
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flexible oxidation of styrene using TBHP over zirconia supported mono-copper substituted phosphotungstate
Rajesh Sadasivan and
Anjali Patel *
*
Polyoxometalates & Catalysis Laboratory, Department of Chemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadooara, Gujarat, India. E-mail: anjali.patel-chem@msubaroda.ac.in
First published on 3rd September 2019
Abstract
A heterogeneous catalyst comprising mono-copper substituted phosphotungstate and hydrous zirconia was synthesized using wet impregnation method, characterized by various physico-chemical techniques and evaluated for solvent-free oxidation of styrene using TBHP as oxidant. Various reaction parameters like time, catalyst amount, amount of TBHP and temperature were optimized with focus on optimum selectivity of styrene-oxide. Further, the catalytic activity was compared with that of unfunctionalized PW11Cu to understand the role of the support. Finally, the role of each component of the reaction was clearly elucidated by a detailed kinetic study of the reaction using both the catalysts.
Introduction
The catalytic oxidation of styrene has gained much attention in industry as well as in academia, as it forms the basis for producing various intermediates, bulk and fine chemicals. The humongous use of benzaldehyde in the flavouring, perfumery, pharmaceutical and dye industries makes it one of the most valuable aromatic aldehydes.1 Moreover, styrene oxide produces styrene glycol and its derivatives which are used in surface coatings and cosmetics.2 It is therefore, not surprising that to date, various homogeneous and heterogeneous catalysts such as metal complexes,3–6 ionic liquids,7,8 metal–organic frameworks (MOFs),9–11 zeolites,12,13 metal nanoparticles (NPs),14,15 metal oxides,16–18 etc. have been used for the oxidation of styrene.1The greatest disadvantage of catalysts in bulk form is their low surface area, which reduces the efficiency of the catalyst.19 Moreover, homogeneous catalysts always have the drawback of limited reusability as they cannot be easily be recycled. In order to overcome these drawbacks, anchoring of catalytic materials on various supports via dative, covalent, acid–basic, or electrostatic interactions has emerged as an upcoming research area.20–23 Amongst all materials, polyoxometalates (POMs) anchored to various supports have received increasing attention as they can undergo fast reversible multi-electron redox transformations under mild conditions. Use of different supports like mesoporous silica, zirconia, zeolites, and alumina to immobilize POMs have been explored for oxidation of styrene.
In 2008, Tangestaninejad et al. synthesized di-vanadium substituted phosphomolybdate supported on mesoporous MCM-41 and used the same for oxidation of various cyclic and aromatic alkenes to their respective aldehydes and ketones under UV radiations.24 In the same year, Guo et al. synthesized periodic mesoporous composite catalysts, [(n-C4H9)4N]4[γ-SiW10O34(H2O)2]/SBA-15], and used for epoxidation of styrene.25 In 2009, various transition metal substituted polyoxotungstates, K10−nXn+MW11O39 (X = P/Si, M = Co/Ni/Cu/Mn), were supported on Schiff base modified SBA-15 by Guo et al. These were further used for selective oxidation of styrene to benzaldehyde.26 Later, in 2010, Kholdeeeva et al. synthesized MIL-101 supported phosphotungstates [(PW4O24)3− and (PW12O40)3−] and used them as sustainable and reusable catalysts for selective epoxidation of styrene.27 In 2013, Balula et al. reported the synthesis of SiW11Co and SiW11Fe immobilized onto an amine functionalized SBA-15 and investigated their catalytic property for oxidation of styrene and geraniol.28 In the following year, the same group reported the synthesis of a zinc substituted polyoxometalate, [PW11Zn-(H2O)O39]5− (PW11Zn), and encapsulation into silica nanoparticles using a cross-linked organic–inorganic core. They further used the same as a versatile catalyst for the oxidation of styrene.29 Selective oxidation of styrene to epoxystyrene was carried out by Zhang et al. in the same year using phosphomolybdic acid supported on 1-triethoxysilylpropyl-3-methylimidazoliumchloride IL modified MCM-41.30 Our group has worked extensively in this field wherein, a wide range of POMs have been anchored on various metal-oxide as well as silica based supports and used for styrene oxidation. Parent as well as mono-lacunary phosphotungstates, -molybdates and silicotungstates anchored to zirconia, alumina, mesoporous silica and zeolites were used for oxidation of styrene with green oxidants like hydrogen peroxide and molecular oxygen.31–37
A literature survey showed that there are no reports available where copper substituted phosphotungstate is supported on zirconia. Further, all the above mentioned reports have used either H2O2 or molecular O2 as oxidants. No accounts are available where tert-butyl hydroperoxide (TBHP) has been used as the oxidant for oxidation of styrene, despite it being an equally green and environmentally benign oxidant.
In the present work, we have anchored mono-copper substituted POM to hydrous zirconia by wet impregnation technique and carried out characterization of the supported catalyst by various physico-chemical techniques. We report a comparative study of the catalytic activity of both, the supported as well as unsupported catalysts for the oxidation of styrene using TBHP as the oxidant and discuss the role of the support. Leaching and heterogeneity tests as well as recycle studies have been carried out. Finally, the kinetics of the reaction using both catalysts has also been studied in detail.
Experimental
Materials
All chemicals used were of A. R. grade. 12-tungstophosphoric acid, copper chloride dihydrate, cesium chloride, styrene, dichloromethane, 70% tert-butyl hydroperoxide, liq. NH3 and sodium hydroxide were obtained from Merck. Zirconium oxychloride was procured from Loba Chemie. All chemicals were used as received.Synthesis of zirconia
Hydrous zirconia was synthesized using a technique previously reported by our group.38 To an aqueous solution of ZrOCl2·8H2O, ammonia solution was added drop-wise upto pH 8.5. This was aged at 100 °C for 1 h in a water bath, filtered, washed and dried at 100 °C for 10 h. The obtained material was designated ZrO2.Synthesis of mono-copper substituted phosphotungstate39
H3PW12O40·nH2O (2.88 g; 1 mmol) was dissolved in 10 mL of water and the pH of the solution was adjusted to 4.8 using supersaturated NaOH solution. The solution was heated to 90 °C with stirring. CuCl2·2H2O (0.17 g; 1 mmol) was dissolved in minimum amount of water and then added drop-wise to the hot POM solution. This was then air-refluxed for 1.5 h at 90 °C, filtered hot, and solid CsCl (0.5 g) was immediately added. The resulting greenish blue precipitates were filtered, dried at room temperature and designated as PW11Cu.Synthesis of mono-copper substituted phosphotungstate supported on zirconia
30% PW11Cu supported over zirconia was synthesized by wet impregnation method. To the aqueous solution of PW11Cu (0.3 g/30 mL), 1 g ZrO2 was added and dried at 100 °C for 10 h and the resulting material was designated 30% PW11Cu/ZrO2. On similar lines, 10%, 20% and 40% PW11Cu/ZrO2 were prepared taking 0.1 g/10 mL, 0.2 g/20 mL and 0.4 g/40 mL aqueous solutions of PW11Cu respectively and designated as 10% PW11Cu/ZrO2, 20% PW11Cu/ZrO2 and 40% PW11Cu/ZrO2 respectively.Acidity determination by potentiometry
The different types of acidic sites were determined by potentiometric titrations using n-butyl amine.40 0.50 g of the synthesized material was suspended in 50 mL acetonitrile and aged at 25 °C. To this, 0.1 mL of 0.05 N n-butyl amine in acetonitrile was added at regular time intervals and the potential (mV) after each addition was recorded.Characterization
The synthesized material was characterized for its acidic strength, and also by thermo gravimetric-differential thermal analysis (TG-DTA), BET surface area analysis, Temperature Programmed Reduction (TPR), FT-IR spectroscopy, FT-Raman spectroscopy, 31P MAS NMR Spectroscopy, Powder XRD and ESR Spectroscopy. Adsorption–desorption analysis for specific surface area calculations was carried out in the Micrometrics ASAP 2010 instrument at −196 °C. TGA was carried out using Mettler Toledo Star SW 7.01 instrument up to 550 °C. The TPR studies were investigated in a self-made reactor set-up with a quartz reactor vessel. 50 mg of sample was taken and heated up to 800 °C and the linear ramping rate was 10 °C min−1 with 5% (35 mL min−1) H2/Ar flow for 60 min. the consumption of H2 gas was monitored using GC instrument equipped with TCD (m/s, CIC Instruments, India). FT-IR spectra of the sample were obtained by using the KBr pellet on the Perkin Elmer instrument. 31P MAS NMR was recorded in JOEL ECX 400 MHz High Resolution Multinuclear FT-NMR spectrometer for solids. ESR spectra were recorded on a Varian E-line Century series X-band ESR spectrometer (liquid nitrogen temperature and scanned from 2000 to 3000 Gauss).Catalytic reaction
Oxidation of styrene was carried out using both, unsupported PW11Cu as well as PW11Cu/ZrO2 as catalyst, in a 50 mL batch reactor attached to a double-walled air condenser on a magnetic stirrer and heating plate. Styrene (10 mmol), catalyst and TBHP were added to the substrate. Due to absence of any aqueous media, the catalyst remained heterogeneous. Dichloromethane was used to extract the products after the reaction, which were then analysed using Shimatzu-2014 Gas Chromatograph, using an RTX-5 capillary column. The products were identified by comparison with authentic samples.Leaching test
Polyoxometalates can be easily characterized by a clear heteropoly blue colour when reacted with a mild reducing agent like ascorbic acid. This method was used to check for leaching of PW11Cu from the support.Results and discussion
Catalyst characterization
Absence of blue colour on reaction with ascorbic acid denoted that leaching of PW11Cu from ZrO2 into the reaction medium does not take place, thereby indicating that there are strong interactions between the POM and support.In order to find out the optimum amount of loading, a preliminary reaction was carried out at varied % loading of active species (10–40%) and the results are presented in Fig. 1. It is seen that with increase in loading amount up to 30%, there is a steady increase in conversion, as expected. But it is interesting to note a decrease in selectivity of benzaldehyde with a steady increase in selectivity of styrene-oxide. Beyond 30% loading, there is a decrease in % conversion of styrene, which may be attributed to blocking of catalytic sites due to excess loading.
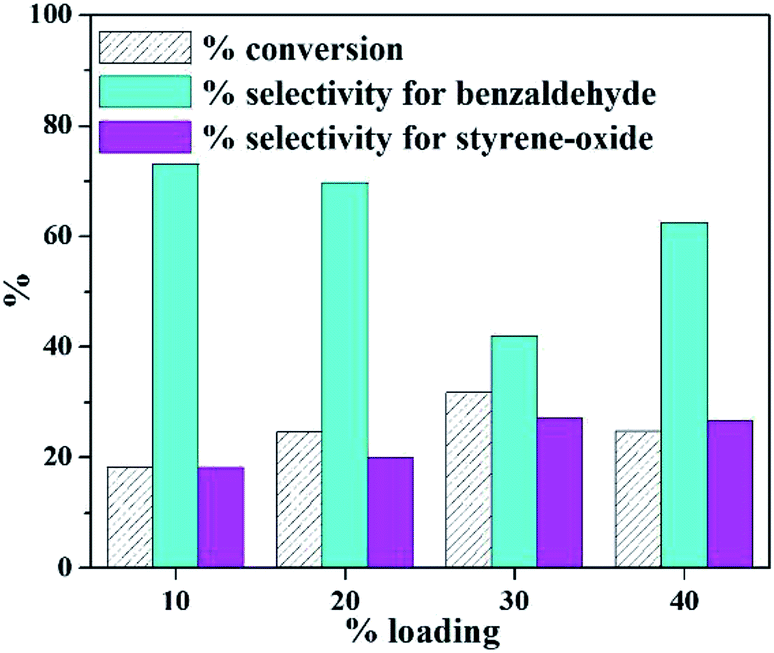 | ||
| Fig. 1 Effect of % loading (catalyst amount – 25 mg; time – 8 h; TBHP – 2 mL; temperature – 60 °C). | ||
It is well known that the completion of the reaction to give benzaldehyde depends on the acidity of the catalyst. In the present case, the acidity of the catalyst decreases with increase in % loading of active species. As a result, the selectivity of the intermediate, i.e., styrene-oxide tends to increase. Keeping in mind the importance of epoxide in the chemical industry, 30% PW11Cu/ZrO2 was optimized and further characterizations as well as optimizations have been carried out using 30% PW11Cu/ZrO2 designated as only PW11Cu/ZrO2.
TGA of PW11Cu/ZrO2 (Fig. 2) shows initial weight loss of 4.4% up to 150 °C, which is attributed to adsorbed water. Further weight loss of 7.2% is noticed between 180–350 °C because of water of crystallization. No other substantial weight loss indicates that the synthesized material is stable up to 550 °C.
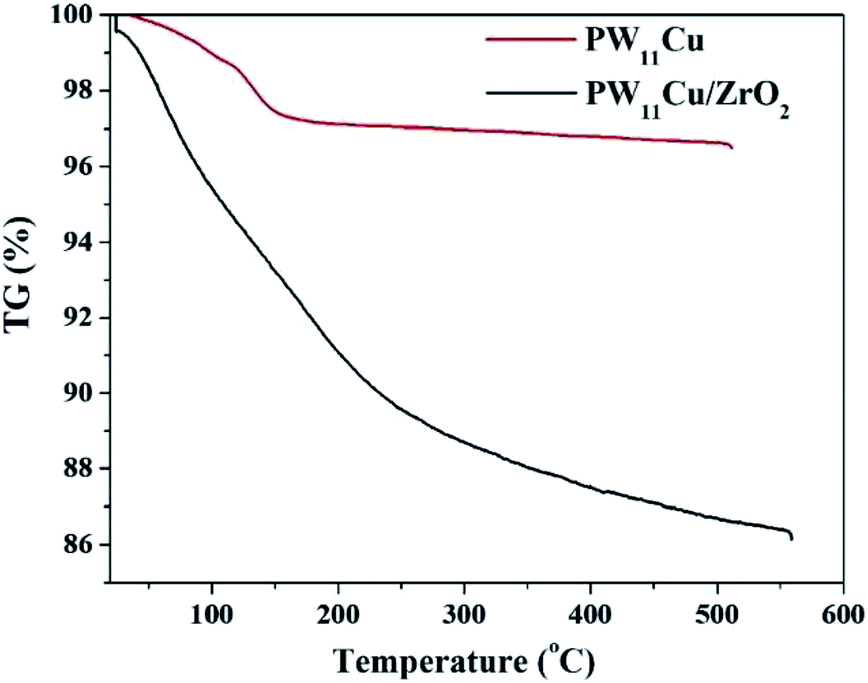 | ||
| Fig. 2 TGA of PW11Cu and PW11Cu/ZrO2. | ||
The FT-IR spectra of ZrO2, PW11Cu and PW11Cu/ZrO2 are shown in Fig. 3. ZrO2 shows bands at 3365, 1625, 1396 and 608 cm−1 which are characteristic of asymmetric O–H stretching, H–O–H and O–H–O bending and Zr–O–H bending vibrations respectively.
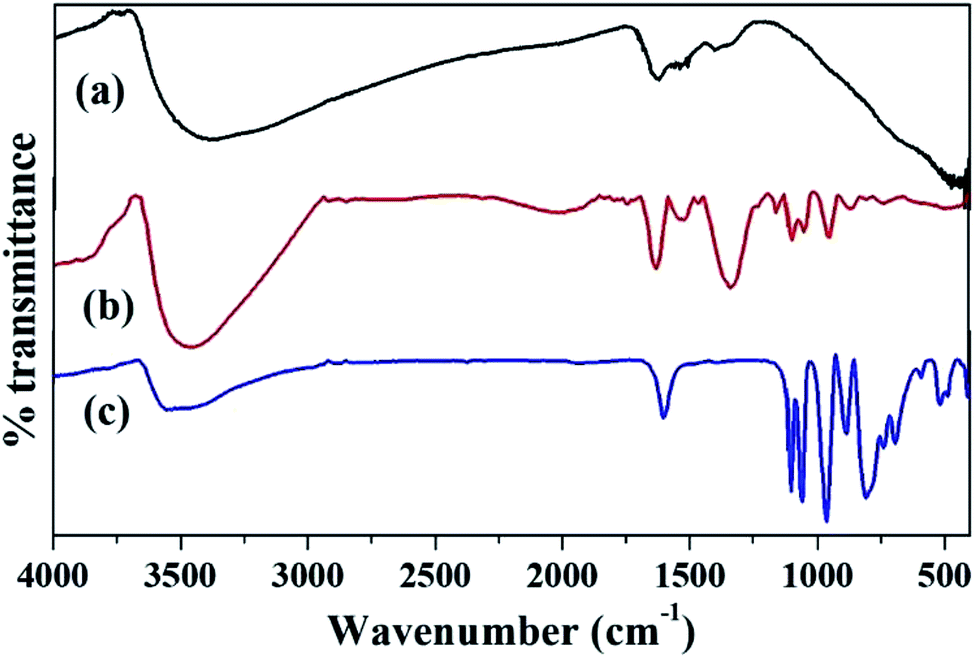 | ||
| Fig. 3 FT-IR spectra of (a) ZrO2, (b) PW11Cu/ZrO2 and (c) PW11Cu. | ||
PW11Cu exhibits bands at 1103 and 1060 cm−1 corresponding to P–O stretching, 964 cm−1 corresponding to W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, 887 and 810 cm−1 corresponding to W–O–W stretching and 516 cm−1 corresponding to Cu–O stretching vibrations respectively. The FT-IR spectrum of PW11Cu/ZrO2 shows characteristic bands of both PW11Cu and ZrO2. Bands at 1103 and 1064 cm−1, 952 cm−1 and 813 cm−1 corresponding to P–O, WO and W–O–W stretching vibrations are similar to PW11Cu. In addition, broad bands at 3417, 1631 and 1402 cm−1 correspond to O–H stretching, H–O–H and O–H–O bending respectively are similar to that of ZrO2. The slight shifts observed in frequencies of supported catalyst is attributed to the chemical interaction of PW11Cu with the support. However, the Cu–O vibration band is not visible, due to overlapping with the Zr–O–H band and hence confirmed by FT-Raman.
O stretching, 887 and 810 cm−1 corresponding to W–O–W stretching and 516 cm−1 corresponding to Cu–O stretching vibrations respectively. The FT-IR spectrum of PW11Cu/ZrO2 shows characteristic bands of both PW11Cu and ZrO2. Bands at 1103 and 1064 cm−1, 952 cm−1 and 813 cm−1 corresponding to P–O, WO and W–O–W stretching vibrations are similar to PW11Cu. In addition, broad bands at 3417, 1631 and 1402 cm−1 correspond to O–H stretching, H–O–H and O–H–O bending respectively are similar to that of ZrO2. The slight shifts observed in frequencies of supported catalyst is attributed to the chemical interaction of PW11Cu with the support. However, the Cu–O vibration band is not visible, due to overlapping with the Zr–O–H band and hence confirmed by FT-Raman.
Fig. 4 shows the FT-Raman spectra of PW11Cu as well as PW11Cu/ZrO2. ZrO2 shows a number of broad peaks from 100–800 cm−1 associated with long range amorphous state disordering.41 The Raman spectra of PW11Cu shows peaks at 996 cm−1 corresponding to WO symmetric stretch; 985, 215 and 153 cm−1 corresponding to W–O symmetric stretch; and 967 and 946 cm−1 corresponding to W–O–W symmetric stretch respectively. Further, an additional peak at 483 cm−1 is incorporation of copper in the lacuna of PW11. Similarly, Raman spectra of PW11Cu/ZrO2 shows peaks at 985 cm−1 corresponding to WO symmetric stretch, 972 and 218 cm−1 corresponding to W–O symmetric stretch, and 964 cm−1 corresponding to W–O–W symmetric stretch respectively. A slight shift as well as decrease in intensity, along with absence of some peaks may be because of the interaction of PW11Cu with the support. Thus, FT-IR and FT-Raman confirm that PW11Cu remains intact even after impregnation on to the support.
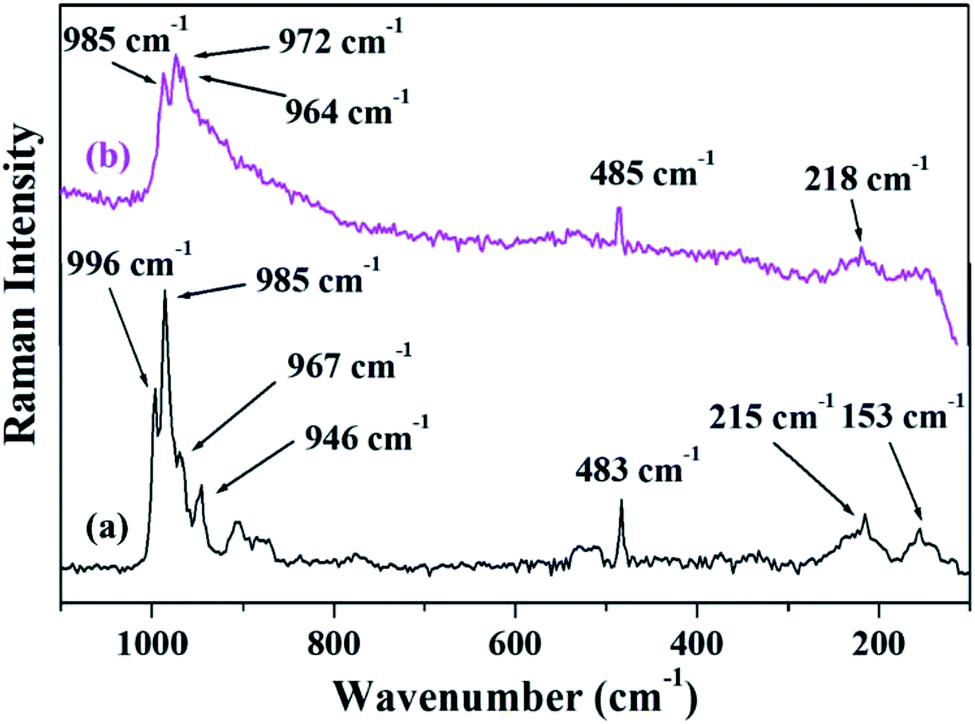 | ||
| Fig. 4 FT–Raman spectra of (a) PW11Cu, (b) PW11Cu/ZrO2. | ||
X-band liquid nitrogen temperature ESR spectra of unsupported PW11Cu, ZrO2 and PW11Cu/ZrO2 are presented in Fig. 5. PW11Cu gives a four line hyperfine spectrum with g = 2.0883 and g = 2.4031, typically found in Cu(II) complexes.39 PW11Cu/ZrO2 retains the hyperfine spectrum of PW11Cu, indicating that copper remains in 2+ oxidation state and the environment around Cu(II) stays intact even after impregnation on to the support. However, the decrease in intensity observed may be due to interaction of PW11Cu with the support.
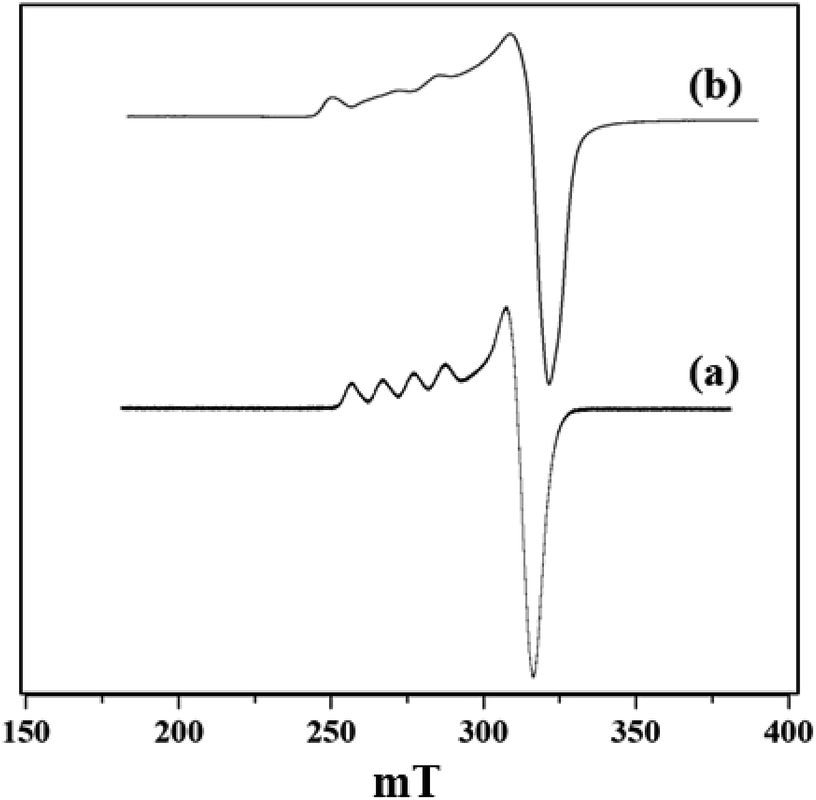 | ||
| Fig. 5 ESR spectra of (a) PW11Cu and (b) PW11Cu/ZrO2. | ||
The H2-TPR spectra of PW11Cu and PW11Cu/ZrO2 are shown in Fig. 6. Romanelli et al. obtained two peaks in the TPR pattern of sodium salt of PW11Cu with maxima at 666 °C and 960 °C which have been assigned to reduction of species after anion decomposition.42
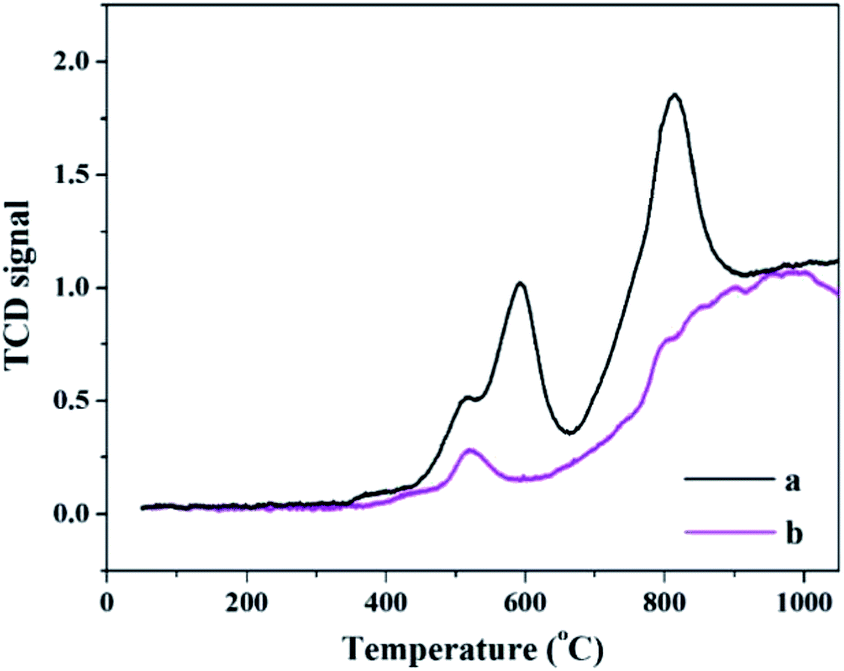 | ||
| Fig. 6 H2-TPR spectra of (a) PW11Cu and (b) PW11Cu/ZrO2. | ||
In the present case, similar maxima are obtained at 592 °C and 819 °C, which is attributed to formation of WO3 species.43 The decrease in reduction temperature is attributed to increase in the consumption of H2 gas due to Cs counter cation.44 The lowering of reduction temperature may also be an indication of enhanced oxidation ability.
The H2-TPR spectra of PW11Cu/ZrO2 has a peak with lesser intensity and shows maxima at 518 °C, while the second peak gets overlapped with the reduction peaks of ZrO2, which fall in the temperature range of 700–900 °C.45 The decrease in intensity as well as reduction temperature may be due to chemical interaction of [PW11Cu]5− anions with the support.42 This is further confirmed by 31P MAS NMR.
31P NMR is an important tool to understand the environment around phosphorus in polyoxometalates as well as the interaction of the anion with support.46 The 31P MAS NMR of PW11Cu and PW11Cu/ZrO2 are presented in Fig. 7. The peak at −10.42 in case of the supported material arises because of the PO4 unit of PW11Cu. The slight downfield shift may be attributed to the hydrogen bonding between the terminal oxygen of the POM and –OH groups of zirconia. The broad peak at −2.35 is attributed to the formation of [![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Zr–OH2]+[Cs4(PW11CuO39)]− species.47–49 This may be explained as follows. Loading of PW11Cu on to ZrO2 is carried out by wet impregnation at 100 °C. Dehydration of water molecules of zirconia occurs, which is followed by subsequent entrapment of PW11Cu anions within the network of zirconia. Further, due to the acidic medium, Zr–OH gets protonated to form [Zr–OH2]+, which acts as a cation for the anionic (PW11CuO39)5− species. As a result, a chemical bond formation occurs between the two as opposed to simple physisorption. This, along with hydrogen bonding, ensures that PW11Cu does not leach during the catalytic reaction and the catalyst remains intact. Intensity of both the peaks in NMR indicates that while majority of PW11Cu form ionic pairs with zirconia, some interact only by hydrogen bonds.
Zr–OH2]+[Cs4(PW11CuO39)]− species.47–49 This may be explained as follows. Loading of PW11Cu on to ZrO2 is carried out by wet impregnation at 100 °C. Dehydration of water molecules of zirconia occurs, which is followed by subsequent entrapment of PW11Cu anions within the network of zirconia. Further, due to the acidic medium, Zr–OH gets protonated to form [Zr–OH2]+, which acts as a cation for the anionic (PW11CuO39)5− species. As a result, a chemical bond formation occurs between the two as opposed to simple physisorption. This, along with hydrogen bonding, ensures that PW11Cu does not leach during the catalytic reaction and the catalyst remains intact. Intensity of both the peaks in NMR indicates that while majority of PW11Cu form ionic pairs with zirconia, some interact only by hydrogen bonds.
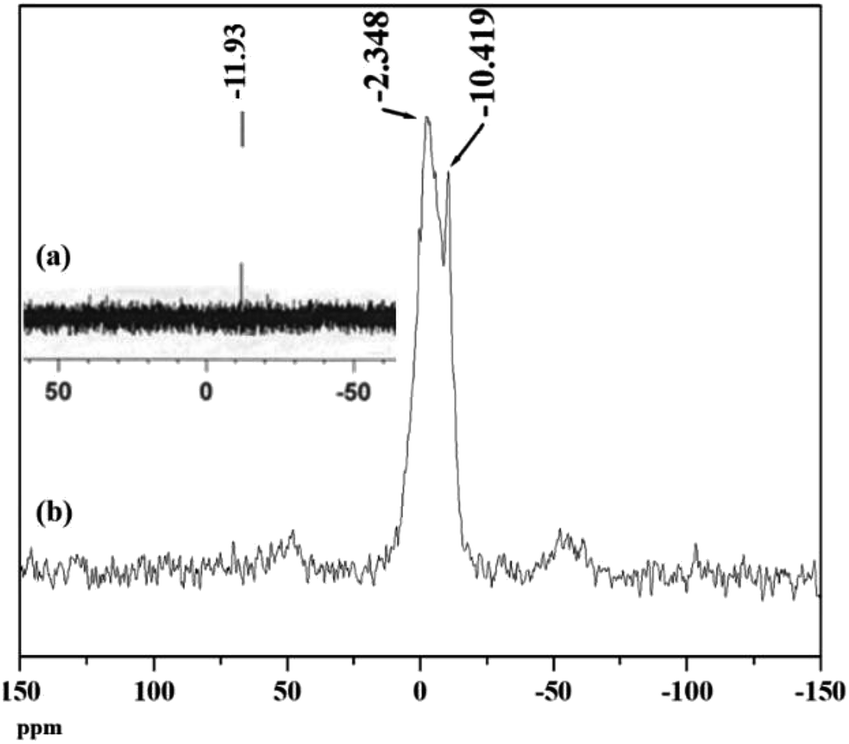 | ||
| Fig. 7 31P MAS NMR of (a) PW11Cu and (b) PW11Cu/ZrO2. | ||
The wide angle powder XRD of unsupported PW11Cu, ZrO2 and PW11Cu/ZrO2 are presented in Fig. 8. PW11Cu shows sharp peaks from 20–30 degrees 2θ characteristic to the Keggin structure, with a slight shift due to incorporation of copper. This is further confirmed by sharp peaks at 48 degrees 2θ attributed to Cu(II).39 The absence of crystalline peaks in case of PW11Cu/ZrO2 indicates complete dispersion of PW11Cu on to the support.
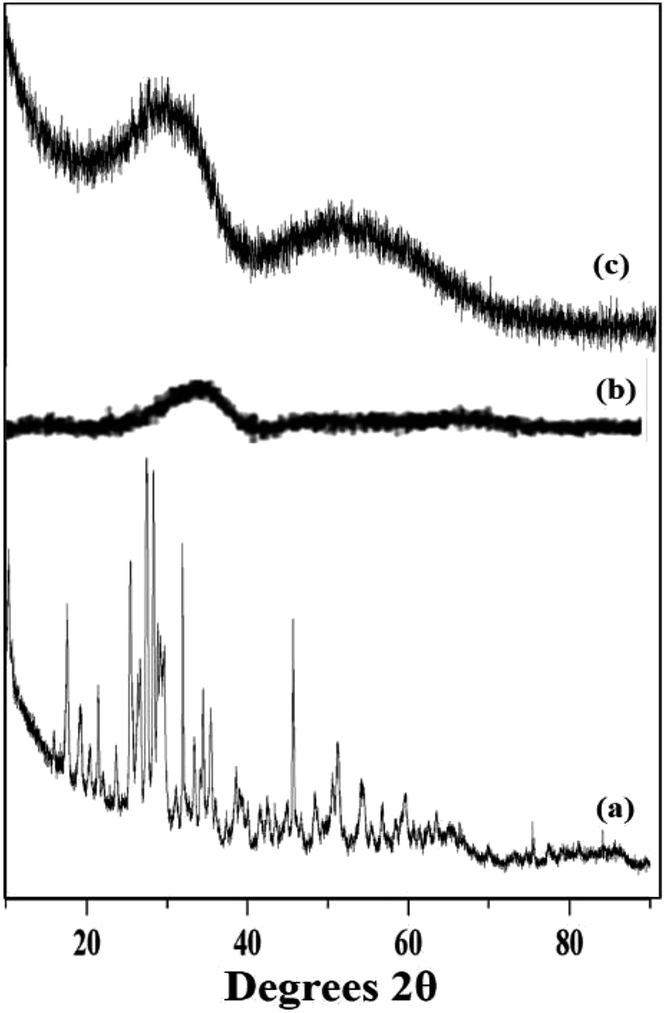 | ||
| Fig. 8 Wide angle powder XRD of (a) PW11Cu, (b) ZrO2 and (c) PW11Cu/ZrO2. | ||
The BET surface area of ZrO2 and PW11Cu/ZrO2 are shown in Table 1. It is clearly seen that the surface area of PW11Cu/ZrO2 (218 m2 g−1) is greater than that of zirconia (170 m2 g−1), because of the supporting, which is as expected. An average pore diameter of 38.25 Å was obtained from the pore size distribution curve (Fig. 9).
| Material | Surface area (m2 g−1) |
|---|---|
| ZrO2 | 170 |
| PW11Cu/ZrO2 | 218 |
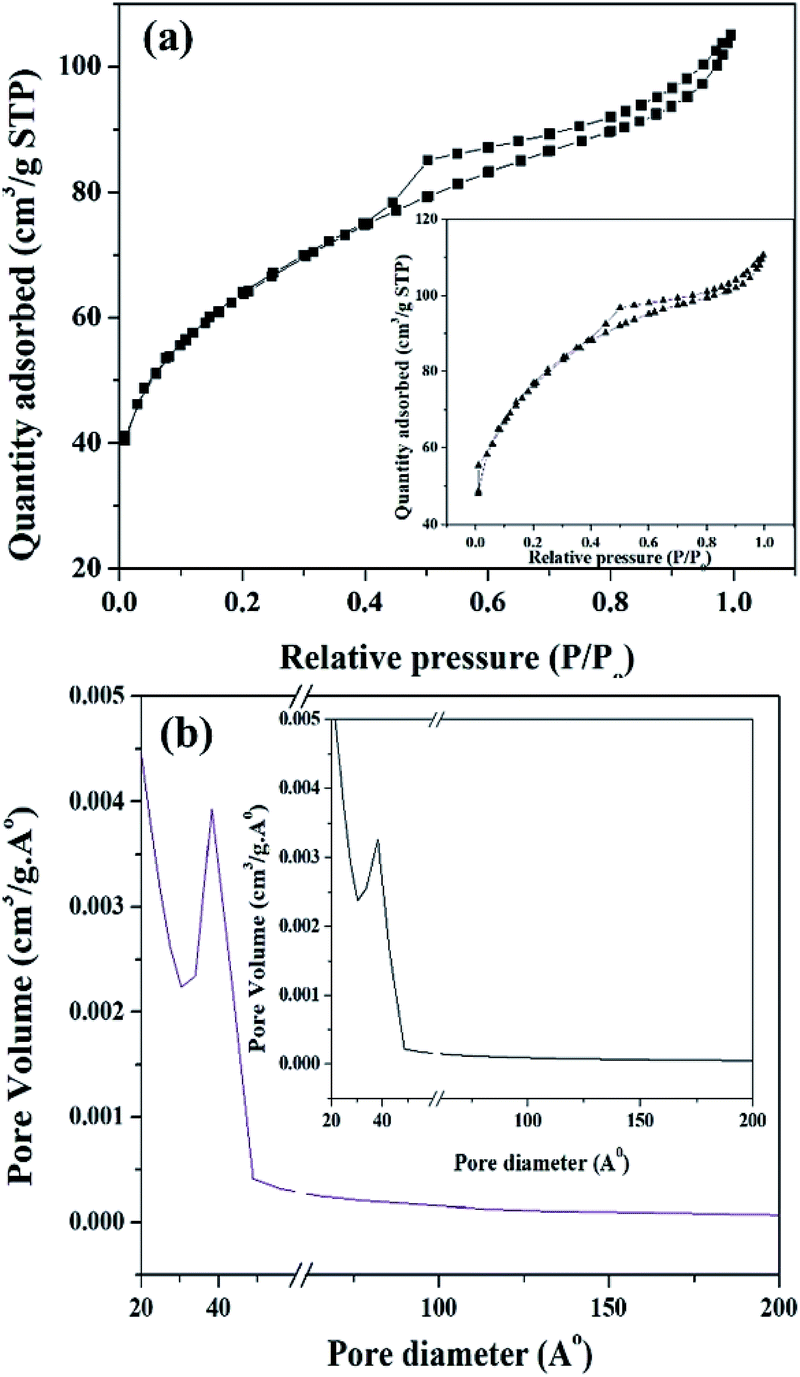 | ||
| Fig. 9 (a) N2 adsorption–desorption isotherm and (b) pore size distribution curve of PW11Cu/ZrO2 and (insets) pure ZrO2. | ||
Catalytic activity – oxidation of styrene
A detailed catalytic study was carried out for the oxidation of styrene using the both, unsupported PW11Cu as well as PW11Cu/ZrO2 for a comparative study on the role of support (Scheme 1). The reaction in the absence of the catalyst gives negligible conversion, indicating the necessity of the catalyst for oxidation reaction. Initially, a preliminary reaction was carried out with H2O2 as the oxidant, and the conversion obtained was insignificant. Hence, further reactions were carried out with TBHP as the oxidant. Reaction parameters like % loading, reaction time, catalyst amount, amount of TBHP and reaction temperature were optimized to give maximum conversion and selectivity of desired product. Each experiment was carried out thrice and the results obtained were reproducible with an error of ±2%. The major products formed were benzaldehyde and styrene-oxide while small amounts of acetophenone and benzoic acid were formed. Small quantities of tert-butyl alcohol was formed as by-product as expected.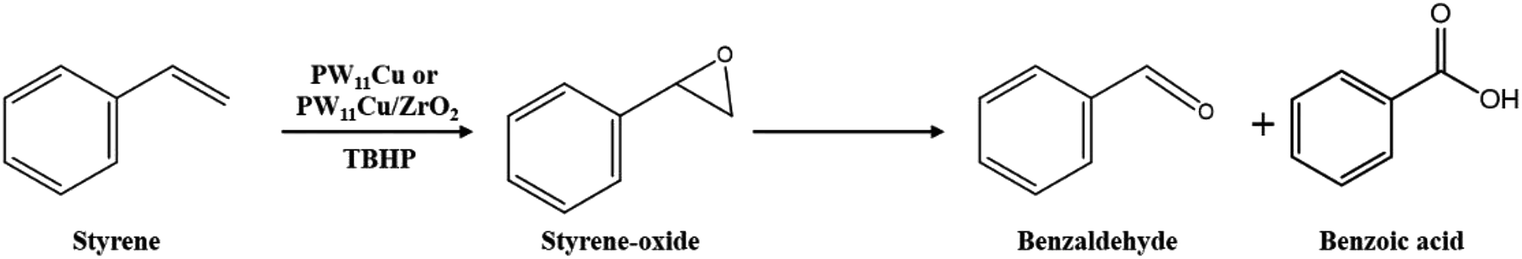 | ||
| Scheme 1 Oxidation of styrene. | ||
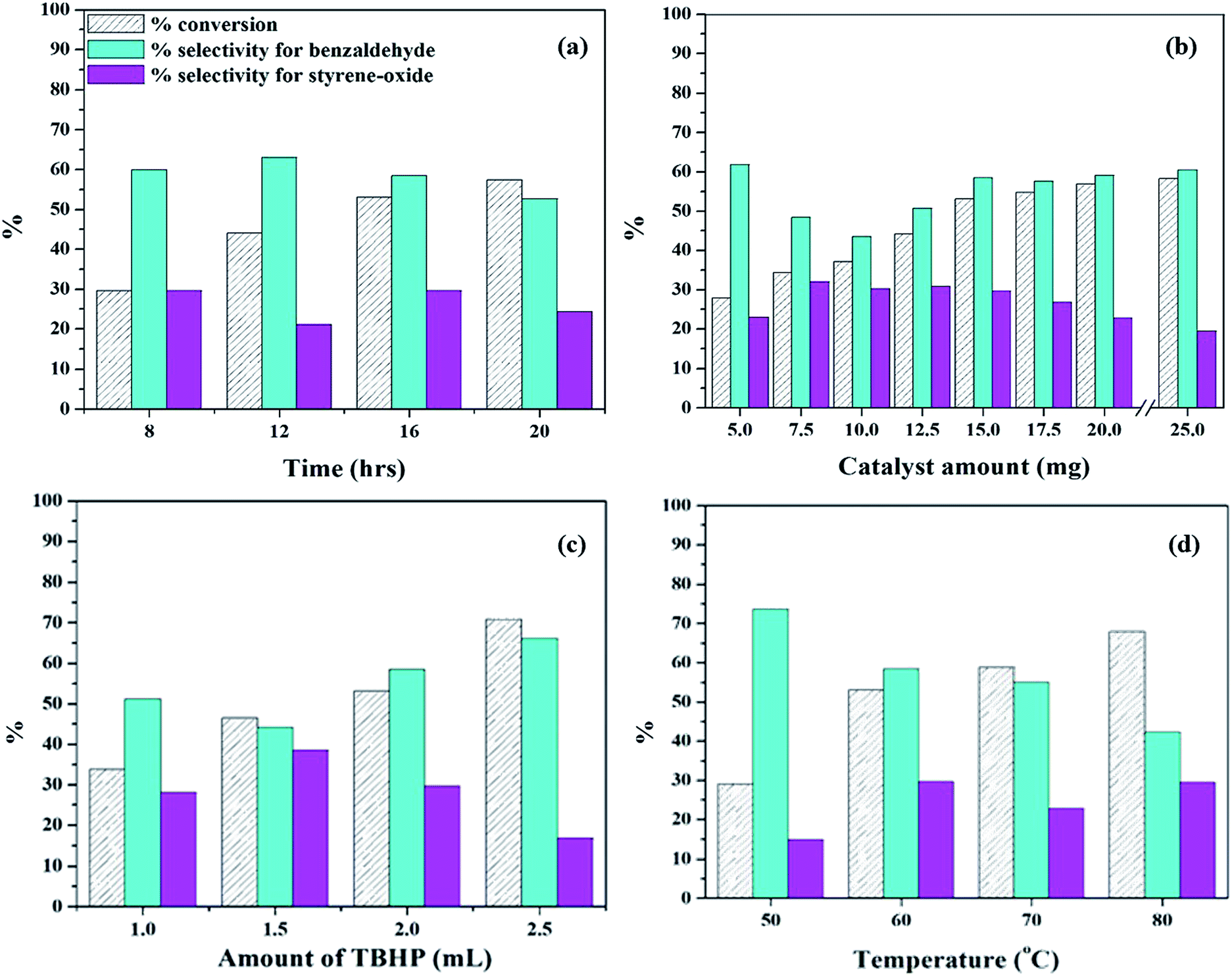 | ||
| Fig. 10 Optimization of parameters for oxidation of styrene (a) effect of time (catalyst amount – 15 mg; temp – 60 °C; TBHP – 2 mL); (b) effect of catalyst amount (time – 16 h; temp – 60 °C; TBHP – 2 mL); (c) effect of amount of TBHP (catalyst amount – 15 mg; temp – 60 °C; time – 16 h); (d) effect of temperature (catalyst amount – 15 mg; time – 16 h; TBHP – 2 mL). | ||
The reaction was then carried out at different catalyst amounts and the results are presented in table 10b. It can be seen that with increase in catalyst amount from 5 mg to 15 mg, there is a steady increase in conversion of styrene. However, the selectivity of benzaldehyde initially decreases up to 10 mg and then increases, while the selectivity of styrene oxide initially increases and then decreases. This may be because with increase in catalyst amount, the reaction tends to move forward towards completion. Beyond 15 mg, the increase in conversion as well as selectivity of benzaldehyde is negligible. Hence, 15 mg was optimized as the catalyst amount.
The amount of TBHP was then varied to study the effect of oxidant and the results are presented in Fig. 10c. With increase in TBHP amount from 1 mL to 2.5 mL, there is an increase conversion and selectivity of benzaldehyde along with decrease in selectivity for styrene-oxide, which is the expected trend. Excess of TBHP would lead to increase in oxygen concentration, and this would lead to further oxidation of benzaldehyde to benzoic acid, as seen in case of 2.5 mL TBHP. Hence, 2 mL TBHP was considered optimum for the reaction.
Finally, the reaction was carried at different temperatures and the results are presented in Fig. 10d. When the temperature was increased from 50 °C to 60 °C, there is an increase in conversion as well as selectivity of styrene-oxide. But on increasing temperature beyond 60 °C, there is a significant decrease in the selectivity of benzaldehyde as well as styrene-oxide. This may be due to the degradation of TBHP at higher temperatures, thereby resulting in polymerization of styrene. Hence, temperature was optimized at 60 °C.
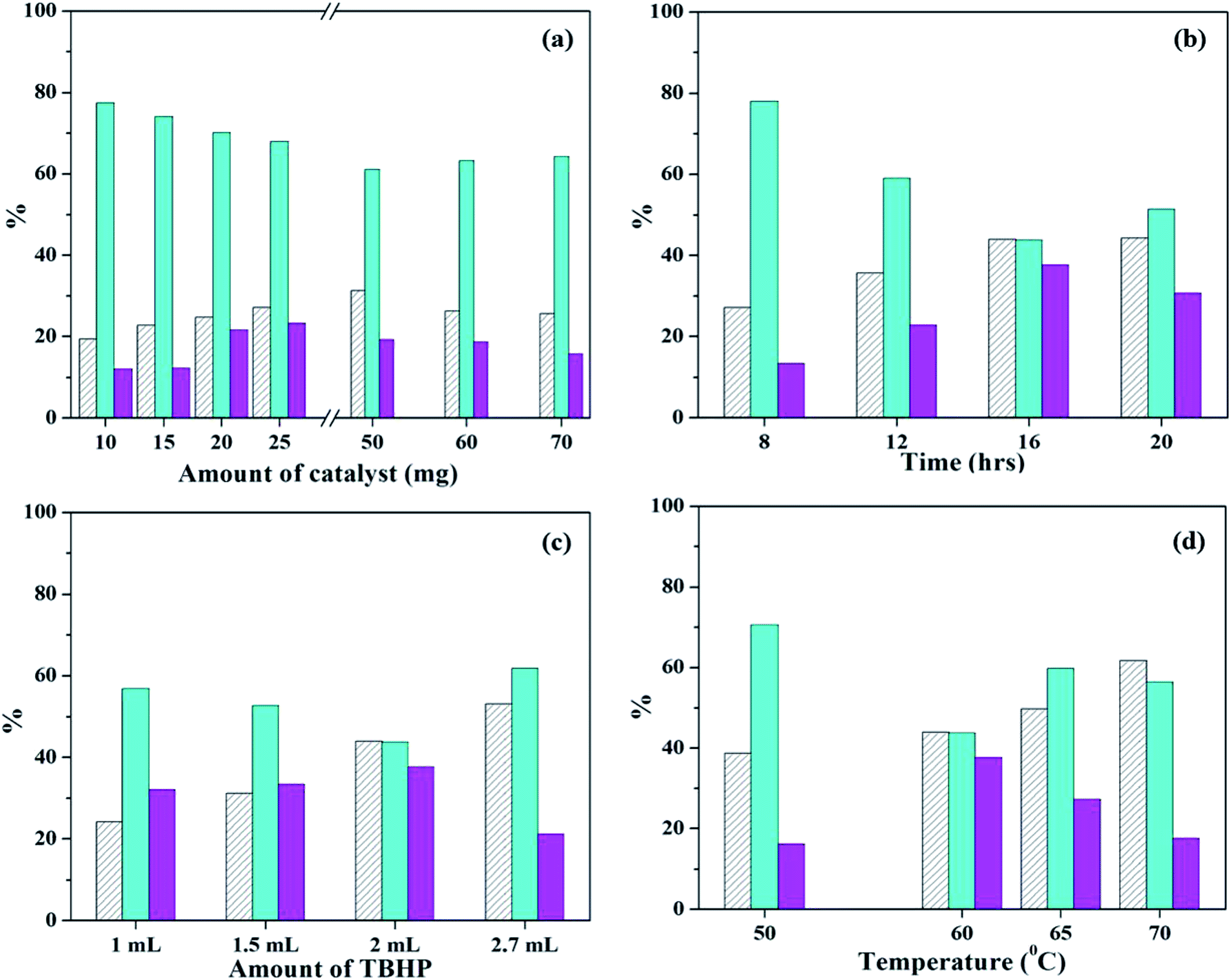 | ||
| Fig. 11 Optimization of parameters for oxidation of styrene (a) effect of catalyst amount (time – 8 h; temp – 60 °C; TBHP – 2 mL); (b) effect of time (catalyst amount – 25 mg; temp – 60 °C; TBHP – 2 mL); (c) effect of amount of TBHP (catalyst amount – 25 mg; temp – 60 °C; time – 16 h); (d) effect of temperature (catalyst amount – 25 mg; time – 16 h; TBHP – 2 mL). | ||
Next, reaction time was optimized keeping all other parameters constant (Fig. 11b). As the time is increased from 8 h to 16 h, steady increase in conversion is observed, along with selectivity of styrene-oxide. Further increase in reaction time shows no significant change in either conversion or selectivity of the required product. Therefore, reaction time was optimized at 16 h.
In order to optimized amount of oxidant, the reaction was carried out by adding different quantities of TBHP (Fig. 11c). With increase in TBHP amount from 1 mL to 2 mL, there is a steady increase in conversionof styrene, with increase in styrene-oxide selectivity and decrease in that of benzaldehyde. On further increase in TBHP amount to 2.7 mL, conversion increases, but selectivity of the epoxide decreases, with an increase in selectivity of benzaldehyde. Hence, 2 mL TBHP was considered optimum for the reaction.
Finally, the reaction temperature was optimized by carrying out the reaction at different temperatures (Fig. 11d). On increasing the temperature from 50 °C to 60 °C, there is increase in conversion and styrene oxide selectivity. But further increase in temperature results in significant decrease of selectivity of both epoxide as well as aldehyde, with formation of unwanted by-products in the form of polymers. This is due to decomposition of TBHP at higher temperatures. Therefore, reaction temperature was optimized at 60 °C.
The reaction conditions for oxidation of styrene using both catalysts were optimized as follows: (I) for PW11Cu: catalyst amount – 15 mg; reaction time – 16 h; TBHP – 2 mL; reaction temperature – 60 °C; (II) for PW11Cu/ZrO2: catalyst amount – 25 mg (active species – 6.25 mg); reaction time – 16 h; TBHP – 2 mL; reaction temperature – 60 °C. It is necessary to bring to notice that in the present systems, optimization has been carried out keeping in priority the selectivity of styrene-oxide. These conditions may be varied by the chemist depending on the requirement of the products.
Test for leaching and heterogeneity
Stability of the catalyst during the course of the reaction is of utmost importance as leaching of active species would make the catalyst unappealing. Hence, PW11Cu was tested for leaching of copper. Absence of characteristic absorption peaks for Cu(II) in the UV-Vis spectrum of reaction mixture confirmed that both catalysts do not leach during the course of the reaction. In case of PW11Cu/ZrO2, absence of blue colour on treating the reaction mixture with ascorbic acid clearly indicated that the catalyst remains intact during the course of the reaction.To check the heterogeneity of PW11Cu, the reaction was first run for 12 h, after which the catalyst was filtered out from the reaction mixture. The reaction was then allowed to proceed further up to 16 h. Similar set of experiments were carried out for PW11Cu/ZrO2. No change in the conversion as well as selectivity of the products (Table 2) indicates that the both catalysts are truly heterogeneous in nature.
Effect of support
A comparison of the activity of unsupported and supported catalysts under optimized conditions as well as the active amount of PW11Cu present in PW11Cu/ZrO2 is shown in Table 3. It can be seen that supported catalyst gives more than double the conversion along with much better selectivity of epoxide, as compared to unsupported one under optimized conditions, which can be explained as follows: (i) homogeneous dispersion of the PW11Cu over ZrO2, due to which more active sites are available for catalysis, and (ii) the overall acidity of the catalyst.| Catalyst | % conversion | % selectivity | Turn over number (TON) | |
|---|---|---|---|---|
| Benzaldehyde | Styrene-oxide | |||
| a Catalyst amount – 15 mg; TBHP – 2 mL; temperature – 60 °C; time – 16 h.b Catalyst amount –6.25 mg; TBHP – 2 mL; temperature – 60 °C; time – 16 h.c Catalyst amount – 25 mg (active amount of PW11Cu-6.25 mg); TBHP – 2 mL; temperature – 60 °C; time – 16 h. | ||||
| aPW11Cu | 44 | 63 | 21 | 1102 |
| bPW11Cu | 21 | 67 | 20 | 1102 |
| cPW11Cu/ZrO2 | 44 | 44 | 37 | 2309 |
It is observed that the conversion for both, supported as well as unsupported catalyst under optimized conditions is the same. However, it is interesting to note that in case of PW11Cu/ZrO2, the active species present is less than half the amount to that of PW11Cu alone. This is the superiority of the supported catalyst.
The total number of acidic sites of the support, PW11Cu and PW11Cu/ZrO2 were calculated by potentiometry and the results are presented in Table 4. There is a phenomenal increase in the overall acidic strength in case of supported catalyst compared to the individual materials, and hence the higher catalytic activity. This is further confirmed by a detailed kinetic study of the reaction using PW11Cu as well as PW11Cu/ZrO2 as catalysts.
| Material | Acidic strength (mV) | Types of acidic sites | Total no. of acidic sites | ||
|---|---|---|---|---|---|
| Very strong | Strong | Weak | |||
| ZrO2 | 53 | 0 | 0.5 | 0.8 | 1.3 |
| PW11Cu | 20 | 0 | 0.1 | 0.3 | 0.4 |
| PW11Cu/ZrO2 | 140 | 0.1 | 0.1 | 1.0 | 1.2 |
Kinetic studies
Kinetic study of the reaction was carried out in detail for both the catalysts in order to comprehend the effect of each component on the rate of reaction. Experiments were carried out with different initial concentrations of styrene and TBHP, keeping the catalyst amount constant. Eqn (1) establishes a relation between the individual concentration of the reactants and time.
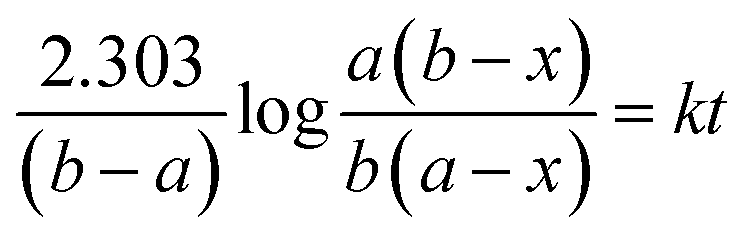 | (1) |
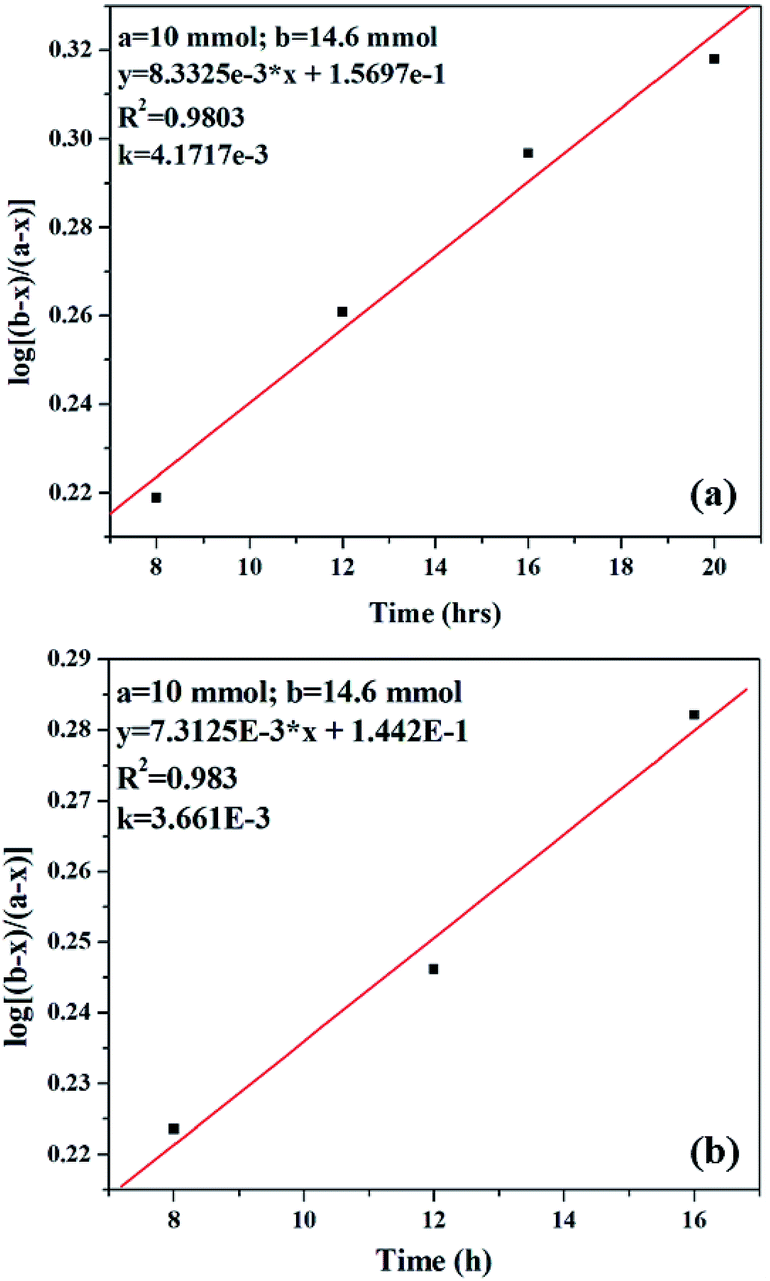 | ||
| Fig. 12 Plot of log[(b − x)/(a − x)] versus time for (a) PW11Cu and (b) PW11Cu/ZrO2. | ||
Alternatively, an experiment was then carried out keeping the concentration of styrene as well as TBHP the same. Eqn (2) establishes a relation between the concentration and time.
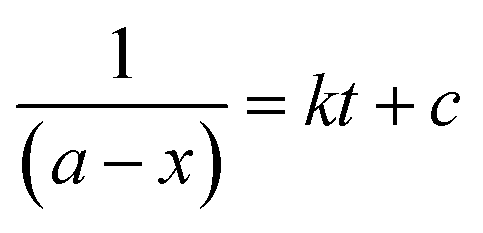 | (2) |
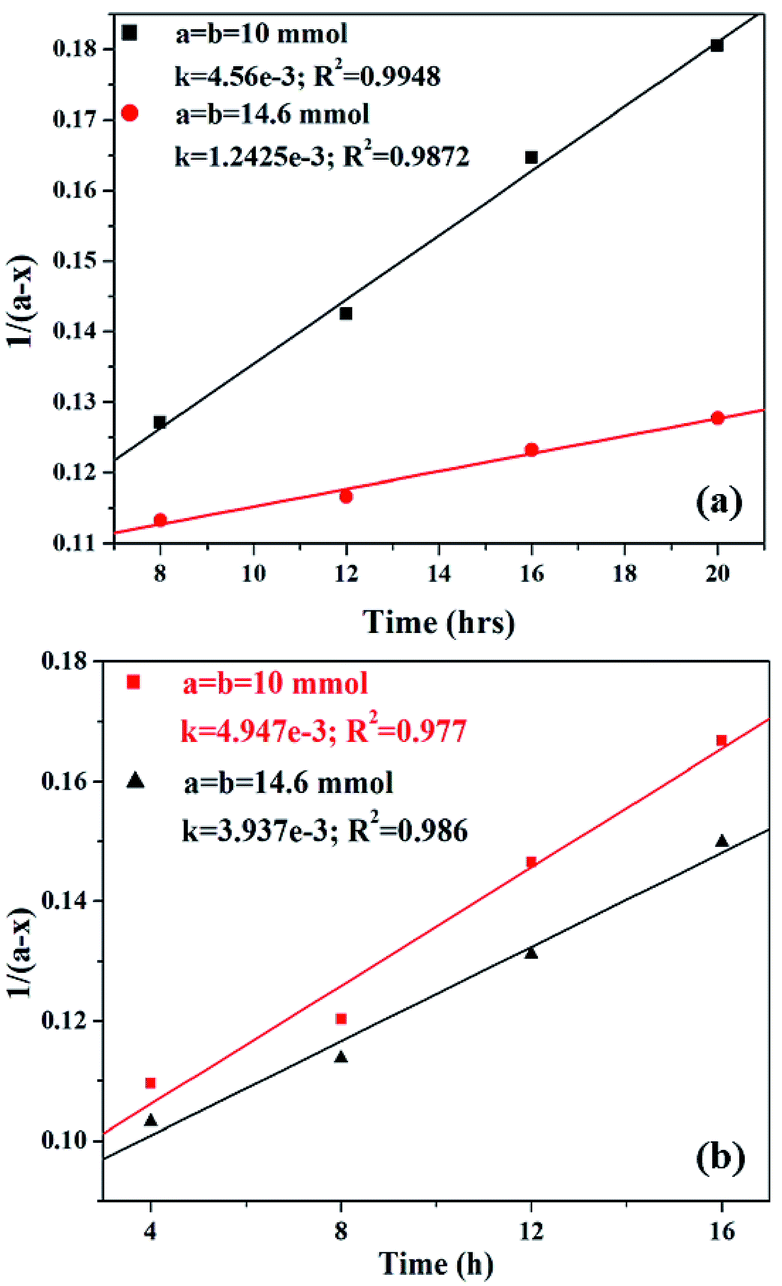 | ||
| Fig. 13 Plot of 1/(a − x) versus time for (a) PW11Cu and (b) PW11Cu/ZrO2. | ||
The effect of reaction rate with respect to catalyst concentration was studied wherein rate constants were plotted at different catalyst concentrations and shown in Fig. 14. When the concentration of PW11Cu is increased from 1.545 × 10−3 mmol to 4.573 × 10−3 mmol, there is a linear increase in rate of the reaction. Similarly, increase in concentration of active species in PW11Cu/ZrO2 from 9.15 × 10−4 mmol to 4.573 × 10−3 mmol, also shows linearity. This indicates that the reaction follows first order kinetics with respect to catalyst concentration as well.50,51
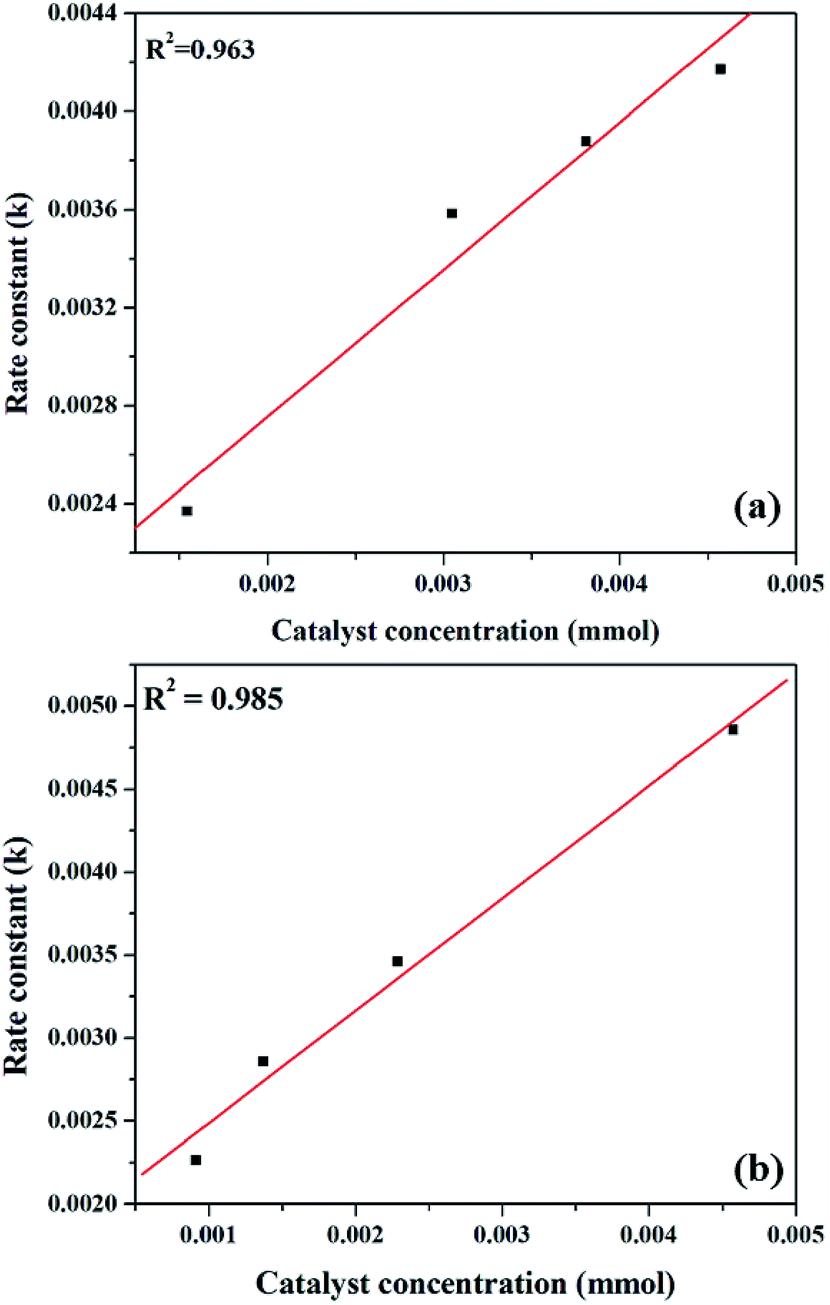 | ||
| Fig. 14 Plot of rate of reaction versus catalyst concentration for (a) PW11Cu and (b) PW11Cu/ZrO2. | ||
Determination of activation energy
As most oxidation reactions are highly sensitive to temperatures, the effect of temperature plays a very significant role. When temperature is increased from 323 K to 353 K, a gradual increase in the conversion of styrene is seen. Thus, 1/T shows a linear relationship with lnk (Fig. 15), and the activation energy was evaluated using the Arrhenius equation.
k = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−Ea/RT e−Ea/RT
| (3) |
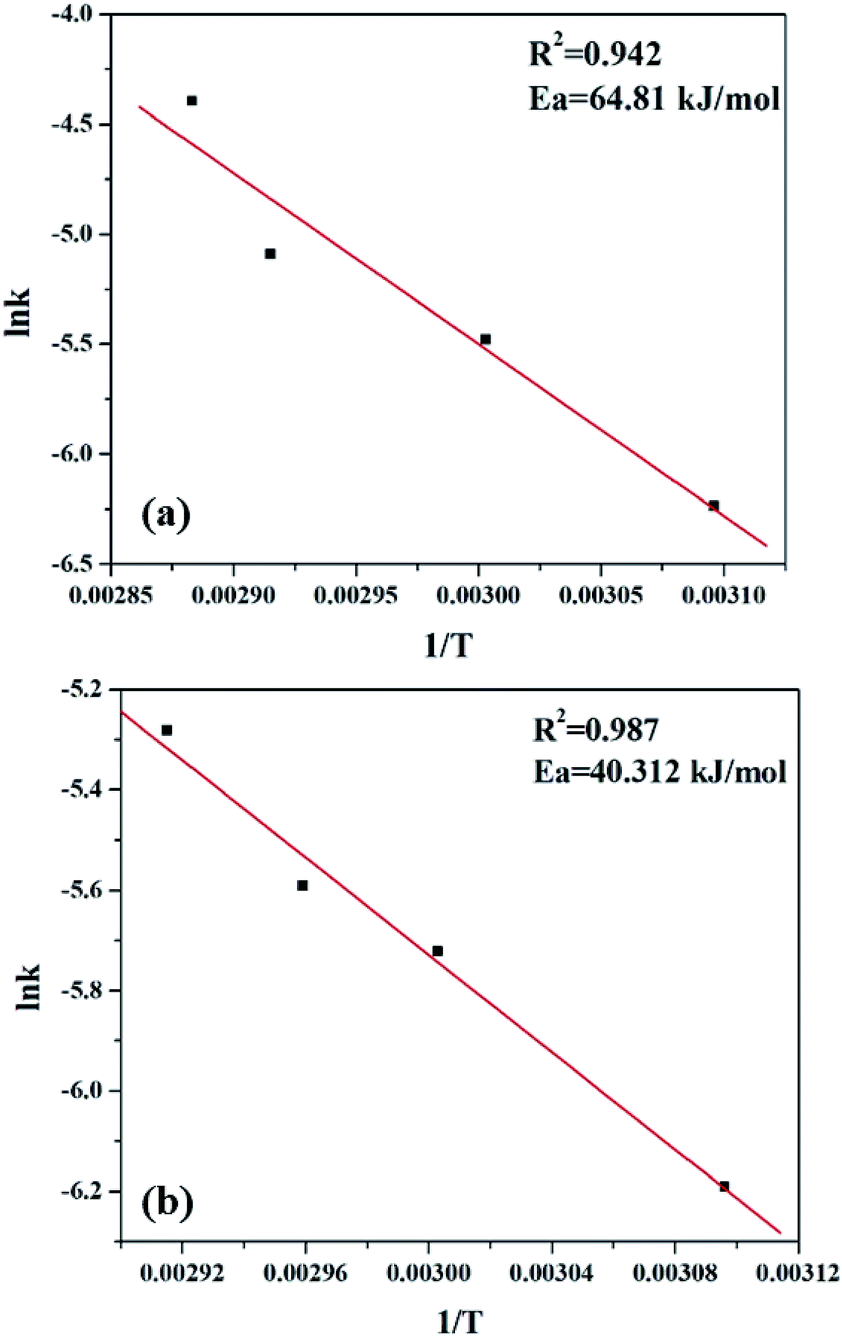 | ||
| Fig. 15 Plot for determination of activation energy for (a) PW11Cu and (b) PW11Cu/ZrO2. | ||
As seen clearly, the present system is a two phase system, and hence it is necessary to clarify whether the reaction is truly governed by a chemical step or it is merely limited to mass transfer or diffusion. Usually, mass transfer limited reactions show Ea in the range of 10–15 kJ mol−1, as opposed to a true chemical reaction which shows Ea > 25 kJ mol−1.50,52 Significantly higher activation energy of 64.81 kJ mol−1 for PW11Cu and 40.31 kJ mol−1 in case of PW11Cu/ZrO2 indicates that the reaction is truly governed by a chemical step and also that both the catalysts have been exploited to its maximum capacity. Interestingly, lower activation energy of PW11Cu/ZrO2 compared to PW11Cu also showcases the superiority of the supported system over unsupported one. Thus, the conclusions obtained from kinetic studies fittingly support that of catalysis that PW11Cu/ZrO2 is a better catalytic system than PW11Cu.
2,6-Di-tert-butyl-4-methyl phenol was used as the radical scavenger to gain an idea on the mechanism and know the intermediate in the reaction for both the systems. The radical scavenger was added 12 h after the reaction was started and then the reaction was allowed to proceed to completion, and the results are present in Table 5. No significant change in the conversion and selectivity in both the cases, indicates that the intermediate formed is a radical. The slight increase in conversion and selectivity of benzaldehyde may be due to presence of the radical intermediate after immediate addition of scavenger, as in the present case, the oxidant used is TBHP, which is a well-known radical initiator. This confirms that the reaction proceeds via radical mechanism, which is as expected.53
Recycle study
In order the regenerate the catalysts, the reaction mixture was centrifuged to separate the catalyst, and this was followed by washing with methanol and drying. The regenerated catalyst was used in the same reaction under optimized conditions and the results are presented in Fig. 16.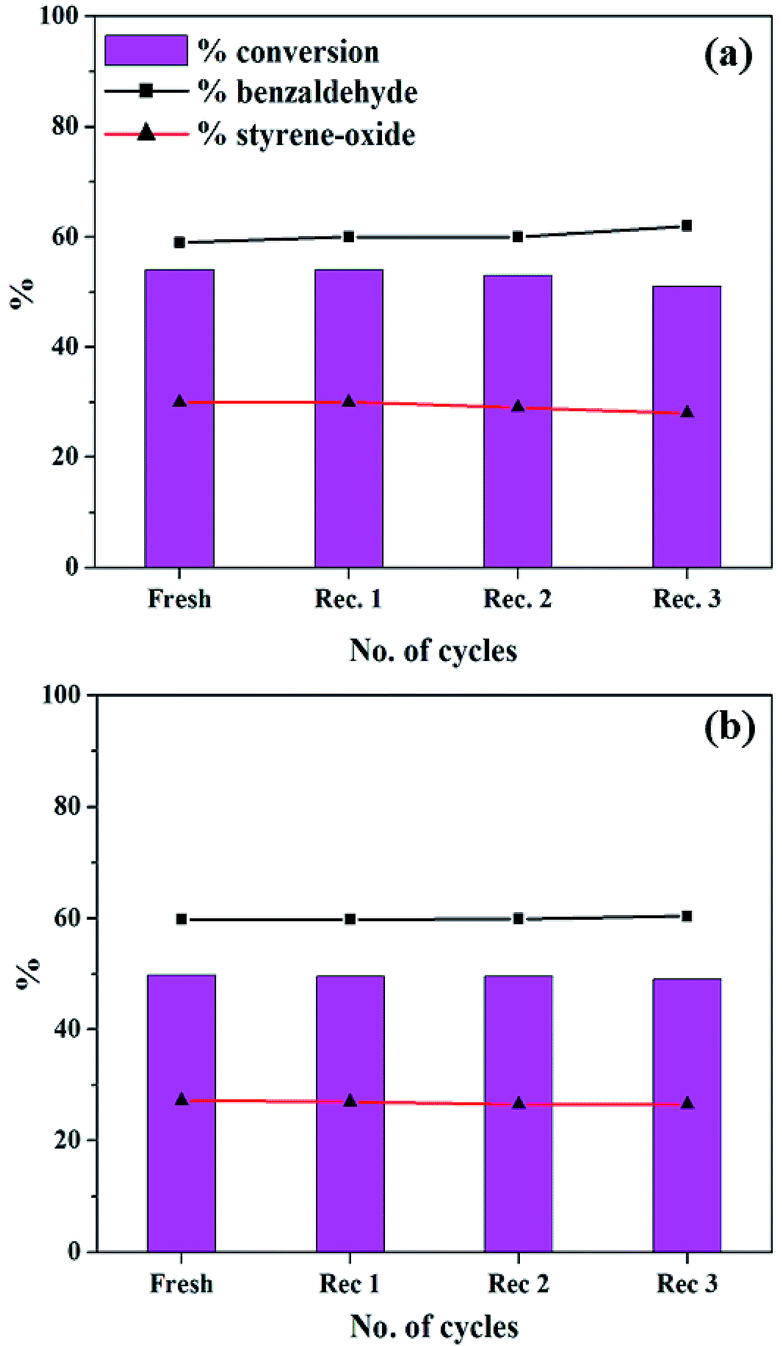 | ||
| Fig. 16 Recycle study for (a) PW11Cu (catalyst amount: 15 mg; temp: 60 °C; TBHP: 2 mL) and (b) PW11Cu/ZrO2 (catalyst amount: 25 mg; temp: 60 °C; TBHP: 2 mL). | ||
In both the cases, same conversion and selectivity for the products are obtained even after multiple cycles indicating that the catalyst remains stable during the course of the reaction and that it can be reused for multiple times. A slight decrease in conversion in the third cycle may be due to loss of the catalyst during the recovery.
Characterization of regenerated catalyst
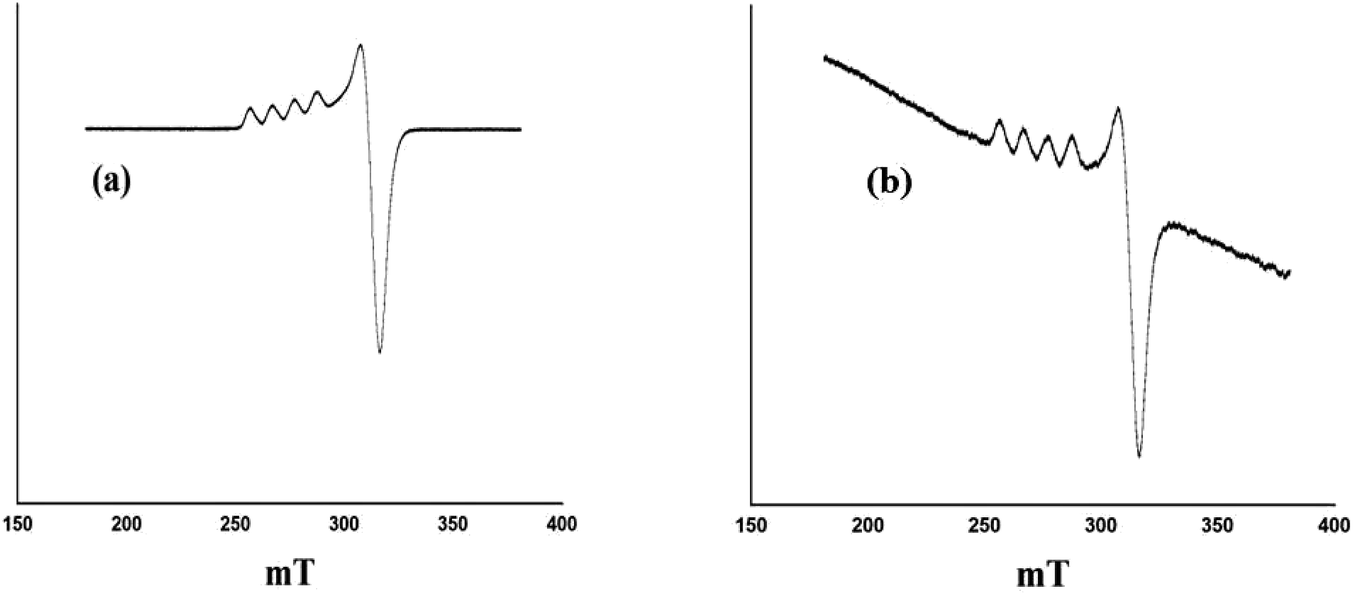
Regenerated PW11Cu was characterized by ESR and FT-IR spectroscopy while regenerated PW11Cu/ZrO2 was characterized by FT-IR, FT-Raman, and powder XRD. Fig. 17 shows the ESR spectra of fresh and recycled PW11Cu. It can be clearly seen that both the spectra are similar, as the five line spectrum is obtained back in the recycled PW11Cu confirming that the environment around Cu remains intact after regeneration.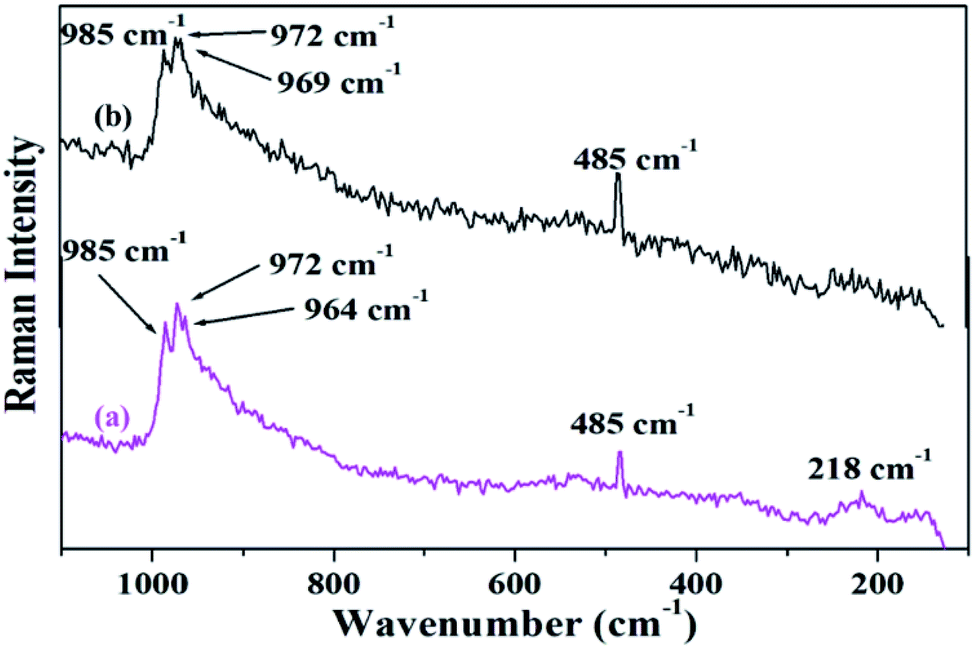 | ||
| Fig. 17 ESR spectra of (a) PW11Cu and (b) Rec. PW11Cu. | ||
The FT-IR spectrum of Rec. PW11Cu (Table 6) shows characteristic bands at 1103, 1056, 964, 887 and 813 cm−1 corresponding to P–O, WO, and W–O–W stretching frequencies respectively. The Cu–O band can be clearly seen at 483 cm−1. No significant shifting of bands clearly indicates that the catalyst does not degrade and the structural morphology of the complex remains unchanged during the course of the reaction. Similarly, Rec. PW11Cu/ZrO2 shows bands at 1101, 1057, 960 cm−1 corresponding to P–O and WO frequencies, and 3417, 1665 and 1413 cm−1 respectively for O–H, H–O–H and O–H–O frequencies of ZrO2. The slight decrease in the sharpness of the bands in recycled catalyst may be due to impurities that remain on the catalyst after recycling.
| Catalyst | FT-IR frequencies (cm−1) | ||||||
|---|---|---|---|---|---|---|---|
| P–O | WO |
W–O–W | Cu–O | O–H | H–O–H | O–H–O | |
| PW11Cu | 1103 | 964 | 887 | 489 | — | — | — |
| 1061 | 810 | ||||||
| Rec. PW11Cu | 1103 | 964 | 887 | 483 | — | — | — |
| 1056 | 813 | ||||||
| PW11Cu/ZrO2 | 1103 | 952 | 813 | — | 3417 | 1631 | 1402 |
| 1064 | |||||||
| Rec. PW11Cu/ZrO2 | 1101 | 960 | — | — | 3417 | 1665 | 1413 |
| 1057 | |||||||
The FT-Raman spectra of fresh and recycled PW11Cu/ZrO2 (Fig. 18) shows that all the characteristic peaks of the fresh catalyst are retained in the recycled one. Thus, FT-IR and FT-Raman spectra indicate that the structure remained intact even after regeneration.
 | ||
| Fig. 18 FT-Raman frequencies of (a) fresh and (b) recycled PW11Cu/ZrO2. | ||
The wide angle powder XRD patterns of fresh and recycled PW11Cu/ZrO2 are shown in Fig. 19. No appreciable change in the powder XRD of regenerated catalyst compared to the fresh one indicates that the structure remains unaffected after the regeneration of catalyst.
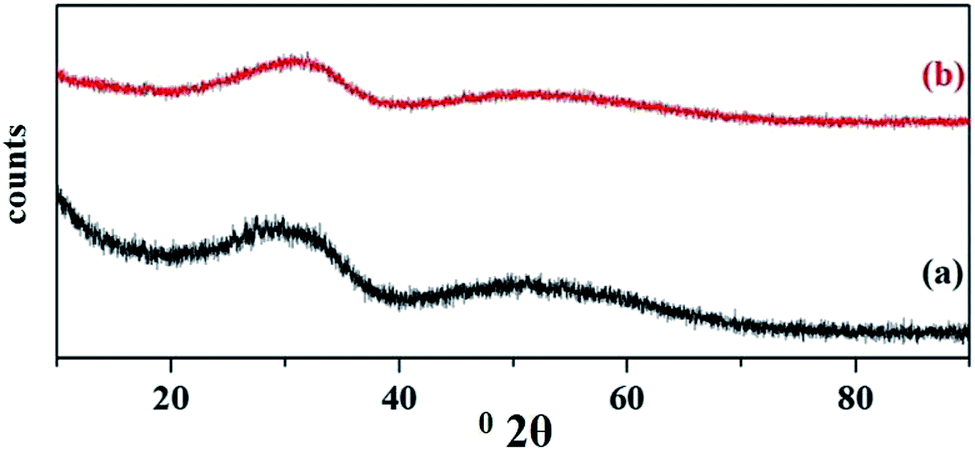 | ||
| Fig. 19 Wide angle powder XRD of (a) fresh and (b) regenerated PW11Cu/ZrO2. | ||
Conclusions
Mono-copper substituted phosphotungstate supported on hydrous zirconia was successfully synthesized by wet impregnation technique and characterized using various techniques. FT-IR and FT-Raman confirmed the intact structure of PW11Cu as well as support even after impregnation, while powder XRD confirmed homogeneous dispersion. TPR indicated strong chemical interactions between PW11Cu and the support, and 31P MAS NMR confirmed the same. The supported catalyst showed better activity as compared to the unsupported one towards oxidation of styrene with TBHP oxidant, where higher conversion (PW11Cu/ZrO2: 44%, PW11Cu: 21%) and higher selectivity towards styrene-oxide (PW11Cu/ZrO2: 37%, PW11Cu: 20%) along with substantially higher TON (PW11Cu/ZrO2: 2309, PW11Cu: 1102) showcased the superiority of supported catalyst. Both the catalysts were recycled and reused up to three cycles with negligible loss in catalytic activity. Detailed kinetic studies showed first order dependence with respect to both substrates individually, and an overall second order dependence. Substantially lower activation energy of PW11Cu/ZrO2 (40.31 kJ mol−1) compared to PW11Cu (64.81 kJ mol−1) further affirmed the superiority of supported catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Department of Science and Technology (DST), Govt. of India, Project No. SR/S1/IC-36/2012 for financial support. Sincere thanks to Department of Chemistry, The Maharaja Sayajirao University of Baroda for BET and Department of Physics, The Maharaja Sayajirao University of Baroda for FT-Raman analyses. We also acknowledge Prof. N. Lingaiah, CSIR-IICT, Hyderabad, for H2-TPR studies. One of the authors, RS is thankful to Council of Scientific and Industrial Research (CSIR), Govt. of India, for award of Senior Research Fellowship (SRF).Notes and references
- A. Wang and H. Jing, Dalton Trans., 2014, 43, 1011–1018 RSC.
- N. Masunga, G. S. Tito and R. Meijboom, Appl. Catal., A, 2018, 552, 154–167 CrossRef CAS.
- J.-M. Brégeault, Dalton Trans., 2003, 3289–3302, 10.1039/b303073n.
- O. Das and T. K. Paine, Dalton Trans., 2012, 41, 11476–11481 RSC.
- A. B. Sorokin and E. V. Kudrik, Catal. Today, 2011, 159, 37–46 CrossRef CAS.
- J.-Y. Liu, X.-F. Li, Y.-Z. Li, W.-B. Chang and A.-J. Huang, J. Mol. Catal. A: Chem., 2002, 187, 163–167 CrossRef CAS.
- G. Liu, M. Hou, J. Song, Z. Zhang, T. Wu and B. Han, J. Mol. Catal. A: Chem., 2010, 316, 90–94 CrossRef CAS.
- Y. Song, H. Jing, B. Li and D. Bai, Chem.–Eur. J., 2011, 17, 8731–8738 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275–11288 RSC.
- Y. Fu, D. Sun, M. Qin, R. Huang and Z. Li, RSC Adv., 2012, 2, 3309–3314 RSC.
- M.-H. Xie, X.-L. Yang and C.-D. Wu, Chem. Commun., 2011, 47, 5521–5523 RSC.
- B. Tang, X. H. Lu, D. Zhou, J. Lei, Z. H. Niu, J. Fan and Q. H. Xia, Catal. Commun., 2012, 21, 68–71 CrossRef CAS.
- B. Du, S.-I. Kim, L.-L. Lou, A. Jia, G. Liu, B. Qi and S. Liu, Appl. Catal., A, 2012, 425–426, 191–198 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20–44 RSC.
- J. Sun, Y. Li, X. Liu, Q. Yang, J. Liu, X. Sun, D. G. Evans and X. Duan, Chem. Commun., 2012, 48, 3379–3381 CAS.
- K. V. Bineesh, D.-K. Kim, M.-I. L. Kim, M. Selvaraj and D.-W. Park, Dalton Trans., 2011, 40, 3938–3945 RSC.
- K. Zhong, J. Xue, Y. Mao, C. Wang, T. Zhai, P. Liu, X. Xia, H. Li and Y. Tong, RSC Adv., 2012, 2, 11520–11528 RSC.
- W. Sun and J. Hu, React. Kinet., Mech. Catal., 2016, 119, 305–318 CrossRef CAS.
- Y. Guo, C. Hu, C. Jiang, Y. Yang, S. Jiang, X. Li and E. Wang, J. Catal., 2003, 217, 141–151 CAS.
- Y. Yang, Y. Guo, C. Hu, C. Jiang and E. Wang, J. Mater. Chem., 2003, 13, 1686–1694 RSC.
- F. Bigi, A. Corradini, C. Quarantelli and G. Sartori, J. Catal., 2007, 250, 222–230 CAS.
- T. Kovalchuk, H. Sfihi, V. Zaitsev and J. Fraissard, J. Catal., 2007, 249, 1–14 CrossRef CAS.
- S. Tangestaninejad, V. Mirkhani, M. Moghadam, I. Mohammadpoor-Baltork, E. Shams and H. Salavati, Ultrason. Sonochem., 2008, 15, 438–447 CrossRef CAS PubMed.
- X. Yu, L. Xu, X. Yang, Y. Guo, K. Li, J. Hu, W. Li, F. Ma and Y. Guo, Appl. Surf. Sci., 2008, 254, 4444–4451 CrossRef CAS.
- J. Hu, K. Li, W. Li, F. Ma and Y. Guo, Appl. Catal., A, 2009, 364, 211–220 CrossRef CAS.
- N. V. Maksimchuk, K. A. Kovalenko, S. S. Arzumanov, Y. A. Chesalov, M. S. Melgunov, A. G. Stepanov, V. P. Fedin and O. A. Kholdeeva, Inorg. Chem., 2010, 49, 2920–2930 CrossRef CAS PubMed.
- S. S. Balula, L. Cunha-Silva, I. C. M. S. Santos, A. C. Estrada, A. C. Fernandes, J. A. S. Cavaleiro, J. Pires, C. Freire and A. M. V. Cavaleiro, New J. Chem., 2013, 37, 2341 RSC.
- L. S. Nogueira, S. Ribeiro, C. M. Granadeiro, E. Pereira, G. Feio, L. Cunha-Silva and S. S. Balula, Dalton Trans., 2014, 43, 9518–9528 RSC.
- B. Wang, J. Zhang, X. Zou, H. Dong and P. Yao, Chem. Eng. J., 2015, 260, 172–177 CrossRef CAS.
- A. Patel and S. Singh, J. Taiwan Inst. Chem. Eng., 2016, 64, 306–313 CAS.
- S. Pathan and A. Patel, Dalton Trans., 2011, 40, 348–355 RSC.
- A. Patel and S. Pathan, Ind. Eng. Chem. Res., 2011, 51, 732–740 CrossRef.
- S. Pathan and A. Patel, Chem. Eng. J., 2014, 243, 183–191 CrossRef CAS.
- S. Singh and A. Patel, Catal. Lett., 2016, 146, 1059–1072 CrossRef CAS.
- N. Narkhede, A. Patel and S. Singh, Dalton Trans., 2014, 43, 2512–2520 RSC.
- S. Singh, N. Narkhede and A. Patel, RSC Adv., 2015, 5, 36270–36278 RSC.
- S. Patel, N. Purohit and A. Patel, J. Mol. Catal. A: Chem., 2003, 192, 195–202 CAS.
- A. U. Patel and R. Sadasivan, ChemistrySelect, 2018, 3, 11087–11097 CrossRef.
- P. Vázquez, L. Pizzio, C. Cáceres, M. Blanco, H. Thomas, E. Alesso, L. Finkielsztein, B. Lantaño, G. Moltrasio and J. Aguirre, J. Mol. Catal. A: Chem., 2000, 161, 223–232 Search PubMed.
- H. Wang, G. Li, Y. Xue and L. Li, J. Solid State Chem., 2007, 180, 2790–2797 CrossRef CAS.
- R. A. Frenzel, G. P. Romanelli, M. N. Blanco and L. R. Pizzio, J. Chem. Sci., 2015, 127, 123–132 CrossRef CAS.
- J. A. Gamelas, F. A. S. Couto, M. C. N. Trovão, A. M. V. Cavaleiro, J. A. S. Cavaleiro and J. D. P. de Jesus, Thermochim. Acta, 1999, 326, 165–173 CrossRef CAS.
- S. Liu, L. Chen, G. Wang, J. Liu, Y. Gao, C. Li and H. Shan, J. Energy Chem., 2016, 25, 85–92 CrossRef.
- M. Shimokawabe, H. Asakawa and N. Takezawa, Appl. Catal., 1990, 59, 45–58 CAS.
- I. V. Kozhevnikov, K. R. Kloetstra, A. Sinnema, H. W. Zandbergen and H. van Bekkum, J. Mol. Catal. A: Chem., 1996, 114, 287–298 CrossRef CAS.
- S. Singh and A. Patel, J. Taiwan Inst. Chem. Eng., 2015, 52, 120–126 CrossRef CAS.
- J. Alcañiz-Monge, B. E. Bakkali, G. Trautwein and S. Reinoso, Appl. Catal., B, 2018, 224, 194–203 CrossRef.
- C. Jiang, Y. Guo, C. Hu, C. Wang and D. Li, Mater. Res. Bull., 2004, 39, 251–261 CrossRef CAS.
- R. Sadasivan, A. Patel and A. Ballabh, Inorg. Chim. Acta, 2019, 487, 345–353 CrossRef CAS.
- Y. Liang, C. Yi, S. Tricard, J. Fang, J. Zhao and W. Shen, RSC Adv., 2015, 5, 17993–17999 CAS.
- G. C. Bond, Heterogeneous catalysis : principles and applications, Clarendon Press, Oxford, 1974 Search PubMed.
- S. Pathan and A. Patel, Catal. Sci. Technol., 2014, 4, 648–656 RSC.
| This journal is © The Royal Society of Chemistry 2019 |