
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-metalated porous organic polymer for Suzuki–Miyaura cross-coupling reaction†
Ying Dong‡
*,
Jing-Jing Jv‡,
Yue Li,
Wen-Han Li,
Yun-Qi Chen,
Qian Sun,
Jian-Ping Ma and
Yu-Bin Dong*
and
Yu-Bin Dong*
College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Centre of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan 250014, P. R. China. E-mail: dongyinggreat@163.com; yubindong@sdnu.edu.cn
First published on 28th June 2019
Abstract
A new Ni(II)-α-diimine-based porous organic polymer, namely Ni(II)-α-diimine-POP, was constructed in high yield via the Sonogashira coupling reaction between the metallo-building block of Ni(II)-α-diimine and 1,3,5-triethynylbenzene. Besides the high thermal and chemical stability, the obtained Ni(II)-α-diimine-POP can be a highly active reusable heterogeneous catalyst to promote the Suzuki–Miyaura coupling reaction. The obtained results indicate that the Ni(II)-α-diimine-POP herein is a promising sustainable alternative to the Pd-based catalysts for catalysing the C–C formation in a heterogeneous way.
Introduction
Homogeneous metal catalysts play a leading role in C–C bond-forming reactions.1a Due to the increasing environmental issues and need for sustainable development, the metal-involving heterogeneous catalysts for C–C coupling such as in the Suzuki–Miyaura cross coupling reaction have drawn more and more attention during the past several decades. Among them, the precious palladium-based catalysts have been the protagonist.1b To date, various solid carriers such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs), carbon-based nanomaterials (CNMs), silica, and so on were employed to support precious Pd complex,2 Pd NP3 and Pd-bimetallic alloy4 for the fabrication of Pd heterogeneous catalysts. In contrast, the first row transition metals such as Ni are rarely used to fabricate the solid carrier-supported heterogeneous catalysts. Compared to Pd, Ni is more reactive, low-cost and earth-abundant. As a sustainable alternative to Pd, the Ni-based catalysts, especially solid carrier supported Ni metal catalysts, for the C–C cross-coupling reactions are extremely appealing.5The α-diimine-based metal catalysts have gained remarkable attention because of their easy synthesis, air stability, and high catalytic activity.6 For example, α-diimine Ni(II) complexes have been widely used in polymerization,7 dry CO2 reforming of methane,8 chemical bond activation,9 bimetallic catalysis,10 reductive cross-coupling reaction,11 and reductive amination.12
On the other hand, porous organic polymers (POPs), as a typical class of porous organic materials, have been broadly applied in the field of gas storage and separation,13 drug delivery,14 water treatment15 and heterogeneous catalysis.16 On account of their high surface area, controllable porosity and ability to be functionalized, both crystalline and amorphous POPs, such as covalent organic frameworks (COFs),17 hypercrosslinked polymers (HCP),18 porous aromatic frameworks (PAFs),19 polymers of intrinsic microporosity (PIMs),20 conjugated microporous polymers (CMP),21 are an promising class of carriers to upload active catalytic species. So far, the metal nanoparticle (M NP) loaded and metalated POPs by post-synthetic approach have been the main theme in the fabrication of POP-supported metal-solid catalysts.22 In principle, the POP-based and metal-involved catalytic materials could also be prepared by in situ one-pot assembly of metal-containing building blocks. By doing so, the POPs with high-density and evenly distributed metal catalytic sites would be generated. So far, the metalated POPs obtained in this way, however, are very rarely reported.23
In this contribution, we report, the first of its kind, a Ni(II)-POP which was generated from the metallo-building block of Ni(II) α-diimine and 1,3,5-triethynylbenzene via Sonogashira cross-coupling reaction under solvothermal conditions. The obtained Ni(II)-α-diimine-POP can be a highly active and reusable heterogeneous catalyst to promote Suzuki–Miyaura cross-coupling reactions.
Results and discussion
Structural and morphological characterization
Ni(II)-α-diimine-POP was successfully synthesized through a Sonogashira coupling reaction following the route outlined in Scheme 1. The diiodine-substituted ligand A was prepared as the bright yellow crystalline solids by double Schiff-base condensation between 4-iodo-2,6-diisopropylbenzenamine and butane-2,3-dione in good yield. The metallo-building block B was synthesized as the brick-red crystalline solids by metalation of A with DME(NiBr2) in moderate yield. Besides routine characterizations, the molecular structure of A was further determined by the X-ray single crystal analysis (CCDC 1906672, Fig. S1, Tables S1 and S2, ESI†). After combination of B and 1,3,5-triethynylbenzene, the Ni(II)-α-diimine-POP was generated through Sonogashira cross-coupling reaction under solvothermal conditions (toluene, 80 °C, 72 h, 86%). After the reaction, the resulting precipitate was treated by Soxhlet extraction with CH2Cl2, methanol and acetone to remove any possible residues. After dried in vacuo at 110 °C for 12 h, the target POP was obtained as deep brown solids (Scheme 1, inset). The obtained POP was the irregular granular particle which was well evidenced by the scanning electron microscopy (SEM, Scheme 1, inset).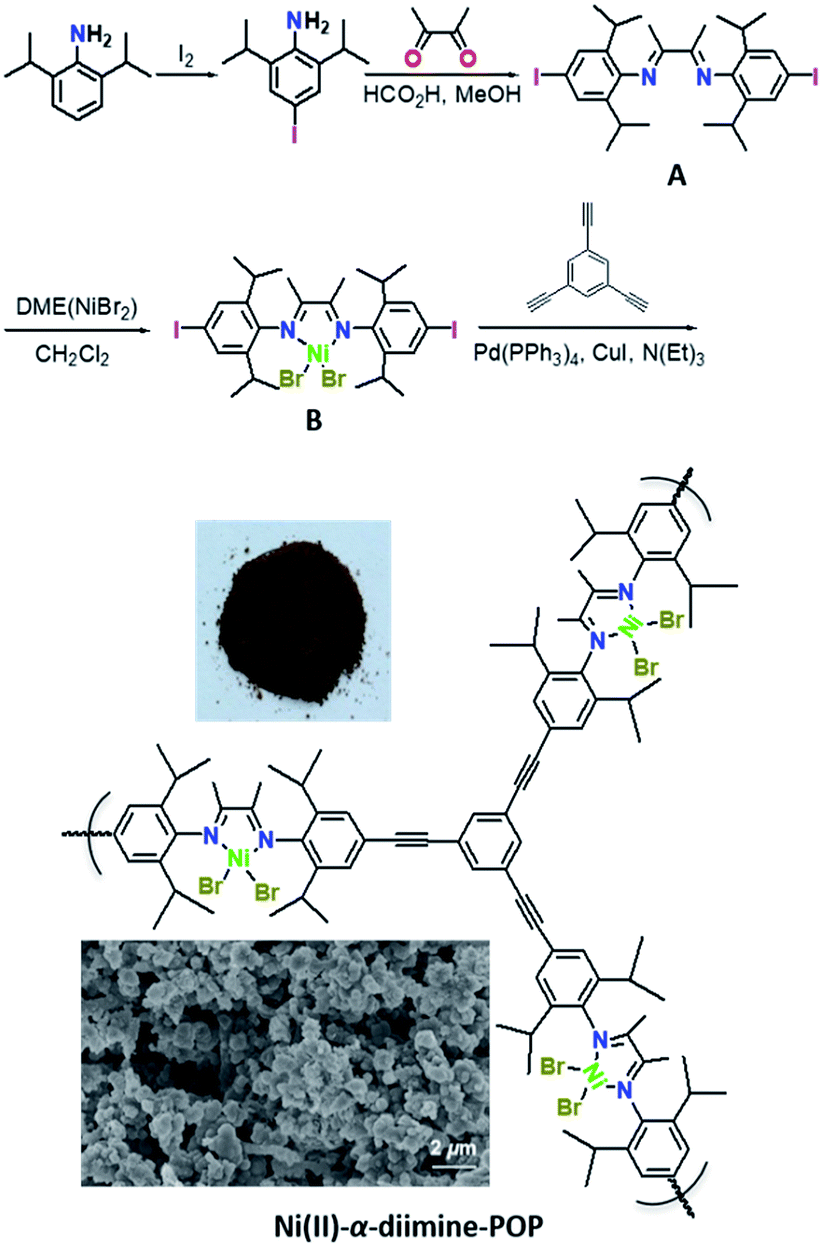 | ||
| Scheme 1 Synthesis of ligand A, metallo-building block B and Ni(II)-α-diimine-POP. The photograph and SEM image of Ni(II)-α-diimine-POP are inserted. | ||
IR spectra (Fig. 1a) showed that the characteristic peaks at 3278 cm−1 attributed to the νC–H (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H) in 1,3,5-triethynylbenzene basically disappeared in Ni(II)-α-diimine-POP after coupling reaction, meanwhile the band at 2109 cm−1 that corresponded to the –CC– in 1,3,5-triethynylbenzene moved to 2206 cm−1, indicating that the precursors of B and 1,3,5-triethynylbenzene in POP were successfully connected to each other via covalent C–C bond. Furthermore, the solid-state 13C NMR also supported this POP formation. As indicated in Fig. 1b, the peak for the aliphatic carbon atoms of –CH3 and –CH(CH3)2 groups was located at 19.03 ppm.24 The broad signals centred at 74.58 and 132.40 ppm were respectively ascribed to the bridging –CC– and phenyl moieties.25 The signal at 163.62 ppm was associated with carbon atom in the imine group.26
C–H) in 1,3,5-triethynylbenzene basically disappeared in Ni(II)-α-diimine-POP after coupling reaction, meanwhile the band at 2109 cm−1 that corresponded to the –CC– in 1,3,5-triethynylbenzene moved to 2206 cm−1, indicating that the precursors of B and 1,3,5-triethynylbenzene in POP were successfully connected to each other via covalent C–C bond. Furthermore, the solid-state 13C NMR also supported this POP formation. As indicated in Fig. 1b, the peak for the aliphatic carbon atoms of –CH3 and –CH(CH3)2 groups was located at 19.03 ppm.24 The broad signals centred at 74.58 and 132.40 ppm were respectively ascribed to the bridging –CC– and phenyl moieties.25 The signal at 163.62 ppm was associated with carbon atom in the imine group.26
 | ||
| Fig. 1 (a) IR spectra of Ni(II)-α-diimine-POP and its precursors. (b) Solid-state 13C NMR of Ni(II)-α-diimine-POP. | ||
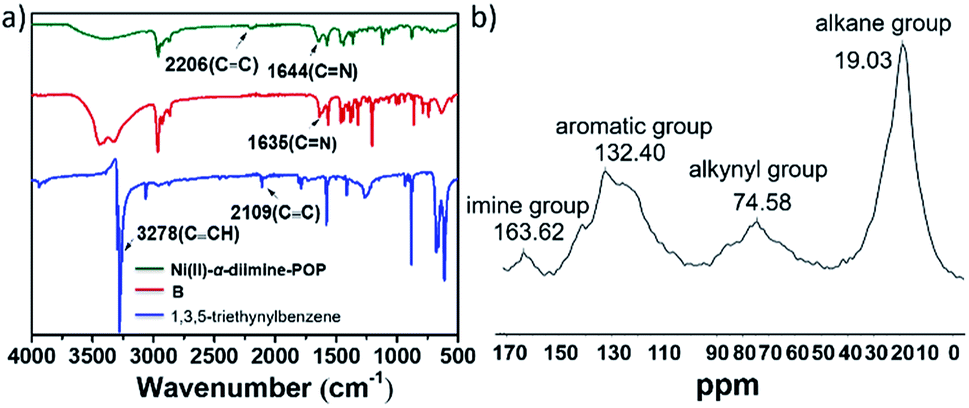
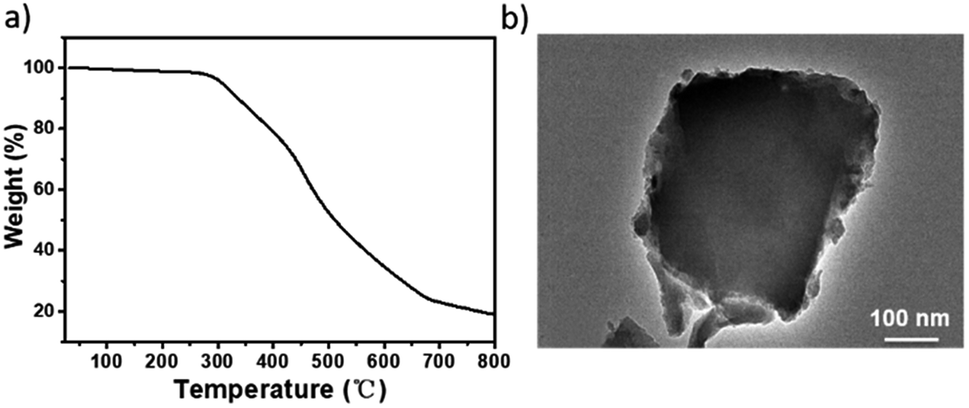
The TGA trace indicated that the obtained Ni(II)-α-diimine-POP remained intact till temperature over 250 °C, implying its good thermal stability (Fig. 2a). Notably, no Ni NP was generated during POP formation process, which demonstrated by the high-resolution transmission electron microscopy (HRTEM, Fig. 2b). The existed Ni species valence was demonstrated by the X-ray photoelectron spectroscopy (XPS) measurement. As shown in Fig. 3a, the observation of 2p3/2 and 2p1/2 peaks at 855.97 and 873.28 eV confirmed that the nickel species in Ni(II)-α-diimine-POP existed as Ni(II).27 The other observed doublet was attributed to intensive shake-up satellites that always occurs for Ni(II) during acquiring XPS.27 On the other hand, the single N 1s peak in B (399.03 eV) and Ni(II)-α-diimine-POP (398.96 eV) shifted to higher binding energy compared to that of A (398.88 eV), suggesting that Ni(II) species in B and Ni(II)-α-diimine-POP was chelated by N donors (Table 1).28 Inductively coupled plasma (ICP) analysis showed that the nickel content in Ni(II)-α-diimine-POP was up to 7.6 wt% (calcd 8.3 wt%).
 | ||
| Fig. 2 (a) TGA trace of Ni(II)-α-diimine-POP. (b) HRTEM image of Ni(II)-α-diimine-POP. | ||
 | ||
| Fig. 3 (a) XPS spectrum of Ni species in Ni(II)-α-diimine-POP. (b) SEM-EDX spectrum of Ni(II)-α-diimine-POP and it after five catalytic runs. (c) N2 sorption isotherm of Ni(II)-α-diimine-POP. | ||
| Compound | N 1s1/2 (eV) | Ni 2p3/2 (eV) | Ni 2p1/2 (eV) |
|---|---|---|---|
| A | 398.88 | — | — |
| NiBr2 | — | 855.40 | 872.98 |
| B | 399.03 | 856.28 | 872.79 |
| Ni(II)-α-diimine-POP | 398.96 | 855.97 | 873.28 |
| Ni(II)-α-diimine-POP after 5 runs | 398.92 | 856.16 | 873.68 |
As mentioned above, the Ni(II)-α-diimine-POP herein was prepared by metallo-building block via in situ one-pot approach, so it should feature the uniform texture. As shown in Fig. 3b, the C, N, Ni and Br species evenly distributed in the POP matrix, as evidenced by the SEM-energy dispersive X-ray (EDX) mapping.
The crystalline nature of the Ni(II)-α-diimine-POP was characterized by PXRD measurement (Fig. S2, ESI†). A observed broad peak from 20 to 25° suggested its amorphous nature. The N2 sorption analysis at 77 K was used to measure its specific surface area with architectural rigidity and permanent porosity (Fig. 3c). Brunauer–Emmett–Teller (BET) analysis of Ni(II)-α-diimine-POP indicated its surface area was up to 265.3 m2 g−1. Also, the pore size of the POP was determined based on the N2 sorption isotherm by employing the nonlocal density functional theory (NLDFT) method, and a series of sharp and broad peaks were observed. It was found to be mostly mesoporous having the major contribution of the pore width at ca. 2.7 nm (Fig. S2, ESI†).
Catalytic properties
Next, we examined the catalytic activity of Ni(II)-α-diimine-POP for the Suzuki–Miyaura cross coupling reactions, in which the iodobenzene and phenylboronic acid coupling was chosen as the model reaction (Fig. S3, ESI†). As shown in Table 2, the solvent screening revealed that toluene is the best one among the other solvents such as N,N-dimethylformamide (DMF), iso-propyl alcohol (IPA), dioxane, acetone, tetrahydrofuran (THF), and CH3CN that we tested (Table 2, entries 1 to 7). In addition, various bases, including K3PO4·3H2O, K2CO3, triethylamine (TEA), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), pyridine, and piperidine, were used to performed the reaction, and we found that K3PO4·3H2O was better than the other inorganic and organic bases (Table 2, entries 8–13). Furthermore, when the reaction was carried out with less catalyst loading, 2 (Table 2, entry 14), 3 (Table 2, entry 15) or 4 mol% (Table 2, entry 16) instead of 5 mol%, the coupled product was obtained in significantly lower 45–89% yields. On the other hand, the reaction temperature appeared to be crucial to the catalytic efficiency. As indicated in Table 2, the catalytic activity of Ni(II)-α-diimine-POP was significantly diminished at lower temperature (from r.t. to 80 °C, Table 2, entries 17–19). Also, shortening reaction time of 4–6 h would also lead to the significantly reduced 58–79% yields under the given reaction conditions (Table 2, entries 20–21). We did not observe any coupled product formation in the absence of the nickel source (Table 2, entry 22), further confirming the loaded Ni served as the catalytic active sites.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (Ni mol% equiv.) | Base | Solvent | T (°C) | T (h) | Yieldb (%) |
| a Reaction conditions: iodobenzene (1.0 mmol), phenylboronic acid (1.1 mmol), base (2.0 mmol), solvent (2 mL), in N2.b Yields were determined by GC analysis (Fig. S4, ESI). | ||||||
| 1 | 5 | K3PO4·3H2O | DMF | 100 | 8 | 41 |
| 2 | 5 | K3PO4·3H2O | IPA | 100 | 8 | 70 |
| 3 | 5 | K3PO4·3H2O | Dioxane | 100 | 8 | 67 |
| 4 | 5 | K3PO4·3H2O | Acetone | 100 | 8 | 27 |
| 5 | 5 | K3PO4·3H2O | Toluene | 100 | 8 | 99 |
| 6 | 5 | K3PO4·3H2O | THF | 100 | 8 | 23 |
| 7 | 5 | K3PO4·3H2O | CH3CN | 100 | 8 | 8 |
| 8 | 5 | K3PO4 | Toluene | 100 | 8 | 94 |
| 9 | 5 | K2CO3 | Toluene | 100 | 8 | 85 |
| 10 | 5 | TEA | Toluene | 100 | 8 | 38 |
| 11 | 5 | DBU | Toluene | 100 | 8 | 12 |
| 12 | 5 | Pyridine | Toluene | 100 | 8 | 0 |
| 13 | 5 | Piperidine | Toluene | 100 | 8 | 0 |
| 14 | 2 | K3PO4·3H2O | Toluene | 100 | 8 | 45 |
| 15 | 3 | K3PO4·3H2O | Toluene | 100 | 8 | 68 |
| 16 | 4 | K3PO4·3H2O | Toluene | 100 | 8 | 89 |
| 17 | 5 | K3PO4·3H2O | Toluene | r.t. | 8 | 12 |
| 18 | 5 | K3PO4·3H2O | Toluene | 50 | 8 | 53 |
| 19 | 5 | K3PO4·3H2O | Toluene | 80 | 8 | 85 |
| 20 | 5 | K3PO4·3H2O | Toluene | 100 | 4 | 58 |
| 21 | 5 | K3PO4·3H2O | Toluene | 100 | 6 | 79 |
| 22 | — | K3PO4·3H2O | Toluene | 100 | 6 | 0 |

Based above result, the optimized reaction conditions for the model coupling reaction were determined as: iodobenzene, 1.0 mmol; phenylboronic acid, 1.1 mmol; Ni(II)-α-diimine-POP, 5 mol% Ni equiv.; K3PO4·3H2O, 2.0 mmol; temperature, 100 °C; reaction time, 8 h; toluene, 2 mL.
To verify the heterogeneous nature of this POP-based catalyst, the hot leaching test was conducted. As shown in Fig. 4a, no further reaction occurred after ignition of the reaction at 4.0 h when it reached the yield of ca. 65%, at which point the heterogeneous particles were centrifuged out of the reaction mixture, and the reaction was performed for additional 4 h. GC analysis indicated that the yield of the reaction did not change during the prolonged reaction time, indicating that the Ni(II)-α-diimine-POP exhibited a typical heterogeneous catalyst nature herein.
 | ||
| Fig. 4 (a) Reaction time examination and leaching test for the model Suzuki cross-coupling reaction. (b) Catalytic cycle for the model Suzuki cross-coupling reaction. | ||
Once we confirmed the heterogeneous nature of the catalyst, we also tested its recyclability. After each catalytic run, the solid catalyst was separated by centrifugation, washed with toluene (3 × 2 mL), CH3CN (3 × 2 mL), and dried at 110 °C for 12 h in vacuum. The recycling catalytic runs were conducted by combining the recovered catalyst with inorganic base, iodobenzene, and phenylboronic acid in toluene. As shown in Fig. 4b, the solid catalyst of Ni(II)-α-diimine-POP still showed excellent activity and the cross-coupling yield was even up to 90% after five catalytic cycles (Fig. S5, ESI†). After multiple catalytic cycles, ICP measurement demonstrated that the Ni content in Ni(II)-α-diimine-POP was 6.3 wt%, suggesting a 17% Ni leaching occurred after five catalytic runs. This catalyst loss might be responsible for this slight yield decrease. On the other hand, no valence change for Ni species was observed (Table 1), implying that the Ni species in POP was stable during the reusable processes under the given conditions. In addition, the POP morphology and elemental distribution were well maintained after the recycle (Fig. 3b and S6†).
To test the available scope of this catalyst, we performed coupling reactions of phenylboronic acid with a series of substituted aryl halides under optimized conditions (Table 3). It is noteworthy that the catalytic system was tolerant to a wide range of functional groups such as –CF3, –NO2, –COCH3, –CH3, –OCH3, –CO2Me, and –CN at different substituted positions. Aryl bromides with both electron-donating and electron-withdrawing groups at para-, meta- or ortho-substituted position afforded cross-coupling products of i–viii (Table 3, entries 1–9) with good-to-excellent yields (90–98%). Compared to aryl bromides, the aryl iodides for the cross-coupling reaction were more active. As shown in Table 3 (entries 10–15), the yields for the target coupled products of i, iv, v and viii–x based on iodobenzene and para- or meta-substituted aryl iodides were more than 99%. Meanwhile, the ortho-substituted iodobenzene provided the products of xi and xii in slightly lower 90–92% yields (Table 3, entries 16 and 17). On the other hand, the crossing-coupling reactions between aryl iodide and phenylboronic acids with both electron- donating and electron-withdrawing groups at para-, meta- or ortho-substituted position still afforded excellent 98 to >99% yields (Table 3, entries 18–22, compounds iii, v, vii, xiii and xiv). The slightly lower 90% yield for iv from para-methoxyphenylboronic acid with aryl iodide was also observed (Table 3, entry 23). However, the chlorobenzene and phenylboronic acid coupling gave a low 26% yield of i in 12 h (Table 3, entry 24), indicating that the Ni(II)-α-diimine-POP cannot significantly activate the ArCl-based Suzuki–Miyaura cross-coupling reactions.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | X | R2 | t (h) | Product | Yieldb (%) |
| a Reaction conditions: aryl halide (1.0 mmol), arylboronic acid (1.1 mmol), K3PO4·3H2O (2 mmol, 0.533 g), Ni(II)-α-diimine-POP (5 mol% Ni equiv.), toluene (2 mL), 100 °C, N2.b Yields were determined by GC analysis (Fig. S7, ESI). | ||||||
| 1 | H | Br | H | 12 | i | 94 |
| 2 | 4-NO2 | Br | H | 12 | ii | 97 |
| 3 | 4-COCH3 | Br | H | 12 | iii | 98 |
| 4 | 4-OCH3 | Br | H | 12 | iv | 92 |
| 5 | 4-CO2CH3 | Br | H | 12 | v | 90 |
| 6 | 2-CN | Br | H | 12 | vi | 90 |
| 7 | 2-CH3 | Br | H | 12 | vii | 93 |
| 8 | 3-NO2 | Br | H | 12 | viii | 92 |
| 9 | H | Br | 4-OCH3 | 12 | iv | 90 |
| 10 | H | I | H | 8 | i | 99 |
| 11 | 3-CH3 | I | H | 8 | ix | >99 |
| 12 | 3-NO2 | I | H | 8 | viii | >99 |
| 13 | 4-OCH3 | I | H | 8 | iv | >99 |
| 14 | 4-CN | I | H | 8 | x | >99 |
| 15 | 4-CO2CH3 | I | H | 8 | v | >99 |
| 16 | 2-OCH3 | I | H | 8 | xi | 92 |
| 17 | 2-CF3 | I | H | 8 | xii | 90 |
| 18 | 3-NO2 | I | 4-F | 8 | xiii | 99 |
| 19 | 4-CN | I | 3-OCH3 | 8 | xiv | >99 |
| 20 | H | I | 2-CH3 | 8 | vii | >99 |
| 21 | H | I | 4-COCH3 | 8 | iii | 98 |
| 22 | H | I | 4-CO2CH3 | 8 | v | 98 |
| 23 | H | I | 4-OCH3 | 8 | iv | 90 |
| 24 | H | Cl | H | 12 | i | 26 |
Compared to the reported Ni-loaded solid catalytic systems (Table 4), which usually contained coexistent such as PPh3 or other metal oxides, for the Suzuki–Miyaura coupling reaction, it exhibited an impressive catalytic performance. On the other hand, we believe the Ni-catalysed Suzuki–Miyaura cross-coupling reaction herein went through the same mechanism as the reported one,33–35 which should involve the initial oxidative addition of the aryl halide to a Ni(0) species, followed by trans-metalation and subsequent reductive elimination step to afford the expected coupling product, meanwhile the catalytically active nickel(0) species was regenerated (Fig. S8, ESI†).
| Cat. (mol%) | Conditions | Substrate/yield (%) | Run | Ref. |
|---|---|---|---|---|
| Ni/Cg (8) | PPh3/dioxane/THF, n-BuLi/KF/LiOH, reflux, 9 h | 4-BrPhMe/PhB(OH)2/87 | 2 | 33 |
| Ni-PVP/TiO2–ZrO2 (20) | MeOH/H2O, K2CO3, 60 °C, 8 h | PhI/PhB(OH)2/97 | 6 | 34 |
| UiO-66(L1)/NiCl2/PPh3 (3) | CH3CN, K2CO3, 65 °C, 12 h | PhBr/PhB(OH)2/71 | — | 35 |
| UiO-66(L2)/NiCl2/PPh3 (3) | CH3CN, K2CO3, 65 °C, 12 h | PhBr/PhB(OH)2/84 | — | 35 |
| UiO-66(L3)/NiCl2/PPh3 (3) | CH3CN, K2CO3, 65 °C, 12 h | PhBr/PhB(OH)2/90 | — | 35 |
| UiO-66(L3)/Ni(COD)2/PPh3 (3) | CH3CN, K2CO3, 65 °C, 12 h | PhBr/PhB(OH)2/96 | 7 | 35 |
| Ni(II)-α-diimine-POP (5%) | Toluene, K3PO4·3H2O, 100 °C, 8 h | PhI/PhB(OH)2/99 | 5 | This work |
| Ni(II)-α-diimine-POP (5%) | Toluene, K3PO4·3H2O, 100 °C, 12 h | PhBr/PhB(OH)2/94 | 5 | This work |
Experimental
Materials and measurements
All chemicals and solvents were at least of analytic grade and employed as received without further purification. The elemental analysis was conducted on a PerkinElmer Model 2400 analyzer. MS spectra were obtained by Bruker maxis ultra-high resolution-TOF MS system. NMR data were collected using an AM-400 spectrometer. 13C CP/MAS NMR experiments were performed on Agilent 600 DD2 spectrometer at a resonance frequency of 150.15 MHz. 13C NMR spectra were recorded on a spinning rate of 15 kHz with a 4 mm probe at room temperature. 13C CP/MAS experiments were performed with a delay time of 5 s and a contact time of 1 ms. Infrared spectra were obtained in the 400–4000 cm−1 range using a Bruker ALPHA FT-IR spectrometer. Powder X-ray diffraction (PXRD) measurements were performed at 293 K on a D8 ADVANCE diffractometer (Cu Kα, λ = 1.5406 Å). ICP analysis was performed on an IRIS InterpidII XSP and NU AttoM. XPS spectra were obtained from PHI Versaprobe II. Thermogravimetric analyses were carried out on a TA Instrument Q5 simultaneous TGA under flowing nitrogen at a heating rate of 10°C min−1. HRTEM (high resolution transmission electron microscopy) analysis was performed on a JEOL 2100 Electron Microscope at an operating voltage of 200 kV. The scanning electron microscopy (SEM) micrographs were recorded on a Gemini Zeiss Supra TM scanning electron microscope equipped with energy-dispersive X-ray detector (EDX). The elemental analysis was conducted on a PerkinElmer Model 2400 analyzer. The crystal data were obtained by Agilent SuperNova X-ray single crystal diffractometer.Synthesis of A29
A mixture of 2,6-diisopropylbenzenamine (2.54 g, 14.3 mmol) and I2 (4 g, 15.7 mmol) was charged in 50 mL round-bottom flask. Then, 10 mL cyclohexane and 4 mL saturated Na2CO3 solution added in succession. After stirred at room temperature for 12 h, the mixture was diluted with EtOAc (20 mL) and washed with saturated Na2S2O3 (3 × 40 mL). The combined organic layer was dried with anhydrous MgSO4. The crude product was purified by column chromatography (petroleum ether/EtOAc = 10/1) to give 4-iodo-2,6-diisopropylbenzenamine as a black liquid (3.90 g, 96%). 1H NMR (400 MHZ, CDCl3) δ: 7.37 (s, 2H), 3.82 (s, 2H), 2.94 (2m, 2H), 1.37 (m, J = 8 Hz, 12 H); 13C NMR (400 MHz, CDCl3), δ: 22.4 (4c), 28.0 (2c), 81.2, 131.8 (2c), 135.1 (2c), 140.2.; IR (KBr): 3486 (w), 3404 (w), 2961 (vs), 2870 (m), 1618 (s), 1570 (w), 1460 (s), 1438 (vs), 1384 (m), 1364 (m), 1350 (m), 1299 (w), 1250 (m), 1208 (m), 1125 (w), 1061 (w), 924 (w), 865 (m), 832 (w), 765 (w), 746 (w), 716 (w), 555 (w). HRMS (ESI-TOF) calcd for C12H19IN ([M + H]+), m/z 304.0517; found, m/z 304.0592.A mixture of 4-iodo-2,6-diisopropylbenzenamine (3.7 g, 14 mmol), butane-2,3-dione (0.603 mL, 7 mmol) and 0.5 mL formic acid in 15 mL methanol was charged in 50 mL Schlenk flask. After stirred at room temperature for 6 h, the precipitate was filtered and recrystallized from ethanol to afford A as a bright yellow solid (4.4 g, 81%). 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 2H), 2.63 (m, 2H), 2.07 (s, 3H), 1.18 (d, 12H); 13C NMR (400 MHz, CDCl3) δ 168.5 (2c), 145.8 (2c), 137.8 (2c), 137.8 (2c), 132.3 (4c), 88.3 (2c), 28.5 (2c), 22.8 (8c), 22.5 (4c), 16.7 (2c). IR(KBr): 3486(w), 3403(w), 2961(vs), 2870(m), 1648(s), 1570(w), 1460(s), 1438(s),1384(w), 1364(s), 1324(m), 1242(s), 1186(s), 1126(s), 1072(w), 937(m), 883(w), 864(w), 819(w), 792(m), 711(m), 638(w), 559(w), 539(w), 467(w). HRMS (ESI-TOF) calcd for C28H39I2N2 ([M + H]+), m/z 658.1191; found, m/z 658.1206.
Synthesis of B30,31
A mixture of A (1.1 mmol, 0.722 g) and DME(NiBr2)32 (1 mmol, 0.308 g) in 50 mL dry dichloromethane was charged in a 100 mL Schlenk flask. After stirred at room temperature for 2 day in N2, the reaction system was filtered though a pad of Celite. The resulting solids were further washed with anhydrous diethyl ether, and then dried in vacuo to afford complex B as brick-red solid (0.59 g, 68.0%). IR(KBr): 3439(s), 3326(s), 2965(s), 2929(m), 2868(w), 1636(w), 1567(m), 1462(m), 1441(m), 1408(w), 1382(m), 1364(w), 1346(w), 1323(m), 1306(w), 1256(w), 1240(w), 1207(s), 1168(w), 1153(w), 1069(w), 1006(w), 985(w), 940(w), 887(w), 863(m), 791(m), 742(w), 636(w), 554(w), 480(w). Anal. calcd: for C28H38Br2I2NiN2, C 37.92, H 4.61, N 3.17, Br 17.31. I 30.4%, Ni 6.6% (I and Ni wt% were determined by ICP measurement).Synthesis of Ni(II)-α-diimine-POP
A mixture of the compound B (0.5 mmol, 0.488 g), 1,3,5-triethynylbenzene (0.5 mmol, 75 mg), Pd(PPh3)4 (0.25 mmol, 30 mg), CuI (0.1 mmol, 20 mg) and triethylamine (25 mL) in 50 mL toluene was heated at 80 °C for 72 h in N2. After cooled to room temperature, the obtained crude product was completely washed with water (30 mL), ethanol (30 mL) and dichloromethane (30 mL) respectively. The resulted solids were further Soxhlet extracted with mixed solution of dichloromethane, methanol and acetone (50 mL: 50 mL: 50 mL) and then dried at 110 °C in vacuo to afford Ni(II)-α-diimine-POP as dark gray solids (0.31 g, 86.4%). IR(KBr): 3390(s), 3030(w), 2924(vs), 2852(s), 2041(w), 1705(s), 1595(m), 1518(w), 1492(m), 1463(s), 1377(m), 1225(m), 1085(m), 1015(m), 830(m), 749(w), 616(w). Anal. calcd: C 58.2, H 4.93, N 3.11, Br 19.62. I 5.67%, Ni 7.6% (I and Ni wt% were determined by ICP measurement).General procedure for the Suzuki–Miyaura cross-coupling reaction between iodobenzene and arylboronic acid
A mixture of iodobenzene (1.0 mmol, 116 μL), phenylboronic acid (1.1 mmol, 0.134 g), K3PO4·3H2O (2 mmol, 0.533 g) and Ni(II)-α-diimine-POP (52 mg) in 2 mL toluene was stirred at 100 °C for 8 or 12 h in N2 to afford the corresponding product. Yield was determined by the GC analysis.General procedure for the Suzuki–Miyaura cross-coupling reaction between bromobenzene and arylboronic acid
A mixture of bromobenzene (1.0 mmol, 104 μL), phenylboronic acid (1.1 mmol, 0.134 g), K3PO4·3H2O (2 mmol, 0.533 g) and Ni(II)-α-diimine-POP (52 mg) in 2 mL toluene was stirred at 100 °C for 8 h or 12 h in N2 to afford the corresponding product. Yield was determined by the GC analysis.General procedures for the recycle of Ni(II)-α-diimine-POP
After each catalytic run, the solid catalyst was recovered by centrifugation, washed with toluene (3 × 2 mL), acetonitrile (3 × 2 mL) and dried at 110 °C for 12 h in vacuum and then was reused for the next catalytic run under the same reaction conditions.Conclusions
In summary, we report herein a new Ni(II)-α-diimine decorated porous organic polymer Ni(II)-α-diimine-POP by assembly of metallo-building block and its polymerized partner via in situ one-pot approach. The resulting polymer is porous, solvent- and thermal-stable. More importantly, the obtained Ni(II)-α-diimine-POP can highly promote the Suzuki–Miyaura cross-coupling reaction in a heterogeneous way with excellent yields and a reasonable scope. The catalyst could be reused at least 5 times without significant loss of the catalytic activity (>90% yield). We expect the presented approach to be viable for the construction of many more new metalated POP-based heterogeneous catalytic materials for various organic transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21671122, 21475078, 21802091), Taishan scholar’s construction project.Notes and references
- (a) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2009 Search PubMed; (b) A. Biffis, P. Centomo, A. D. Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- (a) Y.-L. Wei, Y. Li, Y.-Q. Chen, Y. Dong, J.-J. Yao, X.-Y. Han and Y.-B. Dong, Inorg. Chem., 2018, 57, 4379–4386 CrossRef CAS PubMed; (b) N. Gogoi, U. Bora, G. Borah and P. K. Gogoi, Appl. Organomet. Chem., 2017, 31, e3686 CrossRef; (c) Y.-M. Wang, C.-F. Lu, F.-Y. Wang, G.-C. Yang, Z.-X. Chen and J.-Q. Nie, Mol. Catal., 2017, 437, 89–94 CrossRef CAS.
- (a) M. Azad, S. Rostamizadeh, F. Nouri, H. Estiri and Y. Fadakar, Mater. Lett., 2019, 236, 757–760 CrossRef CAS; (b) P. A. Albinana, J. E. Haskouri, M. D. Marcos, F. Estevan, P. Amoros, M. A. Ubeda and F. Perez-Pla, J. Catal., 2018, 367, 283–295 CrossRef; (c) P. Mondal, P. Bhanja, R. Khatunar, A. Bhaumik, D. Das and S. M. Islam, J. Colloid Interface Sci., 2017, 508, 378–386 CrossRef CAS PubMed; (d) G.-J. Chen, J.-S. Wang, F.-Z. Jin, M.-Y. Liu, C.-W. Zhao, Y.-A. Li and Y.-B. Dong, Inorg. Chem., 2016, 55, 3058–3064 CrossRef CAS PubMed; (e) Y.-A. Li, S. Yang, Q.-K. Liu, G.-J. Chen, J.-P. Ma and Y.-B. Dong, Chem. Commun., 2016, 52, 6517–6520 RSC; (f) H.-C. Ma, J.-L. Kan, G.-J. Chen, C.-X. Chen and Y.-B. Dong, Chem. Mater., 2017, 29, 6518–6524 CrossRef CAS.
- (a) M. Taheri, M. Ghiaci and A. Shchukarev, Appl. Organomet. Chem., 2018, 32, e4338–e4339 CrossRef; (b) S. S. Beigbaghlou, R. J. Kalbasi, K. Marjani and A. Habibi, Catal. Lett., 2018, 148, 2446–2458 CrossRef CAS; (c) D.-X. Han, Z.-G. Zhang, Z.-B. Bao, H.-B. Xing and Q.-L. Ren, Front. Chem. Sci. Eng., 2018, 12, 24–31 CrossRef CAS; (d) Ö. Metin, S. F. Ho, C. Alp, H. Can, M. N. Mankin, M. S. Gültekin, M.-F. Chi and S.-H. Sun, Nano Res., 2013, 6, 10–18 CrossRef; (e) G.-J. Chen, H.-C. Ma, W.-L. Xin, X.-B. Li, F.-Z. Jin, J.-S. Wang, M.-Y. Liu and Y.-B. Dong, Inorg. Chem., 2017, 56, 654–660 CrossRef CAS PubMed.
- (a) J. Zhou and G.-C. Fu, J. Am. Chem. Soc., 2004, 126, 1340–1341 CrossRef CAS PubMed; (b) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- C. Zhou and B. Maurice, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef PubMed.
- (a) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS; (b) I. Kim, J. S. Kim, B. H. Han and C. S. Ha, Macromol. Res., 2003, 11, 514–517 CrossRef CAS.
- Y. Wang, L. Yao, S.-H. Wang, D.-H. Mao and C.-W. Hu, Fuel Process. Technol., 2018, 169, 199–206 CrossRef CAS.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- S. De, J.-G. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC.
- Y.-Q. Li, Y.-H. Fan and Q.-F. Jia, Chin. J. Inorg. Chem., 2019, 39, 350–362 Search PubMed.
- (a) K. Murugesan, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2019, 58, 5064–5068 CrossRef CAS PubMed; (b) G. Hahn, P. Kunnas and N. d. J. R. Kempe, Nat. Catal., 2019, 2, 71–77 CrossRef CAS.
- (a) W. Wang, M. Zhou and D. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC; (b) Y. Yuan, F. Sun, L. Li, P. Cui and G. Zhu, Nat. Commun., 2014, 5, 4260 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS PubMed.
- (a) Q. Sun, Z. Dai, X. Meng, L. Wang and F. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS; (b) M. H. Alkordi, L. J. Weselinski, V. D'Elia, S. Barman, A. Cadiau, M. N. Hedhili, A. J. Cairns, G. AbdulHalim, J. Basset and M. Eddaoudi, J. Mater. Chem. A, 2016, 4, 7453–7460 RSC; (c) Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- (a) E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed; (b) A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- C. Pei, T. Ben and S. Qiu, Mater. Horiz., 2015, 2, 11–21 RSC.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- J. Jiang, F. Su, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak, A. I. Cooper and A. Trewin, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- (a) N. Enjamuri, S. Sarkar, B. M. Reddy and J. Mondal, Chem. Rev., 2018, 18, 1–12 Search PubMed; (b) Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC; (c) W. Sebati and S. S. Ray, Catalysts, 2018, 8, 492–519 CrossRef.
- (a) Z. Xie, C. Wang, K. E. de Krafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS PubMed; (b) J.-X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072–1075 CrossRef CAS PubMed.
- W.-L. Wang, A.-M. Zheng, P.-Q. Zhao, C.-G. Xia and F.-W. Li, ACS Catal., 2014, 4, 321–327 CrossRef CAS.
- (a) B. C. Ma, S. Ghasimi, K. Landfester and K. A. I. Zhang, J. Mater. Chem. B, 2016, 4, 5112–5118 RSC; (b) H. Zhou, Q.-Y. Zhang and X.-B. Lu, RSC Adv., 2016, 6, 44995–45000 RSC.
- J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316–16319 CrossRef CAS PubMed.
- R. A. Jagtap, C. P. Vinod and B. Punji, ACS Catal., 2019, 9, 431–441 CrossRef CAS.
- C.-H. Ke, W.-C. Shih, F.-T. Tsai, M.-L. Tsai, W.-M. Ching, H.-H. Hsieh and W.-F. Liaw, Inorg. Chem., 2018, 57, 14715–14726 CrossRef CAS PubMed.
- J. Kim and S. H. Hong, ACS Catal., 2017, 7, 3336–3343 CrossRef CAS.
- J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316–16319 CrossRef CAS PubMed.
- (a) J. L. Rhinechart, N. E. Mitchell and B. K. Long, ACS Catal., 2014, 4, 2501–2504 CrossRef; (b) L.-H. Guo, K.-B. Lian, W.-Y. Kong, S. Xu, G.-R. Jiang and S.-Y. Dai, Organometallics, 2018, 37, 2442–2449 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- B. H. Lipshutz, T. Butler and E. Swift, Org. Lett., 2008, 10, 697–700 CrossRef CAS PubMed.
- R. J. Kalbasi and N. Mosaddegh, Bull. Korean Chem. Soc., 2011, 32, 2584–2592 CrossRef.
- P. Elumalai, H. Mamlouk, Y. M. Wang, L. Feng, S. Yuan, H. C. Zhou and S. T. Madrahimov, ACS Appl. Mater. Interfaces, 2018, 10, 41431–41438 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: additional characterization of Ni(II)-α-diimine-POP, and product characterization. CCDC 1906672. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03679b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |