups such as chloro, bromo, iodo, ether, ester, aldehyde, and ketone functionalities. Some reported examples are shown in Scheme 20. It was also found that the procedure is suitable for aryl amine substrates and azidation proceeded selectively at the position para to the amine group in MeOH. Mechanistic studies suggested that the catalyst-bound intermediate Rh2(esp)2N3 is the key active species in the reaction.
 |
| | Scheme 20 Selected examples of the Rh-catalyzed direct C–H bond azidation of aromatic rings 31 with Zhdankin's reagent reported by Chen and Wang. | |
2.4. Benzotriazole sulphonyl azide as azide source
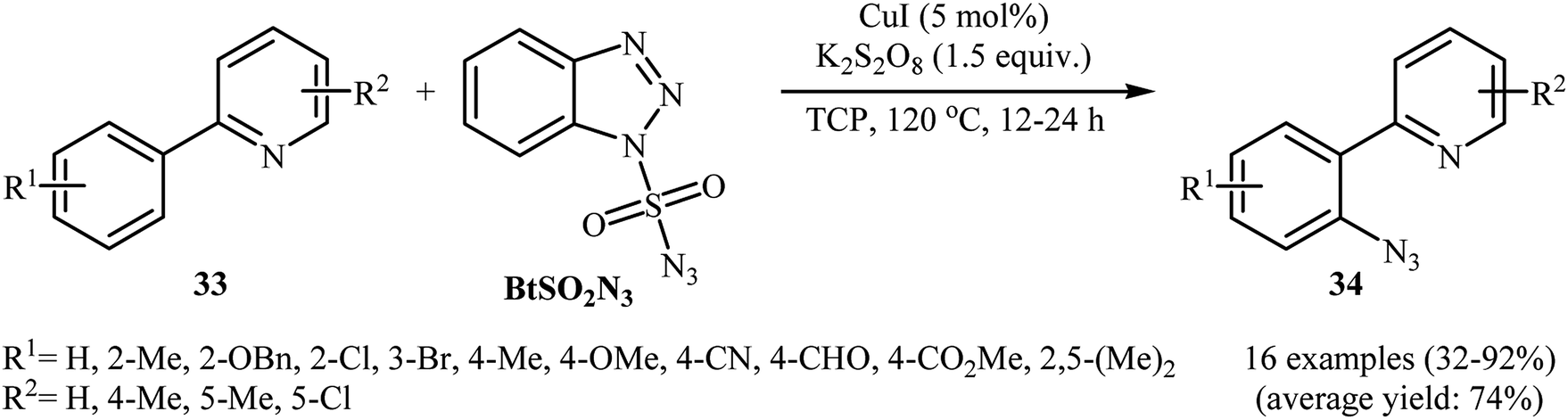
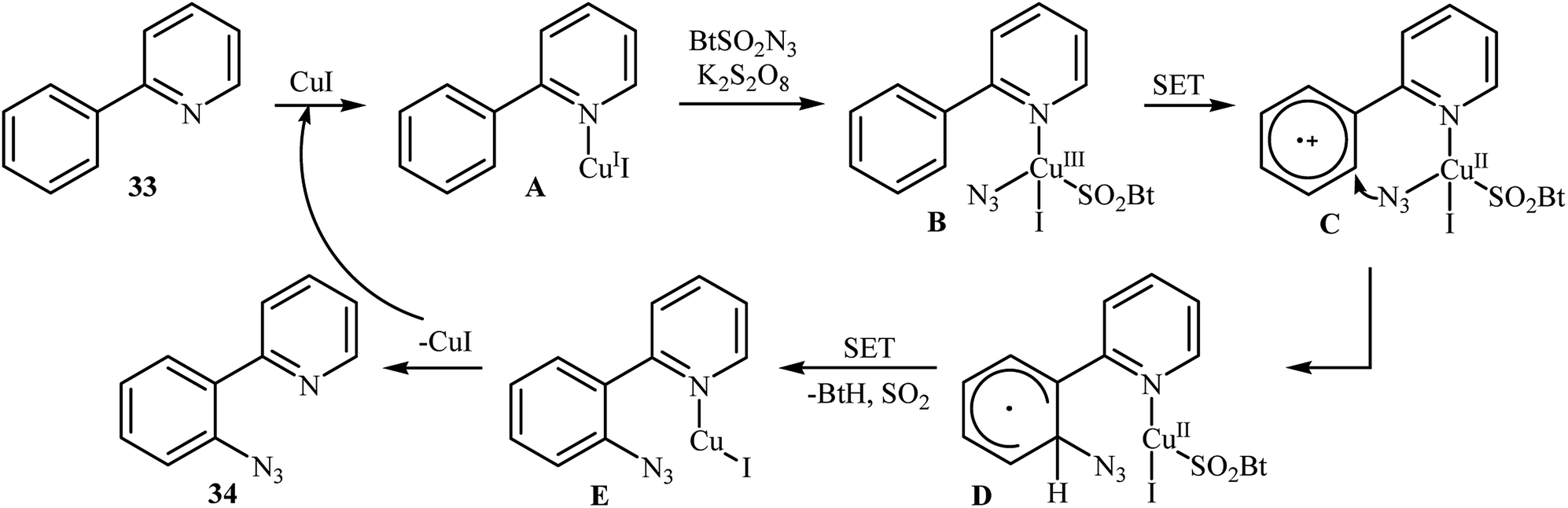
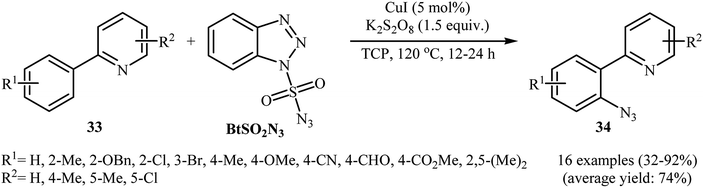
In 2015, Azad and Narula revealed that the treatment of 2-phenylpyridines 33 with benzotriazole sulfonyl azide (BtSO2N3) as a crystalline, safe and easily available azidating agent in the presence of a catalytic amount of CuI and over-stoichiometric amount of K2S2O8 in 1,2,3-trichloropropane (TCP) afforded the corresponding ortho-azide arenes 34 in moderate to excellent yields (Scheme 21).36 The substrate scope was evaluated on 16 functionalized 2-phenylpyridines and proved that both electron-donor and -withdrawing functional groups were tolerated. Of note, other coordinating directing groups, including pyrimidine, pyrazole and thiazole were also found to similarly mediate the ortho-selective C–H azidation of simple arenes under this condition. The mechanistic course suggested by the authors for this azidation reaction is depicted in Scheme 22, and starts with the formation of a CuI complex A by the coordination of the copper catalyst to the 2-phenylpyridine 33 that, after reaction with BtSO2N3 in the presence of K2S2O8 affords intermediate B. Subsequently, a single electron transfer from the metal center to the aromatic ring converts this complex into radical cation C, which undergoes an intramolecular azide transfer to furnish intermediate D. Next, another single electron transfer process along with the elimination of benzotriazole and SO2 converts intermediate D into intermediate E. Finally, demetallation of this intermediate affords the expected azido product 34. To the best of our awareness, this is the only example of direct azidation of aromatic C–H bonds with benzotriazole sulfonyl azide reported so far.
 |
| | Scheme 21 Direct ortho-azidation of arenes 33 using benzotriazole sulphonyl azide as azidating agent. | |
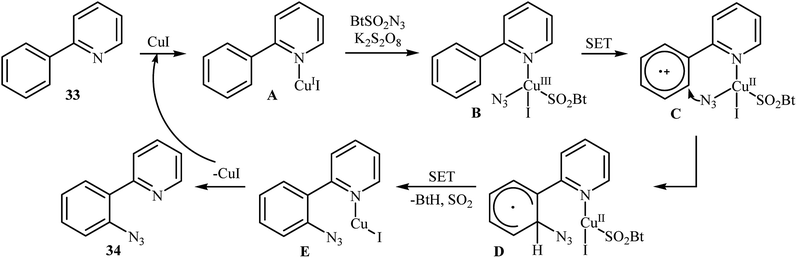 |
| | Scheme 22 Plausible reaction mechanism for the formation of ortho-azide arenes 34. | |
3. Olefinic C–H bonds
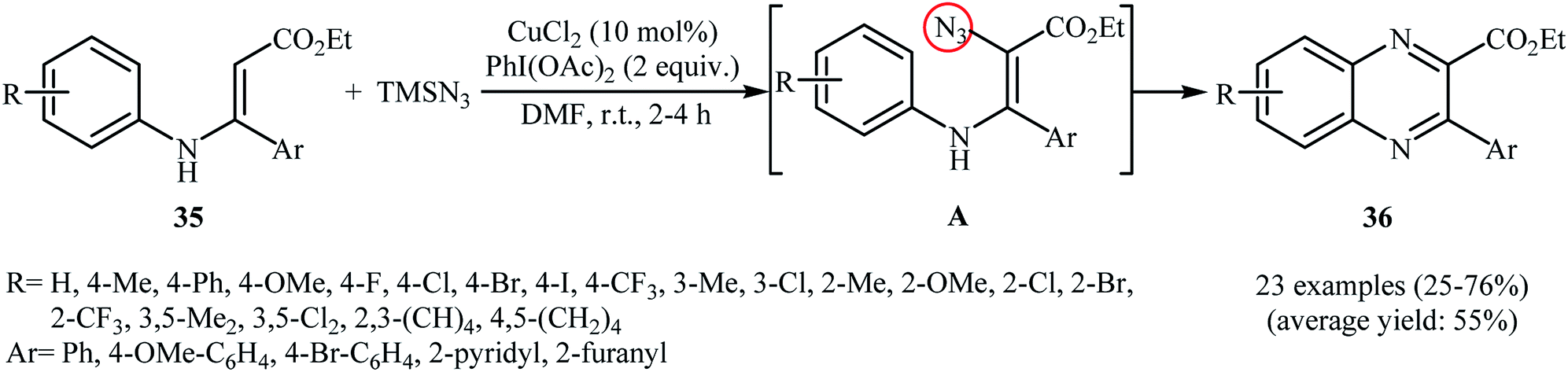
Direct azidation of olefinic C–H bonds has been rarely studied; in fact, only three examples of such a reaction were reported in the literature thus far. In 2016, Ma, Li, and Yu reported an innovative double oxidative C–N bond formation for the synthesis of quinoxaline derivatives.37 They found that N-arylenamines 35 underwent a rapid tandem oxidative azidation/cyclization reaction with TMSN3 in the presence of CuCl2/PhI(OAc)2 combination as a catalytic system in DMF at room temperature. As shown in Scheme 23, a variety of electron-rich and election-poor N-arylenamines were applied to afford the corresponding quinoxalines 36 with yield ranging from 25% to 76%. However, ethyl 3-((2-methoxyphenyl)amino)-3-phenyl acrylate failed to enter into this reaction. It should be mentioned that the presence of electron-withdrawing CO2Et group was crucial for this reaction, while the use of substrates bearing electron-donating groups (e.g., Me) instead of CO2Et did not lead to the desired products under the standard condition. The mechanism suggested by the authors to explain this reaction is based on the formation of vinyl azide intermediate A, followed by its ring closure by elimination of nitrogen gas.
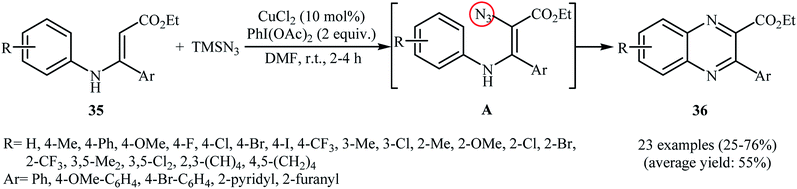 |
| | Scheme 23 Synthesis of quinoxalines 36 through a tandem oxidative azidation/cyclization of N-arylenamines 35 with TMSN3. | |
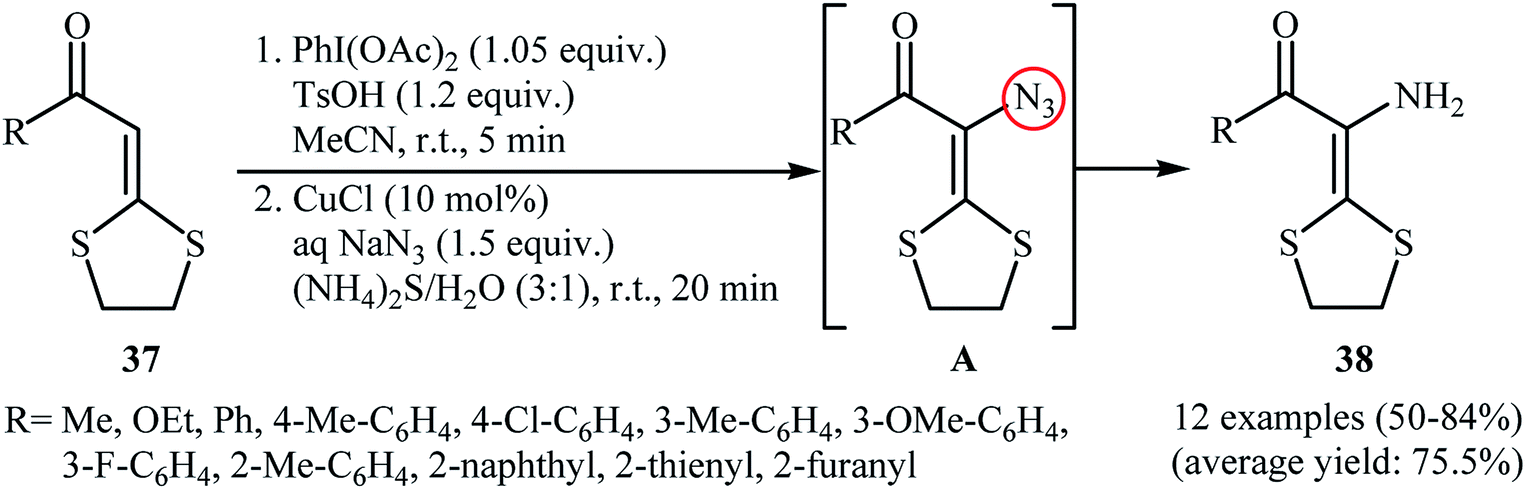
In the same year, Yu and co-workers reported an interesting one-pot procedure for the synthesis of enamines 38 by direct azidation of the corresponding α-oxo ketene dithioacetals 37 with NaN3 followed by reduction of the C–H azidation intermediate A by aqueous (NH4)2S at room temperature (Scheme 24).38 The in situ generated vinyl azide intermediates were captured by phenylacetylene to produce triazoles. Interestingly, 3-amino-1,4-dithian-2-one derivatives were obtained in moderate to good yields when the (NH4)2S was replaced by AcO2. According to the authors proposed mechanism, this reaction proceed through an azidation/ring-expansion rearrangement/thiolactonization sequential process.
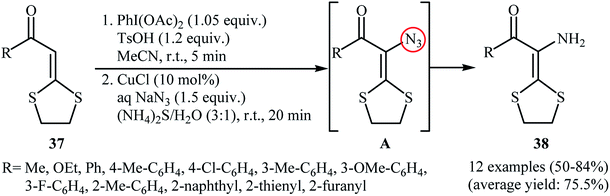 |
| | Scheme 24 Yu's synthesis of enamines 38. | |
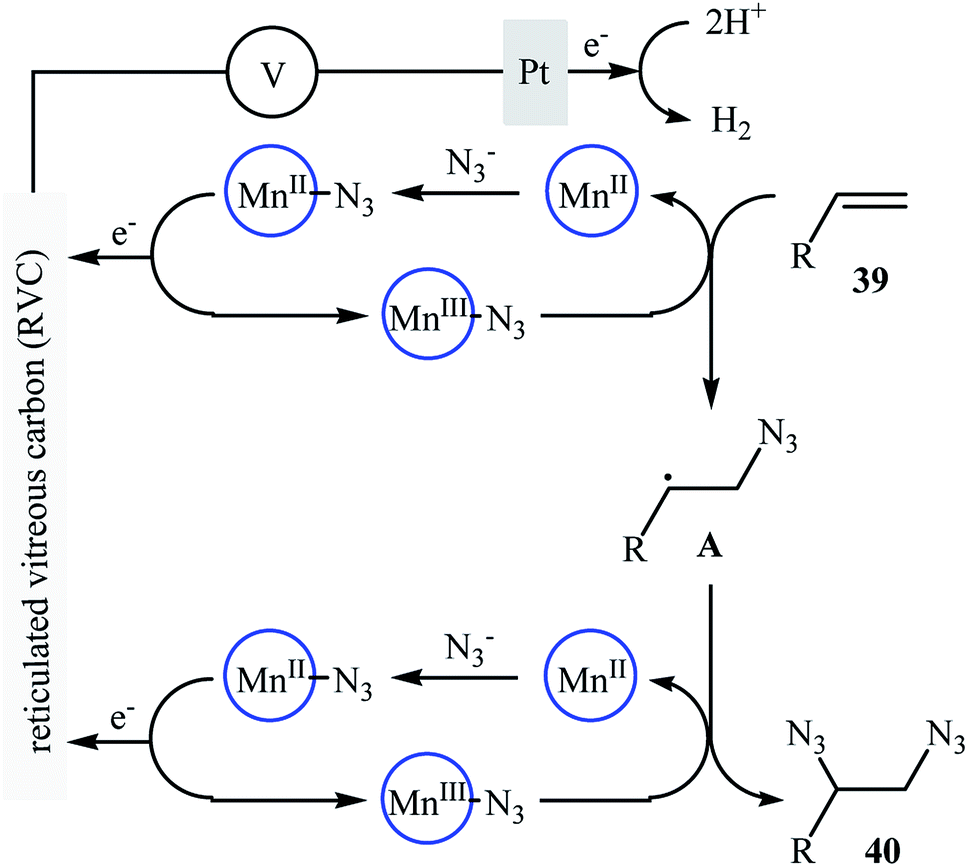
Recently, Lin and co-workers presented an environmentally friendly electrochemical protocol for metal-catalyzed direct diazidation of alkenes 39 which offers a simple, mild and efficient way to synthetically important 1,2-diazides 40.39 The optimal system was identified using NaN3 as the azide source, MnBr2·4H2O as the catalyst, LiClO4 as the electrolyte with an undivided RVC/Pt-cell in MeCN/AcOH (9:1). Under the optimized conditions, various acyclic and cyclic olefins bearing sensitive functional groups such as chloro, bromo, hydroxy, amino, ester, ketone, aldehyde, acid, ether, and thioether showed high yields of the corresponding 1,2-diazide products (Scheme 25). The authors showed that several 1,2-diazides could be chemoselectively converted to the corresponding biologically important 1,2-diamines by reduction under various conditions. Based on the radical clock experiments, they proposed that this azidation reaction might proceed through a radical pathway as depicted in Scheme 26.
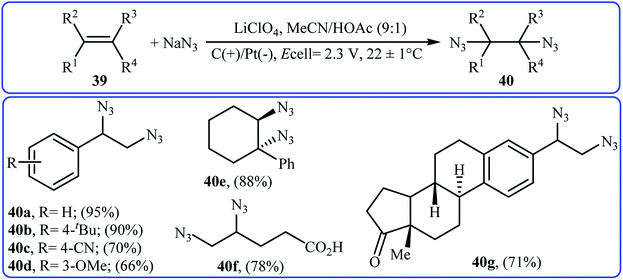 |
| | Scheme 25 Metal-catalyzed electrochemical diazidation of alkenes 39 with NaN3. | |
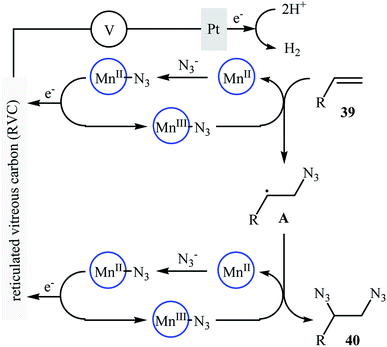 |
| | Scheme 26 Plausible mechanism for the formation of 1,2-diazides 40. | |
4. Aldehydic C–H bonds
4.1. TMSN3 as azide source
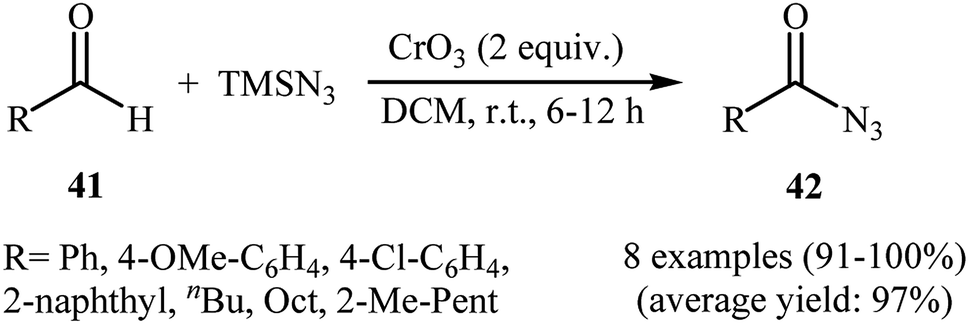
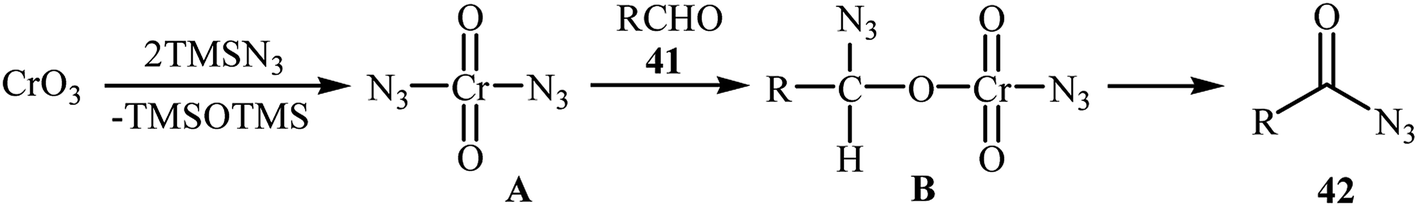
In 1992, Lee and Kwak studied for the first time the direct synthesis of acyl azides from the corresponding aldehydes in a one pot reaction.40 They showed the direct C–H bond azidation of aldehydes 41 with TMSN3 in DCM at room temperature, leading to azidated products 42 in excellent yields (Scheme 27). However, toxic chromium trioxide was used as the terminal oxidant. The reaction is noteworthy in that both aromatic as well as aliphatic aldehydes were tolerated. However, a fair amount of aliphatic acyl azide products underwent rearrangement to the alkyl isocyanates under the standard conditions. The authors elegantly solved this limitation by performing the reaction at −10 °C. A plausible mechanism to explain this azidation reaction is the initial generation of the chromyl azide intermediate A from the reaction of CrO3 with TMSN3, which is subsequently reacted with aldehyde 41 to furnish the chromate ester intermediate B. Finally, the oxidative degradation of the chromate ester B leads to the observed acyl azide 42 (Scheme 28).
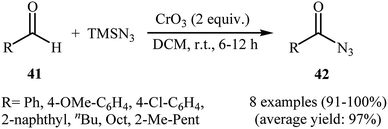 |
| | Scheme 27 CrO3-mediated azidation of aldehydes 41 with TMSN3. | |
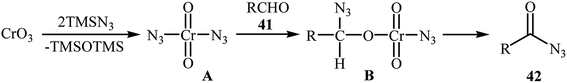 |
| | Scheme 28 Proposed mechanisms for the C–H bond azidation of aldehydes 41 with TMSN3. | |
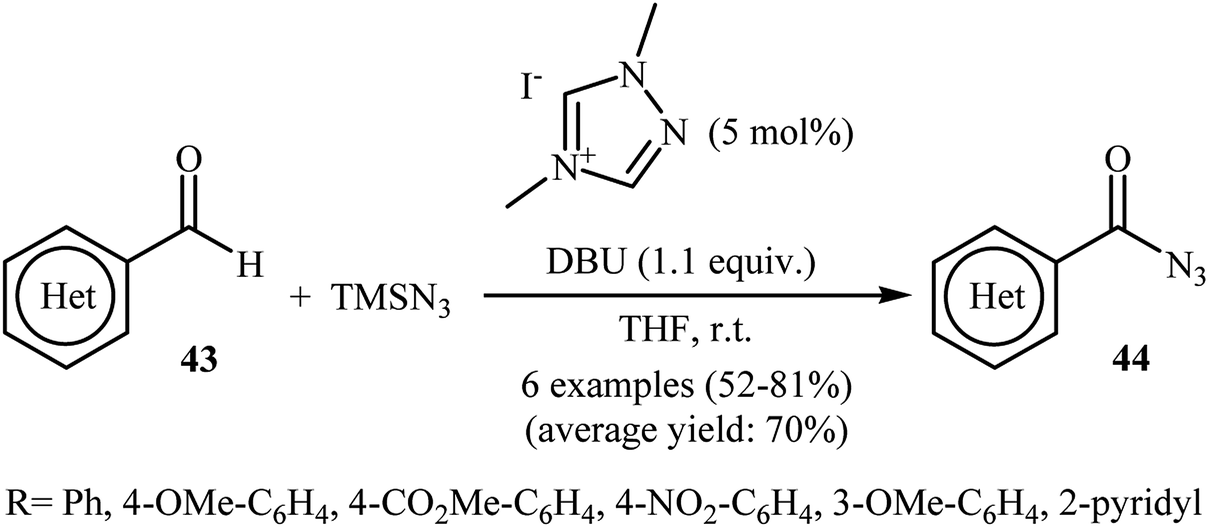
Eighteen years later, with the aim of designing a safer procedure to acyl azide derivatives through direct C–H azidation of aldehydes, Sarkar and Studer were able to demonstrated that a series of (hetero)aromatic acyl azides 44 could be obtained in fair to good yields from the reaction of corresponding (hetero)aromatic aldehydes 43 with TMSN3 employing 1,4-dimethyl-1,2,4-triazolium iodide as a commercially available organocatalyst and DBU as a base in THF (Scheme 29).41 Notably, under similar conditions, performing the reaction with NaN3 and Bu4N+N3− as azide sources gave only traces of the target products.
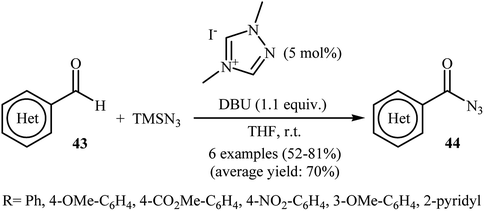 |
| | Scheme 29 NHC-catalyzed azidation of aldehydes 43 with TMSN3. | |
4.2. NaN3 as azide source
After pioneering work by Elmorsy on oxidative azidation of aldehydes with triazidochlorosilane (generated in situ from SiCl4 and NaN3) employing MnO2 as the oxidant,42 the first general report on the synthesis of acyl azids through the direct azidation of aldehydic C–H bonds with NaN3 was published in 2000, by Chen et al.43 They showed that various electron-rich and electron-deficient aromatic aldehydes 45 underwent relatively rapid azidation with NaN3 in the presence of over-stoichiometric amounts of PhI(OAc)2 as a hypervalent iodine reagent in DCM at room temperature. The corresponding aroyl azides 46 were obtained in moderate to excellent yields (Scheme 30). According to the authors proposed mechanism, this reaction proceeds via a free radical pathway similar to that one described in Scheme 16. Later, Bose and Reddy improved the efficiency of this reaction in the terms of reaction time and yields by replacement of PhI(OAc)2 with Dess–Martin periodinane.44
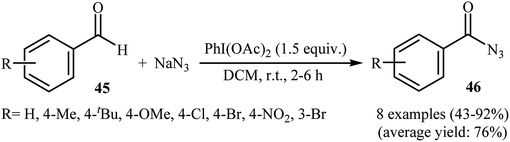 |
| | Scheme 30 Direct conversion of aldehydes 45 to acyl azides 46 using NaN3 and PhI(OAc)2. | |
In 2007, Akamanchi's research group reported the direct conversion of aldehydes 47 to acyl azides 48 using NaN3 as a source of azide and tert-butyl hypochlorite (tBuOCl) as an oxidant.45 In this investigation, a series of common organic solvents (i.e., cyclohexane, tBuOH, MeCN, CHCl3, CCl4, DCM) were used for a comparative study. The best result was obtained with CCl4. In tBuOH the reaction was faster, however, a small amount of the corresponding benzoic anhydride was obtained as a side product. The optimum mole ratio of aldehyde:tBuOCl:NaN3 was found to be 1:2:2. Various aromatic, heteroaromatic, and aliphatic aldehydes 47 were reacted well under the optimized reaction conditions to produce the corresponding acyl azides 48 in good to high yields (Scheme 31).
 |
| | Scheme 31 Ackerman's synthesis of acyl azides 48. | |
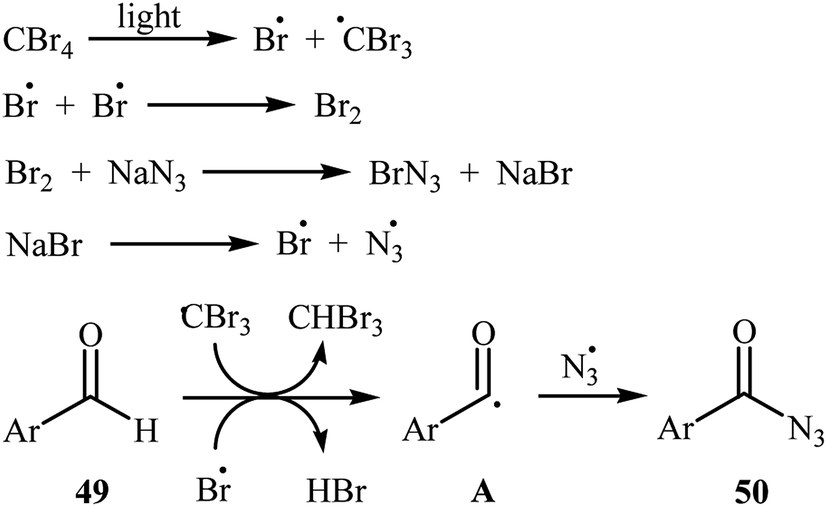
Later, Yadav and co-workers developed an interesting approach for visible light-mediated direct azidation of (hetero)aromatic aldehydes 49 with NaN3 using inexpensive and easily handled CBr4 as a radical generator.46 The reactions proceeded in MeCN under 7 W white LEDs, giving good to excellent yields of acyl azides 50 within 4–6 h (Scheme 32). It was noteworthy that this procedure could be applied to aromatic aldehydes with various functional groups such as OMe, Cl, Br, CO2Et, and NO2 groups, which provided a complementary platform for further manipulation of products to create more complex azido molecules. They nicely applied their methodology in the one-pot synthesis of carbamoyl azides (RNH–CO–N3) by simple heating (80 °C) of acyl azides through Curtius rearrangement. The authors proposed mechanism for this azidation reaction is depicted in Scheme 33. First, Br˙ and ˙CBr3 radicals were generated via the homolytic cleavage of a C–Br bond in CBr4 under irradiation of visible light. Later, abstraction of aldehydic hydrogen atom from the starting aldehyde 49 by these radicals resulted in the formation of the radical species A. Finally, the combination reaction of this radical with the azide radical gave the expected acyl azide 50.
 |
| | Scheme 32 Visible light-mediated azidation of aldehydes 49 with NaN3. | |
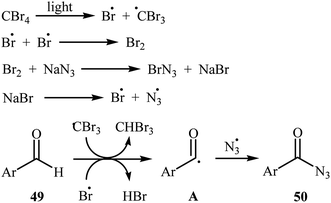 |
| | Scheme 33 Mechanism proposed to explain the synthesis of acyl azides 50. | |
4.3. IN3 as azide source
In 2003, Bols and co-workers introduced iodine azide (IN3) as azidating agent for direct aldehydic C–H azidation.47 Thus, in the absence of any catalyst and additive in MeCN at room temperature, reaction of a small series of aliphatic aldehydes 51 with IN3 furnished the corresponding acyl azides 52 in high yields (Scheme 34). Notably, the synthetically important carbamoyl azides could be directly achieved by performing the process in refluxing MeCN. Twelve years later, a related transformation, using Zhdankin reagent was disclosed by Saito et al.48 Tetrabutylammonium iodide (TBAI) served as catalyst and DCM as solvent. Noteworthy, the in situ generated IN3 by reaction of Zhdankin reagent with TBAI, was acted as the azidating agent in this reaction. Various aliphatic, aromatic, heteroaromatic and bicyclic aldehydes were used to establish the general applicability of the approach.
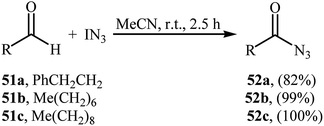 |
| | Scheme 34 Direct azidation of aliphatic aldehydes 51 with IN3. | |
5. Conclusion
This review provides concise overview on the direct azidation of various organic molecules (i.e., arenes, alkenes, aldehydes) via C(sp2)–H activation. This new page to access organic azides has many potential advantages over the traditional approaches, which can be summarized as follows: (i) high pot-, atom- and step-economy; (ii) eco-friendly processes; and (iii) simple operation. Despite the significant achievements during the past few years in this interesting and important research field, many challenges still remain to be overcome: (i) the number of reported examples in the direct azidation of olefinic C–H bonds are scarce and there is still further need to study the scope and limitations of these reactions; (ii) all of the metal-catalyzed C(sp2)–H bonds azidation reactions covered in this review are limited to the use of rhodium and copper catalysts. Thus the exploration of other transition metal catalysts such as iron catalysts and nanosized metal catalysts are highly desirable in term of the cost and maybe the efficiency of the catalysts; (iii) synthetic application of the direct azidation of C(sp2)–H bonds in the preparation of important and commercialized drugs should be explored. It is our hope that this review will be beneficial to elicit further research and thinking in this domain.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS PubMed;
(b) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef;
(c) S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef CAS;
(d) D. Huang and G. Yan, Adv. Synth. Catal., 2017, 359, 1600–1619 CrossRef CAS;
(e) H. Saeidian, H. Sadighian, M. Abdoli and M. Sahandi, J. Mol. Struct., 2017, 1131, 73–78 CrossRef CAS.
-
(a) A. Kamal, N. V. Rao and E. Laxman, Tetrahedron Lett., 1997, 38, 6945–6948 CrossRef CAS;
(b) M. Warrier, M. K. Lo, H. Monbouquette and M. A. Garcia-Garibay, Photochem. Photobiol. Sci., 2004, 3, 859–863 RSC.
-
(a) F. Fazio and C.-H. Wong, Tetrahedron Lett., 2003, 44, 9083–9085 CrossRef CAS;
(b) S. Xie, Y. Zhang, O. Ramström and M. Yan, Chem. Sci., 2016, 7, 713–718 RSC;
(c) F.-L. Zhang, X. Zhu and S. Chiba, Org. Lett., 2015, 17, 3138–3141 CrossRef CAS PubMed.
-
(a) A. Ramesha, S. Bhat and S. Chandrasekaran, J. Org. Chem., 1995, 60, 7682–7683 CrossRef CAS;
(b) L. Benati, G. Bencivenni, R. Leardini, D. Nanni, M. Minozzi, P. Spagnolo, R. Scialpi and G. Zanardi, Org. Lett., 2006, 8, 2499–2502 CrossRef CAS PubMed;
(c) J. H. Lee, S. Gupta, W. Jeong, Y. H. Rhee and J. Park, Angew. Chem., Int. Ed., 2012, 51, 10851–10855 CrossRef CAS PubMed.
-
(a) T.-S. Lin and W. H. Prusoff, J. Med. Chem., 1978, 21, 109–112 CrossRef CAS;
(b) D. B. Smith, G. Kalayanov, C. Sund, A. Winqvist, T. Maltseva, V. J.-P. Leveque, S. Rajyaguru, S. L. Pogam, I. Najera and K. Benkestock, J. Med. Chem., 2009, 52, 2971–2978 CrossRef CAS.
- A. G. Habeeb, P. Praveen Rao and E. E. Knaus, J. Med. Chem., 2001, 44, 3039–3042 CrossRef CAS.
- J. A. Raeburn and J. D. Devine, Scand. J. Infect. Dis., 1973, 5, 135–139 CrossRef CAS.
- P. P. Geurink, W. A. van der Linden, A. C. Mirabella, N. Gallastegui, G. de Bruin, A. E. Blom, M. J. Voges, E. D. Mock, B. I. Florea and G. A. van der Marel, J. Med. Chem., 2013, 56, 1262–1275 CrossRef CAS.
-
(a) P. A. Smith, C. D. Rowe and L. B. Bruner, J. Org. Chem., 1969, 34, 3430–3433 CrossRef CAS;
(b) J. Gavenonis and T. D. Tilley, Organometallics, 2002, 21, 5549–5563 CrossRef CAS.
-
(a) M. Karimzadeh, K. Niknam, N. Manouchehri and D. Tarokh, RSC Adv., 2018, 8, 25785–25793 RSC;
(b) M. A. Karimi Zarchi and Z. Escandari, J. Appl. Polym. Sci., 2011, 121, 1916–1920 CrossRef;
(c) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
-
(a) E. Nyfeler and P. Renaud, Org. Lett., 2008, 10, 985–988 CrossRef CAS PubMed;
(b) Y. Zhu, X. Li, X. Wang, X. Huang, T. Shen, Y. Zhang, X. Sun, M. Zou, S. Song and N. Jiao, Org. Lett., 2015, 17, 4702–4705 CrossRef CAS PubMed.
-
(a) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC;
(b) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC;
(c) M. Pichette Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654–18677 CrossRef CAS PubMed;
(d) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord and T. Besset, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
-
(a) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed;
(b) M. Hamzeloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, Fluorine Chem., 2019, 224, 52–60 CrossRef;
(c) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC;
(d) A. Hosseinian, S. Arshadi, S. Sarhandi, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 289–311 CrossRef CAS;
(e) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(f) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed.
-
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(b) K. Didehban, E. Vessally, A. Hosseinian, L. Edjlali and E. S. Khosroshahi, RSC Adv., 2018, 8, 291–301 RSC;
(c) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(d) K. Nejati, S. Ahmadi, M. Nikpassand, P. D. K. Nezhad and E. Vessally, RSC Adv., 2018, 8, 19125–19143 RSC;
(e) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(f) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC;
(g) E. Vessally, K. Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018, 39, 332–349 CrossRef CAS;
(h) E. Vessally, R. Mohammadi, A. Hosseinian, K. Didehban and L. Edjlali, J. Sulfur Chem., 2018, 39, 443–463 CrossRef CAS;
(i) A. Hosseinian, L. Zare Fekri, A. Monfared, E. Vessally and M. Nikpassand, J. Sulfur Chem., 2018, 39, 674–698 CrossRef CAS;
(j) A. Monfared, R. Mohammadi, S. Ahmadi, M. Nikpassand and A. Hosseinian, RSC Adv., 2019, 9, 3185–3202 RSC;
(k) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS;
(l) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, Fluorine Chem., 2019, 220, 24–34 CrossRef CAS;
(m) A. Monfared, S. Ahmadi, Z. Rahmani, P. D. K. Nezhad and A. Hosseinian, J. Sulfur Chem., 2019, 40, 209–231 CrossRef CAS;
(n) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 8964–8976 RSC;
(o) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(p) A. Hosseinian, Y. J. Sadeghi, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Sulfur Chem., 2019 DOI:10.1080/17415993.2019.1598410;
(q) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed.
- X. Huang and J. T. Groves, ACS Catal., 2015, 6, 751–759 CrossRef.
- D. Lubriks, I. Sokolovs and E. Suna, J. Am. Chem. Soc., 2012, 134, 15436–15442 CrossRef CAS PubMed.
- F. Xie, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2013, 52, 11862–11866 CrossRef CAS PubMed.
- S. Liu, X. Qi, L.-B. Qu, R. Bai and Y. Lan, Catal. Sci. Technol., 2018, 8, 1645–1651 RSC.
- B. Yao, Y. Liu, L. Zhao, D.-X. Wang and M.-X. Wang, J. Org. Chem., 2014, 79, 11139–11145 CrossRef CAS PubMed.
- Y. Dou, Z. Xie, Z. Sun, H. Fang, C. Shen, P. Zhang and Q. Zhu, ChemCatChem, 2016, 8, 3570–3574 CrossRef CAS.
- H. Fang, Y. Dou, J. Ge, M. Chhabra, H. Sun, P. Zhang, Y. Zheng and Q. Zhu, J. Org. Chem., 2017, 82, 11212–11217 CrossRef CAS PubMed.
- P. K. Prasad, R. G. Kalshetti, R. N. Reddi, S. P. Kamble and A. Sudalai, Org. Biomol. Chem., 2016, 14, 3027–3030 RSC.
- M. Jafarzadeh, Synlett, 2007, 2144–2145 CrossRef CAS.
- C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924–18927 CrossRef CAS PubMed.
- F. Stejskal, V. Eigner, H. Dvořáková, P. Cuřínová and P. Lhoták, Tetrahedron Lett., 2015, 56, 5357–5361 CrossRef CAS.
- J. Dhineshkumar, K. Gadde and K. R. Prabhu, J. Org. Chem., 2017, 83, 228–235 CrossRef PubMed.
- Y. Kita, H. Tohma, M. Inagaki, K. Hatanaka and T. Yakura, Tetrahedron Lett., 1991, 32, 4321–4324 CrossRef CAS.
- Y. Kita, H. Tohma, K. Hatanaka, T. Takada, S. Fujita, S. Mitoh, H. Sakurai and S. Oka, J. Am. Chem. Soc., 1994, 116, 3684–3691 CrossRef CAS.
- N. V. Tsarevsky, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 966–974 CrossRef CAS.
- P. Li, J. Zhao, C. Xia and F. Li, Org. Chem. Front., 2015, 2, 1313–1317 RSC.
-
(a) V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, M. S. Formaneck and J. T. Bolz, Tetrahedron Lett., 1994, 35, 9677–9680 CrossRef CAS;
(b) Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679–2683 RSC;
(c) P. T. Rabet, G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2016, 18, 1646–1649 CrossRef CAS PubMed;
(d) B. Zhang and A. Studer, Org. Lett., 2013, 15, 4548–4551 CrossRef CAS PubMed;
(e) Y. Shinomoto, A. Yoshimura, H. Shimizu, M. Yamazaki, V. V. Zhdankin and A. Saito, Org. Lett., 2015, 17, 5212–5215 CrossRef CAS PubMed.
- S. Alazet, J. Preindl, R. Simonet-Davin, S. Nicolai, A. Nanchen, T. Meyer and J. Waser, J. Org. Chem., 2018, 83, 12334–12356 CrossRef CAS PubMed.
- Y. Fan, W. Wan, G. Ma, W. Gao, H. Jiang, S. Zhu and J. Hao, Chem. Commun., 2014, 50, 5733–5736 RSC.
- C. E. Hendrick, K. J. Bitting, S. Cho and Q. Wang, J. Am. Chem. Soc., 2017, 139, 11622–11628 CrossRef CAS PubMed.
- Y. Wang, Z. Fang, X. Chen and Y. Wang, Org. Lett., 2018, 20, 5732–5736 CrossRef CAS PubMed.
- C. S. Azad and A. K. Narula, RSC Adv., 2015, 5, 100223–100227 RSC.
- H. Ma, D. Li and W. Yu, Org. Lett., 2016, 18, 868–871 CrossRef CAS PubMed.
- T. Guo, Q. Jiang and Z. Yu, Adv. Synth. Catal., 2016, 358, 3450–3457 CrossRef CAS.
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed.
- J. G. Lee and K. H. Kwak, Tetrahedron Lett., 1992, 33, 3165–3166 CrossRef CAS.
- S. De Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef CAS PubMed.
- S. S. Elmorsy, Tetrahedron Lett., 1995, 36, 1341–1342 CrossRef CAS.
- D.-J. Chen and Z.-C. Chen, Tetrahedron Lett., 2000, 41, 7361–7363 CrossRef CAS.
- D. S. Bose and A. N. Reddy, Tetrahedron Lett., 2003, 44, 3543–3545 CrossRef CAS.
- N. D. Arote and K. G. Akamanchi, Tetrahedron Lett., 2007, 48, 5661–5664 CrossRef CAS.
- V. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Tetrahedron Lett., 2016, 57, 2502–2505 CrossRef CAS.
- L. Marinescu, J. Thinggaard, I. B. Thomsen and M. Bols, J. Org. Chem., 2003, 68, 9453–9455 CrossRef CAS PubMed.
- Y. Shinomoto, A. Yoshimura, H. Shimizu, M. Yamazaki, V. V. Zhdankin and A. Saito, Org. Lett., 2015, 17, 5212–5215 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *e
*e