
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystallization, rheology and mechanical properties of the blends of poly(L-lactide) with supramolecular polymers based on poly(D-lactide)–poly(ε-caprolactone-co-δ-valerolactone)–poly(D-lactide) triblock copolymers†
Zhanxin Jing *,
Jin Li,
Weiyu Xiao,
Hefeng Xu,
Pengzhi Hong and
Yong Li*
*,
Jin Li,
Weiyu Xiao,
Hefeng Xu,
Pengzhi Hong and
Yong Li*
Department of Applied Chemistry, College of Chemistry and Environment, Guangdong Ocean University, Zhanjiang, Guangdong 524088, China. E-mail: jingzhan_xin@126.com; yongli6808@126.com
First published on 20th August 2019
Abstract
In this study, we investigated the blending of poly(L-lactide) (PLLA) with supramolecular polymers based on poly(D-lactide)–poly(ε-caprolactone-co-δ-valerolactone)–poly(D-lactide) (PDLA–PCVL–PDLA) triblock copolymers as an efficient way to modify PLLA. The supramolecular polymers (SMP) were synthesized by the terminal functionalization of the PDLA–PCVL–PDLA copolymers with 2-ureido-4[1H]-pyrimidinone (UPy). The structure, thermal properties and rheological behavior of the synthesized supramolecular polymers were studied; we found that the formation of the UPy dimers expanded the molecular chain of the polymer and the incorporation of the UPy groups suppressed the crystallization of polymers. In addition, the synthesized supramolecular polymers had a low glass transition temperature of about −50 °C, showing the characteristics of elastomers. On this basis, superior properties such as a fast crystallization rate, high melt strength, and toughness of fully bio-based, i.e., PLA-based materials were achieved simultaneously by blending PLLA with the synthesized supramolecular polymers. In the PLLA/SMP blends, PLLA could form a stereocomplex with its enantiomeric PDLA blocks of supramolecular polymers, and the stereocomplex crystals with the cross-linking networks reinforced the melt strength of the PLLA/SMP blends. The influences of the SMP composition and the SMP content in the PLLA matrix on crystallization and mechanical properties were analyzed. The supramolecular polymers SMP0.49 and SMP1.04 showed a reverse effect on the crystallization of PLLA. Tensile tests revealed that the lower content of the synthesized supramolecular polymers could achieve toughening of the PLLA matrix. Therefore, the introduction of supramolecular polymers based on PDLA–PCVL–PDLA is an effective way to control the crystallization, rheology and mechanical properties of PLLA.
1. Introduction
Poly(lactide) (PLA), which is considered as a sustainable alternative to petrochemical-derived products, is a biodegradable thermoplastic polyester.1,2 PLA has frequently been used for tissue engineering,3 drug delivery4 and packaging5 because of its non-toxicity, biodegradability, and biocompatibility. These properties are attributed to its plant origin; it can be synthesized by the condensation of lactic acid produced from the fermentation of starch. In addition, the mechanical strength of PLA is close to that of polypropylene.6 Therefore, PLA has broad application prospects and potential value. However, some of its properties such as the low crystallization rate7 and poor heat resistance and toughness8 have limited the application of poly(lactide) in some fields. The blending of PLA with soft polymers and the stereocomplex technology have been found to improve its performance. Blending PLA with other soft polymers is a more practical and economical way to improve the brittleness of PLA.9 Many PLLA/soft polymer blends, such as PLLA/PCL, PLLA/PEO, PLLA/PBAT and PLLA/PVA, have been reported. Vilay et al.10 investigated the mechanical and thermal properties of PLLA/poly(ε-caprolactone) (PCL) and PLLA/poly(butylene succinate-co-L-lactate) (PBSL) blends. The results found that the incorporation of PCL and PBSL into the PLLA matrix obviously improved the toughness of the blends, and all the PLLA/PCL and PLLA/PBSL blends were immiscible in the PCL and PBSL phases dispersed in the PLLA-rich phase. Li et al.11 improved the toughness of PLLA via reactive blending with an acrylonitrile-butadiene-styrene (ABS) copolymer and found that the prepared PLLA/ABS blends have a very good stiffness–toughness balance. PLA/poly(butylene adipate-co-terephthalate) blends were studied by Jiang et al.12 The results revealed that the tensile strength and modulus of the PLA/PBAT blends decreased, while their elongation and toughness dramatically increased as the PBAT content increased in the PLA matrix. In addition, the blends comprised immiscible PBAT dispersed in the form of ∼300 nm domains within the PLA matrix. However, the blends prepared with other soft polymers were immiscible, which resulted in weak interfacial adhesion between the PLA matrix and soft polymers due to the insufficient chain entanglement density across the interface.13 Nowadays, several methods such as the fabrication of pre-synthesized blocks or grafted copolymers14–16 and the production of compatibilizers in situ17–19 have been used to improve the toughness of the PLA-based materials via enhancing the interaction between the interfaces. Although toughness can be improved, this would eventually lead to the loss of the mechanical strength of PLA. In addition, the incorporation of flexible polymers can lower the glass transition temperature of PLA, which may deteriorate the heat resistance. Therefore, it is necessary to find new methods to simultaneously improve these characteristics of PLA to expand its application range.The stereocomplex technology is considered to be one of the most effective methods to improve the crystallization rate and heat resistance of PLA, which are important issues in expanding the applications of poly(lactide).20 Poly(L-lactide) (PLLA) and its enantiomer poly(D-lactide) (PDLA) have been reported to show a strong tendency to interact with each other to form stereocomplexes via the strong van der Waals forces of the CH3⋯C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O interactions between enantiomeric molecular chains.21,22 The melting temperature of PLA-based stereocomplexes (sc-PLA) can reach 230 °C, which is higher than that of PLLA or PDLA (about 170 °C). In addition, sc-PLA also has a high crystallization rate and heat resistance compared to PLLA or PDLA. These are mainly attributed to the β-crystal of sc-PLA, which is different from the α-crystal of homo-crystallized PLA.23 To understand the mechanism of the stereocomplexation phenomenon, the effects of many parameters such as molecular weight, L/D ratios, optical purity, preparation method and crystallization conditions on the formation of stereocomplexes have been deeply studied.24–28 Recently, PLA stereocomplexes have been widely used to modify PLA. Our previous studies have reported29,30 that the stereocomplex interface formed between the PLLA matrix and nanoparticles not only promotes the dispersion of nanoparticles, but also increases the melt strength of the polymer matrix. This extensive research has demonstrated that a stereocomplex is an effective nucleating agent and a rheological modifier for PLLA, and it can also improve the thermodynamic properties of PLLA.31,32 However, the stereocomplex technology promotes the crystallization of PLA, which tends to make PLA more brittle. Therefore, this is also a problem that must be considered when the stereocomplex technology is applied to modify PLA.
O interactions between enantiomeric molecular chains.21,22 The melting temperature of PLA-based stereocomplexes (sc-PLA) can reach 230 °C, which is higher than that of PLLA or PDLA (about 170 °C). In addition, sc-PLA also has a high crystallization rate and heat resistance compared to PLLA or PDLA. These are mainly attributed to the β-crystal of sc-PLA, which is different from the α-crystal of homo-crystallized PLA.23 To understand the mechanism of the stereocomplexation phenomenon, the effects of many parameters such as molecular weight, L/D ratios, optical purity, preparation method and crystallization conditions on the formation of stereocomplexes have been deeply studied.24–28 Recently, PLA stereocomplexes have been widely used to modify PLA. Our previous studies have reported29,30 that the stereocomplex interface formed between the PLLA matrix and nanoparticles not only promotes the dispersion of nanoparticles, but also increases the melt strength of the polymer matrix. This extensive research has demonstrated that a stereocomplex is an effective nucleating agent and a rheological modifier for PLLA, and it can also improve the thermodynamic properties of PLLA.31,32 However, the stereocomplex technology promotes the crystallization of PLA, which tends to make PLA more brittle. Therefore, this is also a problem that must be considered when the stereocomplex technology is applied to modify PLA.
Recently, scientists have made many efforts to improve the toughness, crystallinity and melt strength of PLA at the same time. Liu et al.33 prepared PLLA/PDLA–PEG–PDLA blends; they found that the toughness and heat resistance of PLLA are improved due to the synergistic effects of stereocomplexation between enantiomeric PLA and plasticization of PEG blocks. Poly(D-lactide)–Pluronic F68–poly(D-lactide) multiblock copolymers were synthesized by Qi,34 who found that the synthesized multiblock copolymers can act as a tough agent of PLLA and PLLA/multiblock copolymer blends have a continuous amorphous phase with the crystalline regions being the discontinuous portion. In this study, we designed and synthesized supramolecular polymers containing PDLA hard segments and poly(ε-caprolactone-co-δ-valerolactone) (PCVL) soft segments via UPy-functionalized PDLA–PCVL–PDLA copolymers. On this basis, we prepared PLLA/supramolecular polymer blends and investigated their thermal properties, rheological behavior and mechanical properties. This work provides a new approach to prepare PLA-based materials with rapid crystallization capacity, high melt strength and excellent mechanical properties.
2. Experimental
2.1 Materials and reagents
PLLA (Mw = 2.0 × 105 g mol−1) was purchased from Nature Works Company. D-Lactide (optical purity > 99%) was obtained from Jinan Daigang Biomaterial Co., Ltd (Shandong, China). δ-Valerolactone (VL, 98%), ε-caprolactone (CL, 99%), diethylene glycol (DEG, >98%), 2-amino-4-hydroxyl-6-methylpyrimidine and 1,6-hexyldiisocyanate (HDI) were purchased from J&K Chemical (Beijing, China). Dibutyltin dilaurate and stannous octoate (Sn(Oct)2) were acquired from Sinopharm Group Chemical Reagent (China). 2(6-Isocyanatohexylaminocarbonyl-amino)-6-methyl-4[1H]pyrimidinone (UPy-NCO) was synthesized according to a previously reported method35 using 2-amino-4-hydroxyl-6-methylpyrimidine and 1,6-hexyldiisocyanate as raw materials. Toluene was further dried by distillation from sodium prior to use. Other reagents were of analytical grade and used without further purification.2.2 Synthesis of supramolecular polymers based on PDLA–PCVL–PDLA triblock copolymers
Supramolecular polymers based on poly(D-lactide)–poly(ε-caprolactone-co-δ-valerolactone)–poly(D-lactide) triblock copolymers were synthesized as follows: first, a dihydroxyl-terminated poly(ε-caprolactone-co-δ-valerolactone) macroinitiator was synthesized. ε-Caprolactone (10.37 g, 93.31 mmol), δ-valerolactone (2.27 g, 22.67 mmol) and diethylene glycol (120 mg, 1.13 mmol) were added into a Schlenk flask at 120 °C. After mixing evenly, Sn(Oct)2/toluene solution was added. The dosage of Sn(Oct)2 was 0.1 mol% with respect to the amount of ε-caprolactone and δ-valerolactone. The polymerization reaction was allowed to proceed at 120 °C for 24 h. After the reaction, the obtained mixture was dissolved in dichloromethane and precipitated by adding excess of methanol. The obtained precipitate was dried in a vacuum oven at 30 °C for 48 h and abbreviated as PCVL.Then poly(D-lactide)–poly(ε-caprolactone-co-δ-valerolactone)–poly(D-lactide) triblock copolymers (PDLA–PCVL–PDLA) were synthesized. PCVL and D-lactide were added into a Schlenk flask, and the mixture was dried in a vacuum line at 60 °C for 30 min. After being purged with dry nitrogen, the mixture was heated to 130 °C and stirred. Then, Sn(Oct)2/toluene solution was added. The dosage of Sn(Oct)2 was 0.1 mol% with respect to the amount of D-lactide. The polymerization reaction was allowed to occur at 130 °C for 24 h. After the reaction, the obtained mixture was dissolved in dichloromethane and precipitated by adding excess of methanol. The obtained precipitate was dried in a vacuum oven at 30 °C for 48 h, and it was abbreviated as PDLA–PCVL–PDLAx, where x represents the molecular weight of PDLA block calculated using the 1H NMR spectrum.
Eventually, a supramolecular polymer based on PDLA–PCVL–PDLA was synthesized. A PDLA–PCVL–PDLA triblock copolymer and UPy-NCO were added into a dried Schlenk flask. Then, the mixture was further dried in a vacuum line at 60 °C for 30 min. After purging with nitrogen, toluene (50 mL) and Sn(Oct)2 (the dosage of Sn(Oct)2 was 3.0 wt% with respect to the amount of the PDLA–PCVL–PDLA triblock copolymer) were added, and the mixture solution was heated to 110 °C and kept at this temperature for 24 h. Then, toluene was removed by rotary evaporation under reduced pressure. The obtained crude product was dissolved in chloroform (100 mL), and silica and dibutyltin dilaurate were added to the above solution. The mixed solution was stirred at 60 °C for 2 h. The UPy-NCO-absorbed silica was removed through filtration. Chloroform in the obtained transparent solution was eliminated by rotary evaporation under reduced pressure, and the obtained supramolecular polymer was dried in a vacuum oven for 24 h. The synthesized supramolecular polymer was abbreviated as SMPx, where x represents the molecular weight of PDLA block.
2.3 Preparation of poly(L-lactide)/supramolecular polymer blends
The blends of poly(L-lactide)/supramolecular polymers were prepared by solution casting. First, appropriate amounts of PLLA and supramolecular polymers were dissolved in chloroform. Then, the two solutions were mixed by magnetic stirring for 2 h. The resulting mixed solution was poured into a Teflon mould. The mould was placed at room temperature to allow the solvent to evaporate slowly. After solvent evaporation, the obtained films were dried for 24 h at 50 °C in a vacuum oven. The prepared blends were designated as PLLA/SMPx-y%, where x represents the molecular weight of the PDLA block of supramolecular polymers and y% represents the content of supramolecular polymers in the PLLA/SMP blends.2.4 Characterization
3. Results and discussion
3.1 Synthesis and characterization of supramolecular polymers based on PDLA–PCVL–PDLA copolymers
Supramolecular polymers based on PDLA–PCVL–PDLA triblock copolymers were synthesized, as shown in Scheme 1. First, a dihydroxyl-terminated PCVL macroinitiator was synthesized by the ring-opening co-polymerization of ε-caprolactone and δ-valerolactone using diethylene glycol as the initiator and Sn(Oct)2 as the catalyst. Subsequently, a PDLA–PCVL–PDLA triblock copolymer was synthesized via the ring-opening polymerization of D-lactide initiated using the PCVL macroinitiator. Finally, the UPy-functionalized PDLA–PCVL–PDLA copolymer was synthesized by the coupling reaction of the PDLA–PCVL–PDLA triblock copolymer and excess UPy-NCO. Our previous research studies36,37 revealed that UPy dimers can form a self-complementary quadruple hydrogen bond, whose strength is close to the strength of a covalent bond. Therefore, it can promote the expansion of polymer chains (as shown in Scheme 1(b)). To deeply study the formation of self-complementary quadruple hydrogen bonds, NMR and GPC were used to analyze the chemical structure and molecular weight of the supramolecular polymers based on the PDLA–PCVL–PDLA copolymers.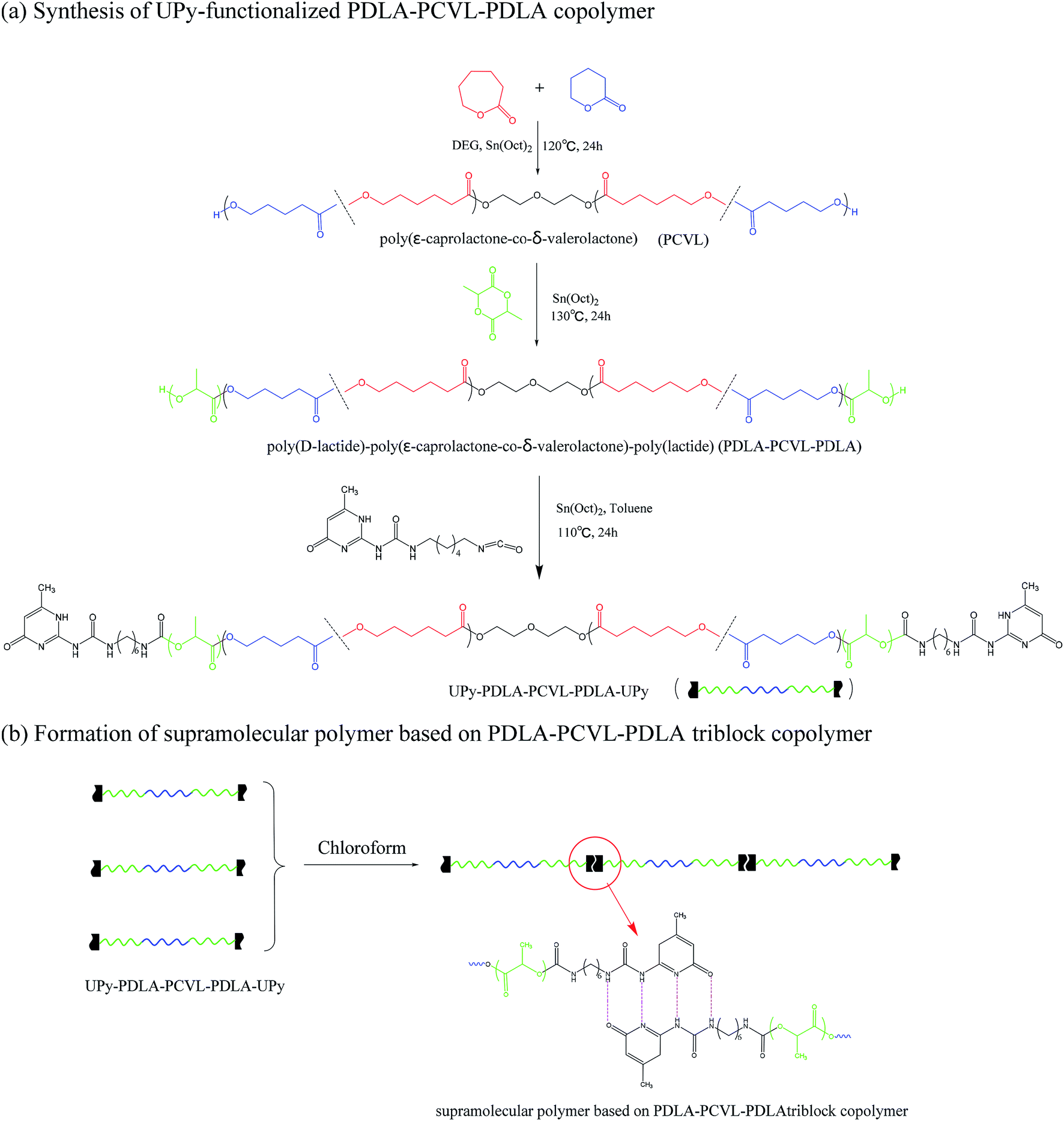 | ||
| Scheme 1 Synthetic route of supramolecular polymers based on PDLA–PCVL–PDLA triblock copolymers. | ||
Fig. 1 displays the 1H NMR spectra of the dihydroxyl-terminated PCVL macroinitiator, the PDLA–PCVL–PDLA triblock copolymer and the corresponding supramolecular polymer. In the 1H NMR spectrum of the PCVL macroinitiator, there are obvious proton peaks at 1.41 ppm, 1.59 ppm, 1.68 ppm, 2.33 ppm and 4.09 ppm, which are attributed to the methylene groups at different positions on the PCVL macromolecular chain. In addition, a weak proton peak is observed at 3.7 ppm, and it can be assigned to the methylene proton linked to the terminal hydroxyl group of PCVL.38 The number-average molecular weight of PCVL can be calculated by NMR, and the obtained result is 6.31 kg mol−1. The GPC curve of PCVL was also measured, as shown in Fig. 2, and it exhibited a single elution peak. The number-average molecular weight and polydispersity index were 7.48 kg mol−1 and 1.38, respectively. For the PDLA–PCVL–PDLA triblock copolymers, proton peaks appeared at 1.61 ppm, 5.18 ppm and 4.37 ppm with respect to PCVL, corresponding to the methyl, methine, and terminal methine protons of the PDLA blocks.36,38 The molecular weight of the PDLA–PCVL–PDLA triblock copolymers was measured by GPC and also calculated by NMR; the results are listed in Table 1. The synthesized PDLA–PCVL–PDLA triblock copolymers displayed molecular weight distribution with lower polydispersity indexes (<1.35), and the molecular weight increased as the D-lactide/PCVL feed ratio increased. These results indicated that the dihydroxyl-terminated PDLA–PCVL–PDLA triblock copolymers with a controlled molecular weight and composition were synthesized. The supramolecular polymer was synthesized by reacting the PDLA–PCVL–PDLA triblock copolymer and excess UPy-NCO. The synthesized supramolecular polymer shows weak peaks at 10.2, 11.9 and 13.1 ppm (Fig. 1(c)), which are the characteristic N–H proton peaks of the UPy motif.36,37,39 In addition, the weak peak (∼4.37 ppm) corresponding to the PDLA terminal methine protons disappeared completely. The GPC curve of the supramolecular polymer was also measured (Fig. 2), revealing that there appears a shoulder at the higher molecular weight side and larger PDI (about 2.0). These results demonstrated that the UPy groups were successfully introduced into the end of PDLA–PCVL–PDLA, and UPy dimerization could expand the polymer chain and increase the molecular weight. Therefore, we successfully synthesized the supramolecular polymer based on the PDLA–PCVL–PDLA triblock copolymer.
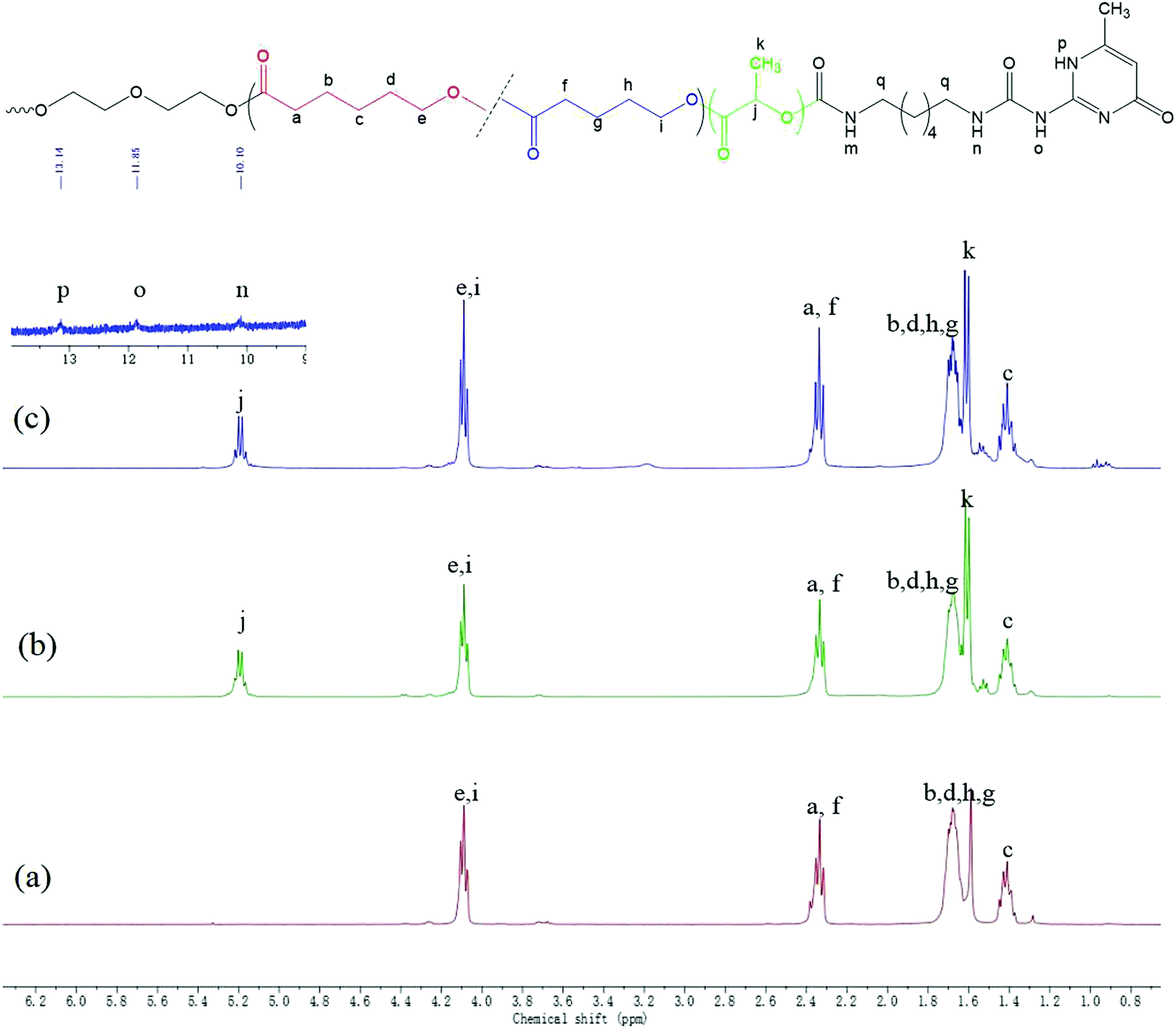 | ||
| Fig. 1 1H NMR spectra of PCVL (a), PDLA–PCVL–PDLA triblock copolymer (b) and the corresponding supramolecular polymer (c). | ||
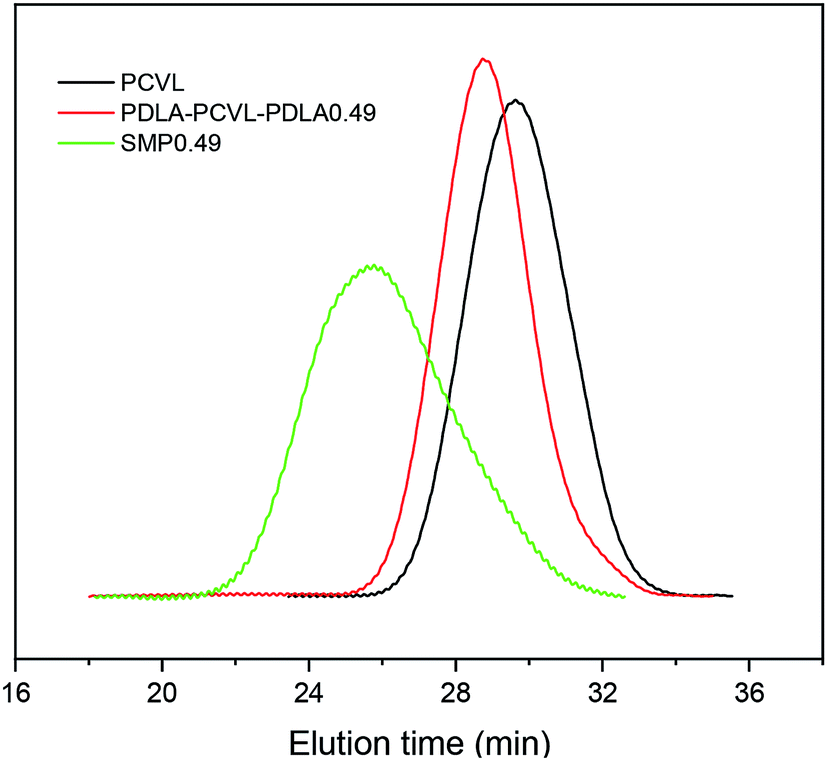 | ||
| Fig. 2 GPC curves of PCVL, PDLA–PCVL–PDLA0.49 triblock copolymer and the corresponding supramolecular polymer (SMP0.49). | ||
| Samples | [LA]/([CL] + [VL])a | Mn of each blockc (kg mol−1) | Mn,NMRb (kg mol−1) | Mn,GPCd (kg mol−1) | PDId | mPLAe (%) | Yield (%) |
|---|---|---|---|---|---|---|---|
| a [LA]/([CL] + [VL]) represents the molar ratio of lactide and the units of ε-caprolactone and δ-valerolactone of the PCVL macroinitiator during the synthesis of PLA–PCVL–PLA triblock copolymers.b Mn acquired from 1H NMR results.c The numerals denote Mn of the corresponding PLA and PCVL blocks, as derived from NMR data.d Mn and polydispersity index (PDI) acquired by GPC.e Mass fractions of PLA in the triblock copolymers calculated from 1H NMR spectroscopy. | |||||||
| PCVL | — | — | 6.31 | 7.48 | 1.38 | — | 79.4 |
| PDLA–PCVL–PDLA0.49 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 :4 |
0.49–6.31–0.49 | 7.29 | 8.30 | 1.35 | 13.4 | 83.2 |
| SMP0.49 | — | — | — | 23.0 | 2.01 | — | 81.4 |
| PDLA–PCVL–PDLA1.04 | 2:4 |
1.04–6.31–1.04 | 8.37 | 8.95 | 1.28 | 32.6 | 66.3 |
| SMP1.04 | — | — | — | 24.6 | 1.81 | — | 91.3 |
The effects of the UPy groups on the crystallization and thermal properties of the PDLA–PCVL–PDLA copolymers were studied by DSC (Fig. 3), and the obtained thermal parameters are listed in Table S1.† It is clear that the DSC cooling curve of the PDLA–PCVL–PDLA0.49 triblock copolymers shows an obvious exothermic peak at ∼5 °C, which is assigned to the crystallization of the PCVL segments. The corresponding reheating curve shows a melting peak of the PCVL segments at ∼30 °C. In the DSC cooling curve of the PDLA–PCVL–PDLA1.04 triblock copolymers, there appear two exothermic peaks at 7.9 and 72.4 °C, which are attributed to the crystallization of the PCVL and PDLA segments, respectively. It can also be observed from the corresponding reheating curve that there are two endothermic peaks at 15–40 and 100–140 °C, which correspond to the melting of the PCVL and PDLA segments. Compared with the result for the PDLA–PCVL–PDLA copolymer, the crystallization peak of the supramolecular polymer based on the PDLA–PCVL–PDLA triblock copolymer reduces or disappears and the corresponding crystallization temperature decreases, as shown in Fig. 3(b). It is clear that the synthesized supramolecular polymers have a low glass transition temperature (about −50 °C), indicating that the supramolecular polymers based on the PDLA–PCVL–PDLA triblock copolymers have good elasticity and can be used for toughening brittle materials. The glass transition temperature of the synthesized supramolecular polymer is related to the length of the PDLA block, and the increase in the length of the PDLA block leads to the increase in the glass transition temperature. In the reheating curve of the supramolecular polymer SMP0.49, there appears a significantly exothermic peak and an obviously endothermic peak at −12.9 and 24.6 °C, which correspond to the cold crystallization and melting of the PCVL segments. The reheating curve of the supramolecular polymer SMP1.04 shows a weak endothermic peak at 117.3 °C assigned to the melting of the PDLA segment. These phenomena revealed that the incorporation of the UPy groups suppressed the crystallization of the polymer. The possible reason is that the formation of self-complementing quadruple hydrogen bonds increases the molecular weight of the polymer. This can drastically restrain the freedom and mobility of the polymer chains, which would increase the difficulty of the regular alignment of polymer molecular chains during crystallization. This enabled the transition of the PDLA–PCVL–PDLA triblock copolymers from turbid, brittle solids to transparent, elastic solids.
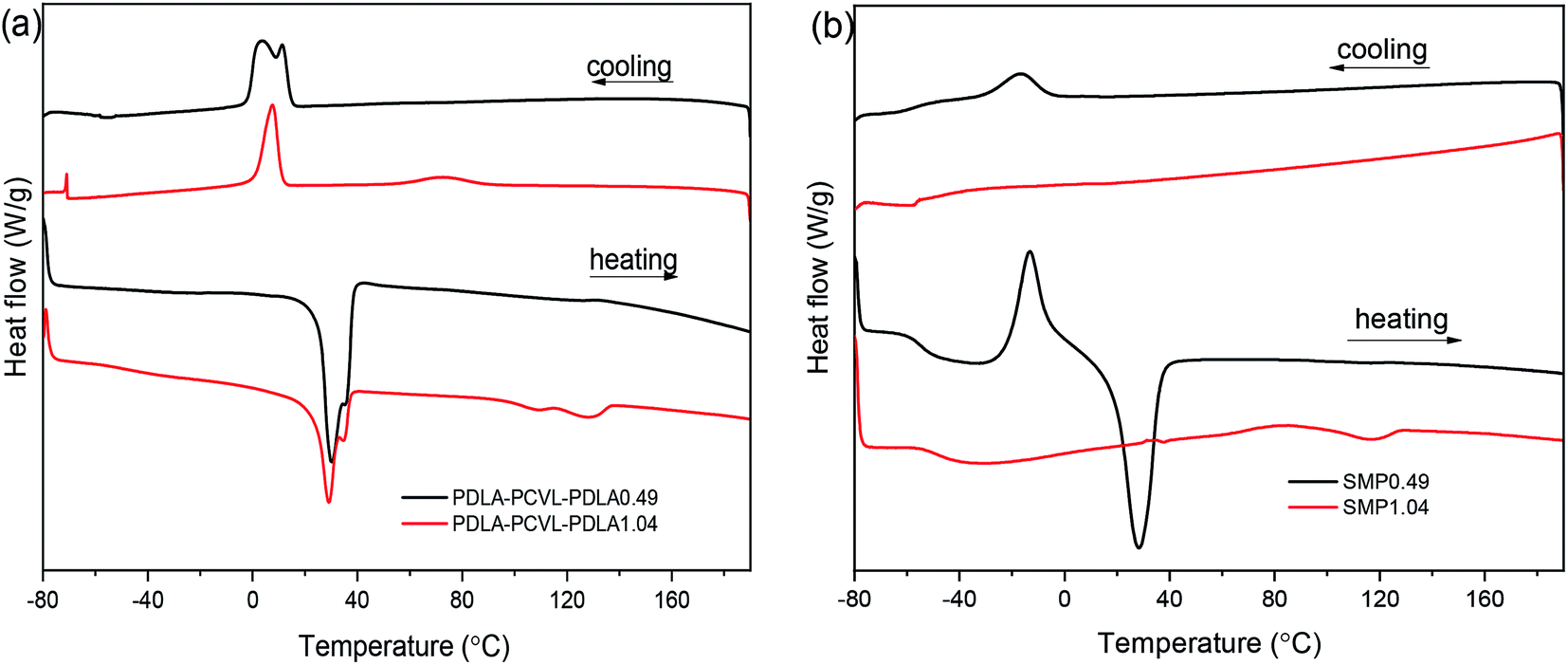 | ||
| Fig. 3 DSC cooling and heating curves of PDLA–PCVL–PDLA triblock copolymers (a) and the corresponding supramolecular polymers (b). | ||
3.2 Rheological behavior of supramolecular polymers based on the PDLA–PCVL–PDLA copolymers
Previous literature has reported that any change in the molecular weight or architecture of polymer chains leads to strong changes in the rheological properties.40–42 Therefore, rheology is well-suited to analyze the association behavior of the PDLA–PCVL–PDLA triblock copolymer and the UPy groups at its chain ends. In particular, temperature-dependent measurements can give significant information of the chain dynamics itself and the temperature-dependent association of the individual UPy group. The variation in the viscoelastic moduli of the synthesized supramolecular polymers as a function of temperature is displayed in Fig. 4. It is clear from Fig. 4(a) that the moduli only drop by several orders of magnitude when the temperature increases to 90 °C. However, with further increase in temperature, the moduli rapidly decreased. When the temperature exceeded 140 °C, which is higher than the melting temperature of the supramolecular polymer SMP0.49, the viscosity of SMP0.49 became very low to give sufficient torque to the rheometer's transducer. The possible reason is that a higher temperature causes the disassociation of the UPy dimers, resulting in decrease in molecular weight. Therefore, the moduli of the synthesized supramolecular polymer quickly decreased in a higher temperature region. When the temperature was gradually decreased, an increase up to the initial moduli was observed. This was attributed to the reversible nature of the UPy dimers. However, it is clear that the heating and cooling processes do not superimpose, and a hysteresis behavior is observed. The moduli in the cooling run are lower than those in the heating run. This may be attributed to the slow crystallization, as confirmed by the results of DSC. A similar result has been reported by Wietor et al.43 for the polycaprolactone supramolecular polymers with ureidopyrimidinone end groups. The supramolecular polymer SMP1.04 showed a similar phenomenon to that of SMP0.49. In particular, for the higher temperature region (>120 °C), the moduli of SMP1.04 were obviously larger than those of SMP0.49. In addition, the moduli in the cooling run could quickly increase to the initial values. This was attributed to the high PDLA content. The DSC results demonstrated that the crystallization capacity of the supramolecular polymer SMP1.04 was superior to that of SMP0.49.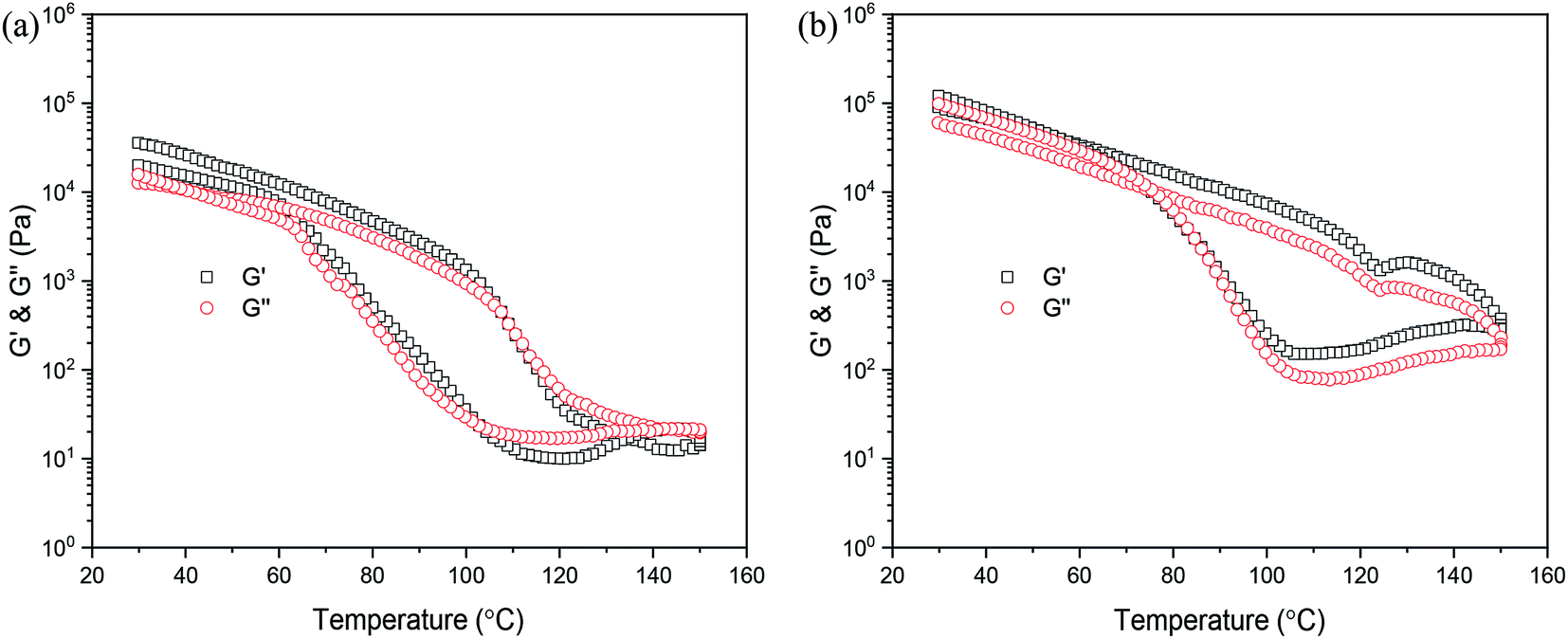 | ||
| Fig. 4 Variation in viscoelastic moduli of supramolecular polymers as a function of temperature: (a) SMP0.49; (b) SMP1.04. | ||
Fig. 5 shows the variation in the storage modulus (G′) and loss modulus (G′′) of the supramolecular polymers melted at 100 °C as a function of frequency. It is clear that the G′ and G′′ values of the supramolecular polymers increase as the frequency increases, and the G′ values are larger than G′′. For an ideal polymer melt, the variation in the moduli as a function of frequency follows the following equations: logG′ ∝ logω and logG′′ ∝ logω. However, it can be observed from Fig. 5 that the moduli of the synthesized supramolecular polymers deviate significantly from the above equation, and the melt exhibits a solid-like viscoelastic behavior. For neat PCVL, its melting temperature is about 30–40 °C. However, the DSC curves of the supramolecular polymers based on the PDLA–PCVL–PDLA triblock copolymers show the melting peak of PCVL, and the melting peak corresponding to the PDLA block is observed in a high-temperature region (Fig. 3). This reveals that the supramolecular polymer still exists in the PDLA crystalline region after melting at 100 °C, which can act as the crosslinking point to restrain the movement of polymer chains. Therefore, the synthesized supramolecular polymers melted at 100 °C showed a solid-like viscoelastic behavior; the higher the PDLA content, the more pronounced the solid-like viscoelastic behavior.
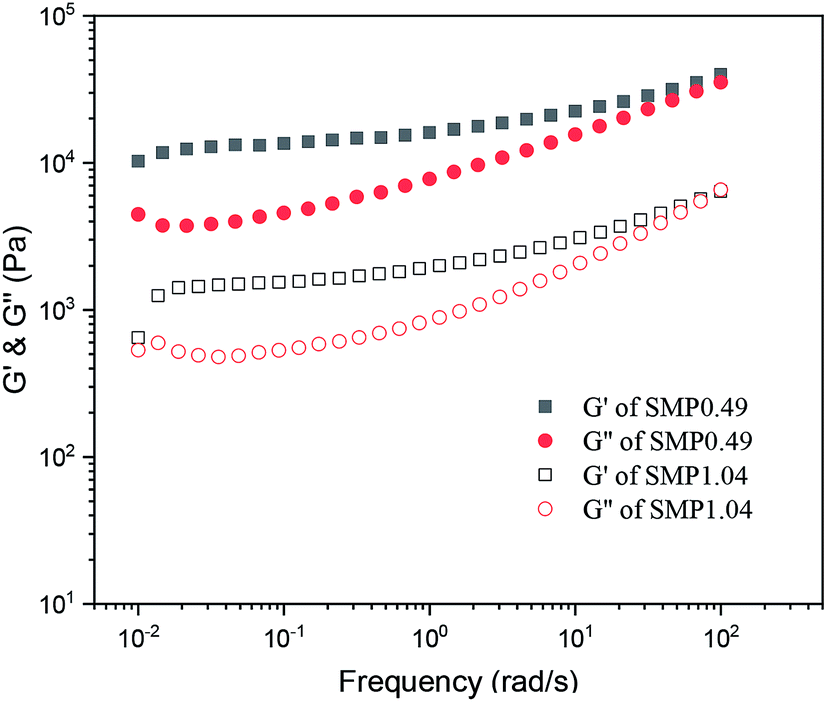 | ||
| Fig. 5 Variation in storage modulus (G′) and loss modulus (G′′) of supramolecular polymers as a function of frequency (100 °C, strain = 1%). | ||
3.3 Crystallization and melting behaviors of PLLA/supramolecular polymer blends
The XRD curves of the PLLA/supramolecular polymer blends prepared by solvent casting were recorded, and the obtained results are shown in Fig. S1.† It is clear that the XRD curve of PLLA shows several obvious diffraction peaks at 14.8°, 16.8° and 19.0°, which correspond to the (010), (110)/(200) and (203) planes of the α crystal formed by the homo-crystallization of PLLA.44 With the increase in the content of the synthesized supramolecular polymer in the PLLA matrix, the diffraction peaks corresponding to α crystal are significantly weakened. For the blend PLLA/SMP1.04-50%, a weak diffraction peak is observed at 24.1°, which is attributed to the stereocomplex formed between PLLA and PDLA blocks of the supramolecular polymer.45The DSC curves of the PLLA/supramolecular polymer blends are displayed in Fig. 6, and the obtained thermal parameters are listed in Table S2.† In Fig. 6(a), neat PLLA shows a weak crystallization peak at 99.4 °C in the DSC cooling curve. The corresponding heating curve presents an exothermic peak at 112.2 °C and an endothermic peak at 155–175 °C (Fig. 6(b)), corresponding to cold crystallization and melting of PLLA. A weak peak is also detected at 60.0 °C, and it is assigned to the glass transition of PLLA. For the blend PLLA/SMP0.49-10%, the cooling curves show weak crystallization of PLLA at 99.3 °C. With the increase in the SMP0.49 content, the crystallization peak of PLLA disappears. As shown in Fig. 6(b), with the increase in the SMP0.49 content, an endothermic peak is observed in a lower temperature region (about 30 °C), which can be attributed to the melting of the PCVL segments of supramolecular polymers. In addition, the glass transition temperature of the PLLA/SMP0.49 blends decreased as the content of SMP0.49 increased. The synthesized supramolecular polymer SMP0.49 contained a higher PCVL content with a low glass transition temperature. When the supramolecular polymer SMP0.49 was introduced into the PLLA matrix, the PLLA/SMP0.49 blends could present a lower glass transition temperature with respect to that of PLLA. It can be observed in Fig. 6(b) that the PLLA/SMP0.49 blends show a significantly exothermic peak at about 122 °C due to the cold crystallization of PLLA. As listed in Table S2,† the cold crystallization enthalpy of the blend PLLA/SMP0.49-10% SMPs is larger than that of neat PLLA, while this area decreases as the SMP content further increases. With the increase in the SMP0.49 content, the melting enthalpy of PLLA decreased, but a weak melting peak assigned to the PLA stereocomplex was observed at about 182 °C.
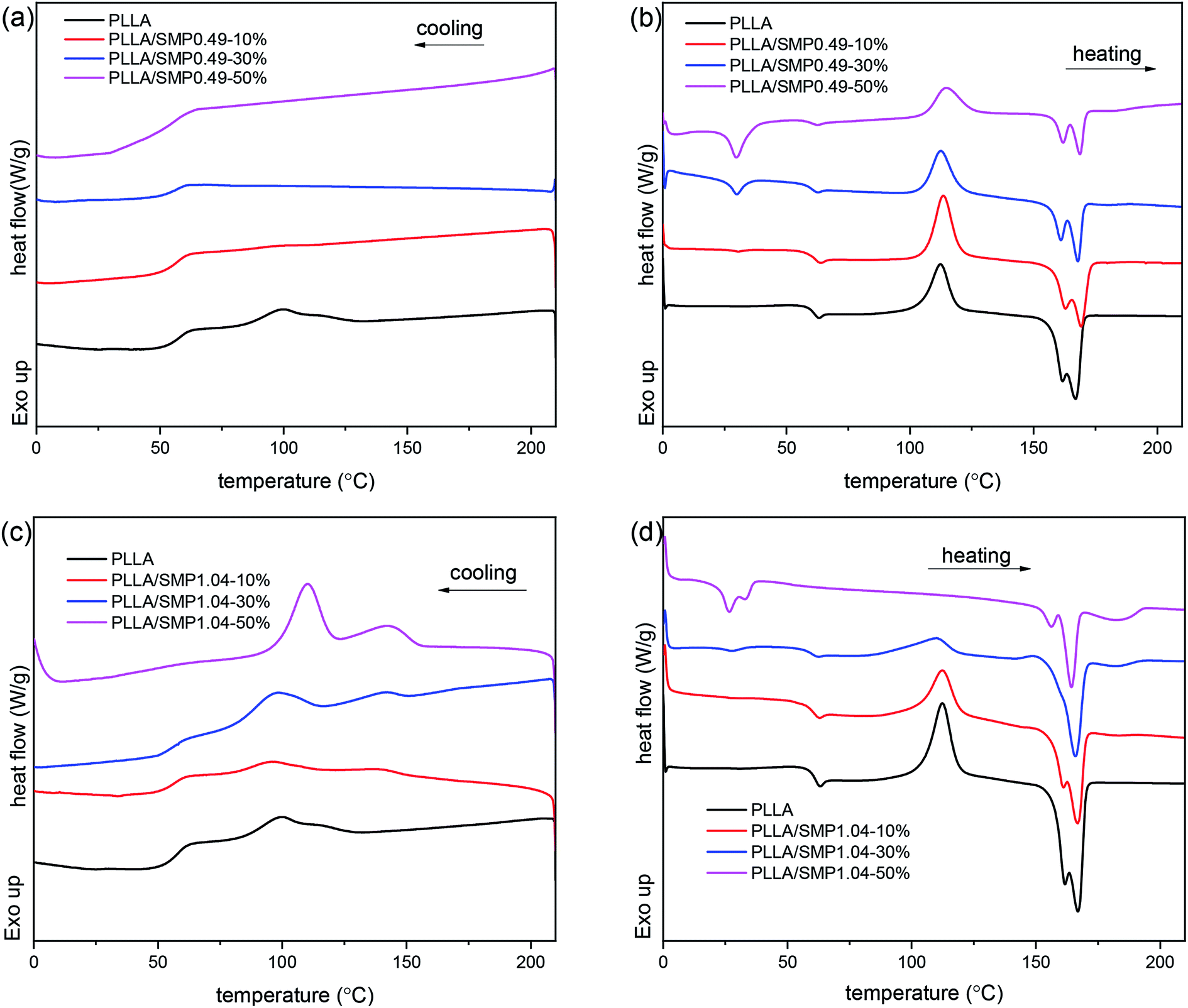 | ||
| Fig. 6 DSC cooling (a and c) and reheating curves (b and d) of poly(L-lactide)/supramolecular polymer blends: (a and b) PLLA/SMP0.49; (c and d) PLLA/SMP1.04. | ||
In Fig. 6(c), the cooling curves of the PLLA/SMP1.04 blends show two weak endothermic peaks: the peak in the higher temperature region is assigned to the stereocomplex crystallization between PLLA and PDLA blocks of supramolecular polymers; the peak corresponding to the lower temperature region is attributed to the homo-crystallization of PLLA. It can also be found from Fig. 6(c) that the area and temperature of the peak corresponding to stereocomplex crystallization increase as the content of SMP1.04 increases. The supramolecular polymer SMP1.04 exhibited a higher PDLA content; thus, the increase in the SMP content led to an increase in the PDLA content in the PLLA/SMP1.04 blends, which was beneficial for stereocomplex crystallization. The peak corresponding to PLLA homo-crystallization also shifted to the higher temperature region as the content of SMP1.04 increased, and the peak area was enhanced. This is because the formed stereocomplex could act as a heterogeneous nucleating agent for PLLA homo-crystallization.46 In Fig. 6(d), the PLLA/SMP1.04 blends show similar reheating curves to that of the PLLA/SMP1.04 blends. The melting of the PCVL block and cold crystallization of PLLA were observed at about 30 °C and 110 °C, respectively. However, with the increase in the SMP1.04 content, the melting peak of the PCVL block and the cold crystallization peak weakened and eventually disappeared. The PLLA/SMP1.04 blends showed multi-step melting at 150–170 °C, which was assigned to the melting–recrystallization–remelting process of PLLA. It is also clear that a weak melting peak of the stereocomplex is observed at about 190 °C, and this melting area increased as the SMP1.04 content increased. Similar results have been reported for the asymmetric PLLA/PDLA blends and the PCL/PLA alternating multiblock supramolecular polymers.37,48,50 It can be found from Table S2† that the crystallinity of PLLA/SMP0.49 decreased with the content of the supramolecular polymers, while the crystallinity of PLLA/SMP1.04 increased. This may be attributed to the different compositions of supramolecular polymers. For the PCVL block, a low glass transition temperature (about −50 °C) was observed and thus, it could accelerate the mobility of polymer chains. The PDLA block could form a PLA stereocomplex, which could act as the heterogeneous nucleating agent for PLLA crystallization. When a supramolecular polymer with a shorter PDLA block is blended with PLLA, it is difficult to achieve stereocomplex formation. Because the supramolecular polymer shows good movement capacity, the short PDLA block is not beneficial to the alternating arrangement of PLLA and PDLA due to the weak hydrogen bond. For the supramolecular polymer with a longer PDLA block, it is easy to form a stereocomplex with PLLA. This is because the longer PDLA block could form strong hydrogen bonds, accelerating the alternating arrangement of the PLLA and PDLA chains. The formed stereocomplex could act as heterogeneous nucleation sites, and the PCVL block would facilitate the favorable arrangement of PLLA around the nucleation sites. Therefore, the synthesized supramolecular polymers SMP0.49 and SMP1.04 showed the reverse effect on the crystallization of PLLA.
3.4 Rheological behaviors of PLLA/supramolecular polymer blends
The low melt strength of PLLA limited its processing conditions. Many methods, such as the nanocomposite technology and cross-linking technology, have been used to solve this problem. Previous investigations found that the stereocomplex crystal is also an effective rheological modifier for the PLLA melt.47–49 In the study, the DSC results demonstrated that the stereocomplex crystals were formed in the PLLA/SMP blends. Therefore, the rheological behaviour of the PLLA/SMP blends was also analyzed, and the obtained results are shown in Fig. 7. Neat PLLA completely melted at 175 °C, which is an ideal polymer melt. Therefore, the G′ and G′′ values of the neat PLLA melt basically obey the following equations: logG′ ∝ 2logω and logG′′ ∝ logω (Fig. S2†). As shown in Fig. 7(a) and (b), the G′ and G′′ values of the blend PLLA/SMPs0.49-10% are lower than those of neat PLLA. This may be attributed to the plasticization of the PCVL block of the supramolecular polymer. With the increase in the SMP content, the G′ and G′′ values of the PLLA/SMP blends increased, and the change in the G′ value was more significant than that in the G′′ value. It is clear that the slopes of the modulus curves of the PLLA/SMP blends decreased at low frequencies, revealing that the PLLA/SMP blends present an obvious nonterminal behavior. The DSC results demonstrated that the PLLA/SMP blends contained stereocomplex crystals, which have a high melting temperature. Therefore, the stereocomplex crystals as the cross-linking point in the melt of the PLLA/SMP blends limited the movement of polymer chains, resulting in a slower relaxation behavior in the melt of the PLLA/SMP blends.
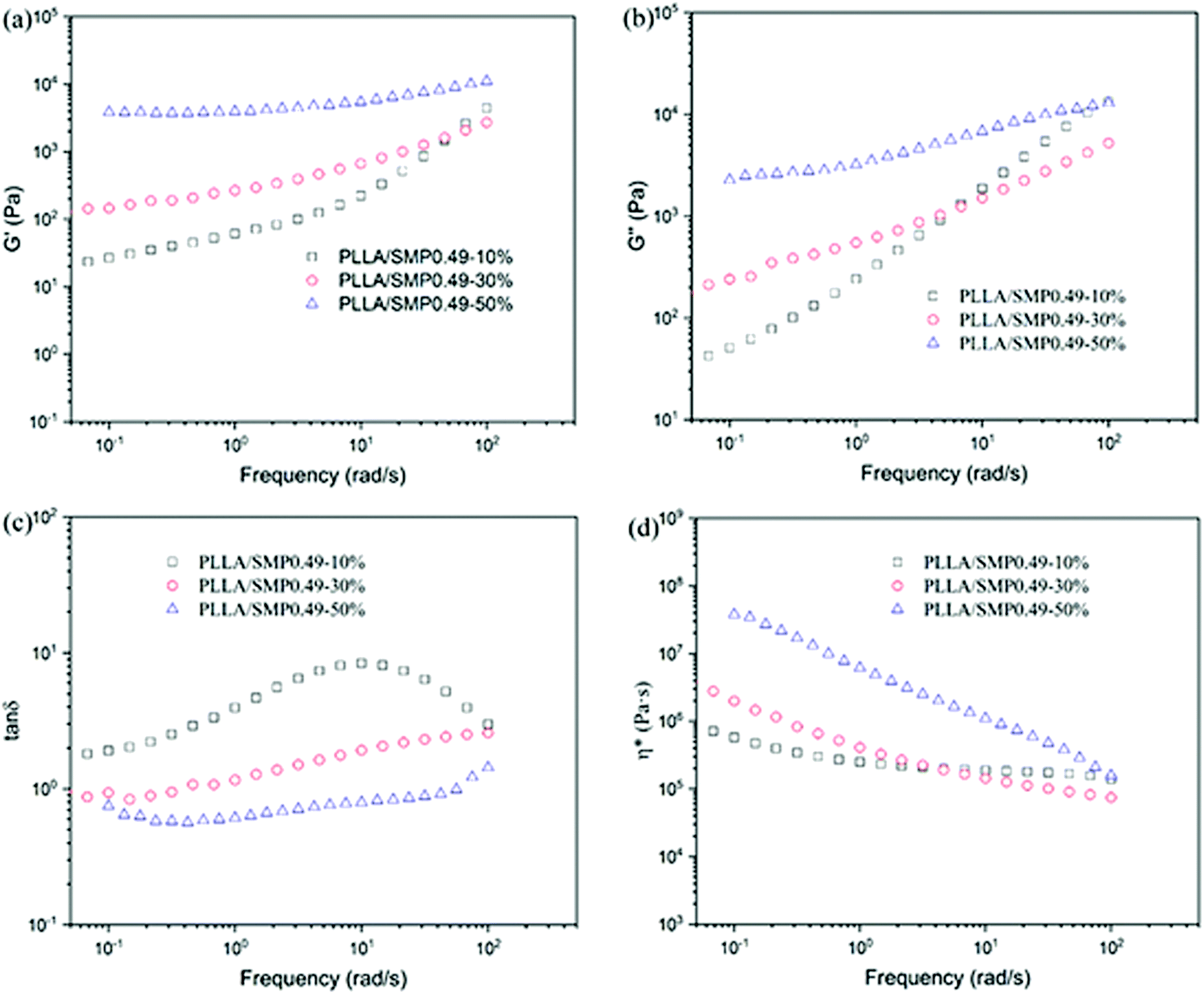 | ||
| Fig. 7 Variation in storage modulus (G′), loss modulus (G′′), loss tangent (tanδ) and complex viscosity (η*) as a function of frequency for PLLA/SMP0.49 blends at 175 °C. | ||
The loss tangent (tanδ) is more sensitive to the relaxation behavior of a polymer melt with respect to G′ and G′′. To further analyze the rheological behavior of the PLLA/SMP blends, the variation in the loss tangent as a function of frequency was also measured, as shown in Fig. 7(c). The curves of neat PLLA show an obvious peak (Fig. S3†), exhibiting a typical behavior of viscoelastic liquids. With the increase in the content of SMP, the value corresponding to this peak obviously decreased, revealing a more significant elastic response. Especially for the blends with more than 30% SMP, this peak completely disappeared. This indicated that the melt of the PLLA/SMP blends achieved a transition from liquid-like to solid-like viscoelastic behaviors when the SMP0.49 content exceeded 30%, which was because the long-range polymer chain movement was restrained by the stereocomplex crystals. The complex viscosity (η*) was also used to investigate the rheological behavior of the PLLA/SMP melt, as shown in Fig. 7(d). For neat PLLA, the complex viscosity (η*) exhibited no tolerance to frequency at a lower frequency (<10 rad s−1) (Fig. S4†), showing a typical Newtonian fluid behavior. At a high frequency (>10 rad s−1), the complex viscosity (η*) was reduced due to shear thinning. However, the complex viscosity curves of the PLLA/SMP melt exhibited a significant slope, and the slope increased as the SMP content increased. This indicates that the PLLA/SMP melt exhibits a non-Newtonian fluid behavior. Therefore, there may be a structure similar to the cross-linked network in the melt of the PLLA/SMP blends.
Our previous studies48,50,51 found that the stereocomplex crystal is different from an inorganic particle, and it has a certain tolerance to temperature, causing the effect of the stereocomplex crystal as a rheological modifier to be affected by the thermal treatment temperature. Therefore, the rheological behavior of the PLLA/SMP blends at different thermal treatment temperatures was measured, and the obtained results are displayed in Fig. 8. For the PLLA/SMP0.49-50% blends, the G′ and G′′ values obviously decreased as the thermal treatment temperature increased. This was mainly because the stereocomplex crystals began to melt as the thermal treatment temperature increased, resulting in the stereocomplex crystals becoming smaller. This not only directly affected the stereocomplex crystals existed in the polymer melt, but also destroyed the network structure based on the stereocomplex crystals in the melt. Therefore, the increased thermal treatment temperature resulted in the weakening of the strength of the PLLA/SMP melt. It could be observed that the G′ and G′′ values of the PLLA/SMP1.04-50% blends melted at 185 °C were larger than those of the PLLA/SMP0.49-50% blends. This was attributed to the high stereocomplex content. The supramolecular polymer SMP1.04 contained more D-lactide repeating units with respect to SMP0.49. This not only led to an increase in the content of the stereocomplex crystals, but also made the formed stereocomplex crystals more perfect. The rheological results demonstrated that a similar cross-linked network was formed in the melt of the PLLA/SMP blends, resulting in a transition from liquid-like to solid-like viscoelastic behaviors, and the network existing in the PLLA matrix was closely related to the content of the PDLA blocks and the thermal treatment temperature.
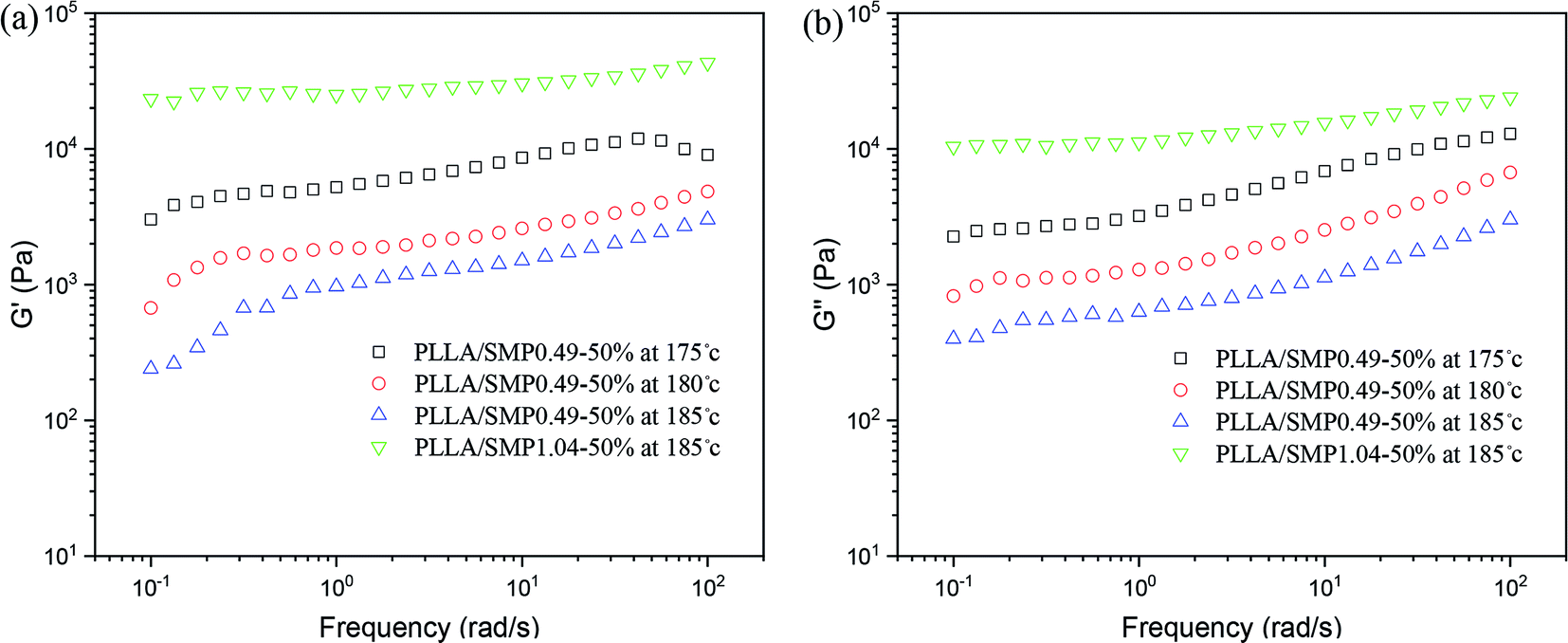 | ||
| Fig. 8 Variation in storage modulus (G′) and loss modulus (G′′) as a function of frequency for PLLA/SMP blends at different temperatures. | ||
3.5 Mechanical properties of PLLA/supramolecular polymer blends
The mechanical properties of the PLLA/supramolecular polymer blends were evaluated by a tensile test. The typical stress–strain curves are shown in Fig. 9, and the obtained data are listed in Table 2. Neat PLLA showed high strengths at yielding (51.3 MPa) and at break (32.8 MPa) and low elongation at break (24.1%). Previous literature33,52 has reported that the elongation at break of neat PLLA does not exceed 15%. The possible reason is that the neat PLLA film prepared by solution casting may have a residual solvent, which can act as a plasticizer. When it is blended with 10 wt% of the synthesized supramolecular polymer SMP0.49, the strengths at yielding and at break decrease to 22.00 MPa and 24.28 MPa, respectively, while the elongation at break increases to 330.4%. This is a reasonable phenomenon of toughening plastics with supramolecular polymers having elastomeric characteristics. A similar result has been reported by Wu et al.52 for the blends of PLLA with an ethylene-vinyl acetate-glycidyl methacrylate elastomer. It is clear from Table 2 that the strengths at yielding and at break of PLLA/SMP0.49-10% are lower than those of PLLA/SMP1.04-10%, while its elongation at break is larger than that of PLLA/SMP1.04-10%. This is closely related to the composition of the synthesized supramolecular polymer. The supramolecular polymer SMP0.49 contained a short rigid PDLA block with respect to SMP1.04. Therefore, the SMP0.49 toughening of PLA was better than SMP1.04. With the increase in the SMP0.49 content, the strength further decreased, and the elongation at break increased. However, when the content of SMP0.49 increased to 50%, the strength and elongation at break decreased. The PLLA/SMP1.04 blends showed a similar phenomenon: the strength decreased as the SMP1.04 content increased, while the elongation at break first increased and then decreased. These results revealed that the lower content of the synthesized supramolecular polymer can achieve toughening of the PLLA matrix, while a higher SMP content results in the poor mechanical properties of the PLLA/SMP blends. For the blends with higher SMP contents, large stereocomplex crystals could be formed. The stereocomplex has two roles. It acts as a heterogeneous nucleating agent to promote the crystallization of PLLA, which is an advantage for maintaining high mechanical strength. However, the stereocomplex played a role of crosslinking point, which would limit the movement of polymer chains. Therefore, the toughening of PLLA can be achieved via introducing a lower content of the synthesized supramolecular polymer with a short PDLA block.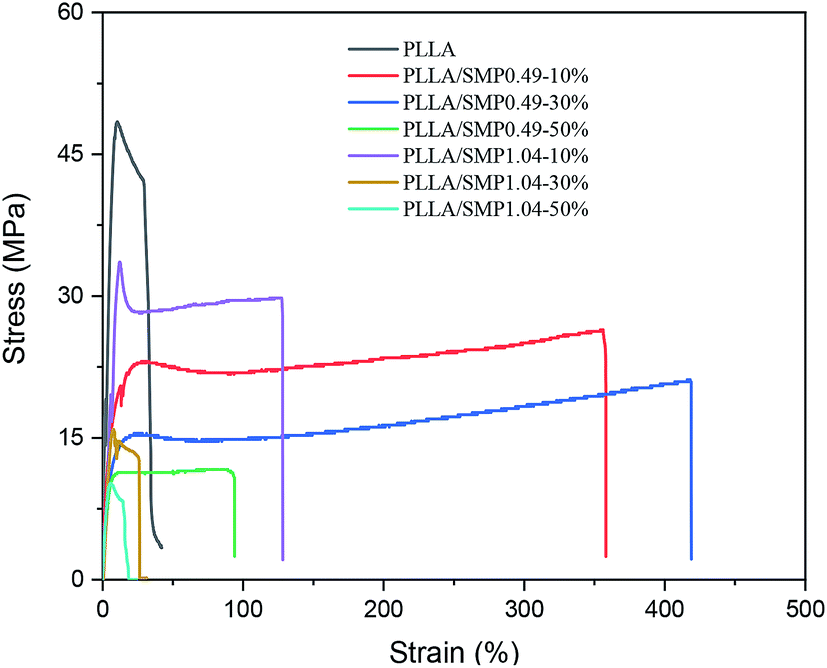 | ||
| Fig. 9 Typical stress–strain curves of the neat PLLA and PLLA/SMP blends. | ||
| Samples | Strength at yielding (MPa) | Strength at break (MPa) | Elongation at break (%) |
|---|---|---|---|
| PLLA | 51.33 ± 4.4 | 32.82 ± 3.1 | 24.1 ± 6.1 |
| PLLA/SMP0.49-10% | 22.00 ± 1.02 | 24.28 ± 2.83 | 330.4 ± 35.3 |
| PLLA/SMP0.49-30% | 15.49 ± 0.97 | 19.90 ± 1.56 | 372.4 ± 76.5 |
| PLLA/SMP0.49-50% | 10.33 ± 2.47 | 9.90 ± 1.45 | 91.4 ± 15.4 |
| PLLA/SMP1.04-10% | 33.62 ± 6.23 | 30.02 ± 5.29 | 143.1 ± 17.3 |
| PLLA/SMP1.04-30% | 16.70 ± 2.45 | 14.13 ± 1.64 | 23.08 ± 2.54 |
| PLLA/SMP1.04-50% | 9.71 ± 0.48 | 9.71 ± 0.48 | 5.38 ± 0.24 |
4. Conclusions
We presented a fully bio-based material by blending PLLA with supramolecular polymers based on the PDLA–PCVL–PDLA triblock copolymers, achieving a fast crystallization rate, high melt strength and excellent mechanical properties. Supramolecular polymers with alternating PDLA/PCVL multi-block structure were synthesized by the self-complementary quadruple hydrogen bonding of the 2-ureido-4[1H]-pyrimidinone (UPy) dimers. The incorporation of the UPy groups inhibited the crystallization of polymers, and the self-complementing quadruple hydrogen bonds formed between the UPy dimers were dynamically reversible and temperature-dependent. On this basis, PLLA/SMP blends were prepared by solution casting and characterized. PLLA could form a stereocomplex with its enantiomeric PDLA blocks of supramolecular polymers, which could reinforce the PLLA melt. The SMP composition and the SMP content in the PLLA matrix significantly affected the crystallization and mechanical properties of the PLLA/SMP blends. SMPs0.49 inhibited the crystallization of PLLA, while SMP1.04 accelerated the crystallization of PLLA. The toughening of PLLA could be achieved by introducing a lower content of the synthesized supramolecular polymer with a short PDLA block. Therefore, this study provides an effective way to control the crystallization, rheology and mechanical properties of PLLA.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Natural Science Foundation of Guangdong Province (2018A030307020), College Youth Innovation Talents Project of Guangdong Province (2017KQNCX089), and Project of enhancing school with innovation of Guangdong Ocean University (Q18304) and Program for Scientific Research Start-up Funds of Guangdong Ocean University (R19010).References
- K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef PubMed.
- X. Pang, X. Zhuang, Z. Tang and X. Chen, Biotechnol. J., 2010, 5, 1125–1136 CrossRef CAS PubMed.
- Q. Zhang, V. N. Mochalin, I. Neitzel, I. Y. Knoke, J. Han, C. A. Klug, J. G. Zhou, P. I. LeIkes and Y. Gogotsi, Biomaterials, 2011, 32, 87–94 CrossRef CAS PubMed.
- X. Niu, Q. Feng, M. Wang, X. Guo and Q. Zheng, J. Controlled Release, 2007, 134, 111–117 CrossRef PubMed.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- A. K. Bledzki and A. Jaszkiewicz, Compos. Sci. Technol., 2010, 70, 1687–1696 CrossRef CAS.
- J. F. Mano, Y. Wang, J. C. Viana, Z. Denchev and M. J. Oliveira, Macromol. Mater. Eng., 2004, 289, 910–915 CrossRef CAS.
- V. Nagarajan, A. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2016, 4, 2899–2916 CrossRef CAS.
- Y. Li and H. Shimizu, Macromol. Biosci., 2007, 7, 921–928 CrossRef CAS PubMed.
- V. Vilay, M. Mariatti, Z. Ahmad, K. Pasomsouk and M. Todo, J. Appl. Polym. Sci., 2009, 114, 1784–1792 CrossRef CAS.
- Y. Li and H. Shimizu, Eur. Polym. J., 2009, 45, 738–746 CrossRef CAS.
- L. Jiang, M. P. Wolcott and J. Zhang, Biomacromolecules, 2006, 7, 199–207 CrossRef CAS PubMed.
- H. Bai, D. Bai, H. Xiu, H. Liu, Q. Zhang, K. Wang, H. Deng, F. Chen, Q. Fu and F. C. Chiu, RSC Adv., 2014, 4, 49374–49385 RSC.
- C. H. Ho, C. H. Wang, C. I. Lin and Y. D. Lee, Polymer, 2008, 49, 3902–3910 CrossRef CAS.
- Y. Wang and M. A. Hillmyer, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2755–2766 CrossRef CAS.
- L. Chen, X. Qiu, Z. Xie, Z. Hong, J. Sun, X. Chen and X. Jing, Carbohydr. Polym., 2006, 65, 75–80 CrossRef CAS.
- H. T. Oyama, Polymer, 2009, 50, 747–751 CrossRef CAS.
- H. Chen, X. Yu, W. Zhou, S. Peng and X. Zhao, Polym. Test., 2018, 70, 275–280 CrossRef CAS.
- Y. Wang, Z. Wei and Y. Li, Eur. Polym. J., 2016, 85, 92–104 CrossRef CAS.
- Y. Yang, L. Zhang, Z. Xiong, Z. Tang, R. Zhang and J. Zhu, Sci. China: Chem., 2016, 59, 1355–1368 CrossRef CAS.
- B. R. Sveinbjornssopn, G. M. Miyake, A. El-Batta and R. H. Grubbs, ACS Macro Lett., 2014, 3, 26–29 CrossRef.
- Z. Jing, X. Shi and G. Zhang, Polymers, 2017, 9, 107 CrossRef PubMed.
- Z. Li, B. H. Tan, T. Lin and C. He, Prog. Polym. Sci., 2016, 62, 22–72 CrossRef CAS.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- K. Fukushima, M. Hirata and Y. Kimura, Macromolecules, 2007, 40, 3049–3055 CrossRef CAS.
- H. Urayama, T. Kannamori and Y. Kimura, Macromol. Mater. Eng., 2002, 287, 116–121 CrossRef CAS.
- R. Y. Bao, W. Yang, W. R. Jiang, Z. Y. Liu, B. H. Xie, M. B. Yang and Q. Fu, Polymer, 2012, 53, 5449–5454 CrossRef CAS.
- P. Purnama and S. H. Kim, Macromolecules, 2010, 43, 1137–11142 CrossRef CAS.
- Z. Jing, X. Shi, G. Zhang and J. Li, Polym. Adv. Technol., 2015, 26, 528–537 CrossRef CAS.
- Z. Jing, X. Shi, G. Zhang and J. Qin, Polym. Adv. Technol., 2015, 26, 223–233 CrossRef CAS.
- J. Narita, M. Katagiri and J. Tsuji, Macromol. Mater. Eng., 2011, 296, 887–893 CrossRef CAS.
- H. Zhao, Y. Bian, Y. Li, Q. Dong, C. Han and L. Dong, J. Mater. Chem. A, 2014, 2, 8881–8892 RSC.
- Y. Liu, J. Shao, J. Sun, X. Bian, L. Feng, S. Xiang, B. Sun, Z. Chen, G. Li and X. Chen, Polym. Degrad. Stab., 2014, 101, 10–17 CrossRef CAS.
- F. Qi, M. Tang, X. Chen, M. Chen, G. Guo and Z. Zhang, Eur. Polym. J., 2015, 71, 314–324 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- Z. Jing, X. Shi, G. Zhang and J. Gu, Polymer, 2017, 121, 124–136 CrossRef CAS.
- X. Shi, Z. Jing, G. Zhang, Y. Xu and Y. Yao, J. Appl. Polym. Sci., 2017, 134, 45575 CrossRef.
- Y. Huang, R. Chang, L. Han, G. Shan, Y. Bao and P. Pan, ACS Sustainable Chem. Eng., 2016, 4, 121–128 CrossRef CAS.
- R. Chang, Y. Huang, G. Shan, Y. Bao, X. Yun, T. Dong and P. Pan, Polym. Chem., 2015, 6, 5899–5910 RSC.
- F. Herbst, K. Schrtöer, I. Gunkel, S. Gröger, T. Thurn-Albrecht, J. Balbach and W. H. Binder, Macromolecules, 2010, 43, 10006–10016 CrossRef CAS.
- J. R. Dorgan and J. S. Williams, J. Rheol., 1999, 43, 1141–1155 CrossRef CAS.
- C. J. Tsenoglou and A. D. Gotsis, Macromolecules, 2001, 34, 4685–4687 CrossRef CAS.
- J. L. Wietor, D. J. M. Van Beek, G. W. Peters, E. Mendes and R. P. Sijbesma, Macromolecules, 2011, 44, 1211–1219 CrossRef CAS.
- H. Xu, C. Teng and M. Yu, Polymer, 2006, 47, 3922–3928 CrossRef CAS.
- J. Sun, H. Yu, X. Zhang, X. Chen and X. Jing, J. Phys. Chem. B, 2011, 115, 2864–2869 CrossRef CAS PubMed.
- H. Tsuji, H. Takai and S. K. Saha, Polymer, 2006, 47, 3826–3837 CrossRef CAS.
- H. Yamane, K. Sasai and M. Takano, J. Rheol., 2004, 48, 599–609 CrossRef CAS.
- Z. Jing, X. Shi and G. Zhang, Polym. Adv. Technol., 2016, 27, 1108–1120 CrossRef CAS.
- X. F. Wei, R. Y. Bao, Z. Q. Cao, W. Yang, B. H. Xie and M. B. Yang, Macromolecules, 2014, 47, 1439–1448 CrossRef CAS.
- Z. Jing, X. Shi, G. Zhang, J. Li, J. Li, L. Zhou and H. Zhang, Polymer, 2016, 92, 210–221 CrossRef CAS.
- X. Shi, Z. Jing and G. Zhang, J. Polym. Res., 2018, 25, 1–16 CrossRef.
- B. Wu, Q. Zeng, D. Niu, W. Yang, W. Dong, M. Chen and P. Ma, Macromolecules, 2019, 52, 1092–1103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04283k |
| This journal is © The Royal Society of Chemistry 2019 |