
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen synthesis of Au decorated CoFe2O4 nanoparticles for catalytic reduction of 4-nitrophenol and dimethylphenylsilane oxidation
Samuel Saire-Sairea,
Eduardo C. M. Barbosa b,
Daniel Garciab,
Leandro H. Andradeb,
Sergi Garcia-Segurac,
Pedro H. C. Camargob and
Hugo Alarcon*a
b,
Daniel Garciab,
Leandro H. Andradeb,
Sergi Garcia-Segurac,
Pedro H. C. Camargob and
Hugo Alarcon*a
aCenter for Development of Advanced Materials and Nanotechnology, Universidad Nacional de Ingeniería, Av. Tupac Amaru 210, Rímac 15333, Lima, Peru. E-mail: halarcon@uni.edu.pe
bDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, 05508-000, São Paulo-SP, Brazil
cNanosystems Engineering Research Center for Nanotechnology-Enabled Water Treatment, School of Sustainable Engineering and the Built Environment, Arizona State University, Tempe, AZ 85287-3005, USA
First published on 16th July 2019
Abstract
Gold nanoparticles (Au NPs) have been widely employed in catalysis. Here, we report on the synthesis and catalytic evaluation of a hybrid material composed of Au NPs deposited at the surface of magnetic cobalt ferrite (CoFe2O4). Our reported approach enabled the synthesis of well-defined Au/CoFe2O4 NPs. The Au NPs were uniformly deposited at the surface of the support, displayed spherical shape, and were monodisperse in size. Their catalytic performance was investigated towards the reduction of 4-nitrophenol and the selective oxidation of dimethylphenylsilane to dimethylphenylsilanol. The material was active towards both transformations. In addition, the LSPR excitation in Au NPs could be employed to enhance the catalytic performance, which was demonstrated in the 4-nitrophenol reduction. Finally, the magnetic support allowed for the easy recovery and reuse of the Au/CoFe2O4 NPs. In this case, our data showed that no significant loss of performance took place even after 10 reaction cycles in the oxidation of dimethylphenylsilane to dimethylphenylsilanol. Overall, our results indicate that Au/CoFe2O4 are interesting systems for catalytic applications merging high performances, recovery and re-use, and enhancement of activities under solar light illumination.
1. Introduction
Green chemistry is a key aspect to ensure sustainable development and manufacturing strategies.1 Catalysts composed of nanoparticles (NPs) contribute to the design of cleaner chemical processes with reduced environmental impact.2 In this framework, nano-engineered catalysts represent a promising avenue to enhanced catalytic activity and selectivity in chemical processes.3 Hybrid nanocomposite materials combine individual properties and synergistically enhance catalytic characteristics leading to multifunctional catalysts.4 The use of metal/metal oxide nanocomposites is promising since it can overcome the barriers to implementation of pure metal nanoparticle catalysts such as enhanced mechanical and chemical stability, increased catalytic activity due to support effects, and may diminish undesired catalyst inhibition.5–7In addition to catalytic properties, metallic nanoparticles such as gold (Au) can be excited under light irradiation of certain wavelengths due to localized surface plasmon resonance (LSPR). Excitation of the LSPR in plasmonic nanoparticles can accelerate and control molecular transformations.8 When the frequency of an incident electromagnetic wave matches that of the oscillation of conduction electrons, the creation of energetic charge carriers are induced on the surface that can participate to enhance the rates of molecular transformations (e.g. charge transfer or excitation processes).8,9 It has been observed that synergies between metallic and plasmonic catalysis resulted in light-driven enhanced catalytic processes.10,11
In this context, excellent opportunities arise for application of hybrid nanocomposites composed of Au with magnetic oxides in catalysis.12 Au nanoparticles supported on magnetic oxides may serve to avoid suspension aggregation and catalytic deactivation.13,14 Meanwhile, the surface coating of magnetic nanoparticles precludes the agglomeration of superparamagnetic nanoparticles enhancing their dispersion in solution.15,16 In addition, the magnetic properties facilitate nanocatalyst recovery by the application of an external magnetic field.16 This noticeable reduces the operational expenditures and allows, at least in principle, an easy reuse of the catalyst.
The main implementation barrier of hybrid nanocatalyst arises from their complex synthesis,17,18 the non-uniform decoration of metallic nanoparticles19,20 and recycling.21 In this paper, a novel green synthesis approach to hybrid magnetic nanocomposite catalyst based on Au NPs decorated on cobalt ferrite (CoFe2O4) is reported. The syntheses of both nanoparticles and their composite were conducted under mild conditions and using water as solvent. The use of CoFe2O4 as stable magnetic substrate enhances catalyst stability and enables reusability after magnetic recovery for several cycles. Homogeneous distribution of Au NPs over the CoFe2O4 magnetic support was obtained due to the successful functionalization of the surface through silanization, which is not attained by conventional in situ synthesis procedures. Then, the catalytic activity was evaluated towards the reduction of 4-nitrophenol to 4-aminophenol and the selective oxidation of dimethylphenylsilane to dimethylphenylsilanol. Moreover, plasmonic enhanced catalysis was observed under visible light irradiation due to the LSPR excitation of Au NPs. Magnetic recovery and reuse of catalyst indicated great reliability and stability of the produced nanocomposite. Our results indicate that nanocatalysts present dual functionality for reduction and oxidation processes, its performance can be enhanced by visible-light illumination, and the magnetic component facilitates recovery from the reaction medium.
2. Experimental methods
2.1 Chemicals
Catalytic activity on reduction reaction was evaluated from the transformation of 4-nitrophenol (p-NP, 99%) supplied by Sigma-Aldrich. Selective oxidation catalysis was evaluated from the oxidation of dimethyl(phenyl)silane (C8H12Si, 98%) supplied by Sigma-Aldrich. Magnetic cobalt ferrite was synthesized using ACS reagent quality iron(II) chloride anhydrous and cobalt chloride hexa-hydrated purchased from Sigma-Aldrich and Merck, respectively. Gold nanoparticles were synthesized using tetrachloroauric(III) acid trihydrate (HAuCl4·3H2O, ≥99%) supplied by Sigma-Aldrich. Other chemicals used in nano-synthesis processes such as trisodium citrate di-hydrate, tannic acid, potassium carbonate, methylamine (40% w/w), (3-aminopropyl)triethoxysilane (APTES), and sodium dodecyl sulfate (SDS) were of analytical grade supplied by Sigma-Aldrich and Merck. Reductant agent sodium borohydride was supplied by Merck. All solutions were prepared with ultrapure water obtained from a Millipore Milli-Q system with resistivity > 18.2 MΩ cm at 25 °C.2.2 Catalyst synthesis
Magnetic catalyst core of CoFe2O4 was synthesized following previous report.22 Briefly, 5 mL of 1 mM FeCl2 solution and 5 mL of 0.5 mM CoCl2 acidic solutions at pH 1.5 adjusted with HCl are added to 18 mL of 4.5 mM SDS solution under continuous magnetic stirring at 25 °C for 30 min. This ensures a Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co molar ratio of 2:1 in the micellar solution. Temperature is increased to 80 °C and 1.8 mL of methylamine solution 8.3 M added to the reactor, which quickly modifies solution coloration to green. After 7–10 min solution becomes brownish due to CoFe2O4 crystallization. Reaction is kept under continuous stirring at 80 °C for 3 h. Nanoparticles were separated using a magnetic field and washed with a 5% NH3 solution and ultrapure water.
:Co molar ratio of 2:1 in the micellar solution. Temperature is increased to 80 °C and 1.8 mL of methylamine solution 8.3 M added to the reactor, which quickly modifies solution coloration to green. After 7–10 min solution becomes brownish due to CoFe2O4 crystallization. Reaction is kept under continuous stirring at 80 °C for 3 h. Nanoparticles were separated using a magnetic field and washed with a 5% NH3 solution and ultrapure water.
Size-controlled synthesis of sub-10 nanometer gold particles were synthesized following a kinetically controlled seeded-growth strategy.23 The use of traces of tannic acid in excess of sodium citrate during gold nucleation allows the formation of size-controlled gold nanoparticles. Stabilization solution was prepared by adding 100 μL of 2.5 mM tannic acid solution was added to 150 mL of 2.2 mM sodium citrate solution in 1.0 mM potassium carbonate prior heating up to 70 °C. Then, dropwise addition of 1.4 mL of 18 mM tetrachloroauric(III) acid solution promoted the nucleation of gold nanoparticles obtaining a reddish-orange solution characteristic of nano-gold colloidal suspensions.
Pristine CoFe2O4 nanoparticles were decorated with Au NPs. The metal oxide CoFe2O4 nanoparticles were functionalized with APTES to enhance coordination of gold nanoparticles to their surface. Nanoparticles of CoFe2O4 (50 mg) were dispersed in 110 mL of ethanol, then 10 mL of APTES solution was added to the suspension. The mixture was sonicated for 10 min under inert atmosphere prior adding 10 mL of 4.0 M acetic acid to attain ca. pH 6.0. After pH adjustment the reaction was kept at 75 °C under mechanical stirring in reflux for 12 h. Functionalized material was magnetically separated and washed with ethanol and ultrapure water. Then, the gold decorated cobalt ferrite nanocomposite catalyst (Au/CoFe2O4) was obtained by dispersing 2 mg of APTES-functionalized CoFe2O4 in 10 mL of colloidal Au nanoparticle suspension and stirred for 30 min. The Au/CoFe2O4 was magnetically separated and washed with ultrapure water.
2.3 Analytical procedures and instruments
Scanning electron microscopy (SEM) images of the catalysts were obtained using a JEOL FEG-SEM JSM 6330F. SEM samples were prepared by drop-casting an aqueous suspension of the particles on a Si wafer, followed by drying under ambient conditions prior analysis. Crystallographic analyses were conducted by X-ray diffractometry (XRD) employing a Bruker D2 Phaser using Cu Kα (λ = 1.5418 Å) radiation source with an scanning window of 20–90°. The IR spectra were collected with an FT-IR spectrophotometer Bruker Alpha in transmittance mode within a wavenumber range of 400–4000 cm−1. Size distribution of synthesized gold nanoparticles was determined by Dynamic Light Scattering (DLS) analyses using a ZetaSizer Nano ZS. Atomic percentage of gold was quantified through total reflection X-ray fluorescence (TXRF) using a S2-PICOFOX with internal standard. Chemical conversion of 4-nitrophenol to p-aminophenol was monitored by UV-vis analysis using an Shimadzu UV-2600 spectrophotometer. Oxidation of dimethylphenylsilane was evaluated from the concentration decay followed by gas chromatography mass spectroscopy (GC/MS) with a Shimadzu GCM-QP2010SE coupled to a RTx®-5MS fused silica capillary column using a temperature ramp of 1 °C min−1 from 80 °C up to 280 °C. Plasmonic-enhanced catalysis experiments were carried out using a solar simulator lamp ORIEL of 100 mW cm−2 in the 315–750 nm range placed 5 cm over the catalytic reactor.3. Results and discussion
3.1 Au/CoFe2O4 catalyst characterization
The SEM images (Fig. 1A) depict the spheroidal structure of the CoFe2O4 magnetic NPs. They displayed an average diameter of 90 nm. Functionalization of the pristine nanoparticles with APTES did not modify the characteristic structure of CoFe2O4, whereas contributed to diminish agglomeration of nanoparticles (Fig. 1B). Fig. 1C and D show the SEM of the CoFe2O4 magnetic NPs after the deposition of Au NPs at their surface (Au/CoFe2O4). It can be observed a homogeneous distribution of spherical Au nanoparticles that are well-dispersed on the CoFe2O4 surface. This result is explained by the role of APTES to act as sites for Au NPs heterogeneous nucleation and growth. Dynamic light scattering analysis of the suspension of Au nanoparticles led to an average size of 7 nm, which is in agreement with the SEM images. | ||
| Fig. 1 Scanning electron microscopy (SEM) images of (A) pristine CoFe2O4, (B) APTES modified CoFe2O4, and (C and D) Au/CoFe2O4 catalysts. | ||
The FTIR spectra for CoFe2O4 is shown in Fig. 2a. It depicts an intense peak at 600 cm−1 characteristic of M–O stretching vibrations previously observed for the tetrahedral sites of cobalt ferrite.24 Characteristic SDS characteristic stretching vibration signals corresponding to C–O at 2915 cm−1, S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 2848 cm−1, and C–H at 1220 cm−1, were not observed.25 The IR spectrum of CoFe2O4 after surface silanization with APTES depicts noticeable differences respect to the pristine surface. A quick comparison with FTIR spectra of APTES allows identifying the presence of intense peaks at 1100 cm−1 and 770 cm−1 corresponding to Si–O and Si–O–C, respectively.26 Furthermore, two additional stretching vibration bands associated to N–C at 1390 cm−1 and C–H at 3000 cm−1 were observed.27 These results suggest the successful functionalization of CoFe2O4, which agrees with the successful attachment of Au NPs observed in Fig. 1C and D.
O at 2848 cm−1, and C–H at 1220 cm−1, were not observed.25 The IR spectrum of CoFe2O4 after surface silanization with APTES depicts noticeable differences respect to the pristine surface. A quick comparison with FTIR spectra of APTES allows identifying the presence of intense peaks at 1100 cm−1 and 770 cm−1 corresponding to Si–O and Si–O–C, respectively.26 Furthermore, two additional stretching vibration bands associated to N–C at 1390 cm−1 and C–H at 3000 cm−1 were observed.27 These results suggest the successful functionalization of CoFe2O4, which agrees with the successful attachment of Au NPs observed in Fig. 1C and D.
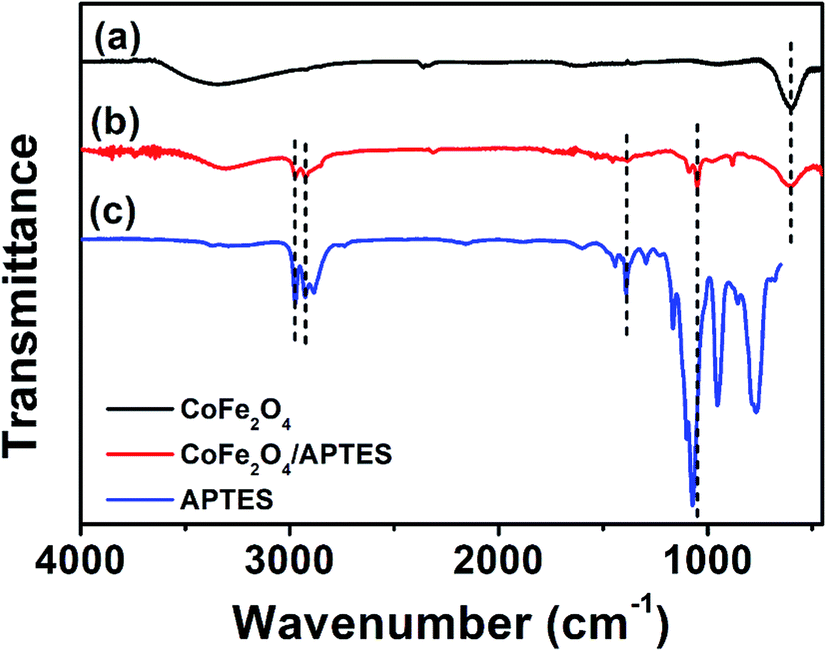 | ||
| Fig. 2 FTIR spectra of (a) CoFe2O4, (b) APTES modified CoFe2O4, and (c) APTES. | ||
The extinction UV-vis spectra of colloidal dispersions of Au NPs (Fig. 3, green trace) depicted a band at 513 nm that corresponds to the dipole mode of LSPR excitation.28 In contrast, absorbance bands were not observed in colloidal dispersions of CoFe2O4 or modified APTES–CoFe2O4 (Fig. 3, black and blue traces). However, the Au nano-enabled composite Au/CoFe2O4 catalyst presented a similar characteristic band than colloidal Au with a maximum at 526 nm (Fig. 3, red trace). This result demonstrate that the band observed in the composite is associated to the characteristic dipole mode of LSPR excitation of gold, which remains photoactive when attached by the APTES linker to the CoFe2O4 surface.
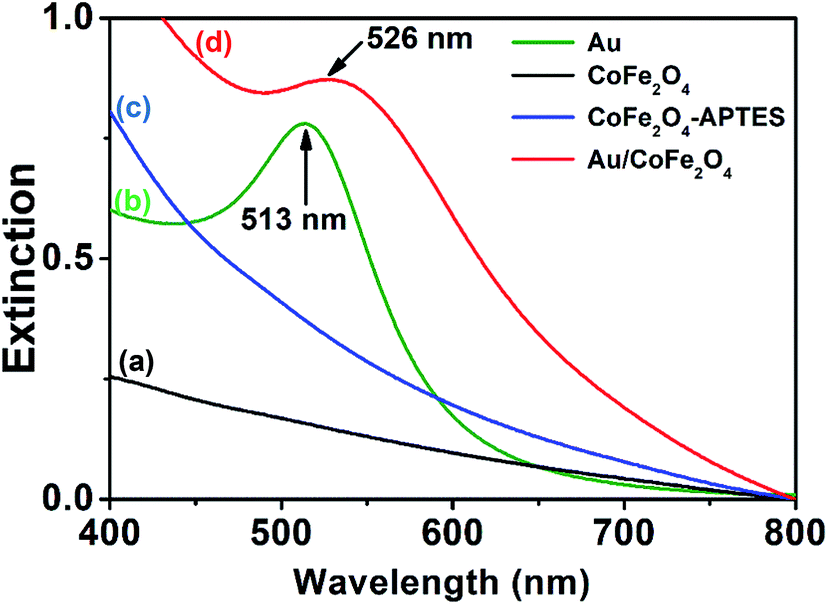 | ||
| Fig. 3 UV-vis extinction spectra from aqueous suspensions containing (a) CoFe2O4, (b) Au nanoparticles, (c) APTES–CoFe2O4, and (d) Au/CoFe2O4. | ||
Fig. 4 shows the X-ray diffractograms of CoFe2O4 and Au/CoFe2O4. Both diffractograms depict characteristic peaks of crystallographic planes of iron spinels (220), (311), (400), (511), and (440); in agreement with the JCPDS cards (PDF 22-1086). These crystallographic planes are characteristic of other spinel structures.29 These results demonstrate the synthesis of defined spinel crystalline structures of CoFe2O4 and not the formation of amorphous oxides. Debye–Scherrer calculations allowed identifying a crystallite size of 15.42 nm for CoFe2O4. It is important to note that identical XRD diffractograms where observed for APTES functionalized CoFe2O4, which surface silanization did not modified the characteristic crystallographic structure of the catalyst. However, surface decorated Au/CoFe2O4 presented additional peaks corresponding to Au as a result of the deposition of Au NPs to the surface. These new signals were associated to crystallographic planes (111), (200), (220), and (311) corresponding to the fcc structure of metallic Au.30,31 This result agrees with the successful formation of Au/CoFe2O4 as observed in the SEM characterization (see Fig. 1).
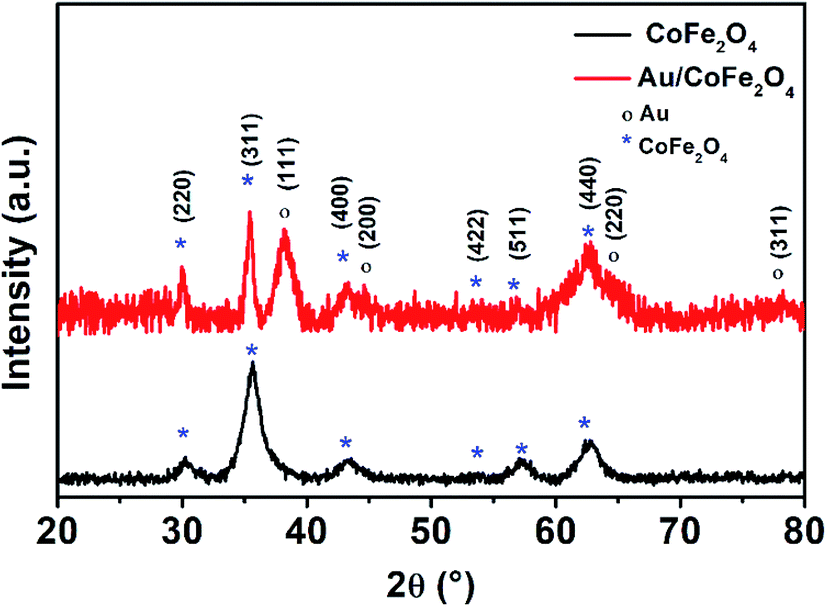 | ||
| Fig. 4 X-ray diffractogram of (black line) CoFe2O4 and (red line) Au/CoFe2O4 NPs. | ||
3.2 Catalytic activity: reduction of 4-nitrophenol
The catalytic activity of the Au/CoFe2O4 material with 2.72% mol Au was initially evaluated towards the reduction of 4-nitrophenol. Fig. 5A illustrates the characteristic UV-vis spectra of p-NP solutions with a maximum absorbance at 317 nm.32 In presence of reductant NaBH4, the solution changes to a yellow coloration due to the deprotonation of the hydroxyl group yielding 4-nitrophenolate ions with a maximum absorbance at 405 nm.33 Fig. 5A depicts the isosbestic point for 4-nitrophenol/4-nitrophenolate at 348 nm. Reduction of 4-nitrophenolate towards 4-aminophenol was not observed in absence of catalyst. This suggests that only with the presence of NaBH4 the reduction reaction does not occur. However, a drastic decrease in the absorbance of the 405 nm band of 4-nitrophenolate was observed along with the formation and increase of 4-aminophenol absorption band at 300 nm, in the presence of the catalyst (see Fig. 5B).34 This result suggests the quick catalytic conversion mediated by Au/CoFe2O4 that acts as a redox mediator for 4-nitrophenolate reduction with NaBH4.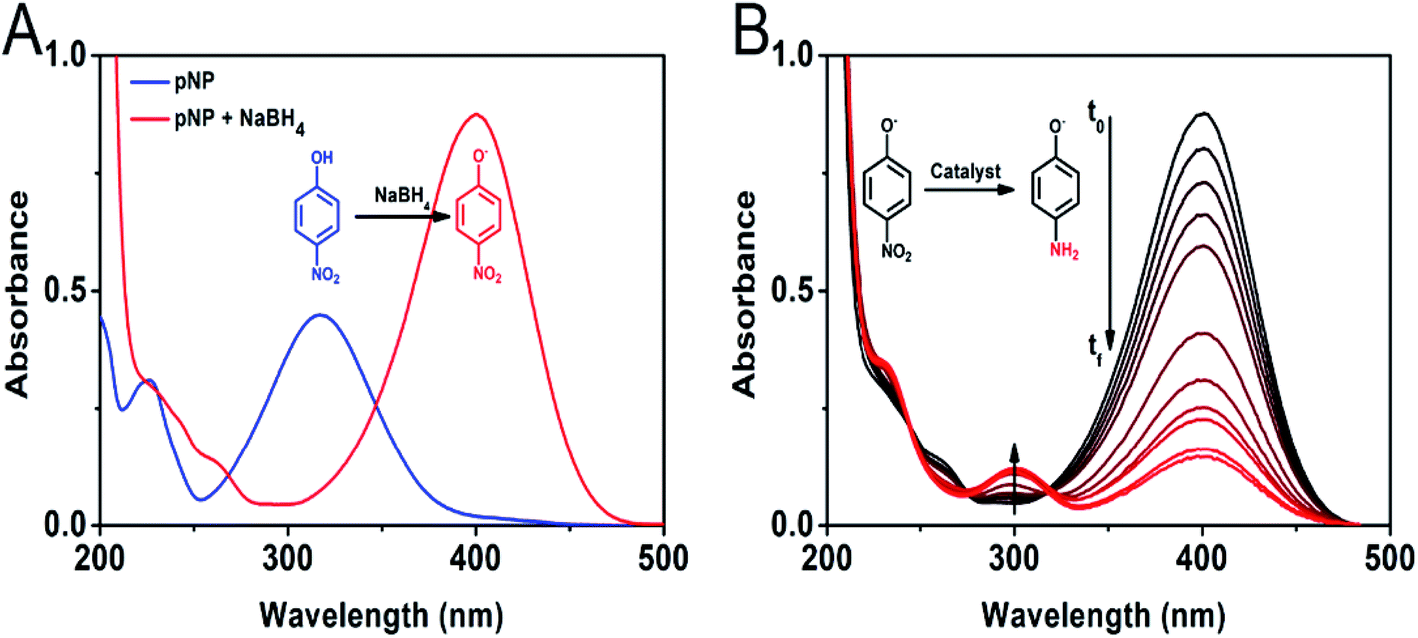 | ||
| Fig. 5 (A) UV-vis spectra for 4-nitrophenol (blue line) and 4-nitrophenoxide (red line) solutions. (B) Time-dependent change in the UV-vis spectra during 4-nitrophenol reduction to 4-aminophenol in presence of NaBH4 and Au/CoFe2O4 catalyst with 2.72% mol Au. | ||
After this initial test, we then employed light excitation to investigate the effect of LSPR over the reaction rates. Fig. 6 shows the 4-nitrophenol reduction employing CoFe2O4 and Au/CoFe2O4 as catalysts in the dark and under light excitation from a solar simulator in the 315–750 nm range. The use of pristine CoFe2O4 as catalyst allowed attaining 94% conversion in 30 min after an initial induction time of 6 min.35 The induction time is not observed when the reduction reaction with CoFe2O4 is conducted under light irradiation. However, similar total conversion of ca. 94% is attained after 30 min of reaction. The pseudo-first order rate constant (k) was calculated from the depletion kinetics of 4-nitrophenol after the induction time. The k in the dark of 1.2 × 10−3 s−1 (R2 = 0.99) was slightly superior (ca. 1.4-fold) to the photo-assisted catalysis with a k of 8.2 × 10−4 s−1 (R2 = 0.91). This result agrees with the identical removal attained after 30 min of reaction (cf. Fig. 6A). It may be inferred that the light irradiation reduces the induction time in detrimental of the charge transfer kinetics.
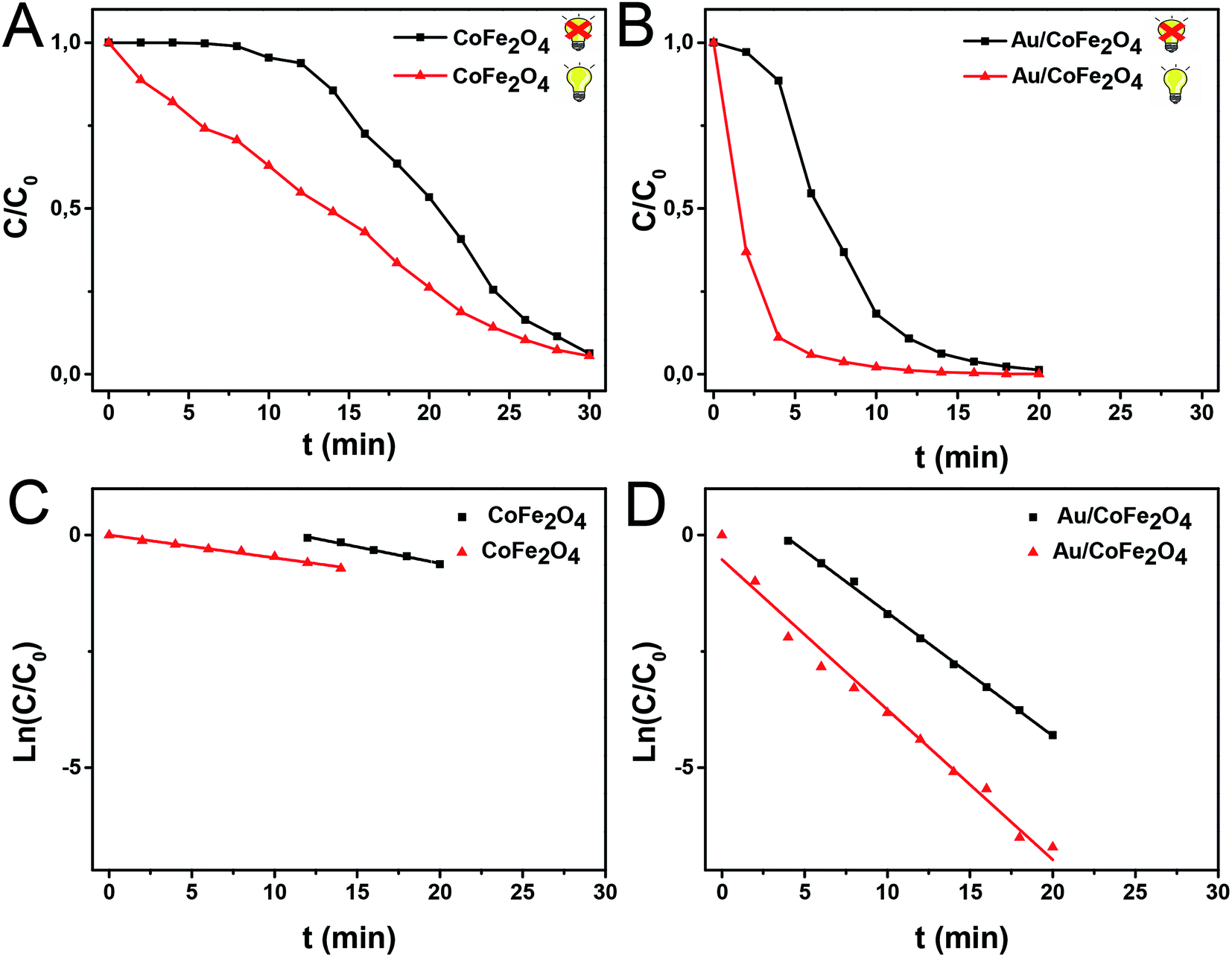 | ||
| Fig. 6 (A and B) 4-Nitrophenol concentration profile as a function of reaction time in the dark (black trace) and under light irradiation (red trace) using CoFe2O4 or Au/CoFe2O4 with 2.72% mol Au as catalysts. (C and D) Kinetics analysis of 4-nitrophenol conversion assuming pseudo-first order kinetics after induction time for (a) CoFe2O4 or (b) Au/CoFe2O4 NPs. | ||
Faster kinetics were described by the nanocomposite Au/CoFe2O4 catalyst with 2.72% mol Au due to the higher catalytic activity of Au NPs relative to CoFe2O4 (see Fig. 6B). Here, a complete conversion of 4-nitrophenol was achieved after 20 min of reaction. Light irradiation enhances the catalytic response due to the synergetic effect between metallic and plasmonic catalysis.36,37 Note that complete conversion was attained in half of the reaction time with high selectivity towards 4-aminophenol. Here, the plasmonic enhancement is well-represented by the acceleration of the conversion kinetics by the values of the kinetic rate constants. The decoration of magnetic CoFe2O4 with Au nanoparticles increases kinetic rates over 5-folds relative to CoFe2O4 due to the higher catalytic activity of Au nanoparticles. Catalytic conversion of 4-nitrophenol with Au/CoFe2O4 in the dark reported k1 values of 4.4 × 10−3 s−1 (R2 = 0.99). Meanwhile the kinetics was increased under light irradiation with k1 values of 5.4 × 10−3 s−1 (R2 = 0.98). Experiments conducted under light irradiation in absence of catalyst showed null reduction, which indicates light–catalyst interaction as key element on the catalytic conversion. Enhancement of the catalytic activity under light irradiation can be associated with the formation of energetic charge carriers (hot electrons) that can be injected into 4-nitrophenolate LUMO orbitals and accelerate the transformation.38 Here, SPR-excited hot electrons can be transferred to the lowest unoccupied adsorbate state that forms an activated transient adsorbate species with weakened bonds, which facilitates the transfer of M–H from the metal surface to the 4-nitrophenol adsorbate accelerating the reduction reaction towards 4-aminophenol.38 The evaluation of the turn over frequency (TOF) shows an almost 6-fold enhancement due to LSPR from 180 h−1 in the dark up to 696 h−1 under light irradiation. Note that these values are higher than previously reported TOF of 0.7 h−1 for Au/boehmite,39 2.5 h−1 Au NPs on mesoporous silica,40 46 h−1 for Au nanoparticles supported on silica nanotubes,41 116 h−1 for Au/g-C3N4,42 or 324 h−1 Au/glass fiber.43 The high catalytic properties can be related to the homogeneous dispersion of nanoparticles that enhances the accessibility to catalytic sites of the nanostructured composite. On top of that, catalyst magnetic recovery allowed reuse in several cycles, which displayed an excellent catalyst reliability after 10 cycles as shown in Fig. 7.
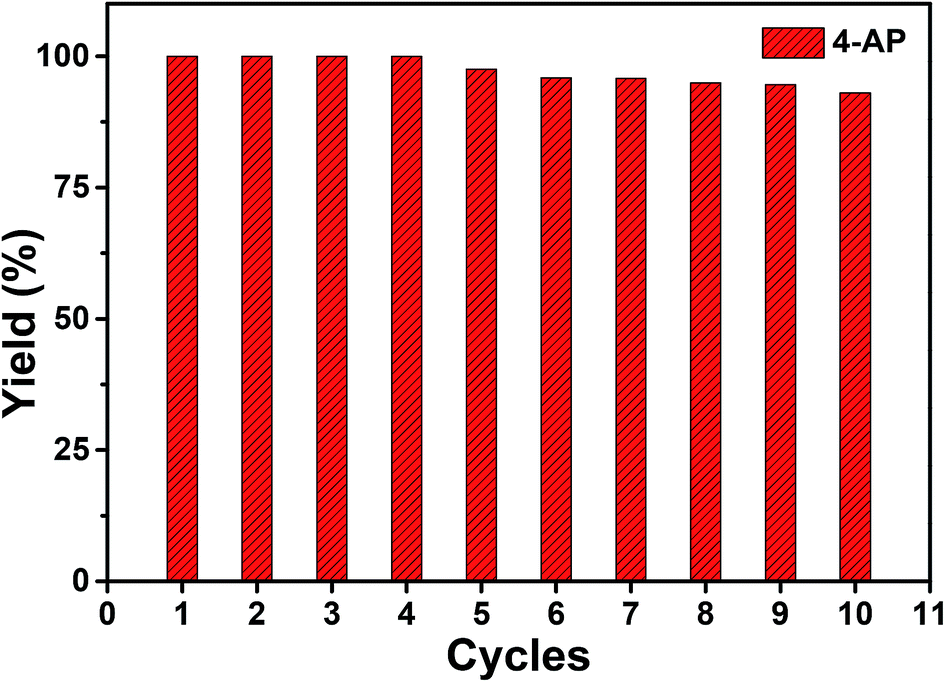 | ||
Fig. 7 Yield percentage of ( ) 4-aminophenol from the catalyzed reduction of 4-nitrophenol by Au/CoFe2O4 with 2.72% mol Au after several cycles of catalyst magnetically recovered reuse. ) 4-aminophenol from the catalyzed reduction of 4-nitrophenol by Au/CoFe2O4 with 2.72% mol Au after several cycles of catalyst magnetically recovered reuse. | ||
3.3 Selective oxidation of dimethylphenylsilane
Organosilanes are widely used in polymeric synthesis,44 antimicrobials,45 and as nucleophilic coupling partners.46 Organosilane manufacturing processes rely on the use of strong oxidants that yield toxic undesired sub-products.47,48 This section studies the catalytic activity of nanocomposite Au/CoFe2O4 as green alternative synthesis route of dimethylphenylsilanol using water and oxygen as oxidants under mild conditions (ambient temperature and 1 atm of pressure).Null oxidation of dimethylphenylsilane was observed using pristine CoFe2O4 as catalyst. However, Au NPs introduces catalytic centers for selective oxidation. However, conversion and selectivity are dependent on the solvent used as media for the catalytic transformation towards dimethylphenylsilanol. Table 1 summarizes dimethylphenylsilane oxidative conversion percentage attained after 10 min of reaction in different solvents. The catalytic activity of the Au/CoFe2O4 NPs after addition of 100 μL of water as oxidant increases from 22% in water, to 83% in tetrahydrofuran (THF), and 96% in acetone. It is important to remark that conversion in acetone was drastically reduced to 54% when 100 μL of water was not added, being the catalytic conversion probably associated to the trace water concentration of the solvent. This behavior allows inferring the role of water as oxidant for dimethylphenylsilanol production. This is in agreement with the identification of the water oxygen as the source of the hydroxyl group addition during the selective oxidation.49,50 In this frame, the low conversion observed in water may be the blocking of catalytic sites by an excess of water molecules, which diminishes the effective contact between reactants on the catalyst surface. This pseudo-poisoning due to the higher affinity of water molecules with the catalyst surface hinders the extension of the reaction resulting in lower catalytic activity. Moreover, the low solubility of silane in neat water may difficult the interaction between silane and catalyst. When acetone and THF (both water-soluble solvents) are employed, the greater solubility of silane facilitates the oxidation process. Acetone was identified as preferred solvent for selective oxidation due to the high conversion percentages attained in short reaction times of 10 min and the selectivity towards dimethylphenylsilanol of 96%, notoriously superior to the 88% observed in THF.
| Solvent | H2O (μL) | Conversion (%) | Selectivity to silanol (%) |
|---|---|---|---|
| H2O | 100 | 22.07 | 100 |
| THF | 100 | 82.78 | 88.68 |
| Acetone | 100 | 96.23 | 96.23 |
| Acetone | — | 53.877 | 100 |
Further analysis of catalytic conversion by GC-MS depicts the reaction kinetics of dimethylphenylsilane that attains a plateau after 8 min of oxidative conversion on Au/CoFe2O4. Fig. 8 depicts the two main products identified as result of the catalytic reaction on the nanocomposite catalyst: (i) dimethylphenylsilanol with 96.23% selectivity, and (ii) disiloxane with 3.77% selectivity at the end of reaction. Interestingly, the time course profile of disiloxane illustrates a maximum concentration of 11.0% at 4 min of reaction that decreases down to 3.77%. This trend can be explained due to the reversibility on the formation of disiloxane that can result in the increased yield of silanol observed at the end of the reaction.51
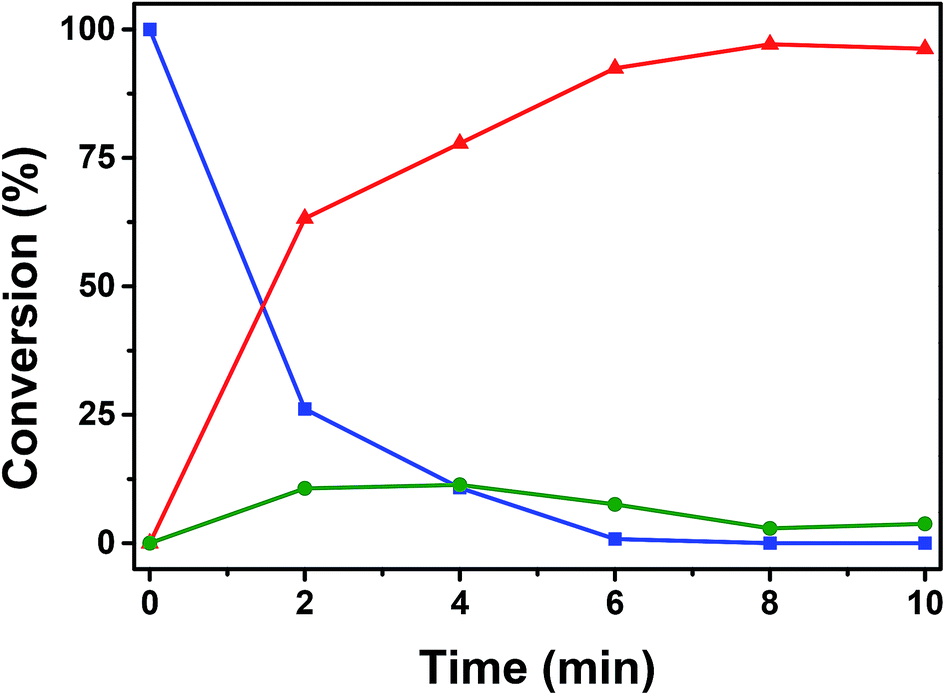 | ||
Fig. 8 ( ) Conversion of dimethylphenylsilane catalyzed oxidation by Au/CoFe2O4 in acetone with 100 μL of H2O. Time course of yielded products ( ) Conversion of dimethylphenylsilane catalyzed oxidation by Au/CoFe2O4 in acetone with 100 μL of H2O. Time course of yielded products ( ) dimethylphenylsilanol, and ( ) dimethylphenylsilanol, and ( ) disiloxane. ) disiloxane. | ||
The effect of the Au loading in the reaction was investigated as shown in Table 2. Lower catalytic oxidation was observed for lower contents of Au below 0.04 mol% due to the null catalytic activity of CoFe2O4 discussed above. The lower activity results also in a higher accumulation of disiloxane, which decreases the desired selectivity towards dimethylphenylsilanol. Maximum conversion and selectivity was observed for 0.04 mol%, where further increases on the content of Au NPs on the nanocomposite would result in a unnecessary higher cost of catalytic material.
| Au loading (mol%) | Conversion (%) | Selectivity to silanol (%) |
|---|---|---|
| 0.01 | 32.35 | 87.45 |
| 0.02 | 45.744 | 91.61 |
| 0.04 | 96.23 | 96.23 |
| 0.06 | 90.777 | 90.777 |
| 0.08 | 89.025 | 89.025 |
| 1.0 | 90.325 | 90.325 |
Even though the CoFe2O4 nanoparticles have null catalytic oxidation activity, their magnetic property can provide a relevant functionality to the composite nanocatalyst. The magnetic properties of the nanocomposite catalyst allow easy and quick separation under application of a magnetic field for catalyst reuse. Catalyst was recovered with a neodymium magnet from solution in less than 15 s, which allowed recovery for re-dispersion and reuse. Fig. 9 shows the highly reproducible conversion of dimethylphenylsilane after 10 cycles with merely a slight decrease in selectivity. Reused catalyst presents higher stabilization of Au–Si bonds which increases the concentration of these adsorbed intermediates on the gold surface.52 Higher ratio of Au–Si over adsorbed water favors the formation of siloxane over oxidation towards silanol. To overcome the preferential competitive reaction towards siloxane higher reaction times are required to enhance the reversible transformation of siloxane towards silanol. In this frame, dimethylphenylsilanol production after 10 min of reaction decreased down to 42% in the 10th cycle. Extending reaction time to 20 min increased dimethylphenylsilanol yield up to 90%.
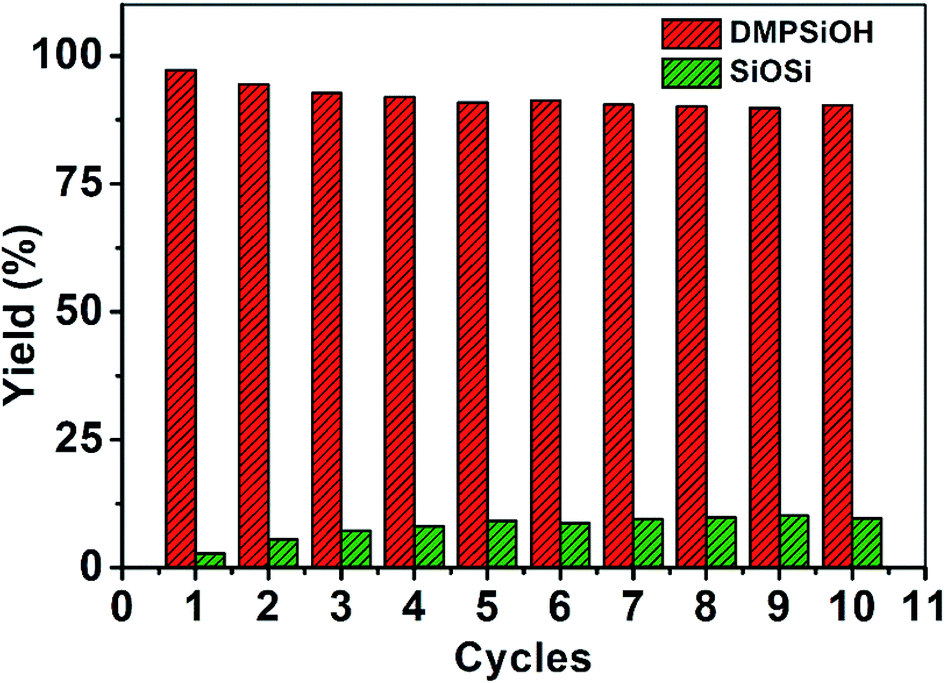 | ||
Fig. 9 Yield percentage of ( ) dimethylphenylsilanol and ( ) dimethylphenylsilanol and ( ) siloxane from the catalyzed oxidation of dimethylphenylsilane by Au/CoFe2O4 in acetone with 100 μL of H2O after several cycles of catalyst magnetically recovered reuse. ) siloxane from the catalyzed oxidation of dimethylphenylsilane by Au/CoFe2O4 in acetone with 100 μL of H2O after several cycles of catalyst magnetically recovered reuse. | ||
4. Conclusion
We reported on the synthesis of nanocomposites comprised of magnetic Au/CoFe2O4 NPs. The efficient deposition of Au at their surface was achieved through CoFe2O4 core functionalization by silanization with APTES. The catalytic activity was evaluated for 4-nitrophenol reduction and organosilanol syntheses by selective oxidation. The Au/CoFe2O4 NPs presented high catalytic activity towards the 4-nitrophenol reduction which could be enhanced under light irradiation as a result of LSPR excitation. We also evaluated the performances towards the silanol oxidation to produce dimethylphenylsilanol as product of interest for chemical industry. Our results demonstrate the role of solvent selection to increase conversion percentage and desired product yield. The magnetic properties of the catalyst allowed easy recovery and re-use. Re-dispersed catalysts reported high reliability in reagent conversion and product selectivity. Therefore, we believe that Au/CoFe2O4 NPs may represent interesting materials for catalytic applications merging high performances, recovery and re-use, and enhancement of activities under solar light illumination.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministerio de Educación (MINEDU) of Peru through the agreement “Inter-institutional Cooperation Agreement between the Ministerio de Educación and the Universidad Nacional de Ingeniería” (No. 401-2017-MINEDU) and through the Master of Science Program (No. 208-2015-FONDECYT). This work was supported by FAPESP (grant number 2015/26308-7; 2017/02854-8). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (CAPES) – Finance Code 001. PHCC thanks the CNPq for the research fellowship. E. C. M. B. thank FAPESP for the fellowship (grant number 2015/11452-5).References
- M. Poliakoff, P. Licence and M. W. George, Curr. Opin. Green Sustain. Chem., 2018, 13, 146–149 CrossRef.
- M. Kaushik and A. Moores, Curr. Opin. Green Sustain. Chem., 2017, 7, 39–45 CrossRef.
- Z. C. Zhang, B. Xu and X. Wang, Chem. Soc. Rev., 2014, 43, 7870–7886 RSC.
- M. N. Chen, L. P. Mo, Z. S. Cui and Z. H. Zhang, Curr. Opin. Green Sustain. Chem., 2019, 15, 27–37 CrossRef.
- J. J. Gao, P. Du, Q. H. Zhang, X. Shen, F. K. Chiang, Y. R. Wen, X. Lin, X. J. Liu and H. J. Qiu, Electrochim. Acta, 2019, 297, 155–162 CrossRef CAS.
- H. O. N. Tugaoen, S. Garcia-Segura, K. Hristovski and P. Westerhoff, Sci. Total Environ., 2017, 599–600, 1524–1551 CrossRef.
- Y. Wei, Z. Zhao, X. Yu, B. Jin, J. Liu, C. Xu, A. Duan, G. Jiang and S. Ma, Catal. Sci. Technol., 2013, 3, 2958–2970 RSC.
- T. P. Araujo, J. Quiroz, E. C. M. Barbosa and P. H. C. Camargo, Curr. Opin. Colloid Interface Sci., 2019, 39, 110–122 CrossRef CAS.
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Nat. Catal., 2018, 1, 656–665 CrossRef.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- K. N. Heck, S. Garcia-Segura, P. Westerhoff and M. S. Wong, Acc. Chem. Res., 2019, 52, 906–915 CrossRef CAS PubMed.
- D. Shi, F. Sheng, X. Zhang and G. Wang, Talanta, 2018, 185, 106–112 CrossRef CAS PubMed.
- E. R. Riva, I. Pastoriza-Santos, A. Lak, T. Pellegrino, J. Pérez-Juste and V. Mattoli, J. Colloid Interface Sci., 2017, 502, 201–209 CrossRef.
- S. Behrens, Nanoscale, 2011, 3, 877–892 RSC.
- M. Nasrollahzadeh, M. Bagherzadeh and H. Karimi, J. Colloid Interface Sci., 2016, 465, 271–278 CrossRef CAS.
- Y. Zhou, Y. Zhu, X. Yang, J. Huang, W. Chen, X. Lv, C. Li and C. Li, RSC Adv., 2015, 5, 50454–50461 RSC.
- M. Nasrollahzadeh, S. Mohammad Sajadi, A. Rostami-Vartooni, M. Alizadeh and M. Bagherzadeh, J. Colloid Interface Sci., 2016, 466, 360–368 CrossRef CAS PubMed.
- P. Dobrowolska, A. Krajewska, M. Gajda-Raczka, B. Bartosewicz, P. Nyga and B. J. Jankiewicz, Materials, 2015, 8, 2849–2862 CrossRef CAS.
- P. Pallavicini, E. Cabrini, A. Casu, G. Dacarro, Y. Antonio Diaz-Fernandez, A. Falqui, C. Milanese and F. Vita, Dalton Trans., 2015, 44, 21088–21098 RSC.
- J. M. Walker and J. M. Zaleski, Nanoscale, 2016, 8, 1535–1544 RSC.
- C. Cannas, A. Ardu, A. Musinu, D. Peddis and G. Piccaluga, Chem. Mater., 2008, 20, 6364–6371 CrossRef CAS.
- J. Piella, N. G. Bastús and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- B. Gillot, Vib. Spectrosc., 1994, 6, 127–148 CrossRef CAS.
- R. P. Sperline, Langmuir, 1997, 13, 3715–3726 CrossRef CAS.
- S. Villa, P. Riani, F. Locardi and F. Canepa, Materials, 2016, 9, 826 CrossRef.
- X.-C. Shen, X.-Z. Fang, Y.-H. Zhou and H. Liang, Chem. Lett., 2004, 33, 1468–1469 CrossRef CAS.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2006, 58, 267–297 CrossRef.
- J. Xiong, Q. Wu, X. Mei, J. Liu, Y. Wei, Z. Zhao, D. Wu and J. Li, ACS Catal., 2018, 8, 7915–7930 CrossRef CAS.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- T. Bhowmik, M. K. Kundu and S. Barman, RSC Adv., 2015, 5, 38760–38773 RSC.
- E. Menumerov, R. A. Hughes and S. Neretina, Nano Lett., 2016, 16, 7791–7797 CrossRef CAS.
- S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, J. Phys. Chem. C, 2007, 111, 4596–4605 CrossRef CAS.
- M. Nasrollahzadeh, S. M. Sajadi, A. Rostami-Vartooni, M. Bagherzadeh and R. Safari, J. Mol. Catal. A: Chem., 2015, 400, 22–30 CrossRef CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CrossRef CAS.
- G. Baffou and R. Quidant, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Publ. Gr., 2011, 10, 911–921 CAS.
- E. C. M. Barbosa, J. L. Fiorio, T. Mou, B. Wang, L. M. Rossi and P. H. C. Camargo, Chem.–Eur. J., 2018, 24, 12330–12339 CrossRef CAS.
- D. Jana, A. Dandapat and G. De, Langmuir, 2010, 26, 12177–12184 CrossRef CAS.
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS.
- Z. Zhang, C. Shao, P. Zou, P. Zhang, M. Zhang, J. Mu, Z. Guo, X. Li, C. Wang and Y. Liu, Chem. Commun., 2011, 47, 3906–3908 RSC.
- Y. Fu, T. Huang, B. Jia, J. Zhu and X. Wang, Appl. Catal., B, 2017, 202, 430–437 CrossRef CAS.
- J. He, W. Ji, L. Yao, Y. Wang, B. Khezri, R. D. Webster and H. Chen, Adv. Mater., 2014, 26, 4151–4155 CrossRef CAS.
- V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847–5910 CrossRef CAS.
- Y. Kim, S. Farrah and R. H. Baney, Int. J. Antimicrob. Agents, 2007, 29, 217–222 CrossRef CAS.
- S. E. Denmark and N. S. Werner, J. Am. Chem. Soc., 2008, 130, 16382–16393 CrossRef CAS PubMed.
- S. M. Sieburth and W. Mu, J. Org. Chem., 1993, 58, 7584–7586 CrossRef CAS.
- P. D. Lickiss and R. Lucas, J. Organomet. Chem., 1995, 521, 229–234 CrossRef.
- K. Mori, M. Tano, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 2002, 26, 1536–1538 RSC.
- C. M. Kisukuri, D. J. Palmeira, T. S. Rodrigues, P. H. C. Camargo and L. H. Andrade, ChemCatChem, 2016, 8, 171–179 CrossRef CAS.
- R. N. Lewis, Ann. N. Y. Acad. Sci., 1969, 159, 291–298 CrossRef CAS.
- S. Yochelis, E. Katzir, Y. Kalcheim, V. Gutkin, O. Millo and Y. Paltiel, J. Nanotechnol., 2012, 2012, 903761 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |