
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSerum metabolomic alterations in multiple myeloma revealed by targeted and untargeted metabolomics approaches: a pilot study†
Venkatesh Chanukuppaab,
Tushar H. Moreab,
Khushman Taunk a,
Ravindra Tawarea,
Tathagata Chatterjeec,
Sanjeevan Sharmad and
Srikanth Rapole*a
a,
Ravindra Tawarea,
Tathagata Chatterjeec,
Sanjeevan Sharmad and
Srikanth Rapole*a
aProteomics Lab, National Centre for Cell Science, Ganeshkhind, Pune-411007, MH, India. E-mail: rsrikanth@nccs.res.in; Fax: +91-20-2569-2259; Tel: +91-20-2570-8075
bSavitribai Phule Pune University, Ganeshkhind, Pune-411007, MH, India
cArmy Hospital (R&R), Dhaula Kuan, New Delhi-110010, DL, India
dArmed Forces Medical College, Pune-411001, MH, India
First published on 18th September 2019
Abstract
Multiple myeloma (MM) is the second most prevalent hematological malignancy characterized by rapid proliferation of plasma cells, which leads to overproduction of antibodies. MM affects around 15% of all hemato-oncology cases across the world. The present study involves identification of metabolomic alterations in the serum of an MM cohort compared to healthy controls using both LC-MRM/MS based targeted and GC-MS based untargeted approaches. Several MM specific serum metabolomic signatures were observed in this study. A total of 54 metabolites were identified as being significantly altered in MM cohort, out of which, 26 metabolites were identified from LC-MRM/MS based targeted analysis, whereas 28 metabolites were identified from the GC-MS based untargeted analysis. Receiver operating characteristic (ROC) curve analysis demonstrated that six metabolites each from both the datasets can be projected as marker metabolites to discriminate MM subjects with higher specificity and sensitivity. Moreover, pathway analysis deciphered that several metabolic pathways were altered in MM including pyrimidine metabolism, purine metabolism, amino acid metabolism, nitrogen metabolism, sulfur metabolism, and the citrate cycle. Comprehensively, this study contributes valuable information regarding MM induced serum metabolite alterations and their pathways, which could offer further insights into this cancer.
1. Introduction
Multiple myeloma (MM) is reported as the second most prevalent hematological malignancy (∼15%) across the world.1 MM is characterized by rapid proliferation of plasma cells and their subsequent accumulation in bone marrow which results in overproduction of antibodies and eventually leads to bone resorption.2 It is one of the most dominant hematological malignancies on the Indian subcontinent, where 4 out of every 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people are detected with this cancer every year.3 Development of MM is a complex process that is associated with cytological abnormalities such as hyperdiploidy and chromosomal translocations.4 Being a heterogeneous disease, MM can be broadly categorized into the following types: monoclonal gammopathy of undetermined clinical significance (MGUS), a benign form of MM; an intermediate stage called as smoldering myeloma and the symptomatic myeloma.5 MGUS could progress into smoldering myeloma, which finally turns into the symptomatic myeloma. Despite recent developments in the field of hematological malignancy research, there is a significant gap regarding the exact nature of molecular changes that occur during malignant evolution leading to MM.
000 people are detected with this cancer every year.3 Development of MM is a complex process that is associated with cytological abnormalities such as hyperdiploidy and chromosomal translocations.4 Being a heterogeneous disease, MM can be broadly categorized into the following types: monoclonal gammopathy of undetermined clinical significance (MGUS), a benign form of MM; an intermediate stage called as smoldering myeloma and the symptomatic myeloma.5 MGUS could progress into smoldering myeloma, which finally turns into the symptomatic myeloma. Despite recent developments in the field of hematological malignancy research, there is a significant gap regarding the exact nature of molecular changes that occur during malignant evolution leading to MM.
It is well-known fact that malignant transformations are associated with altered metabolic pathways mostly associated with biosynthetic and bioenergetic processes.6 Such metabolic rearrangement or switching depicts an adjustment to aid cell survival, tissue remodeling, tumor growth, as well as cancer metastasis. Strikingly, as evident from available literature, suggests that this metabolic adjustment is coordinated by genomic programming and inveigled by the tumor microenvironment. Moreover, in some situations, the alteration in metabolism plays a crucial role in oncogenesis.6,7 Therefore, it is of utmost importance to extract such metabolic deviations by metabolomics approach and understand them in the context of prognosis, therapy response as well as disease pathophysiology.
Metabolomics not only includes the utilization of various analytical methodologies towards identification and quantification of the metabolites pertaining to a biological system, but can monitor the changes at metabolites level in a variety of clinical specimens such as serum, cells, tissue, urine and other biological fluids.8–17 Earlier metabolomics studies in MM were mostly carried out on in vitro models, such as cell lines, to investigate metabolic alterations induced by the drug resistance.18,19 Apart from this, identification of potential targets related to apoptosis20 was also studied in context of MM. Moreover, few clinical studies were also conducted to investigate the serum metabolomics alterations upon disease remission.21 However, to our knowledge, there is no report of comprehensive serum metabolomic investigation using targeted and untargeted metabolomic approaches for MM to decipher the metabolic alterations crucial for understanding the disease pathophysiology, which in future could be translated into disease diagnosis.
Therefore, in the present study, we have carried out an elaborate MM serum metabolic profiling of human patients compared to healthy control subjects using both targeted and untargeted approaches to identify metabolic fingerprint associated with this disease.
2. Materials and methods
2.1 Subject selection, sample collection and processing
The study cohort for targeted metabolomic analysis comprised of 48 volunteers (MM = 24, control = 24) recruited at the Armed Forces Medical College (AFMC), Pune. For untargeted metabolomics analysis, 40 subjects (MM = 21, control = 19) from the same cohort were utilized. As we collected the samples in a long duration of sample collection period because of the reason that the availability of the samples was scarce for this disease, the number of samples for the LC-MS and GC-MS are different. As we performed the GC-MS analysis earlier, the sample cohort for this analysis has 21 healthy control and 19 MM serum samples. Whereas, later we collected some additional samples, which made the total of 24 serum samples each for healthy controls and MM for the LC-MS analysis. The inclusion criteria for MM cohort consisted of only freshly diagnosed patients with minimum 18 years of age and without any past or present anticancer drug interventions. For the control cohort, age and gender matched individuals devoid of hypertension or diabetes was selected. ESI Table S1† summarizes the demographic information of the study cohort used in this study. The study cohort was advised overnight fasting and blood samples were collected the following morning. 5 mL of peripheral blood was collected in serum separator tubes (BD Vacutainer™ SST™ II Advance Tubes). The peripheral blood samples were kept for clotting at room temperature and serum separation from the blood was achieved by centrifuging samples at 1500×g for 5 min at 15 °C. Serum samples were then transferred to cryo-vials, labeled and subsequently liquid nitrogen were used to snap freeze the samples. Post snap freezing, the serum samples were stored at −80 °C ultra-low temperature freezer until further use. All experiments were performed in compliance with guidelines mentioned in Declaration of Helsinki (DoH, 2013). Ethical committees of National Centre for Cell Science (NCCS), Pune and Armed Forces Medical College (AFMC), Pune approved this study (IRB# NCCS/IEC/2016-I/8). The experimental aspects and investigations to be undertaken were clearly explained to each participants and written informed consent was obtained from them. Each subject participated voluntarily for this study.2.2 Targeted LC-MRM/MS based metabolomic profiling
In-house built MRM library of 120 metabolites was used to investigate the metabolic alterations in the serum of MM study cohort. These metabolites were selected based on the previous literature survey of various cancer metabolomic studies.22 The chromatography parameters and details of MRM library are discussed in our previous report23 and the same have been utilized for this study as well. Metabolites extraction was executed by the addition of 200 μL methanol to 25 μL of every serum samples, vortexed vigorously and incubated at −20 °C overnight. Moreover, d2L-phenylalanine (40 ng) was added to the samples prior to extraction, which served as an internal standard. Post incubation, the samples were vortexed vigorously and subjected to 14000×g centrifugation at 4 °C for 10 min. The supernatant was then passed through centrifugal filters (Corning® Costar® Spin-X®) with 0.22 μm nylon membrane. Filtrate obtained was then vacuum dried and reconstituted in 25 μL of sample reconstitution buffer (a mixture of 6.5 mL acetonitrile, 2.5 mL methanol, 1 mL water and 0.2 mL formic acid; all the solvents were LC-MS grade) for acquiring the samples in positive ionization mode mass spectrometry. In case of negative ionization mode, the vacuum dried metabolites were reconstituted in 25 μL of ultrapure LC-MS grade water. Subsequently, 10 μL of each sample was analyzed using a Shimadzu Prominence HPLC system (Shimadzu Corporation, Japan) connected to a triple quadrupole mass spectrometer, 4000 QTRAP (AB SCIEX, USA). For positive mode ionization, metabolites were separated through the XBridge™ HILIC column (5 μm, 4.6 × 150 mm, Waters Corp, USA) and Atlantis™ T3 column (5 μm, 4.6 × 150 mm, Waters Corp, USA) was used for negative ionization mode. QC samples, prepared from the mixture of pure metabolite standards, were run after every 5 serum metabolite sample injections to assess the uniform instrument performance.
The LC buffers and chromatography parameters along with MS sample acquisition parameters were adopted from our earlier studies as mentioned elsewhere.23 Positive ionization chromatographic separation was carried out using 32 min gradient program of mobile phase A (10 mM ammonium formate with 0.1% formic acid in water) and mobile phase B (acetonitrile with 0.1% formic acid) with 700 μL min−1 elution rate. The gradient started with 95% B and further decreased to 40% B over next 15 min and held constant for 6 min, and then the column was gradually returned to 95% B and equilibrated for another 9 min. The negative mode ionization chromatography was carried out with elution rate of 500 μL min−1 and 40 min gradient program consisting of 0% B at initial stage and gradually increased to 98% over next 24 min and maintained for 5 min in same state and eventually returned to 0% B. Finally, the column was equilibrated with 100% solvent A for 9 min. Mass spectra were acquired using following set of parameters; MS source temperature 400 °C with interface heater on, curtain gas: 30 and entrance and exit potentials: 10, two ion source gasses at 45 arbitrary units. LC-MRM/MS data analysis was performed using Analyst 1.5 software (Sciex, USA). Analyst quantitation wizard was used for the peak area integration. For elimination of biasness and to maintain quality control measures, samples were randomized during sample analysis and peak integration performed in a blinded manner. Further, the peak area values obtained after performing the integration analysis were exported to a spreadsheet file for further analysis.
2.3 Untargeted GC-MS-based metabolic profiling
Metabolite extraction was performed as described in LC-MS methodology. Additionally, two-step derivatization method was applied to the dried metabolite extract to produce methyl and silyl derivatives. First step was methylation, which was achieved by addition of 30 μL of methoxyamine hydrochloride solution dissolved in pyridine (20 mg mL−1) and incubating at 37 °C for 90 min. In the second step, silylation reaction was performed by adding 30 μL of BSTFA with 1% TMCS solution followed by 1 h incubation at 70 °C. 1 μL of the derivatized sample was injected into the injection port (250 °C) of the Agilent 5975C GC system (Agilent Technologies, USA) operating under splitless mode and connected to Agilent 5977A mass selective detector. Metabolites were separated on HP-5 MS ultra-inert fused silica capillary column (30 m × 0.25 μm × 0.25 mm, Agilent, USA). UHP grade helium was used as the carrier gas with a flow rate of 1 mL min−1. Following parameters were used to obtain the mass spectra; column inlet temperature 280 °C, MS quadrupole temperature 150 °C, ion source temperature 230 °C. GC oven temperature settings are as follows; the gas chromatography program started with initial oven temperature of 50 °C held for 2 min, then temperature was gradually increased to 100 °C at 3 °C min−1 rate and held for 2 min and the ramped up further to 200 °C at 3 °C min−1 and maintained for 2 min at this temperature. The column temperature was lowered to 50 °C in next 5 min and equilibrated for 2 min. Full scan mode range from m/z 50 to 550 was used for the metabolite data acquisition. Integration and deconvolution of chromatograms were carried out by using the AMDIS algorithm of Agilent Chemstation software. Metabolites identity was confirmed using NIST 11 standard mass spectral database (NIST, Gaithersburg, MD). Peak areas of the metabolites were exported in a matrix format to spreadsheet file after chromatographic integration. Metabolic standards initiative (MSI) guidelines were followed for performing the all metabolomic experiments.242.4 Data pre-processing and statistical analysis
The peak areas of identified metabolites in both targeted and untargeted approaches were subjected to statistical analysis using Metaboanalyst 4.0 and SIMCA 14 (Umetrics, Umea, Sweden). Metabolites having missing value >50% were discarded from the datasets and those metabolites having <50% missing value were subjected to missing value imputation by half of the minimum positive value. Both datasets were normalized to remove the major differences and make metabolic features more comparable to each other.22 The LC-MRM/MS data-set was normalized to median, cube root transformed and range scaling method was used for data scaling. Similarly, GC-MS data was normalized to sum, cube root transformed and auto-scaled.25 Univariate and multivariate statistical analyses were applied to these normalized datasets in order to identify the differentially expressed metabolites that can unambiguously discriminate MM subjects from their respective controls.23,26 Univariate statistical methods such as Wilcoxon rank t-test (p < 0.05) and log2 fold change (≥0.58/≤−0.58) were used to select statistically significant metabolites in both the datasets. Furthermore, multivariate statistical methods like principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA) and orthogonal partial least squares discriminant analysis (OPLS-DA) were employed to visualize the overall distribution of serum metabolites among the study cohort as well as their efficacy in discrimination of MM subjects from the respective controls. Unlike PCA, which is an unsupervised multivariate statistical approach, PLS-DA and OPLS-DA are supervised multivariate statistical methods and a cross validation is needed to avoid over-fitting of the data. The OPLS-DA model was verified by building 200 random permutation models and comparing their performance (R2-goodness of fit and Q2-predictive ability of the model) with the original model.23 Similarly, PLSDA model was also verified. Furthermore, variable importance in projection (VIP) score of OPLS-DA model was used to rank the metabolites as per their contribution in segregation of MM subjects from the controls in the score plot. To select the marker metabolites, receiver operating characteristic (ROC) curve analysis was carried out to identify the features with highest specificity, sensitivity and accuracy to segregate the MM subjects from controls.
2.5 Pathway analysis
Both the datasets, targeted and untargeted were explored towards metabolic pathway analysis for identification of the MM inducible metabolic pathway alterations. The analyses were carried out by MetPA tool of the MetaboAnalyst 3.0 web application (http://metpa.metabolomics.ca). Pathway impact score was used as a measure to identify the altered metabolic pathways. The pathway impact score was calculated based on number of matched metabolites from datasets to a particular metabolic pathway. The hypergeometric test was used for over-representation analysis that tested for its enrichment in a specific pathway and was compared according to random hits. Further the genes associated with the differentially expressed metabolites were identified using Kyoto Encyclopedia of Genes and Genomes (KEGG) database and Human Metabolome Database (HMDB).3. Results
3.1 LC-MRM/MS based targeted metabolomic profiling
In targeted approach, 81 metabolites were consistently detected in all samples out of 120 metabolites targeted. We have built the LC-MRM/MS analysis method for this study by finding the MRM transitions for each metabolite using pure standards purchased from Sigma Aldrich. Representative HILIC and T3 column chromatograms of LC-MRM/MS analysis are presented in ESI Fig. S1.† Univariate statistical methods were applied to identify metabolites that are significantly differing in their concentrations among MM subjects and controls. Further, to extract the MM inducible metabolomic signature, multivariate statistical methods such as PCA, PLS-DA and OPLS-DA were carried out. Modest segregation was found in the 2D score plot of the PCA indicating the presence of concentration differences among the metabolites of MM study cohort (ESI Fig. S2†). Moreover, a supervised multivariate statistical approach such as OPLS-DA was used to sharpen the differences between the MM subjects and their respective controls. Robust group segregation was evident from the OPLS-DA score plot which indicates the metabolic alterations exist in the MM study cohort (Fig. 1a). The model based on OPLS-DA statistics was further authenticated by the permutation test utilizing 200 permutations in between the data groups to avoid biasness due to data over fitting (Fig. 1b). The permutation test yielded R2 value of 0.978 and Q2 value of 0.952 depicting that the original model has performed better than the permutated models and assures of good prediction ability. VIP score was used as criteria to select the metabolites that had highest impact to group separation in score plot of OPLS-DA. Combination of univariate and multivariate statistical approaches (VIP > 1, FDR adjusted p < 0.05, log2 FC > 0.58/<−0.58) were used to propose the panel of metabolites that can discriminate MM subjects from the controls (Table 1). It revealed that 26 metabolites were significantly differed among MM study cohort, out of which 11 metabolites were up-regulated while 15 metabolites were down-regulated in MM subjects as compared to control (Table 1). Hierarchical clustering analysis depicted distinct clusters of MM and control subjects, which demonstrate metabolic adaptation of MM subjects is different when compare to healthy controls (Fig. 1c). Further heat map for the differentially expressed metabolites found through LC-MS analysis were showed in Fig. 2 and S4.†
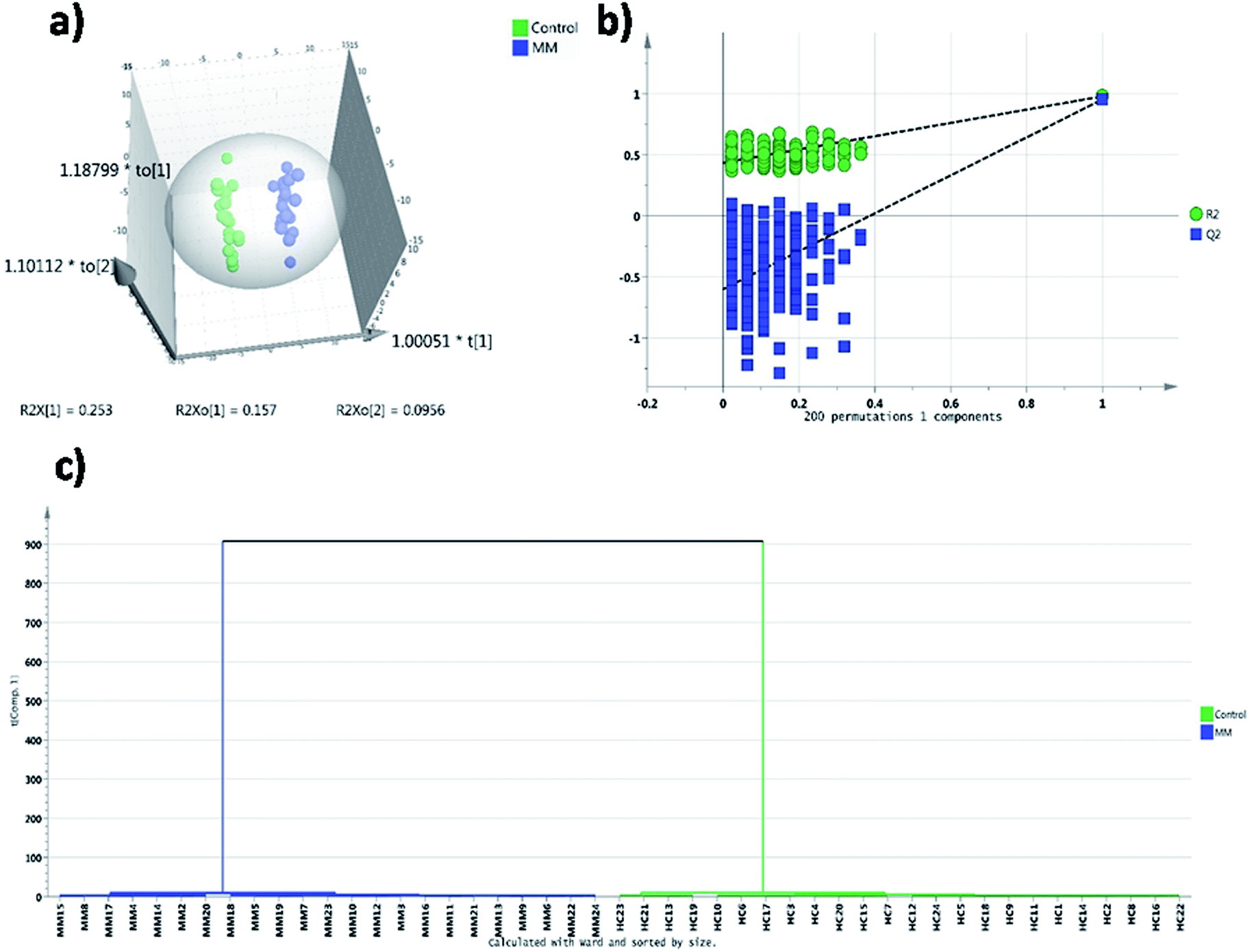 | ||
| Fig. 1 Multivariate statistical analysis of targeted LC-MRM/MS MM serum metabolomics: (a) distribution of MM subjects (n = 24, blue) and healthy controls (n = 24, green) in OPLS-DA score plot, (b) permutation validation of OPLS-DA score plot. Total 200 permutations were carried out on OPLS-DA model and obtained R2 = 0.978 and Q2 = 0.952, where R2 and Q2 indicate the explained variance and predictive ability respectively, (c) hierarchical clustering analysis depicting the segregation of MM (blue) and healthy controls (green). | ||
| Sr. no. | Metabolite | VIP score | p-Value | FDR adjusted p-value | Fold change | AUC | Control CV% | MM CV% |
|---|---|---|---|---|---|---|---|---|
| 1 | NAD | 1.69 | 1.28 × 10−23 | 7.97 × 10−22 | 17.56 | 0.97 | 16.49 | 13.84 |
| 2 | Adenosine | 1.69 | 1.99 × 10−23 | 7.97 × 10−22 | 13.78 | 0.97 | 23.65 | 18.12 |
| 3 | CYTIDINE | 1.63 | 1.78 × 10−16 | 3.56 × 10−15 | 5.56 | 0.97 | 23.68 | 23.49 |
| 4 | Adenine | 1.60 | 9.36 × 10−17 | 2.50 × 10−15 | 3.62 | 0.98 | 27.44 | 17.32 |
| 5 | Oxaloacetic acid | 1.55 | 9.02 × 10−16 | 1.44 × 10−14 | 0.01 | 0.99 | 18.23 | 17.46 |
| 6 | Guanosine | 1.51 | 5.17 × 10−14 | 6.89 × 10−13 | 3.03 | 0.97 | 21.45 | 25.68 |
| 7 | L-Methionine | 1.38 | 2.41 × 10−9 | 2.75 × 10−8 | 0.60 | 0.94 | 24.58 | 27.84 |
| 8 | Mono-ethylmalonate | 1.38 | 3.06 × 10−9 | 3.06 × 10−8 | 0.63 | 0.92 | 27.96 | 21.52 |
| 9 | L-Leucine | 1.34 | 4.65 × 10−8 | 4.13 × 10−7 | 0.67 | 0.91 | 14.94 | 16.75 |
| 10 | L-Homoserine | 1.31 | 2.56 × 10−7 | 2.05 × 10−6 | 0.64 | 0.90 | 22.34 | 25.73 |
| 11 | L-Threonine | 1.30 | 4.42 × 10−7 | 2.95 × 10−6 | 0.64 | 0.89 | 25.62 | 21.98 |
| 12 | Thymine | 1.28 | 3.68 × 10−6 | 1.92 × 10−5 | 0.62 | 0.87 | 18.82 | 16.72 |
| 13 | Methyl malonate | 1.18 | 3.48 × 10−7 | 2.53 × 10−6 | 1.63 | 0.90 | 12.57 | 15.64 |
| 14 | L-Citrulline | 1.17 | 3.02 × 10−6 | 1.86 × 10−5 | 0.52 | 0.86 | 18.93 | 21.88 |
| 15 | Uracil | 1.17 | 2.49 × 10−5 | 9.04 × 10−5 | 0.65 | 0.81 | 28.72 | 24.42 |
| 16 | L-Cysteine | 1.17 | 1.79 × 10−5 | 6.83 × 10−5 | 0.71 | 0.84 | 22.26 | 26.84 |
| 17 | L-Arginine | 1.15 | 1.79 × 10−5 | 6.83 × 10−5 | 0.65 | 0.82 | 15.82 | 19.68 |
| 18 | Xanthosine dihydrate | 1.09 | 0.000619 | 0.001415 | 0.62 | 0.76 | 25.68 | 18.94 |
| 19 | Riboflavin | 1.09 | 4.14 × 10−6 | 1.95 × 10−5 | 2.90 | 0.87 | 19.84 | 23.46 |
| 20 | UTP | 1.08 | 4.98 × 10−5 | 0.000159 | 0.49 | 0.92 | 22.56 | 26.78 |
| 21 | Succinic acid | 1.07 | 4.09 × 10−5 | 0.000136 | 1.50 | 0.84 | 14.98 | 21.36 |
| 22 | L-Glutamine | 1.06 | 6.66 × 10−5 | 0.000197 | 0.67 | 0.81 | 18.96 | 19.14 |
| 23 | Xylitol | 1.04 | 0.000213 | 0.000567 | 1.73 | 0.86 | 22.46 | 21.58 |
| 24 | Adonitol | 1.04 | 0.000124 | 0.000355 | 1.81 | 0.87 | 18.92 | 27.65 |
| 25 | Cytosine | 1.01 | 6.42 × 10−5 | 0.000197 | 2.96 | 0.85 | 19.74 | 23.74 |
| 26 | Thyroxine | 1.00 | 0.000147 | 0.000406 | 0.60 | 0.79 | 29.76 | 19.52 |
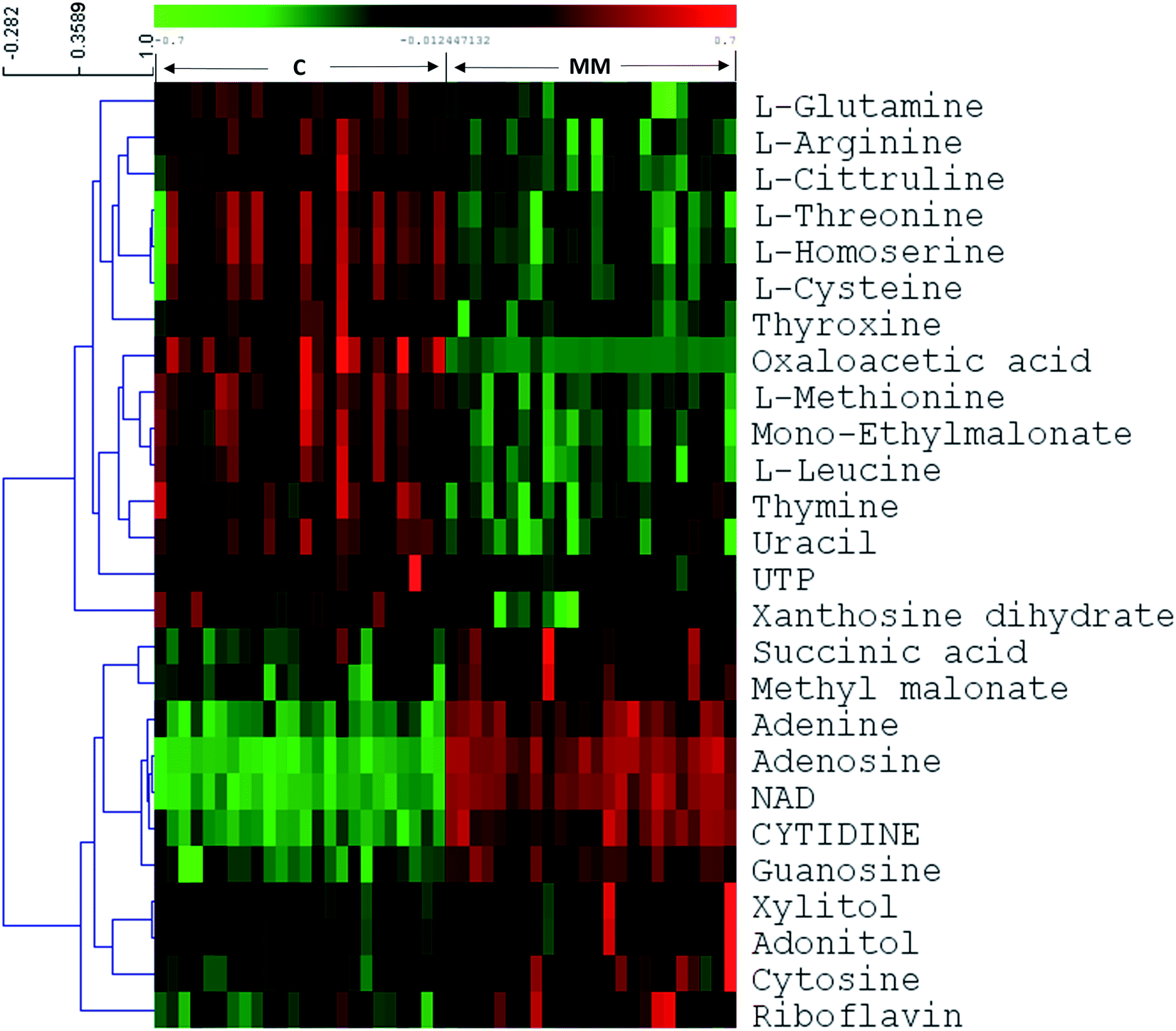 | ||
| Fig. 2 Heat map of LC-MRM/MS based differential metabolites between healthy controls and MM serum samples. The colors from green to red indicate the increased amount of metabolites (C1–C24: healthy controls, M1–M24: multiple myeloma). | ||
3.2 GC-MS based untargeted metabolomic profiling
In untargeted approach, 153 metabolites were detected in MM study cohort. Retention time and NIST library metabolite matching score information of the altered metabolites identified using untargeted GC-MS approach is mentioned in ESI Table S2† as per the MSI guidelines.24,27 For untargeted data analysis, the metabolites that had matching score >80% with the NIST library search were only considered. ESI Fig. S3† depicts a representative GC-MS spectrum for MM serum metabolites. The same set of statistical treatments (univariate and multivariate) mentioned above were performed for the untargeted dataset in order to identify the metabolites that are altered significantly upon MM induction. Multivariate statistical models like PCA and OPLS-DA were built from the untargeted dataset to detect the metabolic fingerprint unique to MM. PCA 2D score plot depicted the fairly segregated MM and control population, which indicates the intrinsic concentration differences exist among the study cohort (ESI Fig. S4†). Furthermore, distinct group separation was observed in the OPLS-DA score plot and metabolic features contributing most to the segregation were identified by the VIP score > 1 (Fig. 3a). A permutation test with 200 permutations was performed to validate the performance of the OPLS-DA model that was built for the MM untargeted metabolite dataset. The permutation test indicates the original model (R2 value = 0.967 and Q2 value = 0.81) is better than permutated models and has not over fitted the data (Fig. 3b). Hierarchical cluster analysis was also in accordance with our earlier findings, revealing clear segregation of study cohort into MM subjects and controls (Fig. 3c). Total 28 most significant metabolites were discovered by using combination of univariate and multivariate approaches (VIP > 1, FDR adjusted p < 0.05, log2 FC > 0.58/<−0.58) of which 16 metabolites were up-regulated and 12 metabolites were down-regulated in MM as compared to control (Table 2). Further, heat map for the differentially expressed metabolites found through GC-MS analysis were showed in Fig. 4 and S5.†
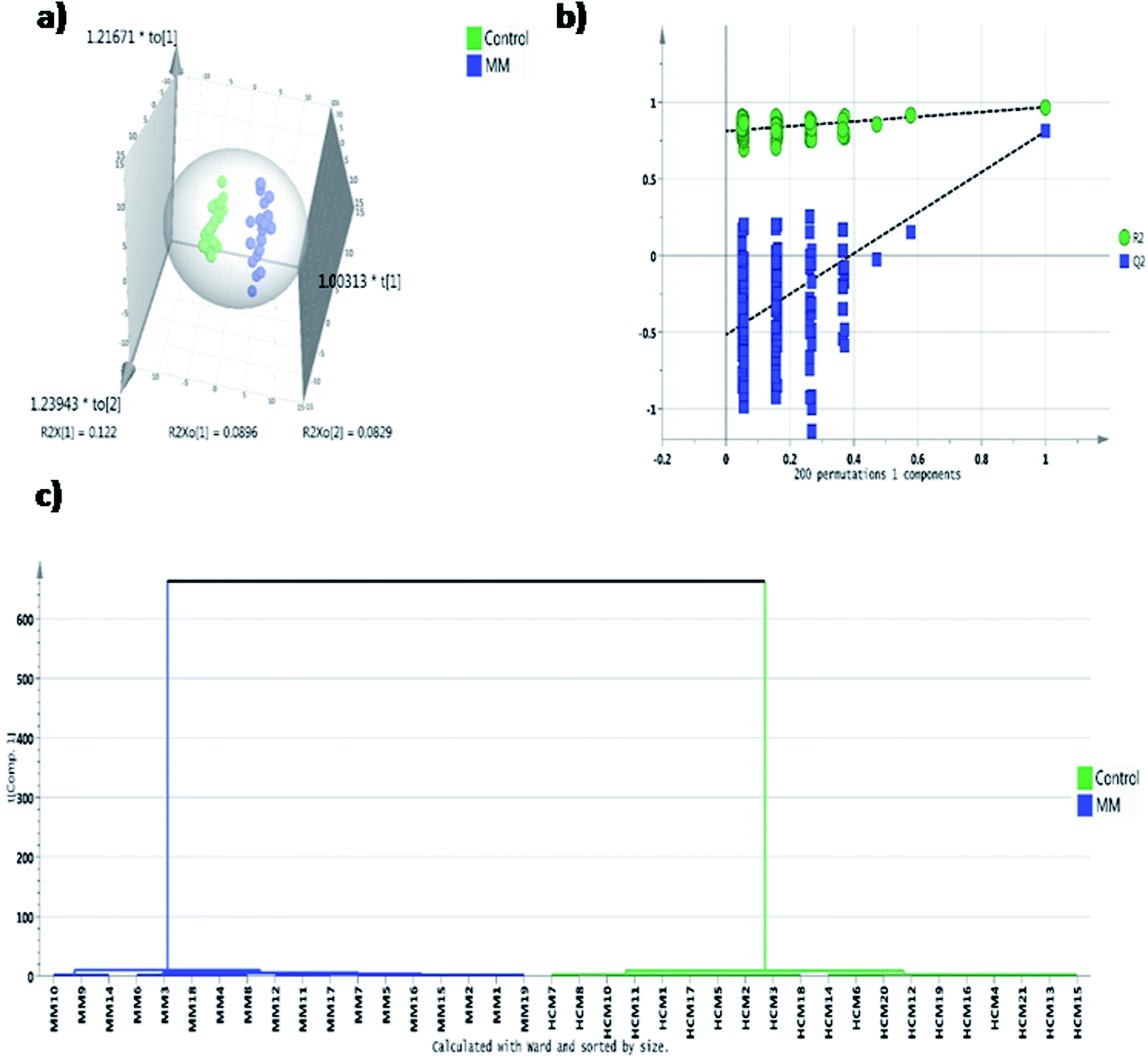 | ||
| Fig. 3 Multivariate statistical analysis of untargeted GC-MS MM serum metabolomics. (a) OPLS-DA score plot depicting the robust separation of MM subjects (n = 19, blue) from healthy controls (n = 21, green), (b) permutation validation of OPLS-DA plot obtained after performing 200 random permutations which yielded R2 = 0.967 and Q2 = 0.81, (c) hierarchical clustering analysis representing good clustering of study subjects. | ||
| Sr. no. | Metabolite | VIP score | p-Value | FDR adjusted p-value | Fold change | AUC | Control CV% | MM CV% |
|---|---|---|---|---|---|---|---|---|
| 1 | D-Ribo-hexitol | 2.30 | 4.20 × 10−20 | 6.43 × 10−18 | 7.08 | 1.00 | 19.36 | 23.58 |
| 2 | Beta-D-glucopyranosiduronic acid | 2.25 | 1.81 × 10−18 | 1.39 × 10−16 | 5.81 | 1.00 | 28.92 | 23.74 |
| 3 | Hexadecanoic acid | 1.93 | 8.82 × 10−8 | 4.50 × 10−6 | 1.80 | 0.95 | 18.62 | 14.85 |
| 4 | 1,5-Anhydro-D-sorbitol | 1.84 | 3.49 × 10−7 | 1.07 × 10−5 | 0.25 | 0.89 | 26.35 | 28.61 |
| 5 | Pseudo uridine | 1.83 | 2.35 × 10−7 | 8.97 × 10−6 | 6.11 | 0.95 | 12.32 | 16.78 |
| 6 | D-Talofuranose | 1.77 | 1.51 × 10−6 | 3.84 × 10−5 | 0.37 | 0.91 | 29.84 | 26.74 |
| 7 | Glucofuranoside | 1.66 | 1.54 × 10−5 | 3.36 × 10−4 | 2.73 | 0.91 | 23.48 | 25.68 |
| 8 | N-Acetyl glucosamine | 1.52 | 2.39 × 10−4 | 3.03 × 10−3 | 0.56 | 0.82 | 19.46 | 23.42 |
| 9 | Purine | 1.51 | 1.15 × 10−4 | 1.88 × 10−3 | 0.45 | 0.84 | 26.59 | 28.32 |
| 10 | Beta-D-galactopyranoside | 1.51 | 1.23 × 10−4 | 1.88 × 10−3 | 0.42 | 0.85 | 21.65 | 25.38 |
| 11 | Pyrimidine | 1.50 | 6.16 × 10−5 | 1.18 × 10−3 | 0.40 | 0.83 | 20.28 | 22.07 |
| 12 | 2-Piperidinecarboxylic acid | 1.50 | 1.60 × 10−4 | 2.22 × 10−3 | 0.44 | 0.81 | 14.63 | 16.09 |
| 13 | D-Lactose | 1.42 | 3.18 × 10−3 | 2.01 × 10−2 | 1.64 | 0.75 | 12.59 | 16.83 |
| 14 | D-Xylofuranose | 1.42 | 4.41 × 10−4 | 4.49 × 10−3 | 2.80 | 0.77 | 15.97 | 16.23 |
| 15 | L-Threonine | 1.42 | 2.06 × 10−3 | 1.50 × 10−2 | 0.20 | 0.69 | 15.86 | 14.93 |
| 16 | Nonadecanoic acid | 1.41 | 8.80 × 10−4 | 8.42 × 10−3 | 2.36 | 0.83 | 24.46 | 26.66 |
| 17 | L-Threitol | 1.40 | 3.58 × 10−4 | 3.92 × 10−3 | 2.24 | 0.81 | 26.32 | 27.39 |
| 18 | D-Glucopyranose | 1.39 | 3.00 × 10−3 | 2.00 × 10−2 | 2.39 | 0.81 | 22.71 | 25.80 |
| 19 | 2-Alpha-mannobiose | 1.38 | 5.87 × 10−3 | 3.10 × 10−2 | 1.50 | 0.73 | 27.28 | 29.84 |
| 20 | Cystathionine | 1.33 | 3.51 × 10−3 | 2.07 × 10−2 | 0.63 | 0.80 | 23.40 | 14.67 |
| 21 | Stearate | 1.27 | 2.82 × 10−3 | 1.96 × 10−2 | 2.11 | 0.87 | 15.82 | 13.26 |
| 22 | Inositol | 1.25 | 1.42 × 10−3 | 1.15 × 10−2 | 1.54 | 0.80 | 18.54 | 19.23 |
| 23 | L-Asparagine | 1.20 | 1.94 × 10−3 | 1.48 × 10−2 | 0.49 | 0.85 | 25.63 | 27.52 |
| 24 | Arachidonic acid | 1.20 | 1.13 × 10−2 | 4.93 × 10−2 | 1.60 | 0.71 | 16.92 | 20.84 |
| 25 | Beta-D-galactofuranose | 1.19 | 7.39 × 10−3 | 3.54 × 10−2 | 2.04 | 0.84 | 24.36 | 19.56 |
| 26 | D-Xylopyranose | 1.18 | 1.31 × 10−3 | 1.15 × 10−2 | 0.23 | 0.79 | 25.87 | 27.32 |
| 27 | Stigmasterol | 1.18 | 1.38 × 10−3 | 1.15 × 10−2 | 0.54 | 0.80 | 28.39 | 27.65 |
| 28 | Maltitol | 1.10 | 7.65 × 10−3 | 3.55 × 10−2 | 3.51 | 0.74 | 26.57 | 28.34 |
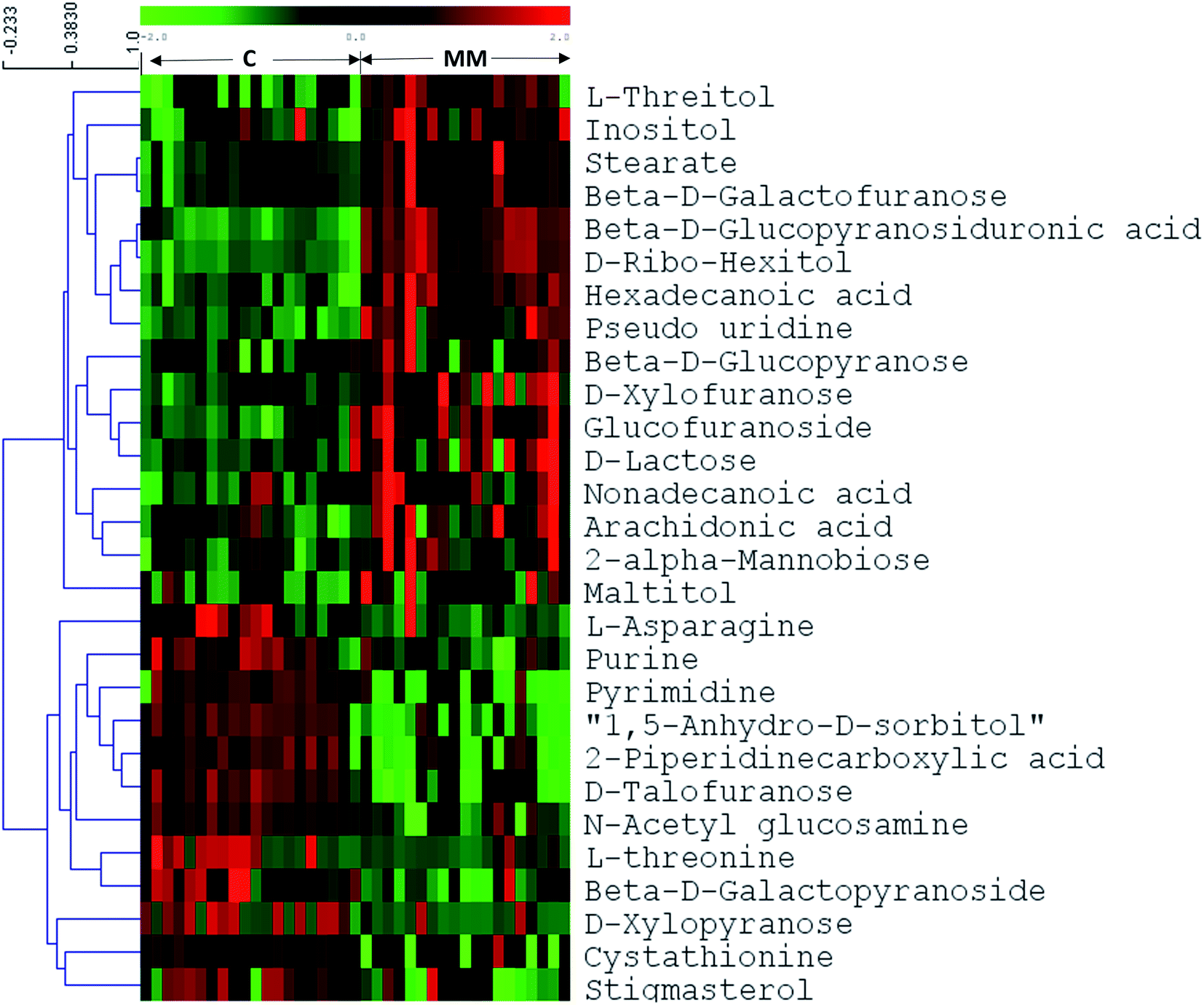 | ||
| Fig. 4 Heat map of GC-MS based differential metabolites between healthy controls and MM serum samples. The colors from green to red indicate the increased amount of metabolites (C1–C21: healthy controls, M1–M19: multiple myeloma). | ||
3.3 Marker metabolite selection from targeted and untargeted datasets
Both targeted and untargeted datasets were subjected to the ROC curve analysis to select the metabolites with the highest specificity, sensitivity and accuracy capable of discriminating the MM subjects from respective controls. The area under the curve (AUC) is used as a measure of the predictive ability of a metabolite in ROC curve analysis. Based on AUC, six metabolites each from both the datasets were projected as marker metabolites to discriminate the MM subjects from their respective controls. In targeted LC-MRM/MS dataset, metabolites such as NAD, adenosine, cytidine, adenine, oxaloacetic acid and guanosine were identified as marker metabolites and their expression pattern along with ROC curve plot is represented in Fig. 5a and b. Likewise, in GC-MS analysis, D-ribo-hexitol, beta-D-glucopyranosiduronic acid, hexadecanoic acid 1,5-anhydro-D-sorbitol, uridine and D-talofuranose are considered as predictive metabolites and their ROC curve plot along with expression pattern is depicted in Fig. 5c and d.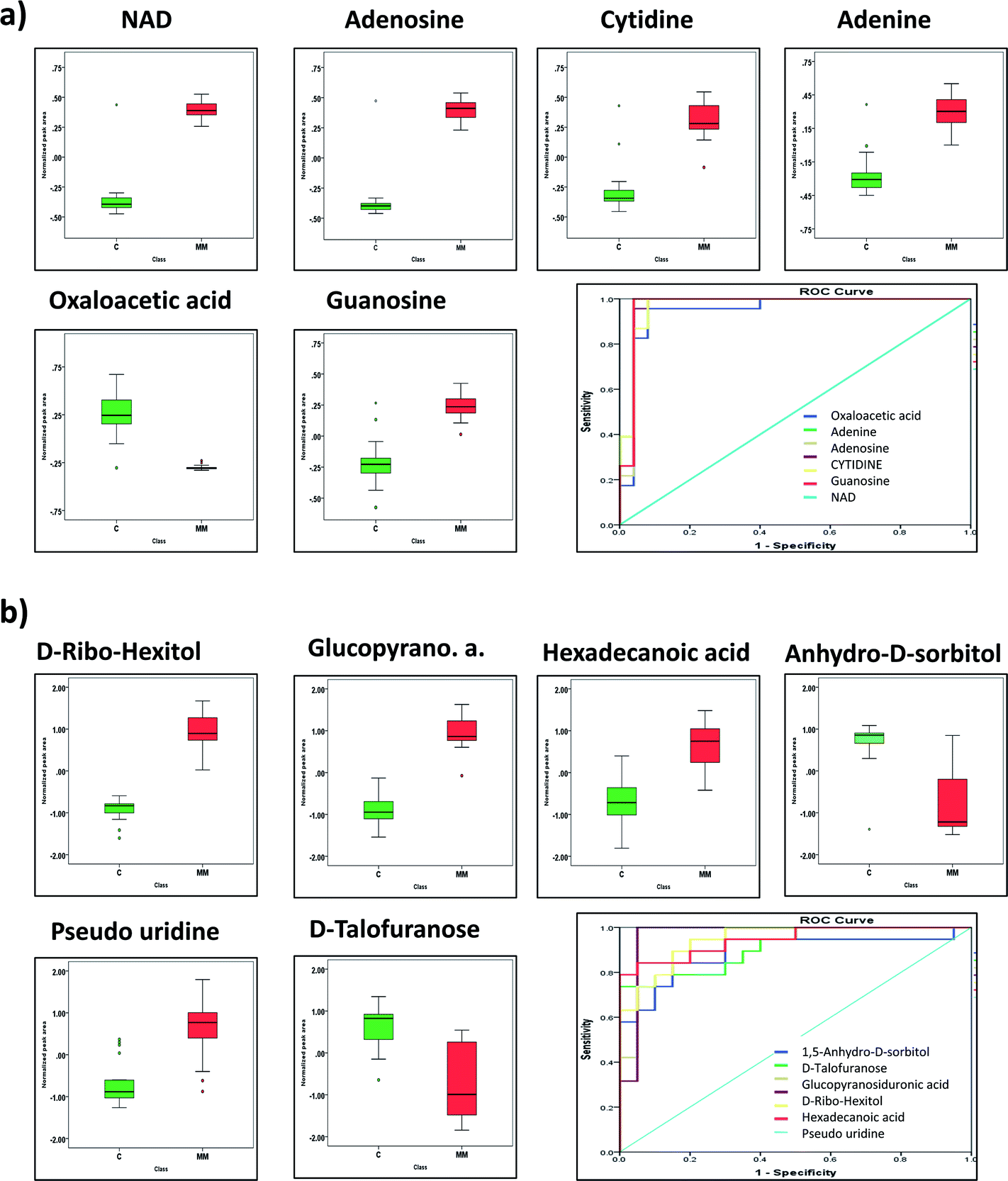 | ||
| Fig. 5 Marker metabolites selected by ROC curve analysis (a) concentration differences between significant metabolites from targeted analysis of MM (red) and healthy control (green) samples demonstrated by box-and-whisker plots, (b) the ROC curve analysis of marker metabolites viz. NAD (AUC = 0.97, sensitivity = 0.95, specificity = 0.96, CI = 95%), adenosine (AUC = 0.97, sensitivity = 0.95, specificity = 0.92, CI = 95%), cytidine (AUC = 0.97, sensitivity = 0.91, specificity = 0.96, CI = 95%), adenine (AUC = 0.98, sensitivity = 0.95, specificity = 0.96, CI = 95%), oxaloacetic acid (AUC = 0.99, sensitivity = 0.92, specificity = 0.97, CI = 95%) and guanosine (AUC = 0.97, sensitivity = 0.87, specificity = 0.88, CI = 95%) from targeted analysis, (c) concentration differences between significant metabolites from untargeted analysis of MM (red) and healthy control (green) samples demonstrated by box-and-whisker plots, (d) the ROC curve analysis of marker metabolites viz. D-ribohexitol (AUC = 1, sensitivity = 0.94, specificity = 0.95, CI = 95%), beta-D-glucopyranosiduronic acid (AUC = 1, sensitivity = 0.94, specificity = 0.95, CI = 95%), hexadecanoic acid (AUC = 0.95, sensitivity = 0.84, specificity = 0.85, CI = 95%), anhydro-D-sorbitol (AUC = 0.89, sensitivity = 0.80, specificity = 0.85, CI = 95%), uridine (AUC = 0.95, sensitivity = 0.84, specificity = 0.90, CI = 95%) and D-talofuranose (AUC = 0.91, sensitivity = 0.75, specificity = 0.79, CI = 95%) from GC-MS analysis. | ||
3.4 Identification of altered MM metabolome associated pathways and genes
To evaluate the impact of MM on metabolic pathways, metabolites from both the MRM based LC-MRM/MS and GC-MS analysis were subjected for pathway analysis using the MetPa tool of MetaboAnalyst 3.0. Pathway analysis results are demonstrated in Fig. 6. Top pathways emerged as pyrimidine metabolism; alanine, aspartate and glutamate metabolism; glycine, serine and threonine metabolism; cysteine and methionine metabolism; purine metabolism; nitrogen metabolism; sulfur metabolism; and citrate cycle (ESI Table S3†). In order to identify the MM differentially regulated metabolites associated genes, 54 serum metabolites were mapped using Kyoto Encyclopedia of Genes and Genomes (KEGG) and human metabolic data base. This analysis resulted in identification of 676 genes which could be related to the MM differentially expressed metabolites (ESI Table S4†).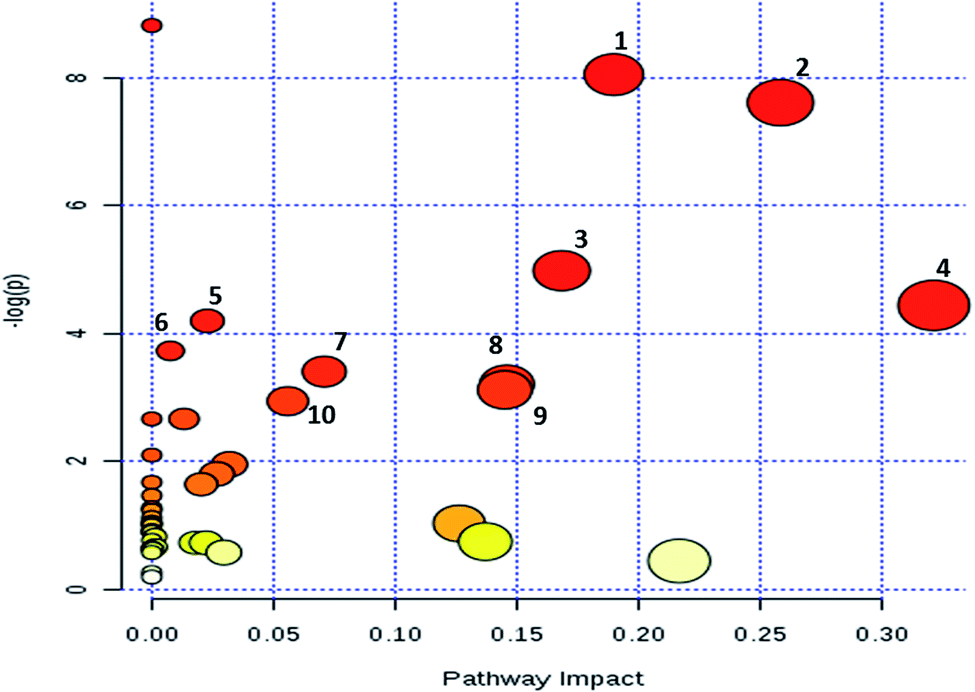 | ||
| Fig. 6 Differentially regulated metabolic pathway analysis of MM serum metabolites. Metabolic pathway map of differentially regulated metabolites generated using MetPa tool of MetaboAnalyst web application. Significant pathways identified includes (1) pyrimidine metabolism, (2) alanine, aspartate and glutamate metabolism, (3) glycine, serine and threonine metabolism, (4) cysteine and methionine metabolism, (5) purine metabolism, (6) nitrogen metabolism, (7) sulfur metabolism, (8) citrate cycle (TCA cycle), (9) riboflavin metabolism (10) pentose and glucuronate interconversions. | ||
4. Discussion
At present, detection of MM is majorly dependent on morphological criteria and its multi-symptomatic nature makes diagnosis and prognosis challenging. This demands the identification of novel clinically relevant molecular markers and potential targets for developing new therapies for MM. Cancer and metabolism share close connectivity as malignant cells undergoes profound metabolic rearrangements to support its proliferative nature and bioenergetics. Hence, in this study, we attempted to explore the serum metabolomic alterations in MM using LC-MRM/MS based targeted as well as GC-MS based untargeted metabolomics. The study showed profound changes in serum metabolites of MM patients as compared to controls. The major metabolic changes identified are discussed further herewith.Targeted LC-MRM/MS analysis unveiled significant alterations of 26 metabolites in MM serum as compared to control. These differentially expressed metabolites consist of 11 up-regulated and 15 down-regulated metabolites. NAD was observed to be up-regulated in MM subjects as compared to the controls, which is considered as a key molecule in various biological processes including energy metabolism and enzymatic regulation.28 It is also the substrate for non-redox reactions that affect gene expression such as Ca2+ transport and apoptosis.29 NAD is involved in the important post-translational modifications of proteins such as acylation and deacylation by SIRTs enzymes.30 Moreover, the aberrant NAD metabolism is reported in various cancers.31 Adenosine, which is found to be increased in MM serum, is an important immunosuppressive molecule.32 Extracellular adenosine bind to adenosine receptors of immune cells suppressing the pro-inflammatory activity of the cells.32 Furthermore, the activation of adenosine receptors increases inflammatory molecules and immunoregulatory cells governing a long-lasting immunosuppressive environment.32 This leads to the discouragement of any antitumor immune responses induced by the body. Oxaloacetic acid (OAA), a key intermediate in the TCA cycle involved in energy production is downregulated in MM subjects when compared with controls. The reason for depleted levels of oxaloacetic acid in serum could be explained by the fact that oxaloacetate along with citrate is used to generate acetyl COA and aspartate that is used in the synthesis of nucleotides and lipids quite extensively in proliferating cells. It is also in agreement with the earlier report that OAA can induce apoptosis in hepatocellular carcinoma cells via inhibition of glycolysis and restoring oxidative phosphorylation.33 Thus, decreased levels of OAA in MM serum samples also suggest a prevalence of glycolysis over oxidative phosphorylation in MM subjects. Guanosine is formed as an intermediate of the nucleic acid breakdown.34 During purine salvage, guanosine can be cleaved further to release guanine, which is salvaged back to GMP by the enzyme hypoxanthine-guanine phosphoribosyl transferase (HGPRT).34 Increased concentrations of guanosine in MM serum imply that the purine salvage is highly active in MM. In addition, elevated levels of cytidine, and adenine in MM represents an overall increase in nucleotide metabolism to support proliferation.34
In untargeted GC-MS analysis, significant changes in 28 metabolites were observed that included 16 up-regulated and 12 down-regulated metabolites. Concentration of hexadecanoic acid (palmitic acid) was found to be increased in MM serum as compared to controls. It is reported that increased palmitic acid level is associated with TL4 mediated invasiveness in pancreatic cancer.35 Moreover, palmitic acid supplementation has shown to rescue adiponectin-induced apoptosis in MM cells.36 Therefore, our observation of increased palmitic acid in MM might be linked with its role in invasiveness and proliferation that needs to be further investigated. Further, decrement in 1,5-anhydro-D-sorbitol was observed in MM serum, which is an indicator of glycaemic control. Its levels manifest the activity of polyol pathways that are mostly functional in hyper glycaemic conditions.37 Increase in uridine levels reflects active involvement of pyrimidine metabolism which is common metabolic alteration observed in cancer.34 It is well-known fact that, N or O linked N-acetylglucosamine (GlcNAc) is one of the prominent post-translational modifications observed on proteins displayed on the cell surface of malignant cells. Moreover, it is now established that GlcNAc modified proteins aid in cell migration and invasion.38 GlcNAc is reported to be mediating cell signaling directly and indirectly.39 The modification of epithelial cadherin (E-cadherin) with O-linked β-GlcNAc is reported to impair cell adhesion and promote tumour cell invasion.40 We have observed decreased levels of GalNAc in MM serum which strengthens the hypothesis of its preferred utilization for post-translational protein glycosylation in malignant condition. For rapid and continuous synthesis of cell constituting basic machinery, proliferating cancerous cells have high demands of nucleotides. This gets achieved by either nucleotide salvage pathways of purines and pyrimidines or de novo synthesis or a combination of both.34 Nucleotide salvage pathway utilizes downstream components of degraded nuclear material like DNA or RNA in the form of purines and pyrimidines to resynthesize the dNTPs for DNA/RNA formation.34 Decrement of both purines and pyrimidines in GC-MS analysis reinforce hyperactivity of the nucleotide salvage pathway to fulfil the proliferative demand.
The significant metabolites obtained from both approaches were further evaluated for their role in various physiological pathways. The metabolic pathway analysis revealed perturbations in alanine, aspartate and glutamate metabolism, pyrimidine metabolism, cysteine and methionine metabolism, glycine, serine and threonine metabolism, purine metabolism, nitrogen metabolism, sulfur metabolism, and citrate cycle (TCA cycle). Several metabolites from purine and pyrimidine metabolism have been deregulated which is in good agreement with previous reports from various cancer metabolomics studies.41 Sulfur metabolism is important in the generation of sulfur-containing amino acids that perform many vital functions.42 Alterations in the TCA cycle, as observed in this study, has been demonstrated to play a vital role in carcinogenesis.43 Our results demonstrate that certain key metabolic pathways including NAD metabolism, TCA cycle, nucleotide metabolism and sulphur metabolism etc. gets altered in MM and targeting these pathways and its intermediary metabolites may help in developing novel targets for treating MM.
5. Conclusions
Overall, the present study demonstrates the potential of serum metabolomics approach towards the segregation of MM cohort from healthy controls. Increased metabolite coverage was obtained with the use of different metabolomics platforms such as LC-MRM/MS and GC-MS. The results obtained are noteworthy and helpful in discrimination of MM individuals from healthy controls. We believe that these results may be useful in future diagnostic, prognostic and therapeutic development. In addition, these results will help to understand the disease pathophysiology of MM in the context of altered cancer metabolism. However, thorough validation of the inferences drawn from this study in a large cohort of samples is required before its implementation in the clinical setting. Validation in large cohort of patients for the metabolite based biosignature, identified in this study, can help diagnose MM in early stages. Early diagnosis will eventually lead to proper on-time therapeutic interventions from the clinicians, which could help in better management of this disease, thereby improving the patient life and survival expectancy.Author contributions
Conceived the study: VC, TC, SS, SR; designed the study: VC, TC, SS, SR; performed the experiments: VC, THM; compiled and analyzed data: VC, THM, KT, RT, SR; statistical analysis: VC, THM, SR; provided clinical samples: TC, SS; provided chemicals and reagents: SR. All authors reviewed the manuscript and contributed in writing.Conflicts of interest
Authors declare that they do not have any conflict of interests.Acknowledgements
General: We thank all the volunteers for participating in this study. Funding: authors would like to acknowledge Department of Biotechnology (DBT), Govt. of India, grant no. BT/PR10855/BRB/10/1330/2014 and NCCS intramural funding. VC and RT acknowledge Council of Scientific and Industrial Research (CSIR), New Delhi, India for fellowship.References
- M. H. Z. Guang, A. McCann, G. Bianchi, L. Zhang, P. Dowling, D. Bazou, P. O'Gorman and K. C. Anderson, Leuk. Lymphoma, 2018, 59, 542–561 CrossRef CAS PubMed
.
- T. Hideshima, P. L. Bergsagel, W. M. Kuehl and K. C. Anderson, Blood, 2004, 104, 607–618 CrossRef CAS PubMed
- O. Landgren and B. Weiss, Leukemia, 2009, 23, 1691 CrossRef CAS PubMed
- V. Scudla, M. Zemanova, J. Minarik, J. Bacovsky, M. Ordeltova, K. Indrak, M. Budikova, L. Dusek and V. Farbiakova, Neoplasma, 2006, 53, 277–284 CAS
- W. M. Kuehl and P. L. Bergsagel, Nat. Rev. Cancer, 2002, 2, 175 CrossRef CAS PubMed
- M. D. Hirschey, R. J. DeBerardinis, A. M. E. Diehl, J. E. Drew, C. Frezza, M. F. Green, L. W. Jones, Y. H. Ko, A. Le and M. A. Lea, Semin. Cancer Biol., 2015, s129–s150 CrossRef CAS PubMed
- P. Mishra and S. Ambs, Mol. Cell. Oncol., 2015, 2, e992217 CrossRef PubMed
- W. B. Dunn, D. Broadhurst, P. Begley, E. Zelena, S. Francis-McIntyre, N. Anderson, M. Brown, J. D. Knowles, A. Halsall and J. N. Haselden, Nat. Protoc., 2011, 6, 1060 CrossRef CAS PubMed
- O. Fiehn, J. Kopka, P. Dörmann, T. Altmann, R. N. Trethewey and L. Willmitzer, Nat. Biotechnol., 2000, 18, 1157 CrossRef CAS PubMed
- E. Zelena, W. B. Dunn, D. Broadhurst, S. Francis-McIntyre, K. M. Carroll, P. Begley, S. O'Hagan, J. D. Knowles, A. Halsall and HUSERMET Consortium, Anal. Chem., 2009, 81, 1357–1364 CrossRef CAS PubMed
- C. Gieger, L. Geistlinger, E. Altmaier, M. H. De Angelis, F. Kronenberg, T. Meitinger, H.-W. Mewes, H.-E. Wichmann, K. M. Weinberger and J. Adamski, PLoS Genet., 2008, 4, e1000282 CrossRef PubMed
- R. Pandher, C. Ducruix, S. Eccles and F. Raynaud, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1352–1358 CrossRef CAS PubMed
- J. Munger, B. D. Bennett, A. Parikh, X.-J. Feng, J. McArdle, H. A. Rabitz, T. Shenk and J. D. Rabinowitz, Nat. Biotechnol., 2008, 26, 1179 CrossRef CAS PubMed
- W. Welthagen, R. A. Shellie, J. Spranger, M. Ristow, R. Zimmermann and O. Fiehn, Metabolomics, 2005, 1, 65–73 CrossRef CAS
- M. R. Pears, J. D. Cooper, H. M. Mitchison, R. J. Mortishire-Smith, D. A. Pearce and J. L. Griffin, J. Biol. Chem., 2005, 280(52), 42508–42514 CrossRef CAS PubMed
- W. B. Dunn, D. Broadhurst, D. I. Ellis, M. Brown, A. Halsall, S. O’hagan, I. Spasic, A. Tseng and D. B. Kell, Int. J. Epidemiol., 2008, 37, i23–i30 CrossRef PubMed
- H. G. Gika, G. A. Theodoridis and I. D. Wilson, J. Chromatogr. A, 2008, 1189, 314–322 CrossRef CAS PubMed
- K. A. Zub, M. M. L. de Sousa, A. Sarno, A. Sharma, A. Demirovic, S. Rao, C. Young, P. A. Aas, I. Ericsson and A. Sundan, PLoS One, 2015, 10, e0119857 CrossRef PubMed
- D. R. Jones, Z. Wu, D. Chauhan, K. C. Anderson and J. Peng, Anal. Chem., 2014, 86, 3667–3675 CrossRef CAS PubMed
- C. Bardeleben, S. Sharma, J. R. Reeve, S. Bassilian, P. J. Frost, B. Hoang, Y. Shi and A. Lichtenstein, Mol. Cancer Ther., 2013, 12(7), 1310–1321 CrossRef CAS PubMed
- L. Puchades-Carrasco, R. Lecumberri, J. Martínez-López, J. J. Lahuerta, M. V. Mateos, F. Prósper, J. San Miguel and A. Pineda-Lucena, Clin. Cancer Res., 2013, 19(17), 4770–4779 CrossRef CAS PubMed
- T. H. More, S. RoyChoudhury, J. Christie, K. Taunk, A. Mane, M. K. Santra, K. Chaudhury and S. Rapole, OncoTargets Ther., 2018, 9, 2678 Search PubMed
- T. H. More, R. Taware, K. Taunk, V. Chanukuppa, V. Naik, A. Mane and S. Rapole, Metabolomics, 2018, 14, 107 CrossRef PubMed
- L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W.-M. Fan, O. Fiehn, R. Goodacre and J. L. Griffin, Metabolomics, 2007, 3, 211–221 CrossRef CAS PubMed
- J. Xia, I. V. Sinelnikov, B. Han and D. S. Wishart, Nucleic Acids Res., 2015, 43, W251–W257 CrossRef CAS PubMed
- H. Flach, M. Rosenbaum, M. Duchniewicz, S. Kim, S. L. Zhang, M. D. Cahalan, G. Mittler and R. Grosschedl, Immunity, 2010, 33, 723–735 CrossRef CAS PubMed
- O. Fiehn, D. Robertson, J. Griffin, M. van der Werf, B. Nikolau, N. Morrison, L. W. Sumner, R. Goodacre, N. W. Hardy and C. Taylor, Metabolomics, 2007, 3, 175–178 CrossRef CAS
- A. Nikiforov, V. Kulikova and M. Ziegler, Crit. Rev. Biochem. Mol. Biol., 2015, 50, 284–297 CrossRef CAS PubMed
- D. Surjana, G. M. Halliday and D. L. Damian, J. Nucleic Acids, 2010, 157591 Search PubMed
- A. R. Stram and R. M. Payne, Cell. Mol. Life Sci., 2016, 73, 4063–4073 CrossRef CAS PubMed
- B. Poljsak, J. Clin. Exp. Oncol., 2016, 5, 4 Search PubMed
- J. Stagg and M. Smyth, Oncogene, 2010, 29, 5346 CrossRef CAS
- Y. Kuang, X. Han, M. Xu and Q. Yang, Cancer Med., 2018, 7, 1416–1429 CrossRef CAS PubMed
- C. K. Mathews, Nat. Rev. Cancer, 2015, 15, 528 CrossRef CAS PubMed
- M. J. Binker-Cosen, D. Richards, B. Oliver, H. Y. Gaisano, M. G. Binker and L. I. Cosen-Binker, Biochem. Biophys. Res. Commun., 2017, 484, 152–158 CrossRef CAS PubMed
- E. Medina, K. Oberheu, S. Polusani, V. Ortega, G. Velagaleti and B. Oyajobi, Leukemia, 2014, 28, 2080 CrossRef CAS PubMed
- A. Ferretti, S. D'Ascenzo, A. Knijn, E. Iorio, V. Dolo, A. Pavan and F. Podo, Br. J. Cancer, 2002, 86, 1180 CrossRef CAS PubMed
- D. J. Gill, K. M. Tham, J. Chia, S. C. Wang, C. Steentoft, H. Clausen, E. A. Bard-Chapeau and F. A. Bard, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3152–E3161 CrossRef CAS PubMed
- J. B. Konopka, Scientifica, 2012, 489208 Search PubMed
- Y. R. Yang, D. H. Kim, Y.-K. Seo, D. Park, H.-J. Jang, S. Y. Choi, Y. H. Lee, G. H. Lee, K. Nakajima and N. Taniguchi, OncoTargets Ther., 2015, 6, 12529 Search PubMed
- A. N. Lane and T. W.-M. Fan, Nucleic Acids Res., 2015, 43, 2466–2485 CrossRef CAS PubMed
- J. T. Brosnan and M. E. Brosnan, J. Nutr., 2006, 136, 1636S–1640S CrossRef CAS PubMed
- S. Cardaci and M. R. Ciriolo, Int. J. Cell Biol., 2012, 161837 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04458b |
| This journal is © The Royal Society of Chemistry 2019 |