
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed synthesis of phenol and diaryl ether derivatives via hydroxylation of diaryliodoniums†
Lianbao Ye *a,
Chao Hana,
Peiqi Shia,
Wei Gaoa and
Wenjie Meib
*a,
Chao Hana,
Peiqi Shia,
Wei Gaoa and
Wenjie Meib
aSchool of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, Guangdong, China. E-mail: yelb7909@163.com; Tel: +86-20-39352139
bGuangdong Province Engineering Technology Center for Molecular Probes & Biomedical Imaging, Guangdong Pharmaceutical University, Guangzhou 510006, Guangdong, China
First published on 10th July 2019
Abstract
A copper-catalysed hydroxylation of diaryliodoniums to generate phenols and diaryl ethers is reported. This method allows the synthesis of diversely functionalized phenols under mild reaction conditions without the need for a strong inorganic base or an expensive noble-metal catalyst. Significantly, convenient application of diaryliodoniums is demonstrated in the preparation of diaryl ethers in a one-pot operation.
Introduction
Phenols and their derivatives are ubiquitous constituents for constructing bioactive natural products, agrochemicals and pharmaceuticals and serve as important motifs of polymers as well as novel materials.1 Currently, many useful methods have been developed to prepare these phenol moieties by synthetic researchers.2 Couplings of aryl halides and nucleophilic hydroxide salts in the presence of palladium or copper complex have been proved to be classical methods to prepare these compounds (Scheme 1a).3i In addition, hydroxylation of boronic acids (Scheme 1b) and hydrolysis of diazonium salts are also efficient strategies.4 Although phenol derivatives are available by different transformations, it is very important to develop novel methodologies with mild reaction conditions, high efficiency and low waste generation. | ||
| Scheme 1 Selected methods for the synthesis of phenols. | ||
Diaryliodoniums are air- and moisture-stable versatile electrophilic arylating reagents, which are easily available when iodoarenes are treated with mild oxidants.5 They have been utilized to arylate a broad scope of nucleophiles in recent years, including aryl/alkyl amines,6 phenols,7 aliphatic alcohol and benzoic acid, which afford the corresponding aromatic compounds.8 Despite this achievement, the use of strong inorganic bases or organic bases is adverse to reaction, especially for basic-sensitive functional groups.9 Recently, We and other groups reported mild and rapid transformations of cyclic hypervalent iodoniums into oxygen-bridged polycyclic heteroarenes and dibenzofuran derivatives.10 Owing to these previous work, we envisage a strategy for the synthesis of phenols via the hydroxylation of diaryl iodoniums. In terms of the oxygen donor, waster is the most environmentally friendly oxygen source.11 The arylation of diaryliodoniums with waster as the nucleophile is believed to be a valuable access to phenols. In this study, we develop a one-pot reaction for the synthesis of a wide range of phenols and diaryl ethers from diaryliodoniums. This versatile method operates under mild reaction conditions, with inexpensive CuCl as the catalyst (Scheme 1c).
Results and discussion
At the outset of our studies, the hydroxylation of diaryliodoniums was conducted by the exposure substrate 1a to Cu(OAc)2 (0.1 equiv.), Na2CO3 (2.0 equiv.), H2O (5.0 equiv.) in DMF at 40 °C for 2 hours, leading to the desired product 2a in a moderate yield (entry 1). Encouraged by this result, we subsequently screened other copper catalysts (entry 2–5), including Cu(OTf)2, CuCl2, CuSO4 and CuI were inferior to Cu(OAc)2, while CuCl showed the best efficiency. Compared with DMF, solvents of toluene, DCE, dioxane and MeCN were not helpful for the reaction conversion (entry 6–9). The examine of bases disclosed that mild NaOAc was ideal choice (entry 10–14). The yields reduced when the reaction was performed in shortened time (entry 15–16). It was observed that only 39% 2a was yielded at rt (entry 17), which indicated that slight heating was needed for the diaryliodoniums reactivity. Further study revealed that it is not fruitful without the addition of water (entry 18), and even no targeted product was isolated in the absence of CuCl (entry 19) (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalysis | Base | Solvent | T (°C) | Yield (%) |
| 1 | Cu(OAc)2 | Na2CO3 | DMF | 40 | 57 |
| 2 | Cu(OTf)2 | Na2CO3 | DMF | 40 | 0 |
| 3 | CuSO4 | Na2CO3 | DMF | 40 | 0 |
| 4 | CuI | Na2CO3 | DMF | 40 | 50 |
| 5 | CuCl | Na2CO3 | DMF | 40 | 85 |
| 6 | CuCl | Na2CO3 | Toluene | 40 | 10 |
| 7 | CuCl | Na2CO3 | DCE | 40 | 21 |
| 8 | CuCl | Na2CO3 | Dioxane | 40 | 0 |
| 9 | CuCl | Na2CO3 | MeCN | 40 | 42 |
| 10 | CuCl | Et3N | DMF | 40 | 77 |
| 11 | CuCl | K2CO3 | DMF | 40 | 85 |
| 12 | CuCl | NaOAc | DMF | 40 | 92 |
| 13 | CuCl | K3PO4 | DMF | 40 | 14 |
| 14 | CuCl | NaOH | DMF | 40 | 0 |
| 15 | CuCl | NaOAc | DMF | 40 | 86a |
| 16 | CuCl | NaOAc | DMF | 40 | 79b |
| 17 | CuCl | NaOAc | DMF | rt | 39(83c) |
| 18 | CuCl | NaOAc | DMF | 40 | 11d |
| 19 | — | NaOAc | DMF | 40 | 0 |
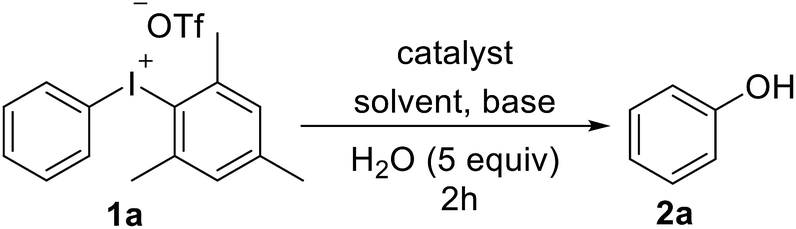
With optimized reaction conditions in hand, diversely functionalized diaryliodoniums were subjected to the established reaction conditions to explore the efficiency and scope of this copper-catalyzed hydroxylation reaction. As summarized in Scheme 2, this protocol afforded phenols bearing electron-rich, electron-poor, or sterically bulky substituted groups in good yields (2a–2l). Electron-neutral phenols were delivered in excellent yields (2a–2c). In terms of electron-deficient substrates, para-, and meta-substituted diaryliodoniums containing –F, –OCF3, –CF3 and –CO2Me were compatible to produce phenols 2d–2g in the yield of 56–87%. The reaction also furnished the electron-rich product efficiently (2h). Notably, 1-naphthol was available in a 74% yield (2i). It is necessary to emphasize that the reaction conditions were tolerant of ortho-functional groups such as methyl and bromo to give 2j and 2k. Hydroxylated heteroarenes are prevalent bioactive intermediates in medicinal chemistry. To our surprise, it was demonstrated that our strategies were suitable for the synthesis of hydroxylated pyridine 2l.
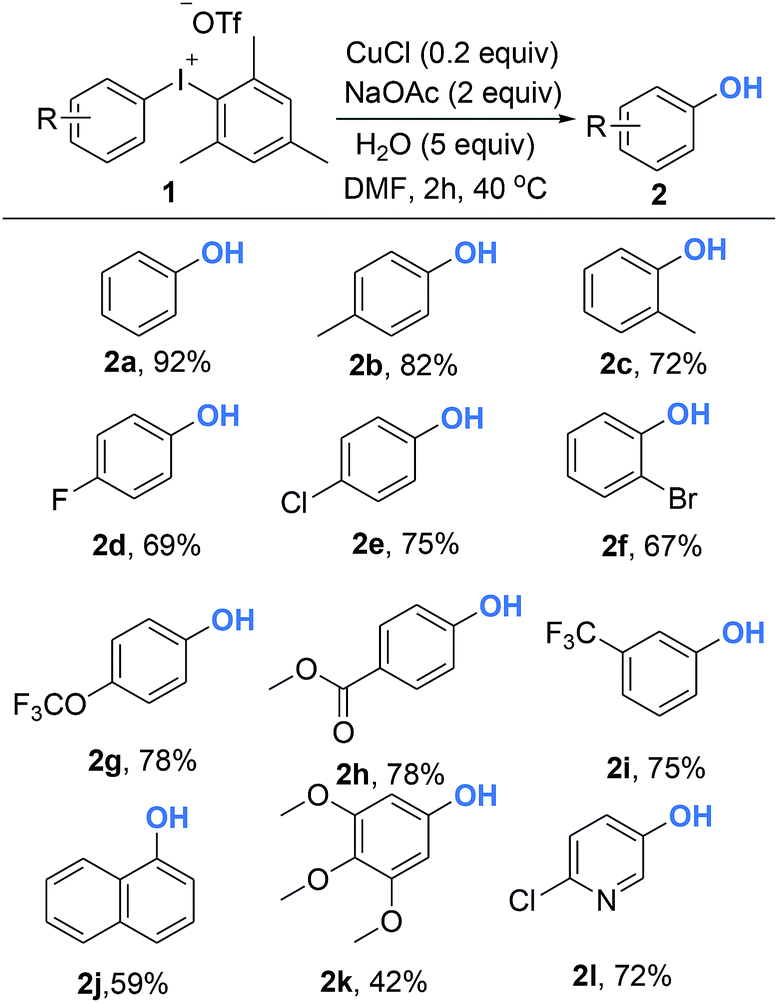 | ||
| Scheme 2 Copper-catalyzed synthesis of phenols from diaryliodoniums. Reaction conditions: 1 (0.2 mmol), CuCl (10 mol%), NaOAc (2 equiv.), H2O (5 equiv.), DMF (1.0 mL), Ar, 40 °C, 2 h. | ||
Diaryl ethers are significant building blocks and have found wide applications in the preparation of natural products and synthetic compounds with various biological properties and functions.12 Although diaryliodoniums have been extensively investigated as versatile arylating reagents in recent years, their poor atom economy limits further applications, as one equivalent of iodobenzene was generated as a waste in most of common reactions.13 Until recently, some examples were reported to overcome the drawback by designing one-pot, tandem reactions that can use the aryl iodide side product in a second arylation process in situ.14 Inspired by this domino arylating strategy, it is appealing to synthesize diaryl ethers directly, which represents a feasibility of using diaryliodoniums in an economic way. As shown in Scheme 3, the access to various aromatic ethers was realized by the addition of Cs2CO3 and dimethylglycine at 130 °C. As expected, symmetric diaryl ethers substituted by both electron-donating and electron-withdrawing groups formed smoothly (4a–4g). It is notable that the intact chloro (4e) or bromo (4f) moieties allow potential modification of polymer materials. Besides, unsymmetric diaryl ethers were synthesized efficiently in our method (4h–4i). Based on our observation, a copper-catalyzed hydroxylation of linear diaryliodoniums was proposed. Initially, oxidative addition of iodonium 1a to copper(I) would provide copper(III) intermediate A. This electrophilic species can be attacked by H2O under basic conditions, and intermediate B and C were formed consequently through this process. Finally, a reductive elimination of C would generate phenol 2a (Fig. 1).
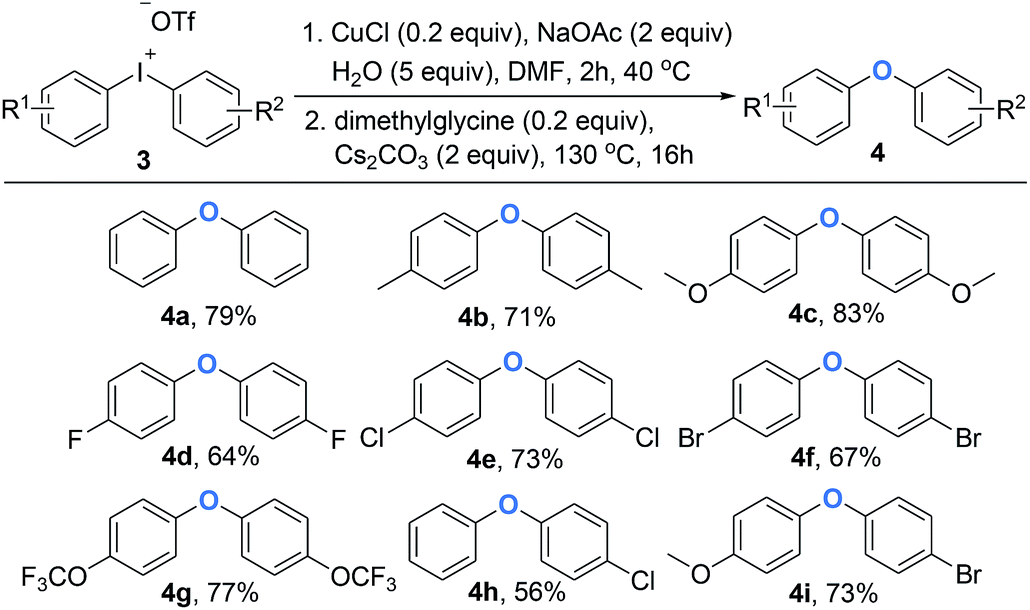 | ||
| Scheme 3 One-pot synthesis of diaryl ethers from diaryliodoniums. Reaction conditions: (1) 1 (0.2 mmol), CuCl (10 mol%), NaOAc (2 equiv.), H2O (5 equiv.), DMF (1.0 mL), Ar, 40 °C, 2 h; (2) Cs2CO3 (2 equiv.) and dimethylglycine (0.2 equiv.) were added, Ar, 130 °C, 16 h. | ||
 | ||
| Fig. 1 The proposed mechanism for the hydroxylation of diaryliodoniums. | ||
Conclusions
In summary, we have developed a copper-catalysed hydroxylation diaryliodoniums in a mild reaction conditions. This protocol enables the synthesis of aryl/heteroaryl phenols with various functionalities. Moreover, the one-pot, tandem process to diaryl ethers realizes convenient application of diaryliodoniums. Together with the use of an inexpensive metal catalyst, simple operation and atom economy, featuring the practical value of our protocol. Further study on utilization of this strategy in medicinal chemistry is underway in our lab.Experimental
General information
All solvents were commercially available and were used without further purification unless stated. The chemicals used were either purchased from commercial sources or prepared according to literature procedures for CDPIs. The 1H, 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance spectrometer 400 at 400 MHz, 100 MHz respectively. Chemical shifts are given in ppm (δ) referenced to CDCl3 with 7.26 for 1H and 77.10 for 13C, and to d6-DMSO with 2.50 for 1H and 39.5 for 13C. In the case of multiplet, the signals are reported as intervals. Signals are abbreviated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants are expressed in hertz. Mass spectra were recorded on a BRUKER VPEXII spectrometer (ESI mode). The progress of the reactions was monitored by thin-layer chromatography on a glass plate coated with silica gel with fluorescent indicator (GF254). Column chromatography was performed on silica gel (200–300 mesh).General procedure to synthesize phenols (2)
To a round-bottom flask was added diaryliodonium (0.2 mmol, 1.0 equiv.), NaOAc (2.0 equiv.), CuCl (0.1 equiv.), 5 (0.1 equiv.), DMF (1.0 mL). Then the flask was sealed, degassed and recharged with argon. The reaction proceeded at 40 °C for 2 h under argon atmosphere. Then, the reaction mixture was extracted with EtOAc, the combined organic layers were washed with H2O and brine and dried over anhydrous Na2SO4, evaporated in vacuo. The residue was purified by column chromatography on a silica gel (PE/EtOAc) to provide compounds 2.General procedure to synthesize diaryl ethers (4)
To a round-bottom flask was added diaryliodonium (0.2 mmol, 1.0 equiv.), NaOAc (2.0 equiv.), CuCl (0.1 equiv.), 5 (0.1 equiv.), DMF (1.0 mL). Then the flask was sealed, degassed and recharged with argon. The reaction proceeded at 40 °C for 2 h under argon atmosphere. Then, Cs2CO3 (2 equiv.) and dimethylglycine (0.2 equiv.) were added to the reaction residue, degassed and recharged with argon again. The reaction proceeded at 130 °C for 16 h under argon atmosphere. The reaction mixture was extracted with EtOAc, the combined organic layers were washed with H2O and brine and dried over anhydrous Na2SO4, evaporated in vacuo. The residue was purified by column chromatography on a silica gel (PE/EtOAc) to provide compounds 4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by Special Innovation Project of Guangdong Education Department (Natural Science) (2017KTSCX107), Medical Scientific Research Foundation of Guangdong province (B2019004), Science and Technology Planning Project of Guangdong Province (2016A020215159, 2017ZC0199), Key Laboratory of New Drug Discovery and Evaluation of Ordinary Universities of Guangdong province (2017KSYS002), Guangzhou Key Laboratory of Construction and Application of New Drug Screening Model Systems (201805010006), Guangdong province Precise Medicine and Big Data Engineering Technology Research Center for Traditional Chinese Medicine, the Open Project Foundation of the Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry (201502).Notes and references
-
(a) Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, Weinheim, 2003 CrossRef
; (b) J. H. P. Tyman, Synthetic and Natural Phenols; Elsevier: Amsterdam, 1996 Search PubMed
-
(a) H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 5th edn, 2010, pp. 359–374 Search PubMed
-
(a) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912 RSC
-
(a) J. J. Molloy, T. A. Clohessy, C. Irving, N. A. Anderson, G. C. Lloyd-Jonesc and A. J. B. Watson, Chem. Sci., 2017, 8, 1551–1559 RSC
-
(a) M. Bielawski and B. Olofsson, Chem. Commun., 2007, 2521–2523 RSC
-
(a) N. Purkait, G. Kervefors, E. Linde and B. Olofsson, Angew. Chem., Int. Ed., 2018, 57, 11427–11431 CrossRef CAS PubMed
- L. Chan, A. McNally, Q. Y. Toh, A. Mendoza and M. J. Gaunt, Chem. Sci., 2015, 6, 1277–1281 RSC
- T. B. Petersen, R. Khan and B. Olofsson, Org. Lett., 2011, 13, 3462–3465 CrossRef CAS PubMed
- M. Reitti, R. Gurubrahamam, M. Walther, E. Lindstedt and B. Olofsson, Org. Lett., 2018, 20, 1785–1788 CrossRef CAS
-
(a) D. Zhu, Z. Wu, B. Luo, Y. Du, P. Liu, Y. Chen, Y. Hu, P. Huang and S. Wen, Org. Lett., 2018, 20, 4815–4818 CrossRef CAS
-
(a) D. Shen, C. Saracini, Y. Lee, W. Sun, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 15857–15860 CrossRef CAS
-
(a) K. C. Nicolaou, C. N. C. Boddy, S. Bräse and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096–2152 CrossRef
-
(a) X. Huang, Q. Zhu and Y. Xu, Synth. Commun., 2001, 31, 2823 CrossRef CAS
-
(a) C. J. Teskey, S. M. A. Sohel, D. L. Bunting, S. G. Modha and M. F. Greaney, Angew. Chem., Int. Ed., 2017, 56, 5263–5266 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04282b |
| This journal is © The Royal Society of Chemistry 2019 |