DOI:
10.1039/C9RA04088A
(Paper)
RSC Adv., 2019,
9, 22817-22822
Vinylpyrroles: solid-state structures and aggregation-induced emission properties†
Received
30th May 2019
, Accepted 16th July 2019
First published on 23rd July 2019
Abstract
An aggregation-induced emission chromophore, vinylpyrrole, was prepared from a formylpyrrole derivative, Meldrum's acid, and 1,3-dimethylbarbituric acid. The optical properties of the chromophore both in the solution and solid states were investigated by UV-vis and fluorescence spectroscopy. Single crystal X-ray diffraction measurements revealed that the dimethylbarbituric acid adduct formed a J-aggregate in the solid and resulted in higher fluorescence quantum yield compared to the Meldrum's acid adduct. Emission enhancement was found to occur by the restriction of molecular rotation in the solid state.
Introduction
Organic luminescence compounds have attracted significant attention due to their importance in applications such as organic light-emitting diodes and fluorescence probes.1,2 In general, the aggregation or strong intermolecular interactions between luminophores causes a loss of the luminescence quantum yield due to energy migration and/or excimer formation, which is otherwise known as aggregation caused quenching (ACQ) or concentration quenching. As such, the majority of luminescence studies are performed in dilute solutions or solid matrices; this is usually accompanied by drawbacks such as difficulties in sealing the solution, or a low luminescence intensity for practical applications. In 2001, an aggregation-induced emission (AIE) dye that exhibits intense luminescence in the solid or aggregated states was reported.3–6 Since the first “AIEgen” was developed, a number of organic compounds, namely tri- and tetraarylethenes,7–13 cyanostilbene,14–16 and carborane,17–19 were found to exhibit AIE properties, and have been utilised in practical light emitting devices and sensors.10,20–23 In the isolated state, AIEgens lose their excitation energy due to intramolecular rotation of their phenyl rings. In contrast, in the aggregated state, the free rotation of these phenyl rings is restricted by intermolecular interactions, and so the phenyl rings can prevent strong interactions between the luminophores. This results in intense emissions in the solid and/or aggregated states. To date, numerous AIE molecules with diverse core structures have been developed, where the majority of quenching mechanisms in solution is dependent on the phenyl group, and investigations into the rotatory motion of aromatic heterocyclic groups are still very limited. For example, pyrrole, an aromatic heterocyclic compound, is found in various natural compounds, and plays important biological roles.24–27 In addition, recent publications reported pyrrole-containing AIE molecules,13,28–31 while in our group, we recently achieved blue to red luminescent bipyrroles through the incorporation of electron-deficient vinyl groups.32–34 The cyclic terminal moieties such as Meldrum's acid and 1,3-dimethylbarbituric acid at the vinyl terminal are advantageous for high fluorescence quantum yield. Thus, herein we report the preparation of vinylpyrroles 1 and 2 (Scheme 1) that exhibit AIE properties in a tetrahydrofuran (THF)/water solvent system. The solid state photophysical properties and crystal structures are also discussed.
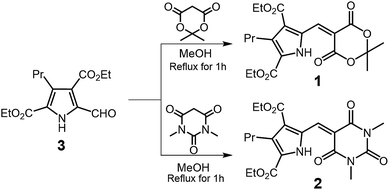 |
| | Scheme 1 Synthetic route to compounds 1 and 2. | |
Experimental section
Materials and instruments
All reagents and solvents were purchased from FUJIFILM Wako Pure Chemical Corporation or Tokyo Chemical Industry Co., Ltd. and used as received unless otherwise stated. Compounds 3 was synthesized according to the literature.35 UV-vis absorption spectra in solution were recorded on a JASCO V-570 spectrophotometer. Diffuse-reflectance spectra were measured by a JASCO V-550 spectrophotometer. Emission spectra were measured by a JASCO FP8500 or Hitachi F7000 spectrophotometer. 1H and 13C NMR spectra were obtained at 25 °C on a JEOL ECS-400 FT-NMR spectrometer with tetramethylsilane as an internal standard of the chemical shift. Infrared (IR) spectra were recorded on a JASCO FT/IR-410 spectrometer. Electrospray ionization time of flight mass (ESI-TOF-MS) spectrometry were carried out with JEOL JMS-T100CS spectrometer using methanol/dichloromethane as a solvent system. CHN elemental analysis was performed at Center for Instrumental Analysis of Kyushu Institute of Technology. Absolute photoluminescence quantum yields were determined by using a Hamamatsu Absolute PL Quantum Yield Spectrometer C11347 Quantaurus-QY (400–1100 nm) which equipped with an integrating sphere.
Synthesis
General procedure. In a round bottomed flask, 70 mg (0.25 mmol) of diethyl 5-formyl-3-propyl-2,4-pyrroledicarboxylate (3) and 2.5 mmol of Meldrum's acid or 1,3-dimethylbarbituric acid were dissolved in 10 mL of methanol. The solution was refluxed for 30 min. The reaction mixture was cooled to room temperature and 20 mL of water was added to the mixture. Resulting yellow precipitates were collected by filtration and washed with water. The product was purified by a neutral alumina.
Diethyl 5-[(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)methyl]-3-propyl-1H-pyrrole-2,4-dicarboxylate (1). Yield: 89 mg (88%). Elemental analysis found: C 59.07, H 6.14, N 3.56, calcd (C20H25NO8) C 58.96, H 6.19, N 3.44; 1H NMR (400 MHz, 298 K, CDCl3, ppm): δ = 13.49 (s, 1H, NH), 9.34 (s, 1H, vinyl-CH), 4.43 (q, J = 7.2 Hz, 4H, –CH2CH3), 3.10 (m, 2H, –CH2CH2CH3), 1.79 (s, 6H, >C(CH3)2), 1.58 (m, –CH2CH2CH3), 1.45 (t, J = 7.2 Hz, 6H, –CH2CH3), 0.98 (t, J = 7.4 Hz, 3H, –CH2CH2CH3); 13C NMR (100 MHz, 298 K, CDCl3, ppm) δ = 163.75, 162.82, 159.61, 143.35, 137.61, 128.56, 126.56, 124.99, 108.48, 105.03, 61.46, 61.18, 27.50, 27.18, 24.32, 14.32, 14.18, 14.15; IR (KBr) wavenumber cm−1: 2979 (νC–H), 2959 (νC–H), 2870 (νC–H), 1748 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1708 (νCO); ESI-TOF-MS (CH3OH/CH2Cl2): m/z = 408.17 ([M + H]+).
O), 1708 (νCO); ESI-TOF-MS (CH3OH/CH2Cl2): m/z = 408.17 ([M + H]+).
Diethyl 5-[(1,3-dimethyl-2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)methyl]-3-propyl-1H-pyrrole-2,4-dicarboxylate (2). Yield: 94 mg (90%). Elemental analysis found: C 56.98, H 5.89, N 9.86, calcd (C20H25N3O7) C 57.27, H 6.01, N 10.02; 1H NMR (400 MHz, 298 K, CDCl3, ppm): δ = 14.16 (s, 1H, NH), 9.41 (s, 1H, vinyl-CH), 4.44, (q, J = 7.2 Hz, 4H, –CH2CH3), 3.47, 3.43 (s, 6H, >NCH3), 3.09 (m, 2H, –CH2CH2CH3), 1.62 (m, 2H, –CH2CH2CH3), 1.47, 1.45 (t, J = 7.6 Hz, 6H, –CH2CH3), 0.98 (t, J = 7.4 Hz, 3H, –CH2CH2CH3), 13C NMR (100 MHz, 298 K, CDCl3, ppm) δ = 163.71, 163.22, 162.30, 160.21, 151.02, 143.02, 137.60, 129.33, 126.22, 124.84, 112.60, 61.43, 61.07, 29.19, 28.85, 27.32, 24.39, 14.26, 14.17; IR (KBr) wavenumber cm−1: 2961 (νC–H), 2933 (νC–H), 2871 (νC–H), 1723 (νCO), 1702 (νCO), 1671 (νCO), 1646 (νCO); ESI-TOF-MS (CH3OH/CH2Cl2): m/z = 420.20 ([M + H]+).
X-ray crystallography
X-ray diffraction data were collected using a Bruker SMART APEX CCD diffractometer (for 1) and Rigaku XtaLAB mini diffractometer (for 2) both equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) from a fine-focus sealed tube. Each single crystals of 1 or 2 was mounted on a glass fiber and the data frames were integrated using SAINT or CrysAlisPro.36,37 The integrated data were merged to give a unique data set for the structure determination. SADABS or CrysAlisPro were used for absorption correction.38 The structure was solved by a direct method and refined by the full-matrix least-squares method on all F2 data using the Olex2 suite of program.39 All non-hydrogen atoms were anisotropically refined. Hydrogen atoms were placed at geometrically idealized positions and constrained to ride on their parent atoms with Uiso(H) = 1.2Ueq(NH, CH and CH2) and Uiso(H) = 1.5Ueq(CH3). Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1911989 and 1911990.†
Computational study
Molecular structures and electronic transitions were evaluated by time-dependent density functional theory (TD-DFT) calculations. The initial coordinates of compounds 1 and 2 used for the TD-DFT calculations were obtained directly from the X-ray crystal structures. The TD-DFT calculations were performed using the Gaussian 09 suite.40 The camB3LYP/6-31G(d, p) was used as the basis set. The solvent effect was considered in THF by the polarisable continuum model using the integral equation formalism variant (IEFPCM).
Aggregation-induced emission behaviour in THF/water mixture
Stock solutions of 1 and 2 in THF with a concentration of 1.0 × 10−3 M were prepared. The aggregates were freshly prepared by adding water into the pyrrole solutions to make a total amount of 5 mL mixture. For example, the water fraction of 80% mixture including 1.0 × 10−4 M of a dye was prepared by 0.50 mL of the stock solution and diluted with 0.50 mL of THF followed by addition of 4.0 mL water. The fluorescence spectra were recorded by Hitachi F7000 spectrophotometer. Fluorescence and excitation slit widths were set to 5.0 nm and 10.0 nm, respectively. All spectra were recorded under 365 nm excitation wavelength.
Viscosity effects on the UV-vis and fluorescence spectra of 2
50 μL of a stock solution of 2 in CH2Cl2 with a concentration of 1.0 × 10−3 M was stored in glass tubes. After removal of the solvent, the residue was re-dissolved in 5 mL of methanol/glycerol solvents (100/0, 50/50, 40/60, 30/70, 20/80, 10/90 and 5/95, v/v). UV-vis spectra indicates there are no precipitates in all of the solutions (Fig. S1 and S2†).
Results and discussion
The target compounds were obtained from diethyl 5-formyl-3-propyl-2,4-pyrroledicarboxylate (3), Meldrum's acid, and 1,3-dimethylbarbituric acid in refluxing methanol (Scheme 1).41 The crude products were precipitated from the reaction mixtures by the addition of water. These precipitates were collected and washed with water to yield analytically pure products 1 and 2 in excellent yields (88 and 90%, respectively). Both compounds were characterised by standard spectroscopic methods and CHN elemental analysis. The 1H NMR spectra of the products recorded in CDCl3 suggested that intramolecular hydrogen bonding between the NH and carbonyl groups stabilised the planar structure of the molecules in solution. We note that the NH singlet peaks of 1 and 2 were observed between 13 and 15 ppm, which is significantly downfield shifted compared with the corresponding peaks for the starting aldehyde. Similar trends can be found in previously reported analogues.33,34
To obtain further structural insights, X-ray diffraction analysis was performed on compounds 1 and 2.‡ For this purpose, single crystals of 1 and 2 were obtained from dichloromethane/n-heptane and dichloromethane/n-hexane solutions with monoclinic C2/c and P21/c symmetries for 1 and 2, respectively. The obtained ORTEP diagrams together with the atom labelling and molecular packing diagrams are displayed in Fig. 1 and 2. Crystallographic details and selected molecular geometries are provided in the ESI.† It was found that in the solid state, compounds 1 and 2 adopted planar structures, as expected from the spectroscopic results. The torsion angles between C1–C5–C6–C7 were only 3.47° and 1.77° for 1 and 2, respectively. Indeed, it was clear that intramolecular hydrogen bonding between N1–H1⋯O1 (dN1⋯O1 = 2.696 Å for 1, and 2.613 Å for 2) gives rise to the planar conformation in the solid-state. The hydrogen bonding interactions are often found in the bipyrrole systems.33,34,42 Although the molecular structures were similar, the intermolecular interactions present in 1 and 2 were different. The shortest inter-layer distance in 1, which is defined as the distance between the mean plane of the pyrrole ring (i.e., N1, C1, C2, C3, and C4), was determined to be 3.50 Å. In addition, as shown in Fig. 2a, two molecules of 1 formed an antiparallel dimer-like aggregate. In contrast, compound 2 formed an infinite π–π stack along the b-axis, with an inter-planar distance of 3.46 Å (Fig. 2b). The slip angle of the π–π interactions was calculated to be 41.5°, which corresponds to the J-aggregate.43
 |
| | Fig. 1 Crystal structures of 1 and 2. The thermal ellipsoids were drawn at 30 and 50% for (a) 1 and (b) 2, respectively. | |
 |
| | Fig. 2 Crystal structures of 1 and 2. The thermal ellipsoids were drawn at 30 and 50% for (a) 1 and (b) 2, respectively. | |
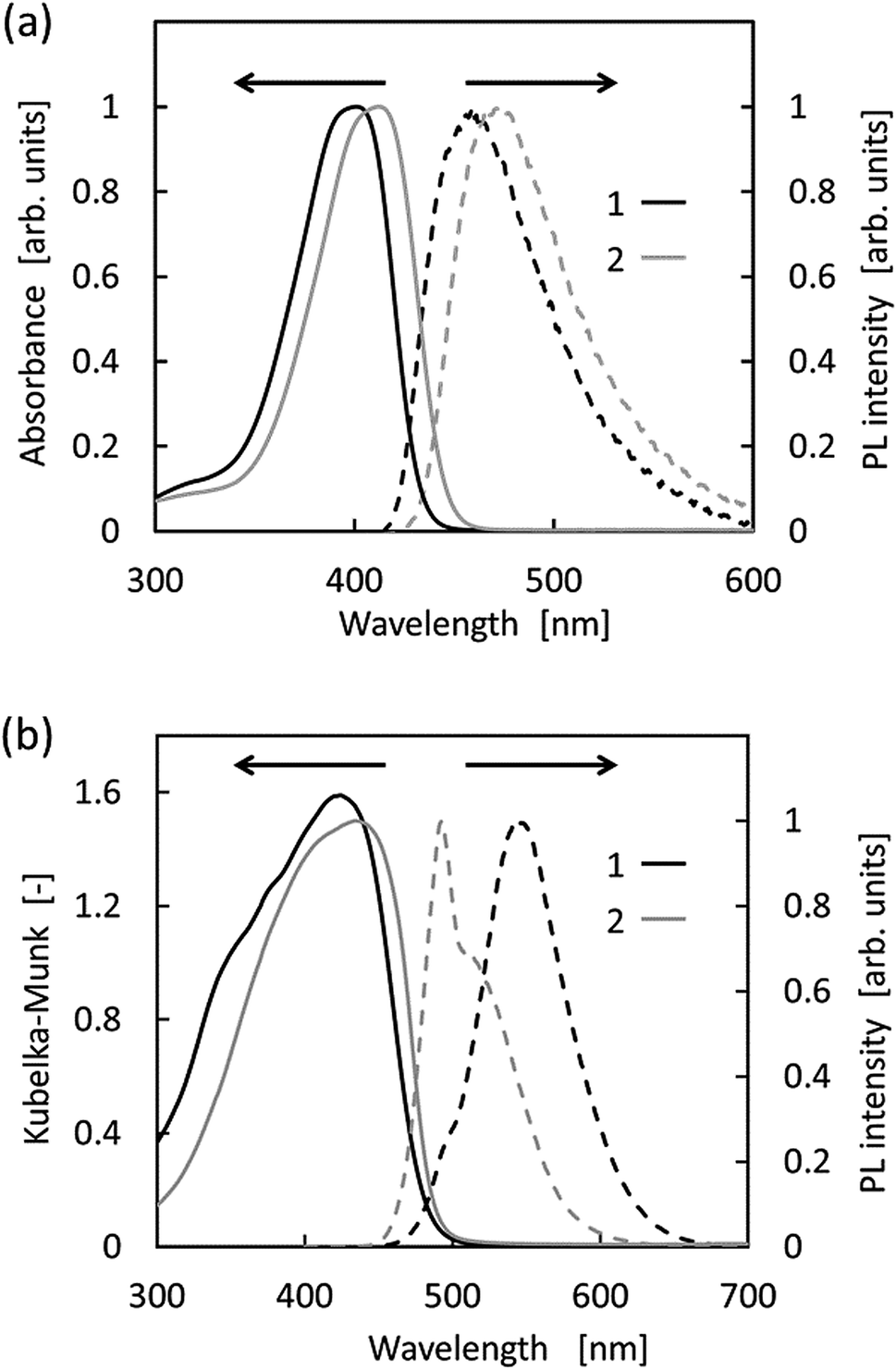
The optical properties of 1 and 2 recorded in both the solution-state (i.e., in THF) and the solid-state are shown in Fig. 3, and the relevant photophysical data are summarised in Table 1. As shown, the absorption bands of 1 and 2 in THF are located at ∼400 nm. TD-DFT calculations revealed that the absorption bands in THF were estimated to appear at 417.76 and 422.14 nm, for 1 and 2, respectively (Table 1). Both bands were derived from the HOMO–LUMO transitions. Intramolecular charge transfer could be ruled out since no obvious correlations were observed in the Mataga–Lippert plot (Fig. S3–S5†).17 Compounds 1 and 2 were virtually nonfluorescent in solution, i.e., the absolute quantum yields were <1%. In addition, compound 2 exhibited a relatively longer absorption and fluorescence due to the π-extension effect caused by the fully sp2-hybridised barbituric moiety.33,34 It is worth noting that the Stokes shift values of 1 and 2 were >3000 cm−1, which is notably larger than those of π-conjugated compounds. This was attributed to an excited-state feature of pyrrole derivatives, i.e., the single bond between the pyrrole ring and the adjacent π-system.44,45 In contrast to the solution properties, the solid-state fluorescence maxima of compounds 1 and 2 are located at a more red-shifted wavelengths in comparison to the emission obtained in the THF solutions (λFL,solid = 544 and 493 nm for 1 and 2, respectively). The large photoluminescence quantum yield of 2 can be explained by the formation of the J-aggregate, as described above. Indeed, it has been reported that this type of aggregation is advantageous for solid-state fluorophores.14,16 In addition, compound 1 exhibited a relatively large Stokes shift owing to weak intermolecular interactions in the solid-state.
 |
| | Fig. 3 Absorption (solid lines) and photoluminescence (PL) (broken lines) spectra of 1 (black) and 2 (grey) in (a) THF (i.e., the solution-state) and (b) the solid-state. | |
Table 1 Photophysical parameters for 1 and 2 under aerobic conditions
| |
|
1 |
2 |
| TD-DFT calculations were performed using the Gaussian 09 suite and the camB3LYP/6-31G(d, p) basis set. The solvent effect was considered in THF by the polarisable continuum model using the integral equation formalism variant (IEFPCM). |
| THF |
λAbs |
401 nm |
412 nm |
| λFL |
459 nm |
473 nm |
| Stokes shift |
3152 cm−1 |
3130 cm−1 |
| ΦFL |
<0.01 |
<0.01 |
| Calcd λAbsa |
417.76 nm |
422.14 nm |
| Transitiona |
108 (HOMO) |
111 (HOMO) |
| |
−109 (LUMO) |
−112 (LUMO) |
| Oscillator strengtha |
0.7541 |
0.9041 |
| Solid |
λAbs |
424 nm |
434 nm |
| λFL |
544 nm |
493 nm |
| Stokes shift |
5203 cm−1 |
4757 cm−1 |
| ΦFL |
0.10 |
0.24 |
As shown in Fig. 4 and 5, the vinylpyrroles exhibited a drastic increase in fluorescence intensity from the molecularly dissolved state in THF to the aggregated state in the THF-water mixtures. Indeed, compounds 1 and 2 exhibited negligible fluorescence in the THF solution, whereas the fluorescence intensity increased significantly with water fractions >80 vol%. Since 1 and 2 are insoluble in water, the increased water fraction leads to their precipitation. Furthermore, we note that the fluorescence spectra of the two compounds in the presence of 90 vol% water were similar to their solid-state PL spectra, although one shoulder peak had disappeared. Moreover, compound 1 showed a relatively large bathochromic shift upon aggregation compared with compound 2. These observations indicate the occurrence of AIE in these vinylpyrroles.
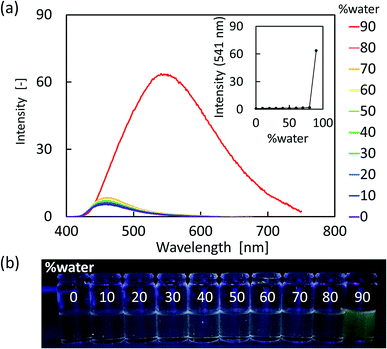 |
| | Fig. 4 (a) PL spectral changes of 1 (1 × 10−4 M) based on the water fraction present in the THF solution. (b) Photographic images of 1 in the THF-water mixtures. | |
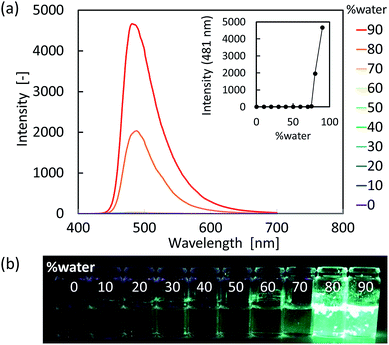 |
| | Fig. 5 (a) PL spectral changes of 2 (1 × 10−4 M) based on the water fraction present in the THF solution. (b) Photographic images of 2 in the THF-water mixtures. | |
We subsequently examined the effect of viscosity on the fluorescence of 2, and thus, the fluorescence intensity as a function of the glycerol (934 mPa s, 25 °C) content in methanol (0.544 mPa s, 25 °C) was plotted as shown in Fig. 6.46 As indicated, log10 I of the fluorescence spectrum increased linearly upon increasing the glycerol content from 50 to 95 vol%. Such fluorescence enhancement at higher glycerol concentrations likely results from the viscosity effect, as no baseline increase was observed for the absorption spectra, i.e., no aggregates and/or precipitates were formed (Fig. S1†). This result indicates that the AIE properties of the vinylpyrroles were predominantly caused by the restriction of intramolecular rotation (RIR) mechanism. We also note that the molecular structures calculated by the TD-DFT (Fig. S16†) were likely planar due to the presence of intramolecular NH⋯O hydrogen bonds. Meanwhile, this hydrogen bond dissociates in the excited state and allows free rotation along the single bond between the pyrrole moiety and the electron-deficient vinyl group.
 |
| | Fig. 6 Photoluminescence peak intensity of 2 vs. the composition of the glycerol-methanol mixtures (1 × 10−5 M). | |
Conclusions
We herein reported the successful syntheses of pyrrole derivatives that exhibited aggregation-induced emission (AIE) behaviour in THF-water mixtures. The prepared compounds (numbered 1 and 2) were virtually nonfluorescent in organic solvents, but were highly luminous in the solid-state. Compound 1 produced dimer-like aggregates in the crystal form while compound 2 gave J-type infinite aggregates. In addition, it was found that the intermolecular interactions dominating in each compound directly influenced their photophysical properties. More specifically, compound 2 exhibited relatively short yet intense photoluminescence due to strong intermolecular interactions and J-aggregation formation in the solid-state. These strong interactions are also likely to be advantageous in terms of decreasing the non-radiative relaxations of the excited state. Furthermore, studies into the effect of viscosity revealed that the aggregation-induced emission behaviour of the compounds can be explained by the restriction of intramolecular rotation mechanism. These results confirm that we successfully proposed a novel molecular design for AIE molecules based on the rotation of aromatic heterocyclic compounds that do not contain phenyl rings. We believe that this result will promote further exploration of AIE-active chromophores. Additional studies of AIE-active aromatic heterocyclic compounds are currently ongoing in our groups.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors appreciate Prof. Dr Yoshio Hisaeda and Prof. Dr Toshikazu Ono for their kind help in fluorescence measurements and the X-ray diffraction study. This work was supported by JSPS KAKENHI Grant Number 15K17848 and the International Collaborative Education and Research Project (No. 4401) from the Toyohashi University of Technology. T. O. is grateful for the Yoshida Foundation for the Promotion of Learning and Education for their financial support.
Notes and references
- Organic Light-Emitting Devices: Synthesis Properties and Applications, ed. K. Müllen and U. Scherf, Wiley-VCH, Weinheim, 2006 Search PubMed.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- J. L. Mullin, H. J. Tracy, J. R. Ford, S. R. Keenan and F. Fridman, J. Inorg. Organomet. Polym. Mater., 2007, 17, 201–213 CrossRef CAS.
- X.-L. Peng, S. Ruiz-Barragan, Z.-S. Li, Q.-S. Li and L. Blancafort, J. Mater. Chem. C, 2016, 4, 2802–2810 RSC.
- T. Sajima, T. Nagamura, T. Okawara and K. Takehara, Sci. Adv. Mater., 2015, 7, 981–984 CrossRef CAS.
- R. Hu, C. F. A. Gómez-Durán, J. W. Y. Lam, J. L. Belmonte-Vázquez, C. Deng, S. Chen, R. Ye, E. Peña-Cabrera, Y. Zhong, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 10099–10101 RSC.
- H. Shi, R. T. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- K. Garg, E. Ganapathi, P. Rajakannu and M. Ravikanth, Phys. Chem. Chem. Phys., 2015, 17, 19465–19473 RSC.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS.
- K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS.
- S.-Y. Kim, Y.-J. Cho, G. F. Jin, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2015, 17, 15679–15682 RSC.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed. Engl., 2016, 55, 7171–7175 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- M. Gao, S. Li, Y. Lin, Y. Geng, X. Ling, L. Wang, A. Qin and B. Z. Tang, ACS Sens., 2015, 1, 179–184 CrossRef.
- L. Zong, H. Zhang, Y. Li, Y. Gong, D. Li, J. Wang, Z. Wang, Y. Xie, M. Han, Q. Peng, X. Li, J. Dong, J. Qian, Q. Li and Z. Li, ACS Nano, 2018, 12, 9532–9540 CrossRef CAS PubMed.
- P. A. Gale, Acc. Chem. Res., 2006, 39, 465–475 CrossRef CAS PubMed.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- W. Zhou, Z. X. Jin and Y. J. Wan, Appl. Microbiol. Biotechnol., 2010, 88, 1269–1275 CrossRef CAS PubMed.
- R. Pérez-Tomás, B. Montaner, E. Llagostera and V. Soto-Cerrato, Biochem. Pharmacol., 2003, 66, 1447–1452 CrossRef.
- G. Liu, D. Chen, L. Kong, J. Shi, B. Tong, J. Zhi, X. Feng and Y. Dong, Chem. Commun., 2015, 51, 8555–8558 RSC.
- Y. Gao, G. Feng, T. Jiang, C. Goh, L. Ng, B. Liu, B. Li, L. Yang, J. Hua and H. Tian, Adv. Funct. Mater., 2015, 25, 2857–2866 CrossRef CAS.
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- X. Feng, B. Tong, J. Shen, J. Shi, T. Han, L. Chen, J. Zhi, P. Lu, Y. Ma and Y. Dong, J. Phys. Chem. B, 2010, 114, 16731–16736 CrossRef CAS PubMed.
- T. Okawara, A. Doi, T. Ono, M. Abe, K. Takehara, Y. Hisaeda and S. Matsushima, Tetrahedron Lett., 2015, 56, 1407–1410 CrossRef CAS.
- R. Kawano, T. Kato, R. Fukuda, T. Okawara, K. Takehara and T. Nagamura, ChemistrySelect, 2016, 1, 4144–4151 CrossRef CAS.
- T. Okawara, R. Kawano, H. Morita, A. Finkelstein, R. Toyofuku, K. Matsumoto, K. Takehara, T. Nagamura, S. Iwasa and S. Kumar, Molecules, 2017, 22, 1816 CrossRef PubMed.
- M. R. Johnson, D. C. Miller, K. Bush, J. J. Becker and J. A. Ibers, J. Org. Chem., 1992, 56, 4414–4417 CrossRef.
- SAINT v8.34A, Bruker AXS Inc., Madison, Wisconsin (US), 2013 Search PubMed.
- CrysAlisPro 1.171.39.34a, Rigaku Oxford Diffraction, Oxford Diffraction Ltd., Abingdon, Oxfordshire, England, 2017 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1997 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., 2009 Search PubMed.
- J. B. Paine and D. Dolphin, J. Org. Chem., 1988, 53, 2787–2795 CrossRef CAS.
- G. Anguera, B. Kauffmann, J. I. Borrell, S. Borros and D. Sanchez-Garcia, J. Org. Chem., 2017, 82, 6904–6912 CrossRef CAS PubMed.
- J. Zhou, W. Zhang, X.-F. Jiang, C. Wang, X. Zhou, B. Xu, L. Liu, Z. Xie and Y. Ma, J. Phys. Chem. Lett., 2018, 9, 596–600 CrossRef CAS PubMed.
- C.-M. Che, C.-W. Wan, W.-Z. Lin, W.-Y. Yu, Z.-Y. Zhou, W. Y. Lai and S. T. Lee, Chem. Commun., 2001, 721–722 RSC.
- D. Birnbaum and B. E. Kohler, J. Chem. Phys., 1991, 95, 4783–4789 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 85th edn, 2004 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopy, crystallographic data, absolute quantum yield, and initial coordinates for the TD-DFT calculations. CCDC 1911989 and 1911990. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04088a |
| ‡ Crystal data for 1: C20H25NO8; monoclinic, C2/c; a = 20.82(11) Å, b = 12.78(6) Å, c = 17.52(9) Å, β = 98.80(4)°, V = 4605(41) Å3, Z = 8, T = 296 K; μ(Mo Kα) = 0.091 mm−1; R1 = 0.077 (I > 2σ(I)); wR2 = 0.231; GOF = 1.05. Crystal data for 2: C20H25N3O7; monoclinic, P21/c; a = 17.799(2) Å, b = 5.2201(7) Å, c = 21.974(3) Å, β = 101.833(13)°, V = 1998.3(5) Å3, Z = 4, T = 123 K; μ(Mo Kα) = 0.106 mm−1; R1 = 0.089 (I > 2σ(I)); wR2 = 0.276; GOF = 1.11. CCDC 1911989 and 1911990 contain the supplementary crystallographic data for this communication. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yurina Matsufujib,
Kouhei Mizunoa,
Kenji Takeharaa,
Toshihiko Nagamura
*a,
Yurina Matsufujib,
Kouhei Mizunoa,
Kenji Takeharaa,
Toshihiko Nagamura