DOI:
10.1039/C9RA04304G
(Paper)
RSC Adv., 2019,
9, 22597-22603
Improving optoelectronic and charge transport properties of D–π–D type diketopyrrolopyrrole-pyrene derivatives as multifunctional materials for organic solar cell applications†
Received
8th June 2019
, Accepted 12th July 2019
First published on 22nd July 2019
Abstract
A series of novel diketopyrrolopyrrole-pyrene-based molecules were designed for small molecule based organic solar cell (SMOSC) applications. Their electronic and charge transfer properties were investigated by applying the PBE0/6-31G(d,p) method. The absorption spectra were simulated using the TD-PBE0/6-31G(d,p) method. The results showed that the frontier molecular orbital (FMO) energy levels, reorganization energy, the energetic driving force, and absorption spectra can be tuned by the introduction of different aromatic heterocyclic groups to the side of diketopyrrolopyrrole fragments' backbones. Additionally, the designed molecules possess suitable FMOs to match those of typical acceptors PC61BM and PC71BM. Meanwhile, the designed molecules can act as good ambipolar charge transport materials in SMOSC applications. Meanwhile, the electron and hole reorganization energies of the designed molecules are smaller than those of the typical electron and hole transport materials, respectively. Moreover, the differences between electron and hole reorganization energies do not exceed 0.046 eV. Our results suggest that the designed molecules can act as promising candidates for donor and ambipolar charge transport materials in SMOSC applications.
Introduction
Organic solar cells (OSCs), as promising candidates to address the global energy crisis, have attracted great attention all over the world owing to their outstanding merits, including low cost, light weight, and flexibility.1–6 In particular, the small molecule based OSCs (SMOSCs) have received tremendous interest due to their prominent advantages over polymer solar cells (PSCs), such as defined structures, relatively easy purification, and less batch to batch variation.7–10 With continuous efforts, significant advances have been demonstrated for OSCs. Recently, the power conversion efficiency (PCE) of PSCs and SMOSCs have exceeded 10%.11–13 However, unfortunately, the overall performance of SMOSCs still lags behind that of PSCs.14,15 Accordingly, it remains a great challenge to achieve high performance SMOSCs. To address this issue, an ideal small molecule used as the donor material should meet several requirements: (i) lower band gap and strong absorption for harvesting the solar light effectively, (ii) high hole mobility for fast charge-carrier transport to lead to high short circuit current (Jsc), (iii) suitable frontier molecular orbitals (FMO) energy levels. A deep highest occupied molecular orbital (HOMO) energy of molecules ensures a high open circuit voltage (Voc).16–18 Meanwhile, another crucial parameter is the energetic driving force (ΔEL–L). The ΔEL–L is determined by the lowest unoccupied molecular orbital (LUMO) energy difference between the donor and acceptor. The ΔEL–L plays a major role in enhancing charge transfer, which should exceed the binding energy.19,20 Generally, the fullerene derivatives PC61BM or PC71BM ([6,6]-phenyl-C61-(or C71)-butyric acid methyl ester) were used as acceptors in OSCs.21,22 With the aim to enhance the PCE of SMOSCs, one efficient method is to design electron donor–acceptor (D–A) molecular structures.23,24 They can efficiently tune the FMOs energy levels through an intramolecular charge transfer, which leads to a narrow band gap, intense light absorption, and high PCEs.25–27 Additionally, the intermolecular stacking, film morphology, and charge transport properties can be determined by the type and length of side chains of D–A molecular structures.28,29 It has been reported that a variety of structures, including A–D–A, D–A–D, D–π–A, D–π–D, A–π–A, and D–π–A–π–D were designed.30,31 Among the various D–A type donor building blocks, diketopyrrolopyrrole (DPP)-based molecules have attracted great attention in terms of their outstanding features, such as good planarity, strong light absorption, high carrier mobility, good photochemical stability, and facile synthesis.32,33 Moreover, pyrene is another promising donor building block. The pyrene derivatives can enhance the absorption due to their large aromatic hydrocarbon ring and planarity.34,35 The planarity of molecules can improve the charge transport and interconnectivity.36 Therefore, the planar DPP and pyrene have combined each other to develop novel donor materials.37 Furthermore, it is worth noting that the charge mobility can been improved through introducing extended aromatic bridges to the DPP derivatives.38 Recently, DPP-based DDP–pyrene–DDP type molecules have been reported for use as donor material for SMOSCs. It was found that these molecules exhibited high hole mobility and PCEs for SMOSCs applications.39,40
Taking into account these merits of DPP and pyrene derivatives mentioned above, we designed eight D–A–D type molecules as the acceptor materials for improving the efficiency of SMOSCs (Scheme 1). In order to study the influence of the DPP moiety on the optical, electronic, charge transfer properties, different aromatic heterocyclic groups have been introduced into the side of DPP in molecules. Applying density functional theory (DFT) approach, the properties of the designed molecules such as HOMO and LUMO energies (EHOMO and ELUMO), HOMO–LUMO gap (Eg), ΔEL–L, the reorganization energy (λ), adiabatic ionization potential (AIP) and electron affinity (AEA) were systematically investigated. The absorption spectra of the designed molecules were predicted using the time dependent DFT (TD-DFT) method. The charge transfer properties (reorganization energy, λ) were also simulated.
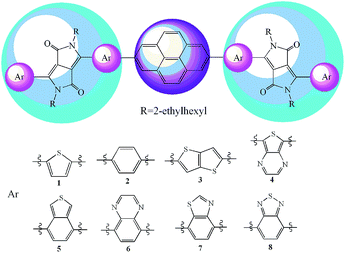 |
| | Scheme 1 Molecular structures of the investigated molecules. | |
Computational methods
All the electronic structure calculations were carried out performed using the Gaussian 09 program suite.41 The molecular structures were optimized by the DFT method. Then the frequency calculations were performed at the same level of theory to confirm that all geometries are stable structures. Under the optimized structures, their absorption spectra were simulated at the TD-DFT level of theory. The all calculations were carried out with the 6-31G(d,p) basis set. The solvent effects on the optical property in chloroform solution (dielectric constant: 4.7113) were also analyzed with the polarised continuum model (PCM).42 For the FMOs energy EHOMO and ELUMO, the optimized geometries were used to obtain EHOMO. However, it is difficult to obtain the ELUMO since the virtual orbitals can not be described definitely by theoretical method.43 Therefore, the ELUMO were be predicted by the sum of the EHOMO and the first vertical excited energy.44,45 It is important to choose an appropriate method in the theoretical investigations. In order to select reasonable method, six representative functionals, B3LYP,46 PBE0,47 LC-ωPBE,48 ωB97XD,49 M062X,50 and CAM-B3LYP51 were used to optimize the geometries of molecule 1, and then using the corresponding TD-DFT method, the absorption spectra were predicted in chloroform, respectively. The calculated data are shown in Table S1 in the ESI.† As showing Table S1,† the calculated longest wavelengths of absorption (λabs) using PBE0 (583 nm) approach compares very satisfactorily with the experimental result (589 nm).39 Furthermore, in order to make further investigation of the validity of the selected approach, both PBE0 and B3LYP methods were also used to obtain the EHOMO and ELUMO of acceptors PC61BM and PC71BM, and the calculated results are listed in Table S2 in the ESI.† The predicted EHOMO and ELUMO of PC61BM (−5.98 and −3.99 eV) and PC71BM (−5.92 and −3.82 eV) using PBE0 approach reproduce well the experimental data (−6.00 and −3.80 eV, −6.00 and −3.95 eV, respectively).52,53 Comprehensively, the PBE0 method arises as the best choice for characterizing the systems studied in our work.
It is worth noting that the λ plays an important role in determining the charge transfer. Usually, lower λ value is beneficial for improving the charge transfer.54,55 Meanwhile, the external λ is much small compared with its internal λ in organic condensed phases.56,57 Therefore, we only consider the internal λ in this work. The electron and hole reorganization energy (λe and λh) were be predicted by:58
| | |
λe = (E−0 − E−−) + (E0− − E00)
| (1) |
| | |
λh = (E+0 − E++) + (E0+ − E00)
| (2) |
where,
E+0(
E−0) is the energy of the cation (anion) structure with the optimized neutral geometry molecule.
E0+(
E0−) corresponds to the energy of the neutral structure with the optimized cation (anion) geometry structure.
E++(
E−−) represents the energy of the cation (anion) calculated with the optimized cation (anion) structure, while
E00 is the energy of the neutral molecule at ground state. Furthermore, it is quite clear that the AIP and AEA play an important role in determining the charge transfer behavior for materials. In this work, the AIP is obtained by the energy difference between cation and neutral molecules. The AEA is determined by the energy difference between neutral and anion molecules. The
λe,
λh, AIP, and AEA were calculated using PBE0/6-31G(d,p) method.
Results and discussion
Match between the donor and acceptor
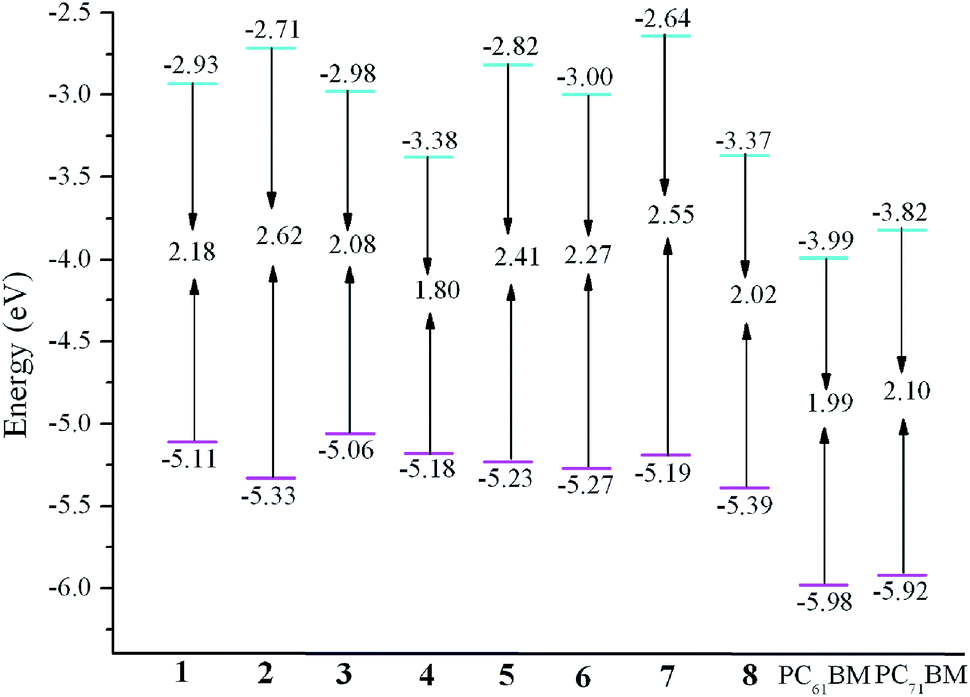
It is noticeable that the electronic energy levels play important role roles to increase the PCE of SMOSCs. Small molecule donors should possess suitable energy levels with the acceptors to ensure efficient charge separation and to maximize the Jsc and Voc. The deep HOMO and high LUMO levels are beneficial for improving the Voc and for efficient exciton dissociation and charge separation, respectively. Moreover, small molecule donors should exhibit lower Eg to guarantee high harvest sunlight efficiency and enhance the Jsc. Moreover, in order to ensure the efficient electron transfer, the ΔEL–L should at least amount to 0.3 eV.59,60 We choose PC61BM and PC71BM as acceptors in our work. Additionally, the explicit molecular orbital contributions from the individual fragments to the FMOs have been investigated by partial density of states (PDOS) analysis and the results are listed in Table 1. With the aim of evaluating whether the FMOs energy of designed donors match with that of the acceptors, the ΔEL–L are estimated. The calculated values of ΔEL–L for 1–8 at the PBE0/6-31G(d,p) level are given in Table 2. The results presented in Table 2 reveal that the ELUMO of 1–8 are positioned above those of PC61BM and PC71BM, respectively. Meanwhile, the ΔEL–L of 1–8 are 0.61–1.35 and 0.45–1.18 eV for PC61BM and PC71BM as acceptors respectively, which are all beyond 0.3 eV. It indicates that the charge transfer from 1–8 to PC61BM and PC71BM efficiently. Additionally, the EHOMO of 1–8 are larger than those of the ELUMO of PC61BM and PC71BM about 1.07–1.40 and 1.24–1.57 eV, respectively (see Table S3 in the ESI†). Both the sequences of difference values are 3 < 1 < 4 < 7 < 5 < 6 < 2 < 8. It suggests that 1–8 possess suitable FMOs to match those of typical acceptors PC61BM and PC71BM for SMOSCs.
Table 1 The HOMOs and LUMOs contributions (%) of 1–8 at the PBE0/6-31G(d,p) level
| Molecules |
HOMOs |
LUMOs |
| PYRa |
DPPb |
PYR |
DPP |
| PYR: pyrene moieties. DPP: diketopyrrolopyrrole moieties. |
| 1 |
6.7 |
93.3 |
11.0 |
89.0 |
| 2 |
3.8 |
96.2 |
6.2 |
93.8 |
| 3 |
4.6 |
95.4 |
6.9 |
93.1 |
| 4 |
8.6 |
91.4 |
9.9 |
90.1 |
| 5 |
3.0 |
97.0 |
5.0 |
95.0 |
| 6 |
1.8 |
98.2 |
3.9 |
96.1 |
| 7 |
1.1 |
98.9 |
4.7 |
95.3 |
| 8 |
3.4 |
96.6 |
4.9 |
95.1 |
Table 2 Evaluation of calculated FMO energies, HOMO–LUMO gap Eg, and energetic driving force ΔEL–L for 1–8 at the PBE0/6-31G(d,p) level
| Molecules |
EHOMO |
ELUMO |
Eg |
ΔEL–La |
ΔEL–Lb |
| Energetic driving force for PC61BM as acceptor. Energetic driving force for PC71BM as acceptor. |
| 1 |
−5.11 |
−2.93 |
2.18 |
1.06 |
0.89 |
| 2 |
−5.33 |
−2.71 |
2.62 |
1.28 |
1.11 |
| 3 |
−5.06 |
−2.98 |
2.08 |
1.01 |
0.84 |
| 4 |
−5.18 |
−3.38 |
1.80 |
0.61 |
0.44 |
| 5 |
−5.23 |
−2.82 |
2.41 |
1.17 |
1.00 |
| 6 |
−5.27 |
−3.00 |
2.27 |
0.99 |
0.82 |
| 7 |
−5.19 |
−2.64 |
2.55 |
1.35 |
1.18 |
| 8 |
−5.39 |
−3.37 |
2.02 |
0.62 |
0.45 |
Frontier molecular orbitals level and band gaps
To gain insight into the influence of the FMO energies on the optical and electronic properties of the designed molecules, the contour plots of HOMOs and LUMOs have been examined (see Fig. 1). Inspection of the distribution of FMO reveals clearly that the HOMOs and LUMOs reveal π characteristic and exhibit strong delocalization features. For molecules 1–5, the electronic cloud distributions of the FMOs spread over the whole molecules with minor contributions from π-bridge fragment (pyrene). For molecules 6 and 7, the electronic cloud distributions of HOMOs are mainly delocalized on the one side DPP fragments, whereas the corresponding the LUMOs mainly reside at another side DPP fragments of molecules. For molecules 8, the HOMO is delocalized throughout the molecule, while the electron density of LUMO is mainly localized on the one side DPP fragment of molecule. This is favorable for the charge transfer across these conjugated donor materials. It suggests that the introduction of the extended aromatic bridges to the DPP derivatives significantly affect on the distributions of FMOs for the molecules.
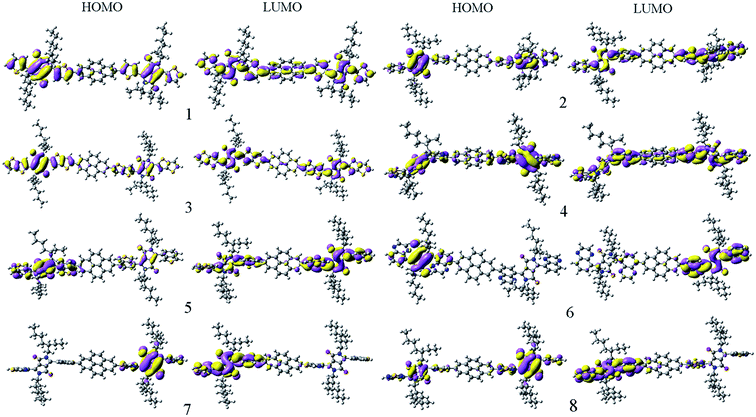 |
| | Fig. 1 The electronic density contours of the frontier orbital for the studied compounds at the PBE0/6-31G(d,p) level. | |
The evaluations of EHOMO, ELUMO, and Eg for the designed molecules as well as PC61BM and PC71BM are plotted in Fig. 2. From Fig. 2 one can find that the EHOMO values are in the order of 3 > 1 > 4 > 7 > 5 > 6 > 2 > 8, and the sequence of ELUMO values is 7 > 2 > 5 > 1 > 3 > 6 > 8 > 4. Clearly, comparing the EHOMO and ELUMO of 2–8 with those of 1, molecules 2, 5, and 7 can lower/raise the EHOMO/ELUMO, whereas molecule 3 can raise/lower the EHOMO/ELUMO, respectively. On the other hand, molecules 4, 6, and 8 can lower both the EHOMO and ELUMO, respectively. The trend of the Eg is 2 > 7 > 5 > 6 > 1 > 3 > 8 > 4. It reveals that molecules 2, 5, 6, and 7 can higher, whereas molecules 3, 4, and 8 can narrower the Eg than that of 1, respectively. The above results suggest that the EHOMO, ELUMO, and Eg can be tuned through the introduction of different aromatic heterocyclic groups to the molecules.
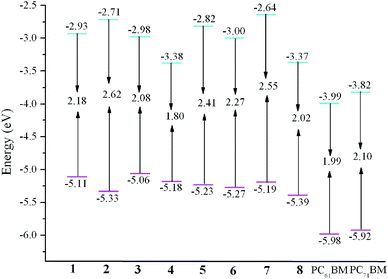 |
| | Fig. 2 Evaluation of calculated FMO energies for investigated molecules as well as FMO energies for PC61BM and PC71BM at the PBE0/6-31G(d,p) level. | |
Absorption and fluorescence spectra
Table 3 collected the longest absorption wavelengths λabs, oscillator strength f, and the main assignments of 1–8. The corresponding simulated absorption spectra are presented in Fig. 3. Inspection of Table 3 reveals clearly that the λabs of molecules 1–5 correspond to the promotion of an electron from the HOMO → LUMO and HOMO−1 → LUMO+1 transitions, respectively. The 530 nm of molecule 6 essentially originates from the coupling of electronic transitions of HOMO → LUMO, HOMO−1 → LUMO, and HOMO → LUMO+1. The shoulder at about 485 and 601 nm of molecules 7 and 8 are mainly derived from HOMO → LUMO, HOMO−1 → LUMO, and HOMO−1 → LUMO+1 transitions, respectively. Comparing the λabs of molecules 2–8 with that of molecule 1, molecules 3, 4, and 8 exhibit bathochromic shifts 31, 98, and 18 nm, respectively. However, molecules 2 and 5–7 show hypsochromic shifts 101, 59, 53, and 98 nm, respectively. The λabs sequence for 1–8 is 4 > 3 > 8 > 1 > 6 > 5 > 7 > 2. It suggests that the λabs of molecules 3, 4, and 8 exhibit bathochromic shifts, whereas the λabs of molecules 2, 5, 6, and 7 show hypsochromic shifts compared with that of molecule 1, respectively. Furthermore, the designed molecules show large f values (0.90–2.33). Molecules 2–5 have slightly larger f values than those of molecules 6–8, respectively. This indicates that molecules 2–5 exhibited slightly larger absorption intensity than those of molecules 6–8. Apparently, through the introduction of different aromatic heterocyclic groups to the side of DPP molecules backbones, their absorption spectrum of the studied molecules can be tuned effectively. With the above considerations, these molecules can act as donor material with strong absorption spectra for SMOSCs.
Table 3 The electronic transition, absorption wavelengths λabs (in nm), the oscillator strength f, and main assignments (coefficient) of 1–8 in chloroform at the TD-PBE0/6-31G(d,p)//PBE0/6-31G(d,p) level, along with available experimental data
| Molecules |
λabs |
f |
Assignment |
| Experimental results of 1 were taken from ref. 39. |
| 1 |
583 |
2.33 |
H → L (0.66) |
| H−1 → L+1 (0.25) |
| 2 |
482 |
1.37 |
H → L (0.58) |
| H−1 → L+1 (0.30) |
| 3 |
614 |
2.93 |
H → L (0.62) |
| H−1 → L+1 (0.32) |
| 4 |
681 |
1.59 |
H → L (0.64) |
| H−1 → L+1 (0.27) |
| 5 |
524 |
1.66 |
H → L (0.49) |
| H−1 → L+1 (0.33) |
| 6 |
530 |
1.00 |
H → L (−0.36) |
| H−1 → L (0.44) |
| H → L+1 (−0.32) |
| 7 |
485 |
0.93 |
H → L (0.33) |
| H−1 → L (0.61) |
| H−1 → L+1 (0.13) |
| 8 |
601 |
0.90 |
H → L (0.56) |
| H−1 → L (−0.36), |
| H−1 → L+1 (0.22) |
| Expa |
589 |
|
|
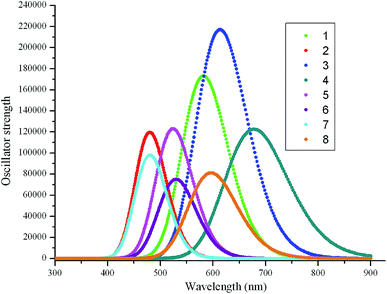 |
| | Fig. 3 The calculated absorption spectra of the investigated molecules (value of full width at half maximum is 3000 cm−1). | |
Charge transfer properties
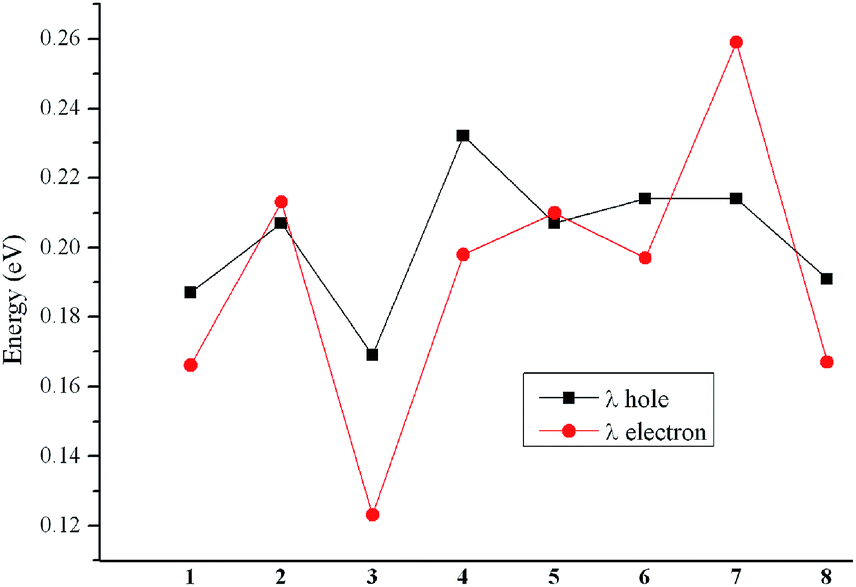
It is commonly known that the AIP, AEA, and λ are three key parameters that determine the charge transfer property. The AIP and AEA are good descriptors to estimate the energy barrier for the injection of holes and electrons, respectively. The λ is the extent to estimate the ability to carry the charge in solid. Usually, lower AIP and higher AEA are favorable for better charge transport ability for hole and electron, respectively.61,62 For λ, the lower λe and λh are beneficial for enhancement the electron and hole transfer rate, respectively.54,55
The predictions of AIP, AEA, λh, and λe of the designed molecules are summarized in Table 4. It is clear that the AIP are in the order of 8 > 2 > 6 > 5 > 7 > 1 > 4 > 3. The AIP of molecules 2 and 5–8 are larger, whereas the AIP of molecules 3 and 4 are smaller than that of molecule 1, respectively. Namely, comparing the AIP of 2–8 with that of 1, molecules 2 and 5–8 can raise the AIP, while molecules 3 and 4 can lower the AIP, respectively. At the same time, the AEA is in the order of 4 > 8 > 3 > 1 > 6 > 5 > 2 > 7. Obviously, comparing the AEA of molecules 2–8 with that of molecule 1, molecules 3, 4, and 8 increase the AEA, whereas molecules 2 and 5–7 decrease the AEA, respectively. It is noticeable that the introduction of different aromatic heterocyclic groups to the molecules affects both their AIP and AEA values.
Table 4 Calculated molecular λe, λh, AIP, AEA and η (all in eV) of 1–8 at the PBE0/6-31G(d,p) level
| Molecules |
λh |
λe |
AIP |
AEA |
η |
| 1 |
0.187 |
0.166 |
5.716 |
2.008 |
1.854 |
| 2 |
0.207 |
0.213 |
5.937 |
1.746 |
2.096 |
| 3 |
0.169 |
0.123 |
5.603 |
2.108 |
1.747 |
| 4 |
0.232 |
0.198 |
5.715 |
2.382 |
1.667 |
| 5 |
0.207 |
0.210 |
5.789 |
1.826 |
1.981 |
| 6 |
0.214 |
0.197 |
5.877 |
1.935 |
1.971 |
| 7 |
0.214 |
0.259 |
5.780 |
1.694 |
2.051 |
| 8 |
0.191 |
0.167 |
5.981 |
2.286 |
1.848 |
A graphical representation of λh and λe has been illustrated in Fig. 4 to represent the results more clearly. Generally, Alq3 (tris(8-hydroxyquinolinato)aluminum(III), λe = 0.276 eV) and TPD (N,N′-diphenyl-N,N′-bis(3-methlphenyl)-(1,1′-biphenyl)-4,4′-diamine, λh = 0.290 eV) are taken as the typical electron and hole transport materials, respectively.63,64 Apparently, the λe and λh of the designed compounds are much smaller than those of Alq3 and TPD, respectively. It indicates that both the electron and hole transfer rates of 1–8 might be higher than those of Alq3 and TPD, respectively. The decreasing sequence of λh is 4 > 6 ≈ 7 > 2 ≈ 5 > 8 > 1 > 3, and the values of λe are in the following order: 7 > 2 > 5 > 4 > 6 > 8 > 1 > 3. Apparently, comparing the λh and λe of molecule 2–8 with those of 1, molecules 2, 4–8 can increase both λh and λe, whereas compound 3 can decrease both λh and λe, respectively. Additionally, molecule 3 has both the smallest λh and λe values, indicating that molecule 3 has the largest hole and electron transport rates among the investigated molecules. Moreover, the differences between λe and λh values of 1–8 do not exceed 0.046 eV. It indicates that they exhibit better hole- and electron-transporting balance. Accordingly, these molecules can act as nice ambipolar charge transport materials for SMOSCs. Therefore, these molecules can be used as good candidates for ambipolar charge transport materials under the proper operating conditions.
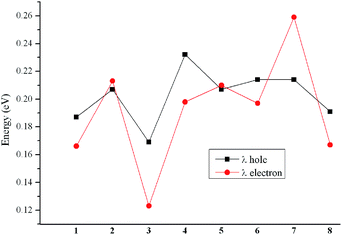 |
| | Fig. 4 Graphical representation of λe and λh calculated at the PBE0/6-31G(d,p) level of theory. | |
Conclusions
In this article, we design a series of novel diketopyrrolopyrrole-pyrene-based molecules as the acceptor materials for SMOSCs applications. The optical, electronic, and charge transporting properties were investigated by applying DFT and TD-DFT methods. The parameters such as ΔEL–L, λ, AIP, and AEA were calculated to preliminarily evaluate the performance of the solar cell. The results show that the EHOMO, ELUMO, Eg, ΔEL–L, AIP, AEA, and λ can be tuned through the introduction of different aromatic heterocyclic groups to the molecules. The absorption spectra were also simulated. Our results suggest that the designed molecules possess suitable FMOs to match those of typical acceptors PC61BM and PC71BM. Additionally, they can be used as ambipolar charge transport materials for SMOSCs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial supports from the NSFC (No. 21563002) are gratefully acknowledged.
Notes and references
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- W. Cao and J. Xue, Energy Environ. Sci., 2014, 7, 2123–2144 RSC.
- K. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- K. H. Hendriks, G. H. L. Heintges, V. S. Wienk, M. M. Gevaerts and R. A. J. Janssen, Angew. Chem., Int. Ed., 2013, 52, 8341–8344 CrossRef CAS PubMed.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS PubMed.
- Q. R. Yin, J. S. Miao, Z. Wu, Z. F. Chang, J. L. Wang, H. B. Wu and Y. Cao, J. Mater. Chem. A, 2015, 3, 11575–11586 RSC.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- Y. Chen, X. Wan and G. Long, Acc. Chem. Res., 2013, 46, 2645–2655 CrossRef CAS PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS PubMed.
- J. H. Kim, C. E. Song, N. Shin, H. Kang, S. Wood, I. N. Kang, B. Kim, B. Kim, J. S. Kim, W. S. Shin and D. H. Hwang, ACS Appl. Mater. Interfaces, 2013, 5, 12820–12831 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed.
- D. Ni, B. Zhao, T. Shi, S. Ma, G. Tu and H. Wu, ACS Macro Lett., 2013, 2, 621–624 CrossRef CAS.
- J. Li, K. H. Ong, S. L. Lim, G. M. Ng, H. S. Tan and Z. K. Chen, Chem. Commun., 2011, 47, 9480–9482 RSC.
- J. E. Coughlin, Z. B. Henson, G. C. Welch and G. C. Bazan, Acc. Chem. Res., 2014, 47, 257–270 CrossRef CAS PubMed.
- Y. Huang, M. Zhang, H. Chen, F. Wu, Z. Cao, L. Zhang and S. Tan, J. Mater. Chem. A, 2014, 2, 5218–5223 RSC.
- M. Knupfer, Appl. Phys. A, 2003, 77, 623–626 CrossRef CAS.
- I. Hill, A. Kahn, Z. Soos and R. Pascal Jr, Chem. Phys. Lett., 2000, 327, 181–188 CrossRef CAS.
- C. He, Q. G. He, X. D. Yang, G. L. Wu, C. H. Yang, F. L. Bai, Z. G. Shuai, L. X. Wang and Y. F. Li, J. Phys. Chem. C, 2007, 111, 8661–8666 CrossRef CAS.
- M. Lenes, G. A. H. Wetzelaer, F. B. Kooistra, S. C. Veenstra, J. C. Hummelen and P. W. M. Blom, Adv. Mater., 2008, 20, 2116–2119 CrossRef CAS.
- M. Cheng, C. Chen, X. Yang, J. Huang, F. Zhang, B. Xu and L. Sun, Chem. Mater., 2015, 27, 1808–1814 CrossRef CAS.
- E. Wang, L. Hou, Z. Wang, S. Hellström, F. Zhang, O. Inganäs and M. R. Andersson, Adv. Mater., 2010, 22, 5240–5244 CrossRef CAS PubMed.
- A. Tang, C. Zhan and J. Yao, Chem. Mater., 2015, 27, 4719–4730 CrossRef CAS.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS PubMed.
- P. Zhou, D. Dang, M. Xiao, Q. Wang, J. Zhong, H. Tan, Y. Pei, R. Yang and W. Zhu, J. Mater. Chem. A, 2015, 3, 10883–10889 RSC.
- H. Qin, L. Li, F. Guo, S. Su, J. Peng, Y. Cao and X. Peng, Energy Environ. Sci., 2014, 7, 1397–1401 RSC.
- S. Chen, L. Xiao, X. Zhu, X. Peng, W. K. Wong and W. Y. Wong, Chem. Commun., 2015, 51, 14439–14442 RSC.
- Y. Li, R. G. Clevenger, L. Jin, K. V. Kilway and Z. Peng, J. Mater. Chem. C, 2014, 2, 7180–7183 RSC.
- J. L. Wang, F. Xiao, J. Yan, Z. Wu, K. K. Liu, Z. F. Chang, R. B. Zhang, H. Chen, H. B. Wu and Y. Cao, Adv. Funct. Mater., 2016, 26, 1803–1812 CrossRef CAS.
- K. Gao, J. Miao, L. Xiao, W. Deng, Y. Kan, T. Liang, C. Wang, F. Huang, J. Peng and Y. Cao, Adv. Mater., 2016, 28, 4727–4733 CrossRef CAS PubMed.
- J. H. Liu, B. Walker, A. Tamayo, Y. Zhang and T. Q. Nguyen, Adv. Funct. Mater., 2013, 23, 47–56 CrossRef CAS.
- X. Liu, G. Luo, X. Cai, H. Wu, S. J. Su and Y. Cao, RSC Adv., 2015, 5, 83155–83163 RSC.
- J. Mei, K. R. Graham, R. Stalder, S. P. Tiwari, H. Cheun, J. Shim, M. Yoshio, C. Nuckolls, B. Kippelen, R. K. Castellano and J. R. Reynolds, Chem. Mater., 2011, 23, 2285–2288 CrossRef CAS.
- O. P. Lee, A. T. Yiu, P. M. Beaujuge, C. H. Woo, T. W. Holcombe, J. E. Millstone, J. D. Douglas, M. S. Chen and J. M. Fréchet, Adv. Mater., 2011, 23, 5359–5363 CrossRef CAS PubMed.
- K. A. Mazzio, M. J. Yuan, K. Okamoto and C. K. Luscombe, ACS Appl. Mater. Interfaces, 2011, 3, 271–278 CrossRef CAS PubMed.
- Q. Wang, J. J. van Franeker, B. J. Bruijnaers, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2016, 4, 10532–10541 RSC.
- H. Huang, L. Xiao, B. Yang, T. Lai, L. Zhang, K. Gao, X. Peng and Y. Cao, RSC Adv., 2016, 6, 59218–59225 RSC.
- J. Chen, L. Duan, M. Xiao, Q. Wang, B. Liu, H. Xia, R. Yang and W. Zhu, J. Mater. Chem. A, 2016, 4, 4952–4961 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. P. Cornard and C. Lapouge, J. Phys. Chem. A, 2006, 110, 7159–7166 CrossRef CAS PubMed.
- J. Wu, Y. H. Kan, Y. Wu and Z. M. Su, J. Phys. Chem. C, 2013, 117, 8420–8428 CrossRef CAS.
- J. Ku, Y. Lansac and Y. H. Jang, J. Phys. Chem. C, 2011, 115, 21508–21516 CrossRef CAS.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554–1561 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- I. Jeon, C. Delacou, T. Nakagawa and M. Y. Yutaka, Chem.–Asian J., 2016, 11, 1268–1272 CrossRef CAS PubMed.
- M. Chandrasekharam, M. A. Reddy, K. Ganesh, G. D. Sharma, S. P. Singh and J. L. Rao, Org. Electron., 2014, 15, 2116–2125 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- S. Di Motta, E. Di Donato, F. Negri, G. Orlandi, D. Fazzi and C. Castiglioni, J. Am. Chem. Soc., 2009, 131, 6591–6595 CrossRef CAS PubMed.
- D. P. McMahon and A. Trois, J. Phys. Chem. Lett., 2010, 1, 941–946 CrossRef CAS.
- M. E. Köse, W. J. Mitchell, N. Kopidakis, C. H. Chang, S. E. Shaheen, K. Kim and G. Rumbles, J. Am. Chem. Soc., 2007, 129, 14257–14270 CrossRef.
- M. C. Scharber, D. Wuhlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. L. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- J. L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- C. C. Liu, S. W. Mao and M. Y. Kuo, J. Phys. Chem. C, 2010, 114, 22316–22321 CrossRef CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- B. C. Lin, C. P. Cheng, Z. Q. You and C. P. Hsu, J. Am. Chem. Soc., 2005, 127, 66–67 CrossRef CAS PubMed.
- N. E. Gruhn, D. A. da Silva Filho, T. G. Bill, M. Malagoli, V. Coropceanu, A. Kahn and J. L. Bredas, J. Am. Chem. Soc., 2002, 124, 7918–7919 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The following are available online at http://www.mdpi.com/xxx/s1, Table S1 calculated the longest absorption wavelengths λabs of parent molecule 1 by various methods with 6-31G(d,p) basis set, along with available experimental data. Table S2 the calculated EHOMO and ELUMO (in eV) for PC61BM and PC71BM at PBE0/6-31G(d,p) and B3LYP/6-31G (d,p) levels, along with available experimental data. Table S3 the differences between the EHOMO of 1–8 and the ELUMO of PC61BM and PC71BM at the PBE0/6-31G(d,p) level. See DOI: 10.1039/c9ra04304g |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Kexin Lia and
Xueli Hana
*ab,
Kexin Lia and
Xueli Hana