
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstraint spaces in carbon materials
Hiroyuki Itoi *a,
Hiroyuki Muramatsub and
Michio Inagakic
*a,
Hiroyuki Muramatsub and
Michio Inagakic
aDepartment of Applied Chemistry, Aichi Institute of Technology, Yachigusa 1247, Yakusa-cho, Toyota, 470-0392 Japan. E-mail: itoi-hiroyuki@aitech.ac.jp
bFaculty of Engineering, Shinshu University, 4-17-1 Wakasato, Nagano, 380-8553 Japan
cProfessor Emeritus of Hokkaido University, 228-7399 Nakagawa, Hosoe-cho, Kita-ku, Hamamatsu, 431-1304 Japan
First published on 23rd July 2019
Abstract
Nano-sized pores in carbon materials are recently known to give certain constraints to the encapsulated materials by keeping them inside, accompanied with some changes in their structure, morphology, stability, etc. Consequently, nano-sized pores endow the constrained materials with improved performances in comparison with those prepared by conventional processes. These pores may be called “constraint spaces” in carbon materials. Here, we review the experimental results related to these constraint spaces by classifying as nanochannels in carbon nanotubes, nanopores and nanochannels in various porous carbons, and the spaces created by carbon coating.
1. Introduction
Recently, carbon materials have attracted attention in a wide range of fields, from materials in scientific research to industrial products for various applications, such as carbon nanotubes, fullerenes, graphene, and carbon nanodots, in addition to carbon fibers, porous carbons, carbon black, high-density graphite blocks, graphite sheets, etc.1,2 One of the characteristics of carbon materials is the formation of pores with a wide range of sizes and morphologies. Carbon nanotubes have channels with well-defined and nano-sized diameters, of which the walls consist of almost perfect graphene layers although they are severely curved. The diameters of the channels are homogeneous when prepared through a batch synthesis and can be tuned by the synthesis conditions. Meanwhile, other porous carbons, such as activated carbon, contain various sizes of pores from micropores with a size of less than 1 nm to macropores with a size of more than 50 nm, with various morphologies. The walls of these pores are composed of aggregates of small crystallites with a graphitic structure and/or a few-layer graphene in some cases, and not only basal planes but also edge sites are exposed on the walls. The development in the techniques of precursor design by blending polymers (polymer-blend method)3 and of carbonization by using various templates (template-assisted carbonization)4,5 can control pore sizes, volumes, and morphologies more easily and with much higher carbon yield than the conventional techniques, such as the so-called activation techniques based on oxidation of the carbon matrix. The formation of channel-like and orderly aligned pores can be realized by selecting appropriate templates, such as zeolites and mesoporous silica.These pores and channels of carbon materials have played important roles in their applications, such as electrochemical capacitor electrodes by forming electric double-layers on a large surface area for energy storage, adsorbents of polluting species in air and water for environment remediation, etc. The interlayer spaces in graphite crystals are also used to accommodate active materials, the products being called graphite intercalation compounds, (e.g., by using lithium ions in rechargeable batteries). In these cases, the pores, channels, and interlayer spaces of graphite work as storage media of functional materials. Recently, however, some researches demonstrated experimentally that the pores and channels of carbon materials work to endow the encapsulated functional materials with performances either improved or different from those of the bulk or prepared by conventional processes (e.g., powder, films), as well as to enable the reactions of encapsulated materials, which are impossible outside the pores or channels. In other words, these pores and channels give certain constraints to the materials by keeping them inside, accompanied by some changes in their structure, morphology, stability, properties, etc., and therefore can be called “constraint spaces” in carbon materials. In the present review, we try to summarize the experimental results supposedly supported by these constraint spaces of carbon nanotubes and porous carbons. In addition to the pores and channels mentioned above, carbon coating processes of functional materials are included here, because some experimental results suggest certain constraints from the carbon walls to the coated materials.
2. In carbon nanotubes
Carbon nanotubes (CNTs) provide long and straight channels with well-defined and uniform diameters. CNTs have been reported to work as constraint spaces for collapsing encapsulated fullerenes to form the wall of CNTs inside and improving the electrochemical activities of encapsulated alkaline metal-doped fullerenes, iodine, sulfur, and organic molecules (e.g., quinones). In addition, water molecules undergo structural change or phase transition in the constraint spaces of CNTs.(a) Fullerenes
Encapsulation of fullerenes, mostly C60 and C70, into single-wall carbon nanotubes (C60@SWCNT and C70@SWCNT) was experimentally observed in the products of laser ablation of graphite impregnated by Co/Ni catalyst and in those of carbon arc discharge using Ni/Y catalyst,6,7 the product being called “bucky-peapod” or simply “peapod”. By using diameter-selected SWCNTs, encapsulations of C60 and C70 were performed by heat treatment at 650 °C to yield high density fullerene chains in each SWCNT, with filling factors of 85 and 72%, respectively.8 The distance between C60 cages in the CNTs was 0.95 nm, which was shorter than that in the crystal with face-centered cubic closest packing (1.00 nm) and longer than that in the polymer of C60 (0.92 nm). On the other hand, two different distances of 1.00 and 1.10 nm between C70 cages were observed for C70@SWCNTs, which were due to morphologies of standing and lying C70 cages, respectively. Encapsulations of C60 and C70 into double-walled and triple-walled carbon nanotubes (DWCNTs and TWCNTs, respectively) were facilitated by reacting CNTs with fullerene vapor at 500 °C.9 The filling factors of both C60 and C70 peapods were about 70%. Theoretical calculations based on the local density approximation in the density-functional theory showed that the encapsulating process of C60 into SWCNT is exothermic for the (10,10) CNT, whereas endothermic for the (8,8) and (9,9) CNTs, suggesting that the minimum radius of the nanotube for the encapsulation is 0.64 nm.10 Structural stability of C60@SWCNTs was examined under high pressures and high temperatures by using in situ synchrotron X-ray diffraction.11,12 The diffraction peak at 25.23 keV (corresponding to the spacing of 0.956 nm (2θ of 2.946°)) reflecting one-dimensional C60 crystals was intensified with increasing pressure up to 10.7 GPa at room temperature and the C60–C60 distance was progressively shortened from 0.956 nm at 0.1 MPa down to 0.892 nm at 10.7 GPa.11 By heating up to 1023 °C under 4 GPa, the C60–C60 distance further reduced to 0.87 nm as a result of C60 polymerization.12 SWCNT peapods of C60 and C70 were synthesized in a high yield using a sublimation method by contacting SWCNTs with fullerene vapors.13The coalescence of C60 inside SWCNTs proceeds at high temperatures. C60@SWCNTs were prepared by reacting SWCNTs with C60 vapor at 400 °C in dry air, and then treated at high temperatures in vacuo (<10−6 Torr).14 Encapsulated C60 molecules are self-assembled to make a chain with nearly uniform center-to-center distances, as shown TEM image in Fig. 1a. C60 molecules remain mostly unchanged in CNTs up to 800 °C and start to coalesce with adjacent ones to form linked beans and/or short nanotube above 800 °C, as shown on 1000 oC-treated one in Fig. 1b. At ∼1200 °C, most C60 molecules coalesce to form tube wall, making this part DWCNTs by retaining the empty single-wall tube partly, as shown in Fig. 1c. After 1200 °C treatment, no C60 molecules remain and most of tubes are partly changed to double-walled, and in some cases, the inner tubes are terminated by caps (Fig. 1c). The thermal treatment of bucky-peapods is the most effective way for the fabrication of catalyst-free high-purity DWCNTs among the methods including arc-discharge and chemical vapor deposition (CVD) methods using catalysts, particularly to form small-diameter inner tubes (0.4–0.7 nm). The diameter of the inner tube synthesized from the peapods of SWCNTs with filling factor of 60–80% depends on the temperature of synthesis, as shown in Fig. 2.15 Almost the same diameter distribution of the inner tubes is achieved by the coalescence of fullerenes up to 1800 °C, but it becomes broad after the heat treatment at 2000 °C, including enlarged diameters due to coalescence of adjacent tubes.
 | ||
| Fig. 1 TEM images of C60-encapsulated SWCNT: (a) as-prepared, (b) after heat treatment at 1000 °C, and (c) after 1200 °C in vacuo. Adapted from ref. 14 with permission from Elsevier. | ||
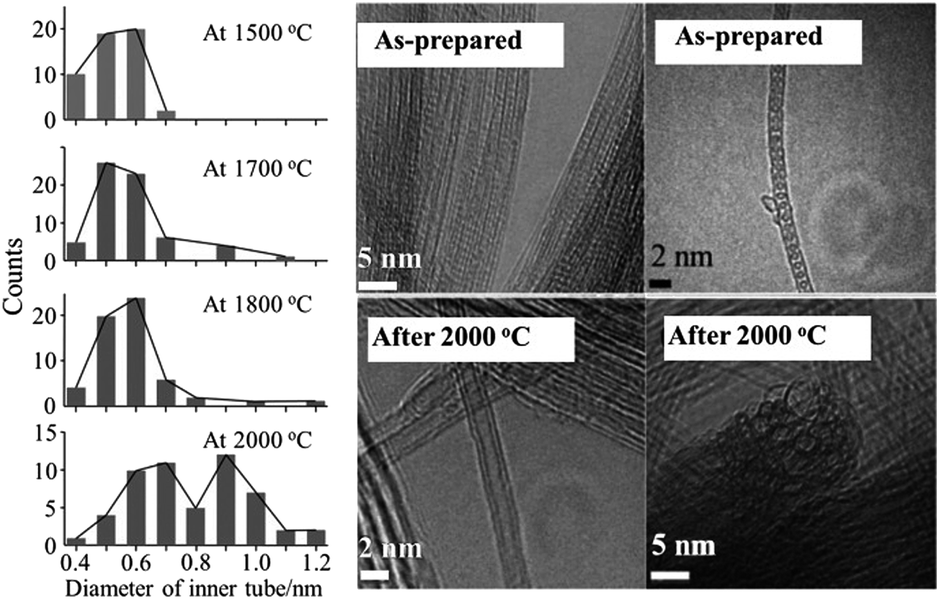 | ||
| Fig. 2 Distribution of the diameter of inner tube for DWCNTs synthesized at different temperature and high-resolution TEM images of as-prepared peapods and DWCNTs synthesized at 2000 °C. Adapted from ref. 15 with permission from John Wiley and Sons. | ||
Coalescence of fullerenes in SWCNTs is induced also by electron irradiation to form DWCNTs, which was followed by in situ TEM observations.16 DWCNTs are mechanically, thermally, and structurally more stable than SWCNTs, probably because of the buffer-like function of the outer tubes.17 The inner tube of DWCNT exhibits unique transport and optical properties such as extremely sharp Raman lines for the radial breathing mode (RBM) of the inner tube, suggesting high crystallinity of the inner tube and highly-unperturbed environment in the interior of the tubes.18 Highly crystalline and uniform TWCNTs were also obtained from the heat treatment of DWCNTs-peapods at 2000 °C in Ar.19 The DWCNTs were prepared by catalytically grown method, and their diameters were enlarged to the enough interior spaces of 1.2–1.6 nm for the encapsulation of C60 through the heat treatment at 2400 °C in Ar atmosphere. Such a high temperature triggers diameter enlargement process via the coalescence between DWCNTs.
Two adjacent DWCNTs (Fig. 3a) coalesce into off-centered coaxial carbon nanotubes (Fig. 3c) by annealing above 2100 °C.20,21 DWCNTs first merge into bi-cable carbon nanotubes by coalescence of the outer tubes (Fig. 3b), and the resulting inner tubes further coalesce inside a large diameter tube (Fig. 3c).21 Eventually, symmetric enlarged DWCNTs appear by more structural rearrangement of encapsulated off-centered inner tube along with the outer tube. The process of coalescence of two DWCNTs and merging into bi-cable CNT were discussed on the bases of detail TEM analyses and molecular dynamic calculations. Coalescence of SWCNTs to multi-walled CNTs was also reported.22
 | ||
| Fig. 3 TEM images on the gradual changes from (a) adjacent DWCNTs, (b) bi-cable structure and (c) off-centered DWCNT during heat treatment at 2100 °C. Adapted from ref. 21 with permission from Elsevier. | ||
Encapsulation of Gd-metallofullerene cages into SWCNTs, which were previously heated at 420 °C in dry air for 20 min to open the tube ends, was carried out in a sealed glass ampoule at 500 °C for 24 h.23 The TEM images of fully encapsulated SWCNTs demonstrated that fullerene molecules were oriented randomly in respect to the tube axis, which could be supposed by observing dark spots due to Gd atoms in the fullerene cages.
K-doping into C60@SWCNTs was performed by exposing C60@SWCNTs to potassium vapor at 473 K over 50 h.24 TEM images before and after K-doping are shown in Fig. 4. After doping, some dark spots in the figures, which are reasonably supposed to be individual K atoms, are clearly observed inside CNTs and are allocated at the interfullerene sites. Electrochemical insertion/de-insertion of Li into C60@SWCNT was carried out in 1 M LiClO4/(EC + DEC) over the potential range of 0–3.0 V,25 and the cell gave the reversible capacity of 550–610 mA h g−1, which is slightly larger than the pristine SWCNT (460–490 mA h g−1), although C60@SWCNT and DWCNT showed a large irreversible capacity. The enhancement in the capacity is explained by the following two possibilities, the change in the electronic structure of the SWCNTs and a steric effect from the encapsulated C60. The encapsulated C60 stabilizes the Li ion desolvated at the entrance of the tubes.
 | ||
| Fig. 4 TEM images of (a) the pristine C60@SWCNTs and (b–d) after K-doped ones. Adapted from ref. 24 with permission from the American Physical Society. | ||
Zn-diphenylporphyrin (Zn-DPP) is not a spherical but a flat molecule, of which size is about 1.0 nm and somewhat larger than C60 and C70, and possible to be encapsulated into SWCNTs with a diameter distribution from 1.25 to 1.47 nm at 400 °C.8 In the resultant Zn-DPP@SWCNTs, both Zn-DPP and SWCNT were supposed to be deformed from the experimental facts of a redshift of the π → π* transition in Zn-DPP and a drastic decrease of the radial breathing mode Raman spectrum intensity of SWCNTs.
(b) Iodine
Encapsulation of iodine into SWCNTs (I@SWCNTs) was shown to be effective to improve their galvanomagnetic properties.26 It was performed by heating the mixture of SWCNTs, which were synthesized by arc-discharge method using Ni/Y catalyst, with iodine at 150 °C in an evacuated glass tube. I-encapsulation was confirmed by the shift of Raman G-band from 1594 cm−1 for the pristine SWCNT to 1597 cm−1 for I@SWCNT and an increase in the intensity ratio of D-band to G-band (ID/IG), suggesting the introduction of high structural disorders by I-doping. The replacement of inorganic transparent conductive film, such as indium tin oxide, may be possible with flexible transparent conductive films of SWCNTs by alkali-metal (K, Rb) or halogen-atom (Br, I) doping, which can improve electrical conductivity of SWCNTs. Electrochemical I-doping in SWCNTs was performed in a NaI aqueous electrolyte.27 The G-band of in situ Raman spectrum for the SWCNTs shifts gradually with an increasing amount of doped I, while tends to be saturated, indicating full I-doping level, as shown in Fig. 5. The electrochemical I-doping level can easily be controlled by tuning the applied potential and time. By reversing the polarity in an electrochemical cell, doped-iodine molecules could be removed completely. To understand the structure of polyiodide ions encapsulated into CNTs and the charge-transfer from CNTs to iodine molecules, in situ Raman spectrum measurements were performed on SWCNTs having different tube diameters at low temperatures down to −100 °C.28 The amounts of the encapsulated iodine molecules estimated by TG analysis were 36.6, 49.0 and 50.0 wt% for the SWCNTs having the averaged diameters of 1.0, 1.5 and 2.5 nm, respectively. The Raman G-band peak position of the SWCNTs shifted toward the higher wavenumber side with decreasing temperature, suggesting that the charge transfer from SWCNTs to encapsulated I molecules increased with decreasing temperature to form polyiodide ions such as I3−, I5− and I7−. | ||
| Fig. 5 G-band shift of SWCNTs as a function of applied potential. Adapted from ref. 27 with permission from the PCCP Owner Societies. | ||
Crystalline KI nanoparticles were grown in the nanospaces of single-wall carbon nanohorns (SWCNHs) by heating the mixture at 1073 K in a sealed quartz tube.29 The nanospaces of SWCNHs, internal tubular spaces and interparticle micropores, were measured as the volume of 0.50 and 0.11 cm3 g−1, respectively. The structure of KI crystals encapsulated in SWCNHs had the same as that formed under the pressure above 1.9 GPa in the bulk crystal, suggesting highly constrained state of KI crystals in the nanospaces of SWCNHs.
(c) Sulfur
In a sealed glass tube at 873 K, sulfur can be encapsulated into SWCNTs and DWCNTs as monoatomic chains with the length up to 160 nm, forming S@SWCNTs and S@DWCNTs.30 In Fig. 6a, b and c, high-resolution TEM images are shown: two straight chains in SWCNT, one zigzag chain and one straight chain in DWCNT, respectively. Both straight and zigzag chains were encapsulated inside SWCNT and DWCNT, respectively. The distance (d) between two neighboring S atoms in both straight and zigzag chains increases gradually with increasing temperature in SWCNTs, as shown in Fig. 6d and e. In DWCNTs, however, d in the chains is slightly smaller than that in SWCNTs at the temperature below 450 K and it approaches that in SWCNT abruptly above 650 K, suggesting that the S chains are immobilized in the host SWCNTs up to 800 K, in other words, SWCNTs provide the most suitable space for stabilizing the S chains. These thermodynamic behavior of S chains in SWCNT and DWCNT shown in Fig. 6d and e, respectively, are quite different from the bulk arrays of S8 molecules, which has the melting point at about 393 K and boiling point at about 718 K. The sulfur atoms in CNTs were supposed to be bound with covalent character, forming one-dimensional crystalline phase, as proved by XRD, and the sulfur chains constrained in the nanotubes were electro-conductive under ambient pressure and temperature; the electric resistivity ρ at 300 K of SWCNTs decreased by the encapsulation of S chains from 1.1 × 10−3 to 5.0 × 10−4 Ω cm. By taking into consideration the fact that the bulk S becomes metallic under ultrahigh pressure more than 90 GPa,31 encapsulated S chains are strongly constrained in the spaces of SWCNT.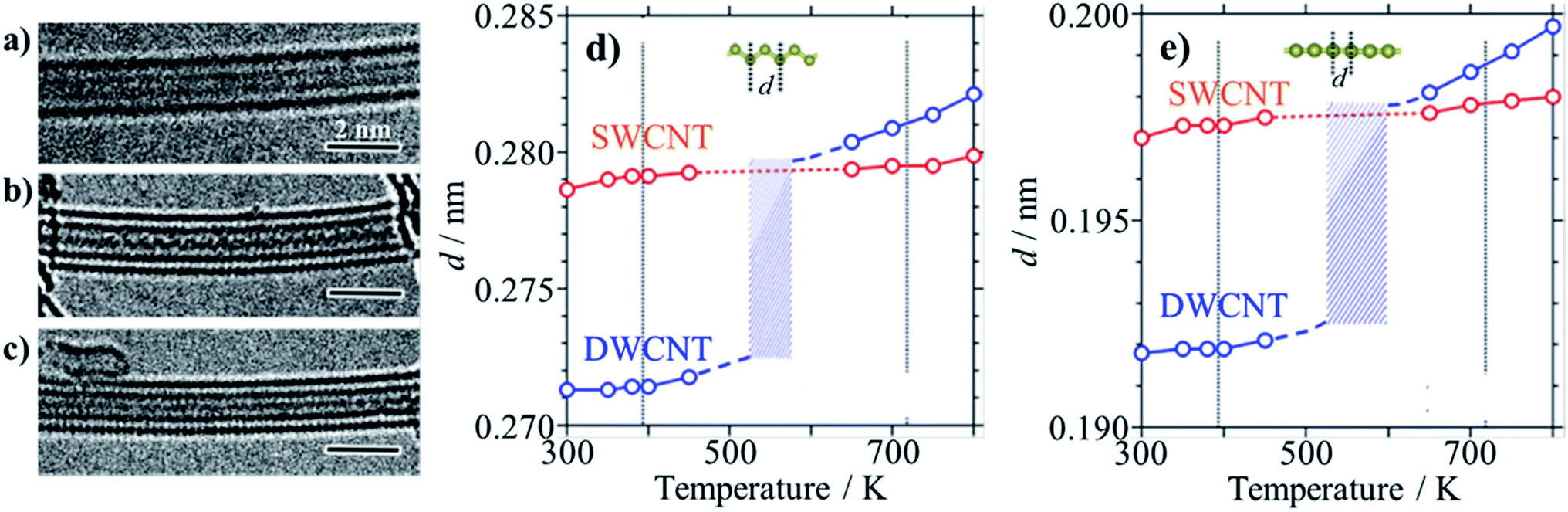 | ||
| Fig. 6 Sulfur encapsulation into carbon nanotubes: TEM images of (a) two straight S-chains in SWCNT, (b) zigzag and (c) straight S-chains in DWCNTs, and temperature dependences of interatomic distance d in SWCNT and DWCNT of (d) zigzag and (e) straight S-chains. Melting and boiling points for bulk sulfur (∼393 and ∼718 K, respectively) are indicated by vertical lines for comparison. Adapted from ref. 30 with permission from Nature Publishing Group. | ||
(d) Organic molecules
Various organic molecules, tetrakis(dimethylamino)ethylene (TDAE), tetramethyl-tetraselenafulvalene (TMTSF), tetrathiafulvalene (TTF), pentacene, anthracene, 3,5-dinitrobenzonitrile, tetracyano-p-quinodimethane (TCNQ) and tetrafluorotetracyano-p-quinodimethane (F4TCNQ), were encapsulated into SWCNTs.32 Encapsulation was performed simply by heating SWCNTs, which were purified and de-capped with H2O2, HCl, and NaOH, with these organic molecules just above their sublimation temperatures in vacuum. Encapsulation (confinement) of organic molecules results in the injection of amphoteric carriers into SWCNTs, which can be controlled by the ionization energy and electron affinity of the encapsulated molecules. As shown in the inset of Fig. 7a, optical absorption spectrum changes by the encapsulation of organic molecules into SWCNT. The intensity of the absorption band at 0.68 V was normalized by that of the pristine SWCNT, and the intensity ratio, Idoped/Ipristine, was plotted against ionization energy and electron affinity of the molecules in Fig. 7a and b, respectively, showing amphoteric carrier doping characteristics of encapsulated organic molecules. The field-effect-transistor devices of SWCNTs encapsulated by organic molecules revealed that TTF- and TMTSF-confined SWCNTs showed n-type behavior, whereas TCNQ-confined SWCNTs showed p-type behavior. These SWCNT semiconductors by confinement of organic molecules have the following advantages: air stability, controllable carrier doping, and simple process for preparation.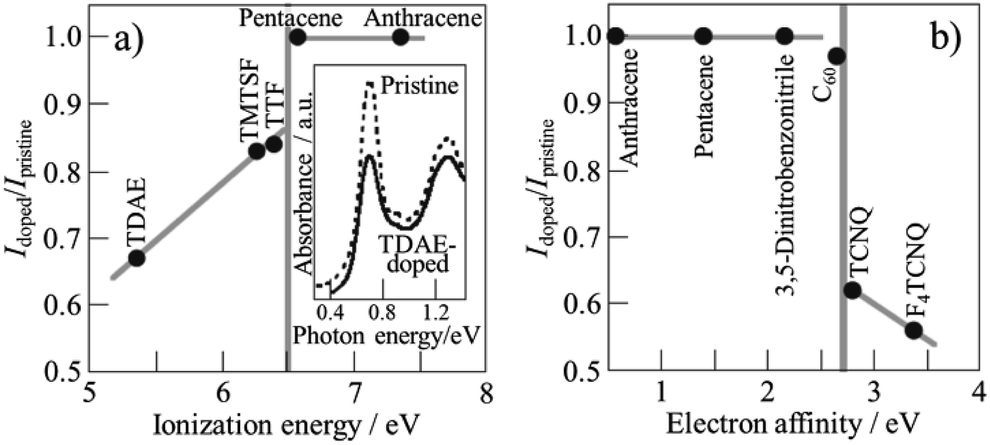 | ||
| Fig. 7 Relations of absorption intensity ratio Idoped/Ipristine to ionization energy and electron affinity of encapsulated molecules. Adapted from ref. 32 with permission from Nature Publishing Group. | ||
Encapsulation of 9,10-anthraquinone (AQ) and 9,10-phenanthrene quinone (PhQ) molecules into SWCNTs was performed by heating their mixture at 200 °C in an evacuated glass-tube.33,34 After the heat treatment, AQ@SWCNTs and PhQ@SWCNTs were recovered by washing with organic solvents (N,N-dimethylformamide for AQ and acetone for PhQ) to remove the excess AQ and PhQ deposited on the outer surface of the SWCNTs. Self-supported films of AQ@SWCNTs and PhQ@SWCNTs were obtained in this step. Electrochemical measurements for lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs) were performed in 1 M LiClO4/(EC + DEC) and 1 M NaClO4/PC electrolytes, respectively. LIB performances of AQ@SWCNT and PhQ@SWCNT were compared with the mechanical mixtures of AQ and PhQ with acetylene black (Fig. 8). These results demonstrated the reversible Li-ion storage of almost all quinone (AQ and PhQ) molecules encapsulated in the SWCNTs. The capacity fading observed on the mechanical mixtures of quinones with acetylene black, which came from the dissolution of quinone molecules into the electrolyte, was markedly suppressed by the encapsulation.33 The effect of the tube diameter of SWCNT on the Li-ion storage was studied using two SWCNTs with different diameters of 1.5 and 2.5 nm (SWCNT-1.5 and -2.5, respectively), of which encapsulated PhQ amount was determined by thermogravimetric measurement to be 22 and 38 wt%, respectively.34 The charge–discharge profiles and the reversible capacities of PhQ@SWCNTs at room temperature were quite similar for Li and Na ions, associating with two steps at 2.8 and 2.4 V for Li and those at 2.3 and 1.9 V for Na. Because the tube diameters of both SWCNTs are larger than the Li and Na ions and do not discriminate between these two ions. The reversible capacity of LIB using PhQ@SWCNT-1.5 was determined from the charge–discharge curves to be very close to the theoretical capacity of the PhQ molecule (258 mA h g−1), but that using PhQ@SWCNT-2.5 was about a half of the theoretical one, suggesting that only a half of the encapsulated PhQ worked as electrode materials. This is explained by the strong interaction between SWCNTs and PhQ for narrower SWCNTs, forming a current flow path at the interface. The reversible LIB capacities of PhQ@SWCNT-1.5 and PhQ@SWCNT-2.5 at 0 °C were much smaller than those at room temperature probably due to kinetic problems, which was supported by the fact that the capacity measured at a very slow charging rate was as high as the theoretical one. Meanwhile, the capacities in SIBs at 0 °C for both electrodes slightly decreased in comparison with those at room temperature. Particularly, the reversible capacity of PhQ@SWCNT-2.5 at 0 °C was almost the same as that at room temperature, probably because of facile Na ion diffusion in SWCNT-2.5 even at a low temperature.
 | ||
| Fig. 8 Charge–discharge curves collected at a current density of 100 mA g−1 at room temperature: mechanical mixture of (a) AQ with acetylene black, (b) PhQ with acetylene black; quinone-encapsulated SWCNTs of (c) AQ@SWCNT and (d) PhQ@SWCNT. Adapted from ref. 33 with permission from the PCCP Owner Societies. | ||
Encapsulation of β-carotene (C40H56) which consists of eight isoprene units having beta-rings at both ends of the molecule, was shown to be very efficient to suppress its degradation under UV irradiation.35 The light degradation of β-carotene is induced by the reaction with radical species (e.g., singlet oxygen) and isomerization; however, a surrounding tube wall prevents β-carotene from such reactions inside the constraint space. Encapsulation of β-carotene, 9,10-dichloroanthracene, and coronene into SWCNTs resulted in the enhancement of LIB capacity.36 Reversible capacity of coronene-encapsulated SWCNTs (7.2 wt%) was calculated to be 793 mA h per tube weight at 100 mA g−1 in 1 M LiClO4/(EC + DEC) electrolyte solution, whereas pristine SWCNTs exhibited reversible capacity of 316 mA h g−1. Although both electrodes showed a high irreversible capacity, but the enhancement in the reversible capacity observed for organic molecule-encapsulated SWCNTs is attributed to the increase in the Li ion storage sites in the tubes.
Encapsulation of metallocene compounds into SWCNTs was studied on ferrocene37–40 and cobaltocene.41 Encapsulation of ferrocene, FeCp2, was performed by contacting purified SWCNTs with ferrocene vapor at 300 °C under vacuum, followed by washing with diethyl ether to remove ferrocene deposited on the surface of tubes.37 The resultant FeCp2@SWCNTs retained the redox activity of FeCp2 itself in 0.1 M Bu4NClO4/acetonitrile electrolyte. FeCp2 confined in SWCNTs could be converted to metallic Fe nanoparticles by heating up to 700 °C under vacuum to form Fe-confined SWCNTs (Fe@SWCNT).38 Fe nanoparticles formed in SWCNTs have two morphologies, as shown TEM images with illustrations in Fig. 9: individual Fe particles with the diameter of 0.5–0.7 nm, suggesting several Fe atom aggregation (Fig. 9a) and somewhat linear alignment of Fe particles (Fig. 9b). According to the TEM observation, the filling yield (ratio of filled SWCNTs to empty SWCNTs) was estimated to be more than 80%. Encapsulation of FeCp2 into DWCNTs (FeCp2@DWCNT) resulted in marked change in electronic properties probably due to the charge transfer between FeCp2 molecules and DWCNTs, and Fe@DWCNT prepared from FeCp2@DWCNT was unipolar n-type semiconductive.39 FeCp2@SWCNT could change to DWCNT by high temperature annealing.40 By annealing at 1150 °C, all Fe atoms were released from the tube, while the annealing at 600 °C led to the formation of metastable iron carbide, which was supposed to act as a reactor by absorbing carbon atoms from one side of the tube and generating an inner tube to another side, forming DWCNT. Once the inner tubes had grown, the iron atoms diffused out of the tubes and aggregated into iron nanoparticles on the outside of DWCNTs, not in the channel. The intensity distributions of RBM Raman spectra for the inner-tube of FeCp2-derived DWCNTs were significantly different from those of C60-peapod-derived DWCNTs. Bis(cyclopentadienyl) cobalt (cobaltocene, CoCp2) could be confined only in the SWCNTs with a tube diameter of ∼1 nm under vacuum at 100 °C, whereas bis(ethylcyclopentadienyl) cobalt (Co(EtCp)2) confinement was possible in a range of tube diameters greater than ∼1 nm.41
 | ||
| Fig. 9 TEM images and illustrations for two morphologies of Fe nanoparticles in Fe@SWCNT. Adapted from ref. 38 with permission. Copyright (2006) the Japan Society of Applied Physics. | ||
(e) Water molecules
Encapsulation of water into strongly hydrophobic CNTs had not been expected, but computer simulations predicted certain possibility,42,43 even the formation of ice nanotubes inside of SWCNTs.44 Water molecules were experimentally shown to be adsorbed into de-capped SWCNTs at room temperature, and reversibly desorbed and re-adsorbed above room temperature.45,46 On water-confined SWCNTs, a structural change of adsorbed water at low temperatures was studied by using X-ray diffraction (XRD).45 XRD patterns of water-confined SWCNT at different temperatures are shown in Fig. 10a. A new diffraction peak appears at Q = 22 nm−1 at the temperature below 235 K, and the intensity of the both peaks at Q = 22 and 7.2 nm−1 increases with decreasing temperature (Fig. 10b), which is explained by solidification of water molecules as nanotube (ice nanotube), as shown schematically in Fig. 10c. By using SWCNTs having different tube diameter (D) in the range of 1.17–2.40 nm, temperature–diameter (T–D) phase diagram of water confined into SWCNTs was determined on the bases of experimental results of XRD, 1H and 2D NMR, and electrical resistance measurements.46 In thin SWCNTs (1.17 < D < ∼1.3 nm), water inside SWCNTs undergoes a liquid-solid-like transition and forms hollow ice nanotube structures at low temperature. For the large diameter SWCNTs (∼1.6 < D < 2.40 nm), on the other hand, water cannot be retained inside the SWCNTs. In the intermediate diameter range (D ∼1.4 nm), hollow ice nanotubes are formed at low temperature, while further increase in the water content leads to filled structures like ice nanotubes containing a one-dimensional water chain inside. | ||
| Fig. 10 Encapsulated water in SWCNT: (a) XRD profiles at different temperatures, (b) temperature dependence of the intensity of the peaks at 22 and 7.2 nm−1, and (c) a schematic illustration of the ice nanotube inside a SWCNT. Adapted from ref. 45 with permission. Copyright 2002 the Physical Society of Japan. | ||
Loading and releasing processes of water molecules into SWCNTs were studied by X-ray diffraction analysis and hybrid reverse Monte Carlo simulations.47 Water loading was performed at vapor pressures of 3.1 and 3.8 kPa, corresponding to water filling rates of 50 and 100%, respectively, and then water was released up to a vapor pressure of 2.3 kPa (corresponding to water filling of 50%). In the loading process, water molecules formed nanoclusters which were well stabilized in the channel of SWCNTs, while less stable water layers than water nanoclusters were formed in the releasing process.
3. In carbon nanopores
Nanopores, micropores, mesopores, and even macropores in carbon materials have been studied as adsorption sites for electrochemically active materials, such as Li for Li-ion batteries and different ions of aqueous and non-aqueous electrolytes for electrochemical capacitors, and also for various molecules and ions in gases and liquids, such as desalination of water, pollutant removals from wastes, etc. Recently, some of functional materials stored into these pores of carbon materials are experimentally demonstrated to show unique properties, as explained below, which are reasonably supposed to be caused by some constraints from nanopores of carbon materials. Functional materials can be encapsulated in these carbon nanopores, where functional materials are stably supported by constraint spaces and have a large contact area with the conductive carbon surface, which endows the functional materials with different characteristics from those in a bulk state.(a) Electrochemically-active organics
Confinement of polyaniline (PANI) into the micropores of activated carbon (AC) was shown to be effective to enhance the volumetric capacitance of electrochemical capacitors while retaining high power density, which is characteristic of electric double-layer capacitors.48,49 Aniline (ANI) was adsorbed at 25 °C into a commercially available AC, followed by electrochemical polymerization in an aqueous 1 M H2SO4 electrolyte.48 The AC employed was KOH-activated and microporous with a Brunauer–Emmett–Teller specific surface area (SBET) of 2070 m2 g−1, of which maximum adsorption capacity of ANI was 48 wt%. As shown the cyclic voltammograms in an aqueous 1 M H2SO4 electrolyte in Fig. 11a, the PANI-confined AC composites exhibit distinct anodic and cathodic peaks at 0.4–0.6 V due to the redox reaction of PANI, in addition to a rectangular voltammogram of the pristine AC, typical electric double-layer behavior. Fig. 11b shows the gravimetric capacitances measured by galvanostatic charge/discharge as a function of current density (rate performance). As shown in the figure, the confinement of PANI into the micropores of AC up to about 30 wt% is effective to enhance the gravimetric capacitance by pseudocapacitance of PANI. The synthesis of PANI in the micropores is not accompanied by the volumetric expansion of the AC particles. Therefore, the capacitance per unit mass of AC is proportionally correlated with the volumetric capacitance for the PANI-confined AC composites. As shown in Fig. 11c, their volumetric capacitances are enhanced with increasing amount of pseudocapacitive PANI. Of note in the figure is that the PANI-confined AC composites exhibit higher rate performance than the AC, i.e., the pseudocapacitance of PANI showed superior rate performance over the double-layer capacitance. This is realized by utilizing constraint spaces of AC, where PANI has a large contact area with conductive carbon surfaces. In addition, the pore confinement was found to be effective for improving the cycle lifetimes by absorbing the swelling of PANI during the charging process.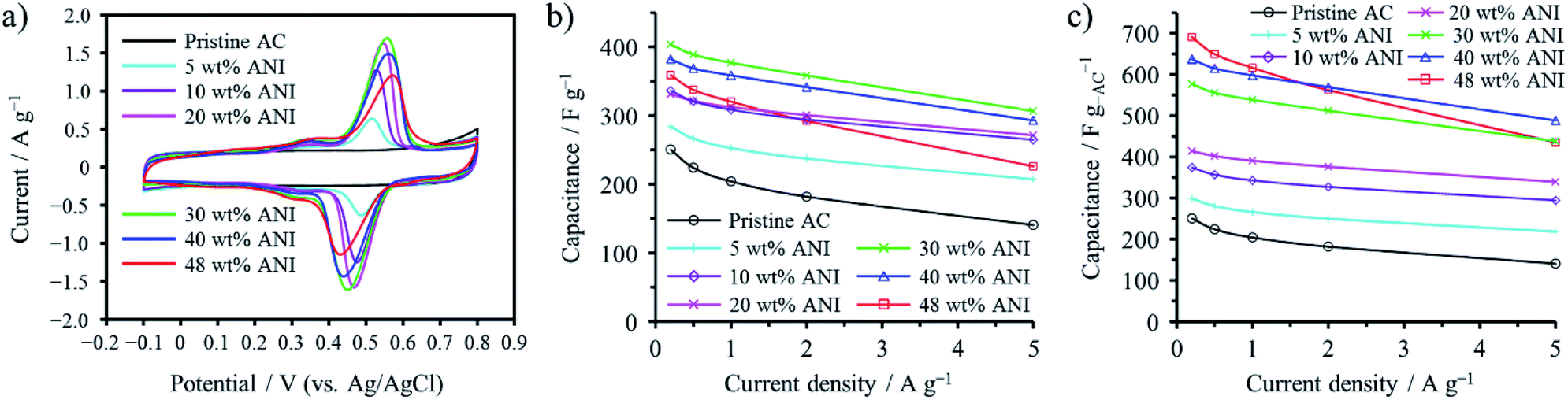 | ||
| Fig. 11 Electrochemical capacitor behaviors of PANI-confined ACs: (a) cyclic voltammograms collected at 1 mV s−1, (b) gravimetric and (c) volumetric capacitance dependences on the current density. Adapted from ref. 48 with permission from the Royal Society of Chemistry. | ||
Polypyrene (PPY) and PANI were confined into not only micropores but also mesopores of a commercial AC (MSC30), which had a large SBET of 3160 m2 g−1, micropore volume (Vmicro) of 0.99 cm3 g−1, and mesopore volume (Vmeso) of 0.60 cm3 g−1. The vapor of pyrene (PY) and ANI were adsorbed in the AC at 150 and 25 °C, respectively, followed by electrochemical polymerization in an aqueous 1 M H2SO4.49 The adsorption saturations of PY and ANI occurred at 60.2 and 62.4 wt%, respectively, for this AC. The capacitive performances are shown on the ACs with different amounts of PPY in Fig. 12. Anodic and cathodic peaks at 0.3–0.4 V due to the redox reaction of PPY confined into the AC pores are clearly observed in their cyclic voltammograms (Fig. 12a), as observed on the PANI-confined ACs (Fig. 11a), which gave the pseudocapacitance to the capacitors. By the confinement of 30–50 wt% PPY, gravimetric and volumetric capacitances of the cells are markedly enhanced, as shown in Fig. 12c and d. The maximum volumetric capacitance for the PPY-confined ACs and the PANI-confined ACs reached 314 and 299 F cm−3, respectively. Although the increment in the observed volumetric capacitances are reasonably supposed to be due to the pseudocapacitance of PPY, the rate performances of the cells are also improved, as shown in Fig. 12b, suggesting that the redox reaction of PPY occur more rapidly than the double-layer formation inside the nanopores. These results revealed that a large contact area between conductive polymers and carbon surfaces enables compatibility of high volumetric energy and high power densities for electrochemical capacitor electrodes.
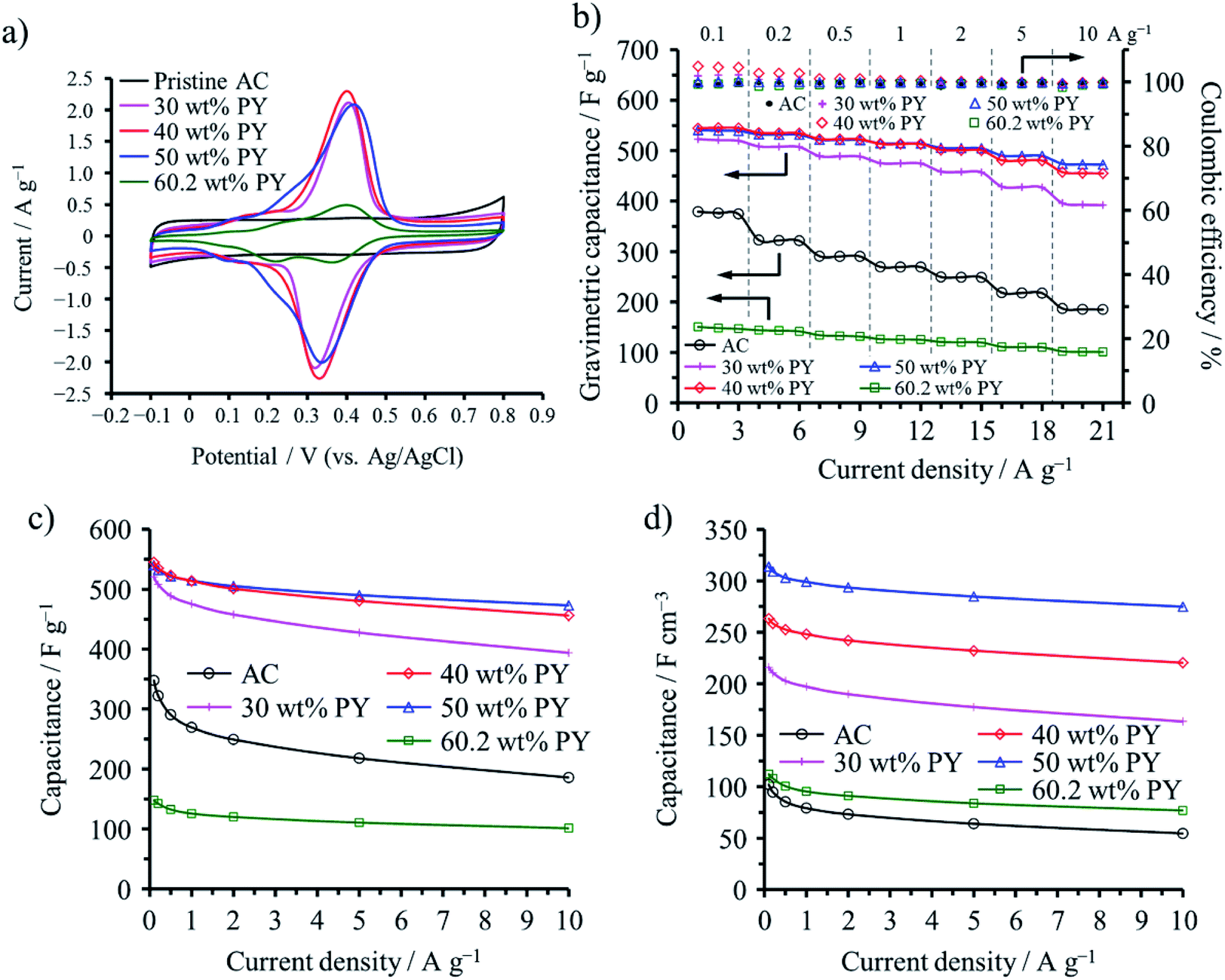 | ||
| Fig. 12 Electrochemical capacitor behaviors of PPY-confined ACs: (a) cyclic voltammograms with 1 mV s−1 rate, (b) dependences of gravimetric capacitance and coulombic efficiency on the cycle number, (c) rate performances of the gravimetric capacitance, and (d) those of the volumetric capacitance. Adapted from ref. 49 with permission from the Royal Society of Chemistry. | ||
2,5-Dichloro-1,4-benzoquinone (DCBQ) was confined into the pores of Ketjenblack (KB; SBET: 1340 m2 g−1, Vmicro: 0.48 cm3 g−1, Vmeso: 1.24 cm3 g−1) at 100 °C (a little higher than the sublimation temperature of DCBQ, 92 °C).50 The weight content of DCBQ in the DCBQ-confined KB was tuned to be 5, 10, 20, and 40 wt%. In addition, KB was saturated using excess amount of DCBQ, with the saturation amount reaching 60.1 wt%. N2 adsorption–desorption curves shown in Fig. 13a suggest that DCBQ was adsorbed into both micropores and mesopores, Vmicro and Vmeso being reduced to 0.17 and 0.71 cm3 g−1, respectively, with a 40 wt% DCBQ content; and giving Vmicro of 0.03 cm3 g−1 and Vmeso of 0.15 cm3 g−1 upon saturation. The DCBQ-confined KBs exhibit clearly the presence of redox reaction of DCBQ by a plateau in galvanostatic charge–discharge curves in an aqueous 1 M H2SO4 electrolyte, as shown in Fig. 13b, although the pristine KB shows almost linear characteristics (i.e., electric double-layer behavior). In Fig. 13c and d, gravimetric and volumetric capacitances are plotted against current density, revealing that excellent rate performance of the pristine KB is retained even in DCBQ-confined KBs. DCBQ has poor electrical conductivity by itself, like most organic compounds, but a large contact area between finely dispersed DCBQ in the pores of KB and conductive carbon surfaces enables fast charge transfer at the interface. Gravimetric capacitance reaches a maximum with a DCBQ content of 40 wt%, but the KB saturated with DCBQ gives a maximum volumetric capacitance. The resulting capacitor was characterized by high capacitances, particularly high volumetric capacitances as 4.7 times higher than that of the pristine KB, along with high rate capability up to 5 A g−1 and excellent cycle lifetimes up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
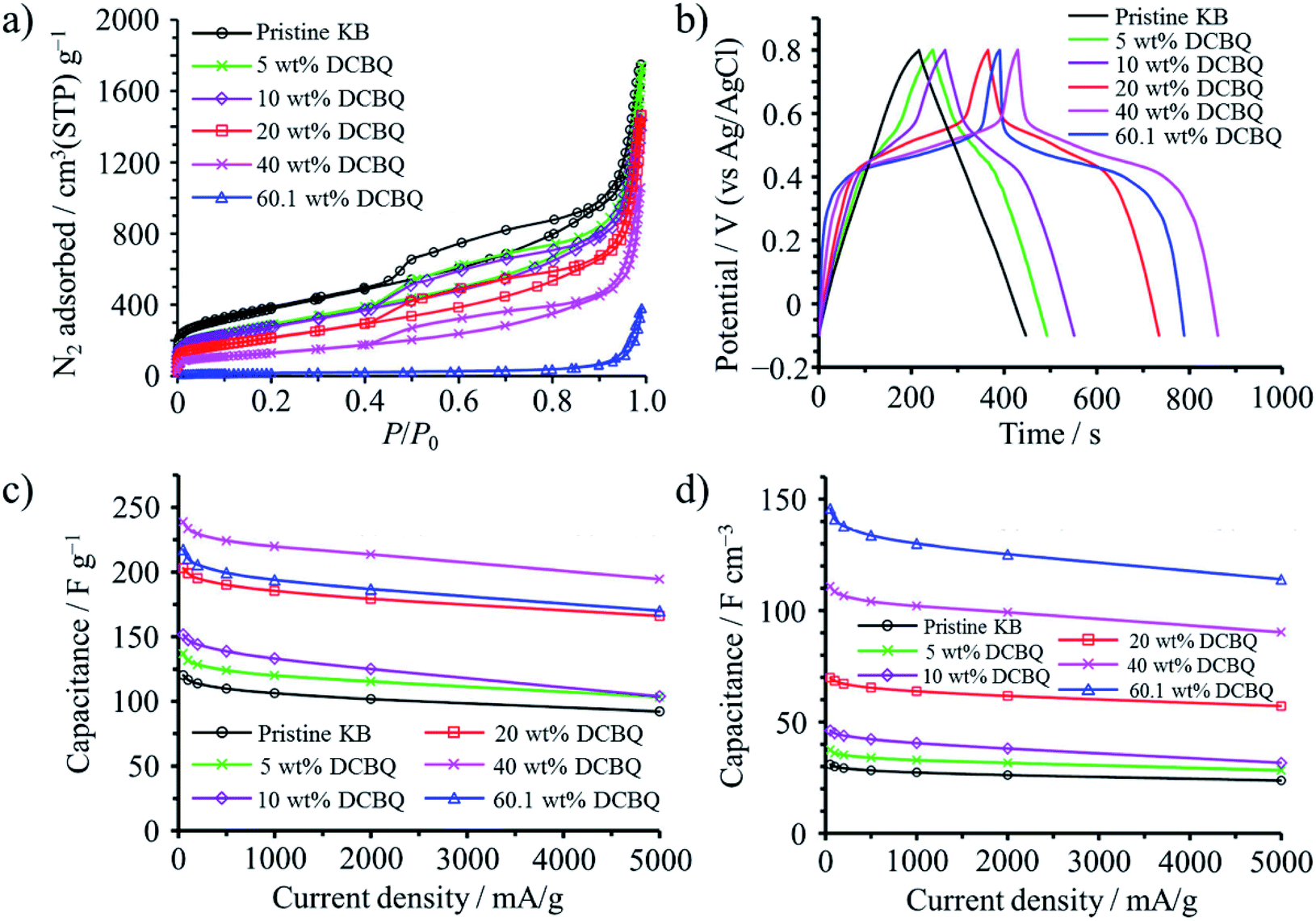 | ||
| Fig. 13 DBCQ-confined Ketjenblack: (a) N2 adsorption–desorption curves at 77 K, (b) galvanostatic charge–discharge curves in 1 M H2SO4 electrolyte measured at a current density of 50 mA g−1, (c) rate performances of the gravimetric capacitance, and (d) those of the volumetric capacitance. Adapted with permission from ref. 50. Copyright 2016 American Chemical Society. | ||
One of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) derivatives, 4-hydroxy-TEMPO benzoate (HTB), was confined into the nanopores of the AC (MSC30; SBET: 3160 m2 g−1, Vmicro: 0.99 cm3 g−1, Vmeso: 0.60 cm3 g−1) through its vapor at 130 °C, and the capacitive performances of the resultant HTB-confined ACs with different HTB contents (20–50 wt%) were studied in an aqueous 1 M H2SO4 electrolyte.51 A marked reduction in pore parameters with increasing adsorbed amount of HTB suggested the confinement of HTB molecules into nanopores of AC, with the SBET, Vmicro, and Vmeso reducing to 250 m2 g−1, 0.07 cm3 g−1, and 0.05 cm3 g−1, respectively, by adsorption of HTB up to 50 wt%. Anodic and cathodic peaks derived from the redox reaction of HTB were clearly observed at around 0.7 V. The redox potential of HTB is higher than those of many organic redox compounds and conductive polymers, which is advantageous to enhance the energy density of the HTB-confined ACs. The composite with 30 wt% HTB exhibited higher volumetric energy and power densities than the pristine AC: 39.5 W h kg−1 at 16.5 W kg−1 and 14.1 W h kg−1 at 8.2 kW kg−1 for the composite in contrast to 18.0 W h kg−1 at 16.5 W kg−1 and 1.1 W h kg−1 at 8.2 kW kg−1 for the pristine AC. Volumetric comparison shows a further enhancement in the volumetric energy and power densities of the composite with 30 wt% HTB: a 3.1 times enhancement (16.5 W h L−1) at 6.9 W L−1 and a 18 time enhancement (5.9 W h L−1) at 3.5 kW L−1. The high redox potential of HTB and a large contact area between the conductive carbon surface and HTB realized compatibility between the enhancements in both volumetric energy and power densities. The confinement of HTB molecules into the nanopores of AC was effective to prevent dissolution of HTB in an aqueous electrolyte with the aid of a hydrophobic group in HTB and to improve markedly the contact of HTB with a conductive carbon surface, which was supposed to allow for fast redox reactions of HTB, in addition to fine dispersion of minute aggregates of HTB molecules.
(b) Inorganics
MnO2 nanocrystals confined into nanopores of porous carbon nanofibers (CNFs) delivered high electrochemical capacitance in 1 M Na2SO4 aqueous electrolyte, the electrodes of the MnO2/CNF composite giving the capacitance of 1282 F g−1 with the current density of 0.2 A g−1.52 Porous CNFs were synthesized from the mixture of polyimide with polyvinylpyrrolidone (PVP) via electrospinning, followed by carbonization. In order to confine MnO2 nanocrystals preferentially in the nanopores, the porous CNFs had to be immersed into the KMnO4 aqueous solution with the concentration of less than 0.024 g L−1, while higher concentration of KMnO4 solution resulted in MnO2 deposition mostly on the external surface of the CNF. The MnO2 nanocrystals in the inner part of CNF were grown along with (200) crystal faces under nanopore constraint and therefore had ultrathin structures, whereas the nanocrystals in the external part of CNF were grown along with (211) crystal faces (Fig. 14). The former nanocrystals enable the shortened diffusion path for electrolyte and a large active surface area. A symmetrical capacitor of the MnO2/CNF composite electrode prepared in the optimized condition could deliver the energy density of 36 W h kg−1 with a power density of 39 W kg−1 and maintained 7.5 W h kg−1 at 10.3 kW kg−1. | ||
| Fig. 14 TEM images of MnO2 nanocrystals confined in the pore (c and d) and deposited on the surface (b) of the porous carbon nanofiber (a). Adapted from ref. 52 with permission from Nature Publishing Group. | ||
The electrochemical cell consisting of the same KOH-activated porous carbon (SBET of 2181 m2 g−1) worked as a hybrid cell, battery-type behavior at the positive electrode and capacitor-type one at the negative electrode in an aqueous electrolyte containing 2 M MnSO4 and 0.5 M KI, although it worked as a capacitor in 2 M MnSO4 electrolyte.53 This hybrid behavior in alkali metal iodide-based aqueous electrolytes was concluded to be owing to the confinement of polyiodides in the porosity of the positive carbon electrode creating a so-called carbon/iodide interface,54 the confinement of polyiodides being confirmed by the measurement of Raman spectra.53 Effectiveness of the confinement of electrochemically-active materials was reported on Sn55,56 and Ge.57,58
BaTiO3 rod-like crystals were synthesized in the nanopores of single-wall carbon nanohorns (SWCNHs).59 TiO2 was firstly encapsulated into the nanopores of SWCNHs by impregnating an aqueous TiCl4 solution and following annealing at 373 K. The resulting TiO2 was reacted with Ba(OCH2CH3)2 at 400 K in an autoclave of N2 atmosphere, and the resultant BaTiO3 rods having high aspect ratio (2–4 nm in diameter and 40–60 nm length) were obtained. By impregnation of SWCNHs in methanol/2-methoxyethanol mixed solution of Ba(OCH2CH3)2 and Ti(OCH(CH3)2)4 and subsequent annealing at 400 K in an autoclave, spherical BaTiO3 particles with the diameters of 2–4 nm were synthesized in the nanopores of SWCNHs, whereas the BaTiO3 particles with the diameters of 20–40 nm were synthesized via the same process without using SWCNHs.
(c) Platinum
Loading of subnanometer-sized Pt clusters (Pt4–5) onto carbon materials was shown to be possible by confining a Pt precursor into their micropores.60 An organometallic precursor, (COD)PtMe2 (COD: 1,5-cyclooctadiene) was selected as a Pt precursor, which was strongly adsorbed in the micropores of carbon substrates by a wet impregnation technique and easily reduced by a thermal treatment. Ketjenblack (KB; SBET: 1290 m2 g−1, Vmicro: 0.46 cm3 g−1) and zeolite-templated microporous carbon (ZTC; SBET: 3680 m2 g−1, Vmicro: 1.55 cm3 g−1) were employed as carbon substrates. The carbon substrate was refluxed in dehydrated acetonitrile solution of the Pt precursor under N2 atmosphere to adsorb the Pt precursor, followed by filtering, washing with acetonitrile, and then washing with diethyl ether to remove the precipitated Pt precursor on the outer surface of the carbon substrate particles. The Pt precursor was reduced to metallic Pt by heat treatment at 300 °C. The contents of the Pt-precursor after wet impregnation and that of metallic Pt after the heat treatment were 6.6 and 3.96 wt%, respectively for KB, and 20.0 and 12.8 wt%, respectively for ZTC. The ratio of the Pt content for KB to that of ZTC substrate was roughly 1/3, being almost the same as the ratio of Vmicro in the two substrates. The TEM images after the Pt-loading on KB and ZTC were shown in Fig. 15a and b, respectively. Even after impregnation and heating at 300 °C, no crystalline phase of the Pt-precursor nor the metallic Pt was detected by XRD, and the sizes of the Pt particles were supposed to be less than 1–2 nm from TEM images. X-ray absorption spectroscopy (XAS) analysis revealed that Pt4–5 subnano-sized clusters were supported on KB and ZTC, and their Pt–Pt bond length were substantially contracted (∼2.8%) in comparison with that of bulk Pt because of the substantial surface tension in the unsaturated coordination environment of the Pt atoms. The adsorption of the Pt-precursor into micropores was supposed to be crucial to prevent agglomeration and/or sintering of Pt nanoparticles. On ZTC with a low Pt content, Pt was dispersed as a single atom, as shown in Fig. 15c. The dispersion of Pt single atoms required the strong interaction between Pt atoms and oxygen-containing functional groups in ZTC, which was supported by the results of XAS analysis. | ||
| Fig. 15 TEM images of Pt-loaded carbon materials: (a) Ketjenblack and (b and c) ZTC. Adapted with permission from ref. 60. Copyright 2017 American Chemical Society. | ||
Pt nanoparticles were loaded on an Ar+-ion irradiated glassy carbon plate by radio-frequency magnetron sputtering with plasma output of 20 W for 60 s at fluences up to 1.0 × 1016 ions per cm2.61 Pt was supposed to be confined at the defects created by the irradiation of 380 keV Ar+ as minute clusters with an average size of about 5 nm. A strong interfacial interaction between Pt nanoparticles and the glassy carbon substrate was estimated from its Pt 4f7/2 and C 1s XPS spectra.
(d) Graphitic carbon nitride
Graphitic carbon nitride (g-C3N4), which has high activity of oxygen reduction reaction (ORR),62 was confined into a mesoporous carbon, CMK-3, by impregnating cyanamide as a precursor, followed by calcination at 550 °C in Ar atmosphere. The resulting g-C3N4@CMK-3 delivered activity comparable to a commercially available Pt/C catalyst, while showed much improved tolerance for methanol contamination.63 As shown in Fig. 16, the characteristic mesoporous structure due to the ordered nanochannels of the host CMK-3 was kept even after confinement of g-C3N4, with the g-C3N4 content being 27.1 wt%. Before the preparation of the composite, the host CMK-3 rods were treated with HNO3 to make the host surfaces hydrophilic. The ORR activity of the composite was much better than the bulk g-C3N4 and the mechanical mixture of g-C3N4 with CMK-3. Since the CMK-3 has a high surface area and high electrical conductivity, CMK-3 not only serves as a support for the g-C3N4 but also confines the g-C3N4 catalyst inside its nanospaces, which facilitates the electron transport necessary for an efficient 4e− ORR pathway. | ||
| Fig. 16 g-C3N4/CMK-3 composite: (a) TEM image, (b) SEM image, and (c) N2 adsorption/desorption isotherm. Adapted with permission from ref. 63. Copyright 2011 American Chemical Society. | ||
In lithium–sulfur (Li–S) batteries, anchoring lithium polysulfides (Li2Sx) in the cathode materials is strongly demanded to inhibit their shuttling between electrodes during charge/discharge cycles, which would lead to the dissolution of Li2Sx (4 ≤ x ≤ 8) into commonly used electrolytes, resulting in the short cycle lifetimes of the electrodes and lithium anode contamination. Codoping of N and S into mesoporous carbons was demonstrated to be effective to enhance the affinity for Li2Sx, preventing their dissolution into an electrolyte.64 The composite of conductive vanadium nitride (VN) with reduced graphene oxide (rGO) was also reported to deliver good rate and cycling performances, with the initial capacity being 1471 mA h g−1 with only 15% capacity reduction after 100 cycles at 0.2C rate.65 The conductive and porous structure of the VN/rGO composite facilitates the electron transportation and Li ion diffusion. In addition, since the VN has not only high electrical conductivity but also catalytic activity, it shows an anchor effect on the polysulfides and accelerates their redox reactions. g-C3N4 nanodots with the sizes less than 5 nm were confined into a MOF-derived N,S-codoped hollow porous carbon shell, and the resulting composite demonstrated excellent performances in Li–S batteries as cathode.66 As shown in Fig. 17, the composite exhibits much higher capacity and much better rate and cycle performances than neat g-C3N4 and the pristine porous carbon. The composite delivered 1447 mA h g−1 at 0.2C rate and 387 mA h g−1 at 5C, with excellent cycling stability as only 0.048% capacity reduction at 1.0C after 500 cycles. The synergetic effect of hollow carbon structure, N,S-codoping, and encapsulation of g-C3N4 facilitates the electron transfer and endows the composite with strong adsorption affinity for polysulfides to suppress the polysulfide shuttling.
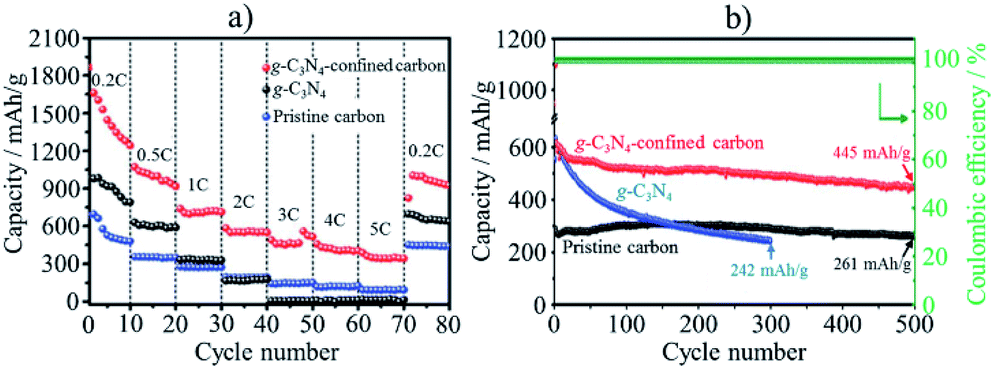 | ||
| Fig. 17 Li–S battery performances of g-C3N4-confined porous carbon in comparison with g-C3N4 and pristine porous carbon: (a) rate performances and (b) cycle performances at 1C rate. Adapted from ref. 66 with permission from the Royal Society of Chemistry. | ||
(e) Methane hydrate
Water molecules confined into nanopores of AC could adsorb methane under the pressure above 3.5 MPa at 2 °C to form methane hydrate.67 The AC used was prepared from mesophase-pitch at 450 °C, followed by the activation using KOH at 800 °C. The AC had a SBET of 3670 m2 g−1, Vmicro of 1.20, and Vmeso of 1.24 cm3 g−1. In Fig. 18, methane adsorption/desorption isotherms at 2 °C are shown for the AC with different pre-adsorbed water contents (Rw in g g−1). Their methane adsorption capacity depended strongly on Rw and was much larger than the dry AC (Rw = 0). At low methane pressure, the wet ACs show the lower adsorption capacity than the dry one, probably due to pore blocking by pre-adsorbed water. Above 3 MPa pressure, however, the adsorption capacity of methane shows sigmoidal increases, particularly for the AC with the high Rw value. The formation of methane hydrate was proved by inelastic neutron scattering and synchrotron X-ray powder diffraction. This condition of methane hydrate formation (3.5 MPa and 2 °C) in nanopores of the AC is much milder, and hydration proceeds much faster (finishing within minutes) than the occurrence of methane hydrate in nature (more than 6 MPa and slightly below room temperature). The ACs with high water contents, Rw of 2.9 and 4.1 (over-saturated), give two-step adsorption isotherms with pronounced hysteresis (Fig. 18b), suggesting that adsorption/desorption mechanisms for methane for wet ACs are different in the dry one, and the methane hydrate formed in carbon nanopores is stable. | ||
| Fig. 18 Methane adsorption/desorption isotherms of the activated carbon having different pre-absorbed water content Rw. Adapted from ref. 67 with permission from Nature Publishing Group. | ||
4. Carbon coating
Carbon coating has been performed on different functional materials to improve their performances, such as a marked improvement in photocatalytic activity of anatase-type TiO2, a performance enhancement of electrode materials for LIBs and electrochemical capacitors, as reviewed before.68 Recent experimental results on electrode materials also suggest that carbon coating may give certain constraints on the functional materials, similar to the carbon walls of nanopores explained above.(a) Metallic silicon
Carbon coating is very effective to exert the intrinsic functionality of metallic silicon as the anode material for LIBs, such as a high theoretical capacity (more than 3.5 A h g−1), by absorbing its large volume change (more than 300%) during lithiation/de-lithiation and improving its relatively low electrical conductivity.Particle sizes of Si as anode materials exhibited marked influences on rate and cycle performances of LIBs with 1 M LiPF6/(EC + DEC).69 Si particles with the particle size of ca. 1 μm (micron-sized Si) showed abrupt decrease in capacity with the cycle number even at a small current density of 0.2 A g−1 due to the pulverization of Si particles. The poor cycle lifetimes of the micron-sized Si particles were attributed to the typical bulk like behavior. On the other hand, nano-sized Si particles with the size of 82 nm exhibited the remarkably enhanced coulombic efficiency at the 1st and following cycles. The capacity retention of the nano-sized Si particles was enhanced by carbon coating with a thickness of ca. 10 nm through a pressure-pulsed chemical vapor deposition (P-CVD) method; the carbon coating decreased the inner resistance. The particle morphology of the nano-sized Si particles and the carbon-coated ones changed from spherical particles to nano-sized wrinkled structure after 20 charge/discharge (lithiation/de-lithiation) cycles (Fig. 19a and b).69,70 At the same time, the coated carbon was also deformed together with wrinkled Si, as evidenced by scanning transmission electron microscopy (STEM) imaging and energy dispersive X-ray spectroscopy (EDS) analysis.69 After 100 cycles, the wrinkled structure of both samples further transformed into an aggregated lump due to the repeated volumetric expansion and the subsequent agglomeration of pulverized Si particles (Fig. 19c).70 The capacity fading accompanied by the structural deformation of the wrinkled Si was suppressed by limiting the capacity up to 1500 mA h g−1 during lithiation because of restricting the volumetric expansion to some extent. Similar structural transformation through wrinkled one and improved battery performances were observed on ball-milled Si, which is more inexpensive than Si nanoparticles.70 On the ball-milled Si consisting of weakly aggregated primary particles with the size of about 47 nm, the initial reversible capacity was improved reaching ca. 3000 mA h g−1 by carbon coating, as shown in Fig. 20a. By restricting the discharge capacity up to 1500 mA h g−1, cycling with different current densities of 0.2–5.0 A g−1 was possible on the carbon-coated Si up to at least 100 cycles, whereas ball-milled Si without carbon coating showed a marked capacity fading (Fig. 20b). Flaky Si nanoparticles (thickness of ca. 16 nm and lateral size of 0.2–1 μm) were produced from Si sawdust by beads-milling in isopropyl alcohol. Since the Si sawdust contained 4 wt% graphite, which came from the graphite substrate used for the cutting process of Si ingots, the graphite was homogeneously dispersed over the Si particles after a beads-milling process.71 Although their structure was converted into nanoflake morphology through a milling process, they could be transformed to wrinkled morphology by cycling of lithiation/de-lithiation and demonstrated better performances as an anode of LIBs. The CVD of the nanoflake Si was also examined, and the resulting material exhibited the enhanced rate capability at 5 A g−1 under the limiting capacity of 1500 mA h g−1 during lithiation, reaching almost the same value as that at 0.2 A g−1. The enhanced rate capability was attributed to the large continuous structure formed by carbon coating. The carbon layer on Si particles is supposed to work as a buffer between neighboring Si nanoparticles even during marked morphology change from spherical or flaky to wrinkled, in other words, by confining Si nanoparticles within coating carbon.
 | ||
| Fig. 19 TEM images and morphology illustrations of nano-sized Si after cycling of lithiation/de-lithiation. Adapted from ref. 70 with permission from Elsevier. | ||
 | ||
| Fig. 20 Capacity changes with cycling with different current densities on pristine ball-milled Si and carbon-coated one: (a) without and (b) with the capacity restriction to 1.5 A h g−1. Adapted from ref. 70 with permission from Elsevier. | ||
Thin Si flakes were synthesized by CVD of silane (SiH4) gas on the cubic crystals of NaCl at 550 °C, followed by dissolution of NaCl. The thickness and lateral size of the Si flakes was controlled to be 50 nm and a micrometer, respectively.72 Carbon coating of the Si flakes was performed by heating in acetylene gas at 900 °C, resulting in the carbon content of 7 wt% and the thickness of about 10 nm. The non-porous structure of the pristine and carbon-coated Si flakes with a low surface area of ca. 10 m2 g−1 suppressed excessive SEI formation and reduced initial irreversible capacity (Fig. 21a), in comparison with that of the commercially available Si nanoparticles. Their initial reversible capacities reached 2943 and 2255 mA h g−1, corresponding to the initial coulombic efficiency of 87.2% and 92.3%, respectively in 1.3 M LiPF6/(EC + DEC). As shown in Fig. 21b, the carbon-coated Si flakes delivered a steady rate performance around 2000 mA h g−1 up to 10C rate, whereas the pristine Si flakes show the inferior rate capability. The full cell coupled with a LiCoO2 cathode shows a steady cycle performance at 0.5C rate for the carbon-coated Si flakes, while the reversible capacity of the pristine Si flakes decayed drastically (Fig. 21c). The carbon layer on Si flakes works not only to improve the electrical conductivity in the anode but also to buffer the large expansion of Si during lithiation.
 | ||
| Fig. 21 Carbon-coated Si flakes in comparison with the pristine Si flakes: (a) the first charge/discharge curves, (b) rate capability at different C-rates and (c) cycle performance of the cell coupled with a LICoO2 cathode at 0.5C rate. Adapted from ref. 72 with permission from Nature Publishing Group. | ||
The structural deformation of the carbon-coated Si flake was monitored by in situ TEM under a bias of −3 V and 3 V, respectively, where a flake was mounted on a Pt wire connected to the Li2O/Li probe; the Li metal and the LiO2 layer serve as the counter electrode and the solid electrolyte, respectively.72 The structural deformation of the flake in the first two lithiation/de-lithiation cycles was schematically illustrated in Fig. 22. During the first lithiation, the flake showed anisotropic swelling, with only 9.7% expansion along lateral direction but about 200% in thickness upon full lithiation (Li15Si4), giving ∼270% volumetric swelling. By de-lithiation, the flake rippled along the lateral direction of the flake and the flake itself was significantly buckled. The second lithiation smoothed out the ripples and the de-lithiation gave severe buckling to the flake, while maintaining its 2D morphology without fracture. In contrast, the pristine Si flake without carbon coating swelled into the lateral direction as much as 31.9% after the first lithiation and could not recover its original dimensions during de-lithiation, preserving the swollen morphology and sharp edges generated during lithiation. The fact that lithiation of the pristine Si took longer times under higher bias than those of the carbon-coated Si flake indicates the relatively poor rate capability. The distinct difference in morphology change during the lithiation/de-lithiation cycle between the pristine and carbon-coated flakes is reasonably supposed to be due to the carbon layer coating, suggesting certain mechanical constraint of the carbon layers on the deformation of Si flakes.
 | ||
| Fig. 22 Schematic illustration of morphology change during lithiation/de-lithiation process. Adapted from ref. 72 with permission from Nature Publishing Group. | ||
B-doped Si/SiO2 composites were prepared from the mixture of ball-milled SiO2 and B2O3 powder (20:1 by mole) at 950 °C in Ar, where B-doped crystalline Si nanoparticles with the size of about 15 nm were dispersed in amorphous SiO2 particles (particles size of about 3 μm).73 Carbon coating of the composite by CVD of acetylene at 700 °C resulted in high cyclic performance of a hybrid cell coupled with mesoporous carbon spheres as a cathode using a 1 M LiPF6/(EC + DEC + DMC) electrolyte (lithium-ion capacitor, LIC), which delivered an energy density of 128 W h kg−1 at a power density of 1.23 kW kg−1 and even 89 W h kg−1 at 9.7 kW kg−1 with the voltage window of 2.0–4.5 V. Amorphous SiO2 matrix of the composites was supposed to work as a buffer layer for a large volume change of Si during lithiation/de-lithiation. In addition, boron-doping into Si increases the electrical conductivity of Si and enables fast charge transfer, necessary for high power density. Furthermore, the electrical conductivity and cycle lifetimes of Si were markedly improved by carbon coating. Si-included carbon composites were prepared from wasted glass (Si source) and wasted polyvinyl butyral (carbon source), both of which were derived from windshields with a polyvinyl butyral layer sandwiched by two glasses.74 The wasted glass separated from the polyvinyl butyral was reduced to Si by Mg powder as a reducing reagent at 650 °C for 6 h under Ar atmosphere, and the byproducts (Mg2Si and Mg2SiO4) and the unreacted SiO2 were removed by HNO3 and HF etching, respectively. The carbon coating was performed by mixing the obtained Si nanoparticles and the polyvinyl butyral and their subsequent heat treatment at 500 °C for 4 h. Its LIB performance was measured in 1 M LiPF6/(EC + PC) electrolyte solution. The reversible capacity of the non-carbon-coated Si in the first charge/discharge cycle reached 3336 mA h g−1 with irreversible capacity of 489 mA h g−1, but it decayed rapidly with the number of cycling measured at 420 mA g−1, down to less than 1000 mA h g−1 after 50 cycles. In contrast, the carbon-coated Si with a carbon content of 60 wt% showed the initial reversible capacity of 856 mA h g−1, while its capacity retention reached 84% at the 300th cycle. Although its initial reversible capacity was less than 1000 mA h g−1, the capacity per unit mass of Si is calculated to be over 2000 mA h g−1. The carbon matrix stabilized the Si nanoparticles and enhanced electrical conductivity of the electrode, which was supported by the result of the impedance analysis. The composite of Si/rGO was prepared from a colloidal mixture of wasted Si sludge containing SiC from the wafer-slicing saw with GO by aerosol spray pyrolysis with thermal reduction of GO.75 The colloidal mixture was sprayed into a tubular furnace by ultrasonic nebulizer, and at the same time, unnecessary SiC was separated from the droplet because the Si particles have a lighter density and smaller sizes than the SiC particles. The as-prepared composite has ball-like morphology and the Si particles are located inside the rGO shell. The composite prepared from the colloidal mixture with 0.1 wt% GO delivered the reversible capacity of about 1600 mA h g−1 in the initial charge/discharge cycles by retaining the capacity of about 1000 mA h g−1 after 50 cycles. The composite prepared from the mixture with 0.4 wt% GO gave much better cyclability although the capacity became smaller (ca. 700 mA h g−1).
Carbon-coated Si nanowires were synthesized from the mixture of bis(bis(trimethylsilyl)amino)tin, Sn(HMDS)2, and monophenylsilane (MPS) with different mole ratios (Sn/Si of 1/16 to 1/64).76 The reaction proceeded under supercritical fluid condition in anhydrous toluene solution at 490 °C and 10.3 MPa, followed by the carbonization at 900 °C under N2/H2 gas flow. At first, MPS decomposed to atomic Si and high order phenylsilane through disproportionation reaction, while Sn(HMDS)2 decomposed to Sn nanoparticles which worked as a seed to form eutectic with Si for growing Si nanowires. The high order phenyl silane byproduct polymerized on the surface of the Si nanowires and the resulting polyphenylsilane was converted to carbon shells during the heat treatment at 900 °C. The morphology of the carbon-coated Si nanowires highly depends on the Sn/Si molar ratio, and the nanowires prepared below a Sn/Si molar ratio of 1/32 before the heat treatment were heavily kinked and covered by thick polyphenylsilane shell. The carbon-coated Si nanowires prepared with a Sn/Si molar ratio of 1/22 showed the markedly straighter and thinner shell, and the carbon shell retained the uniform thickness after the heat treatment (Fig. 23). This composite exhibited a high capacity over 2000 mA h g−1, with nearly 96% coulombic efficiency when cycled at a slow rate of 0.1C for 100 cycles. Moreover, a stable capacity of about 1300 mA h g−1 with more than 98% coulombic efficiency was attained after 100 cycles when measured at 1C. In contrast, the nanowires without the heat treatment exhibited no capacity after 20 cycles at 1C rate, although the capacity at the first cycles was about 300 mA h g−1. The in situ TEM observation of the lithiation of the carbon-coated Si nanowires indicated that uniform carbon coating on the Si nanowires prevented full lithiation while nonuniform and incomplete carbon coating allowed the Si nanowire to be fully lithiated, relieving the stress created by expanding Si nanowire.
 | ||
| Fig. 23 Carbon-coated Si nanowires prepared from the mixture of Sn(HMDS)2 and MPS in Sn/Si mole ratio of 1/22 after carbonization at 900 °C: (a) SEM, (b) TEM and (c) HRTEM images. Adapted with permission from ref. 76. Copyright 2013 American Chemical Society. | ||
(b) Lithium compounds
LICs with two configurations were constructed by using carbon-coated Li3V2(PO4)3 (LVP) as either anode or cathode by coupling with AC as the counter electrodes using 1 M LiPF6/(EC + DEC) electrolyte,77 because a monoclinic LVP can serve as both an anode (V3+/V2+) and as a cathode (V3+/V5+).78,79 In the cell I (carbon-coated LVP(anode)//AC), the principal reactions during charge/discharge are the insertion/de-insertion of Li+ at the carbon-coated LVP as the anode and the adsorption/desorption of PF6− at the AC as the cathode. In the cell II (AC//carbon-coated LVP (cathode)), however, the de-insertion/re-insertion of Li+ at the carbon-coated LVP as the cathode and the adsorption/desorption of Li+ at the AC anode. Charge/discharge curves at a current density of 100 mA g−1 exhibit the multiple plateaus, corresponding to the redox reactions on the carbon-coated LVP (Fig. 24a and d). The cycle performances measured at 100 mA g−1 (Fig. 24b and e) and rate performances at different C rates (Fig. 24 c and f) of the two cells were compared. Even though the principal electrochemical reactions at the electrodes are different between the two cells, their performance are very similar. The energy and power densities for the two cells were not so much different. The presence of the phosphate group increases the redox potential but decreases the electrical conductivity. The carbon coating improves the electrical conductivity of the LVP and facilitates the charge transfer between the LVP and the current collector, realizing high rate performance. | ||
| Fig. 24 Results of galvanostatic charge/discharge analysis of carbon-coated LVP (anode)//AC (cell I) and AC//carbon-coated LVP (cathode) (cell II) in 1 M LiPF6/(EC + DEC) electrolyte: (a and d) charge/discharge curve at 100 mA g−1 rate. (b and e) Cyclic performance at 100 mA g−1 rate, and (c and f) rate performance (1C = 131 mA h g−1). Adapted from ref. 77 with permission from Elsevier. | ||
Carbon-coated Li2MnSiO4 was synthesized by solid-state reaction of the mixture of LiOH, MnCO3 and SiO2 with adipic acid at 900 °C. Its LIC cell coupled with AC anode in 1 M LiPF6/(EC + DMC) delivered the capacitance of 43.2 F g−1 at 1 mA cm−2 and the energy density of 54 W h kg−1 at 0.15 kW kg−1 with the retention of about 85% after 1000 cycles.80 Carbon-coated Li2FeSiO4 was also synthesized as anode materials for LICs by the same procedure and exhibited almost the same LIC performances as the carbon-coated Li2MnSiO4.81 Adipic acid used in these studies is the carbon source to improve the electrical conductivity between the particles of active materials by carbon coating, thus achieving high-power density cathodes for hybrid super capacitors. Carbon coating of H2Ti12O25, which was prepared from Na2CO3 and TiO2 at a molar ratio of 1:3, was performed by mixing with beta cyclodextrin (4.5 wt%) as a carbon source and subsequent calcination at 800 °C for 2 h. The thickness of the carbon layer was determined to be 3.09 nm by TEM observation, and the carbon coating was effective to improve LIC performance, giving negligible capacitance decrease after 100 cycles and energy density of 38.8 W h kg−1 at a power density of 0.18 kW kg−1 in 1.5 M LiBF4 (EC + DMC).82 The carbon layer was supposed to not only enhance the electrical conductivity but also suppress the swollen phenomenon, which was caused by the reduction decomposition between H2Ti12O25 and the electrolyte solution, generating C2H4, CO, and CH4 gases.
(c) Reduced graphene oxides
Coating of N-doped porous carbon on rGO was performed by hydrothermal reaction of aqueous suspension of GO containing glucose and pyrrole at 180 °C, followed by KOH activation at 900 °C.83 The rGO was sandwiched by N-doped porous carbon on both sides of the rGO flakes and the resulting composite had a porous sheet-like structure. The composite had a high SBET of 2680 m2 g−1 and a Vtotal of 1.82 cm3 g−1, possible to adsorb sulfur up to 70 wt% for a cathode material of Li–S batteries. The hybrids with 60 and 70 wt% S showed the initial discharge capacity of 1236 and 1099 mA h g−1, with coulombic efficiencies of 94.5 and 89.2%, respectively (Fig. 25a). The relatively low capacity and coulombic efficiency of the latter hybrid (70 wt% S) is attributed to the shuttling of the sulfide outside the pores. Moreover, the excess sulfur content decreased the cycle stability of the electrode (Fig. 25b) and increased the diffusion resistance due to the hindered ion transfer, which was supported by the results of their impedance analysis. The former hybrid (60 wt% S) could tightly hold polysulfide particles to avoid their shuttling between electrodes (in other words, confining polysulfide particles), as well as to improve electrical conductivity of the cathode. | ||
| Fig. 25 Li–S batteries composed of carbon-coated rGO flakes: (a) charge/discharge curves and (b) rate performances for the cathodes with different S-contents. Adapted from ref. 83 with permission from the Royal Society of Chemistry. | ||
5. Concluding remarks
To understand the details on the constraint spaces formed in carbon materials, many problems remain unsolved, because most papers mentioned in the present review taught us just the experimental results: the procedures of material modifications, the improved functions of the resulting composites, etc. For further development of this research field, understanding the fundamentals of constraint spaces is crucial. For examples, what kind of constraint is effective, whether constraint is due to the limitation in size of the encapsulated materials, mechanical constraint due to unbalance in thermal expansion between the materials and matrix carbon walls, chemical interaction of the materials with edges and/or functional groups on carbon walls or others. In addition, we have to know how these constraints can be controlled and designed, how the constraint spaces intrinsically influence the functions of the materials, etc.Carbon nanotubes provide us well-defined spaces with various controllable parameters for matrix carbon, including their diameter and length, layer number of the wall, etc., in addition to the possibility for the nanotubes after confinement of the material to be directly observed by microscopic techniques. However, it has to be pointed out that the nanotube wall is limited to an almost perfect but highly-curved layer of carbon hexagons (we may say “graphene” layer, but strongly curved), in addition to the high cost and limited amount of carbon nanotubes. Zeolite-templated carbons are composed of ordered micropores with walls of defective graphene-like layers, but they are not commercialized yet and their synthesis procedure is complicated. Also the carbons prepared by using either mesoporous silica or block copolymer surfactants as the templates provide us ordered channels with mesopore-sized diameters, but the walls of these mesoporous carbons consist of amorphous carbon because carbon precursors for these templates are almost limited to phenolic resins. It is not certain whether only carbon coating of the materials can create constraint space or not, however, carbon coating techniques give us variable carbon walls on the target materials; dense carbon films with controlled thickness can be deposited on the materials by CVD, and porous carbon films can form the mixture of particles of the materials with various resins (carbon precursors), such as thermosetting and thermoplastic resins. Since carbon materials are endowed with the structural and textural diversities, as pointed out above, there are infinite combinations for synthesizing the composites using versatile porous carbons. Moreover, the electrical conductivity of carbon materials can widen their applications toward electronic devices and electrocatalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. M. acknowledges the financial support from JSPS KAKENHI grant number 18K04697.References
- M. Inagaki, F. Kang, M. Toyoda and H. Konno, Advanced Materials Science and Engineering of Carbon, Tsinghua University Press and Elsevier, 2014 Search PubMed.
- M. Inagaki and F. Kang, Materials Science and Engineering of Carbon: Fundamentals, Tsinghua University Press and Elsevier, 2nd edn, 2014 Search PubMed.
- Y. Yang, T. Le, F. Kang and M. Inagaki, Carbon, 2017, 111, 546–568 CrossRef CAS.
- M. Inagaki, H. Orikasa and T. Morishita, RSC Adv., 2011, 1, 1620–1640 RSC.
- M. Inagaki, M. Toyoda, Y. Soneda, S. Tsujimura and T. Morishita, Carbon, 2016, 107, 448–473 CrossRef CAS.
- B. W. Smith, M. Monthioux and D. E. Luzzi, Nature, 1998, 396, 323–324 CrossRef CAS.
- B. W. Smith and D. E. Luzzi, Chem. Phys. Lett., 2000, 321, 169–174 CrossRef CAS.
- H. Kataura, Y. Maniwa, M. Abe, A. Fujiwara, T. Kodama, K. Kikuchi, H. Imahori, Y. Misaki, S. Suzuki and Y. Achiba, Appl. Phys. A, 2002, 74, 349–354 CrossRef CAS.
- G. Ning, N. Kishi, H. Okimoto, M. Shiraishi, T. Sugai and H. Shinohara, J. Phys. Chem. C, 2007, 111, 14652–14657 CrossRef CAS.
- S. Okada, S. Saito and A. Oshiyama, Phys. Rev. Lett., 2001, 86, 3835–3838 CrossRef CAS PubMed.
- S. Kawasaki, Y. Matsuoka, T. Yokomae, Y. Nojima, F. Okino, H. Touhara and H. Kataura, Carbon, 2005, 43, 37–45 CrossRef CAS.
- M. Chorro, S. Rols, J. Cambedouzou, L. Alvarez, R. Almairac, J. L. Sauvajol, J. L. Hodeau, L. Marques, M. Mezouar and H. Kataura, Phys. Rev. B, 2006, 74, 205425 CrossRef.
- H. Kataura, Y. Maniwa, T. Kodama, K. Kikuchi, K. Hirahara, K. Suenaga, S. Iijima, S. Suzuki, Y. Achiba and W. Krätschmer, Synth. Met., 2001, 121, 1195–1196 CrossRef CAS.
- S. Bandow, M. Takizawa, K. Hirahara, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2001, 337, 48–54 CrossRef CAS.
- H. Muramatsu, T. Hayashi, Y. A. Kim, D. Shimamoto, M. Endo, V. Meunier, B. G. Sumpter, M. Terrones and M. S. Dresselhaus, Small, 2009, 5, 2678–2682 CrossRef CAS PubMed.
- E. Hernández, V. Meunier, B. W. Smith, R. Rurali, H. Terrones, M. Buongiorno Nardelli, M. Terrones, D. E. Luzzi and J. C. Charlier, Nano Lett., 2003, 3, 1037–1042 CrossRef.
- Y. A. Kim, H. Muramatsu, T. Hayashi, M. Endo, M. Terrones and M. S. Dresselhaus, Chem. Phys. Lett., 2004, 398, 87–92 CrossRef CAS.
- R. Pfeiffer, H. Kuzmany, C. Kramberger, C. Schaman, T. Pichler, H. Kataura, Y. Achiba, J. Kürti and V. Zólyomi, Phys. Rev. Lett., 2003, 90, 225501 CrossRef CAS PubMed.
- H. Muramatsu, D. Shimamoto, T. Hayashi, Y. A. Kim, M. Terrones, M. Endo and M. S. Dresselhaus, Adv. Mater., 2011, 23, 1761–1764 CrossRef CAS PubMed.
- M. Endo, T. Hayashi, H. Muramatsu, Y.-A. Kim, H. Terrones, M. Terrones and M. S. Dresselhaus, Nano Lett., 2004, 4, 1451–1454 CrossRef CAS.
- H. Muramatsu, T. Hayashi, K. Y. Ahm, M. Terrones and M. Endo, Chem. Phys. Lett., 2006, 432, 240–244 CrossRef CAS.
- K. Méténier, S. Bonnamy, F. Béguin, C. Journet, P. Bernier, M. Lamy de La Chapelle, O. Chauvet and S. Lefrant, Carbon, 2002, 40, 1765–1773 CrossRef.
- K. Hirahara, K. Suenaga, S. Bandow, H. Kato, T. Okazaki, H. Shinohara and S. Iijima, Phys. Rev. Lett., 2000, 85, 5384–5387 CrossRef CAS PubMed.
- L. Guan, K. Suenaga, Z. Shi, Z. Gu and S. Iijima, Phys. Rev. Lett., 2005, 94, 045502 CrossRef PubMed.
- S. Kawasaki, Y. Iwai and M. Hirose, Mater. Res. Bull., 2009, 44, 415–417 CrossRef CAS.
- S. Ahn, Y. Kim, Y. Nam, H. Yoo, J. Park, Y. Park, Z. Wang, Z. Shi and Z. Jin, Phys. Rev. B, 2009, 80, 165426 CrossRef.
- H. Song, Y. Ishii, A. Al-zubaidi, T. Sakai and S. Kawasaki, Phys. Chem. Chem. Phys., 2013, 15, 5767–5770 RSC.
- Y. Yoshida, Y. Ishii, N. Kato, C. Li and S. Kawasaki, J. Phys. Chem. C, 2016, 120, 20454–20461 CrossRef CAS.
- K. Urita, Y. Shiga, T. Fujimori, T. Iiyama, Y. Hattori, H. Kanoh, T. Ohba, H. Tanaka, M. Yudasaka, S. Iijima, I. Moriguchi, F. Okino, M. Endo and K. Kaneko, J. Am. Chem. Soc., 2011, 133, 10344–10347 CrossRef CAS PubMed.
- T. Fujimori, A. Morelos-Gómez, Z. Zhu, H. Muramatsu, R. Futamura, K. Urita, M. Terrones, T. Hayashi, M. Endo, S. Young Hong, Y. Chul Choi, D. Tománek and K. Kaneko, Nat. Commun., 2013, 4, 2162 CrossRef PubMed.
- H. Luo, S. Desgreniers, Y. K. Vohra and A. L. Ruoff, Phys. Rev. Lett., 1991, 67, 2998–3001 CrossRef CAS PubMed.
- T. Takenobu, T. Takano, M. Shiraishi, Y. Murakami, M. Ata, H. Kataura, Y. Achiba and Y. Iwasa, Nat. Mater., 2003, 2, 683–688 CrossRef CAS PubMed.
- Y. Ishii, K. Tashiro, K. Hosoe, A. Al-zubaidi and S. Kawasaki, Phys. Chem. Chem. Phys., 2016, 18, 10411–10418 RSC.
- C. Li, Y. Ishii, S. Inayama and S. Kawasaki, Nanotechnology, 2017, 28, 355401 CrossRef.
- K. Yanagi, Y. Miyata and H. Kataura, Adv. Mater., 2006, 18, 437–441 CrossRef CAS.
- S. Kawasaki, Y. Iwai and M. Hirose, Carbon, 2009, 47, 1081–1086 CrossRef CAS.
- L. Guan, Z. Shi, M. Li and Z. Gu, Carbon, 2005, 43, 2780–2785 CrossRef CAS.
- Y. Li, R. Hatakeyama, T. Kaneko and T. Okada, Jpn. J. Appl. Phys., 2006, 45, L428–L431 CrossRef CAS.
- Y. F. Li, R. Hatakeyama, T. Kaneko, T. Izumida, T. Okada and T. Kato, Nanotechnology, 2006, 17, 4143–4147 CrossRef CAS PubMed.
- H. Shiozawa, T. Pichler, A. Grüneis, R. Pfeiffer, H. Kuzmany, Z. Liu, K. Suenaga and H. Kataura, Adv. Mater., 2008, 20, 1443–1449 CrossRef CAS.
- L.-J. Li, A. N. Khlobystov, J. G. Wiltshire, G. A. D. Briggs and R. J. Nicholas, Nat. Mater., 2005, 4, 481–485 CrossRef CAS PubMed.
- M. S. P. Sansom and P. C. Biggin, Nature, 2001, 414, 157–159 CrossRef PubMed.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188–190 CrossRef CAS.
- K. Koga, G. T. Gao, H. Tanaka and X. C. Zeng, Nature, 2001, 412, 802–805 CrossRef CAS PubMed.
- Y. Maniwa, H. Kataura, M. Abe, S. Suzuki, Y. Achiba, H. Kira and K. Matsuda, J. Phys. Soc. Jpn., 2002, 71, 2863–2866 CrossRef CAS.
- H. Kyakuno, K. Matsuda, H. Yahiro, Y. Inami, T. Fukuoka, Y. Miyata, K. Yanagi, Y. Maniwa, H. Kataura, T. Saito, M. Yumura and S. Iijima, J. Chem. Phys., 2011, 134, 244501 CrossRef PubMed.
- T. Ohba, S.-i. Taira, K. Hata and H. Kanoh, J. Phys. Chem. Lett., 2013, 4, 1211–1215 CrossRef CAS PubMed.
- H. Itoi, S. Hayashi, H. Matsufusa and Y. Ohzawa, Chem. Commun., 2017, 53, 3201–3204 RSC.
- H. Itoi, S. Maki, T. Ninomiya, H. Hasegawa, H. Matsufusa, S. Hayashi, H. Iwata and Y. Ohzawa, Nanoscale, 2018, 10, 9760–9772 RSC.
- H. Itoi, Y. Yasue, K. Suda, S. Katoh, H. Hasegawa, S. Hayashi, M. Mitsuoka, H. Iwata and Y. Ohzawa, ACS Sustainable Chem. Eng., 2017, 5, 556–562 CrossRef CAS.
- H. Itoi, H. Hasegawa, H. Iwata and Y. Ohzawa, Sustainable Energy Fuels, 2018, 2, 558–565 RSC.
- T. Le, Y. Yang, L. Yu, Z.-h. Huang and F. Kang, Sci. Rep., 2016, 6, 37368 CrossRef CAS PubMed.
- P. Przygocki, Q. Abbas, P. Babuchowska and F. Béguin, Carbon, 2017, 125, 391–400 CrossRef CAS.
- G. Lota, K. Fic and E. Frackowiak, Electrochem. Commun., 2011, 13, 38–41 CrossRef CAS.
- T. Morishita, T. Hirabayashi, T. Okuni, N. Ota and M. Inagaki, J. Power Sources, 2006, 160, 638–644 CrossRef CAS.
- F. Sun, J. Gao, Y. Zhu, X. Pi, L. Wang, X. Liu and Y. Qin, Sci. Rep., 2017, 7, 40990 CrossRef CAS.
- Y. Xiao, M. Cao, L. Ren and C. Hu, Nanoscale, 2012, 4, 7469–7474 RSC.
- J. Hwang, C. Jo, M. G. Kim, J. Chun, E. Lim, S. Kim, S. Jeong, Y. Kim and J. Lee, ACS Nano, 2015, 9, 5299–5309 CrossRef CAS.
- D. Hoshi, T. Watanabe and T. Ohba, Tanso, 2017, 280, 198–202 CrossRef.
- H. Itoi, H. Nishihara, S. Kobayashi, S. Ittisanronnachai, T. Ishii, R. Berenguer, M. Ito, D. Matsumura and T. Kyotani, J. Phys. Chem. C, 2017, 121, 7892–7902 CrossRef.
- T. Kimata, S. Kato, T. Yamaki, S. Yamamoto, T. Kobayashi and T. Terai, Surf. Coat. Technol., 2016, 306, 123–126 CrossRef CAS.
- M. Inagaki, T. Tsumura, T. Kinumoto and M. Toyoda, Carbon, 2019, 141, 580–607 CrossRef CAS.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith, M. Jaroniec, G. Q. Lu and S. Z. Qiao, J. Am. Chem. Soc., 2011, 133, 20116–20119 CrossRef CAS PubMed.
- J. Balach, H. K. Singh, S. Gomoll, T. Jaumann, M. Klose, S. Oswald, M. Richter, J. Eckert and L. Giebeler, ACS Appl. Mater. Interfaces, 2016, 8, 14586–14595 CrossRef CAS PubMed.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H.-M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed.
- H. Zhang, Z. Zhao, Y.-N. Hou, Y. Tang, Y. Dong, S. Wang, X. Hu, Z. Zhang, X. Wang and J. Qiu, J. Mater. Chem. A, 2018, 6, 7133–7141 RSC.
- M. E. Casco, J. Silvestre-Albero, A. J. Ramírez-Cuesta, F. Rey, J. L. Jordá, A. Bansode, A. Urakawa, I. Peral, M. Martínez-Escandell, K. Kaneko and F. Rodríguez-Reinoso, Nat. Commun., 2015, 6, 6432 CrossRef CAS PubMed.
- M. Inagaki, Carbon, 2012, 50, 3247–3266 CrossRef CAS.
- S. Iwamura, H. Nishihara and T. Kyotani, J. Power Sources, 2013, 222, 400–409 CrossRef CAS.
- T. Kasukabe, H. Nishihara, S. Iwamura and T. Kyotani, J. Power Sources, 2016, 319, 99–103 CrossRef CAS.
- T. Kasukabe, H. Nishihara, K. Kimura, T. Matsumoto, H. Kobayashi, M. Okai and T. Kyotani, Sci. Rep., 2017, 7, 42734 CrossRef CAS PubMed.
- J. Ryu, T. Chen, T. Bok, G. Song, J. Ma, C. Hwang, L. Luo, H.-K. Song, J. Cho, C. Wang, S. Zhang and S. Park, Nat. Commun., 2018, 9, 2924 CrossRef.
- R. Yi, S. Chen, J. Song, M. L. Gordin, A. Manivannan and D. Wang, Adv. Funct. Mater., 2014, 24, 7433–7439 CrossRef CAS.
- M. Choi, J.-C. Kim and D.-W. Kim, Sci. Rep., 2018, 8, 960 CrossRef PubMed.
- S. K. Kim, H. Kim, H. Chang, B.-G. Cho, J. Huang, H. Yoo, H. Kim and H. D. Jang, Sci. Rep., 2016, 6, 33688 CrossRef CAS PubMed.
- T. D. Bogart, D. Oka, X. Lu, M. Gu, C. Wang and B. A. Korgel, ACS Nano, 2014, 8, 915–922 CrossRef CAS.
- R. Satish, V. Aravindan, W. C. Ling and S. Madhavi, J. Power Sources, 2015, 281, 310–317 CrossRef CAS.
- S.-C. Yin, H. Grondey, P. Strobel, H. Huang and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 326–327 CrossRef CAS PubMed.
- S. C. Yin, H. Grondey, P. Strobel, M. Anne and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 10402–10411 CrossRef CAS PubMed.
- K. Karthikeyan, V. Aravindan, S. B. Lee, I. C. Jang, H. H. Lim, G. J. Park, M. Yoshio and Y. S. Lee, J. Power Sources, 2010, 195, 3761–3764 CrossRef CAS.
- K. Karthikeyan, V. Aravindan, S. B. Lee, I. C. Jang, H. H. Lim, G. J. Park, M. Yoshio and Y. S. Lee, J. Alloys Compd., 2010, 504, 224–227 CrossRef CAS.
- J.-R. Yoon, E. Baek, H.-K. Kim, M. Pecht and S.-H. Lee, Carbon, 2016, 101, 9–15 CrossRef CAS.
- S. Niu, W. Lv, C. Zhang, F. Li, L. Tang, Y. He, B. Li, Q.-H. Yang and F. Kang, J. Mater. Chem. A, 2015, 3, 20218–20224 RSC.
| This journal is © The Royal Society of Chemistry 2019 |