
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expanding the chemical diversity of an endophytic fungus Bulgaria inquinans, an ascomycete associated with mistletoe, through an OSMAC approach†
Ni P. Ariantari ab,
Georgios Daletos‡
a,
Attila Mándic,
Tibor Kurtánc,
Werner E. G. Müllerd,
Wenhan Line,
Elena Ancheeva*a and
Peter Proksch*a
ab,
Georgios Daletos‡
a,
Attila Mándic,
Tibor Kurtánc,
Werner E. G. Müllerd,
Wenhan Line,
Elena Ancheeva*a and
Peter Proksch*a
aInstitute of Pharmaceutical Biology and Biotechnology, Heinrich Heine University Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany. E-mail: Elena.Ancheeva@uni-duesseldorf.de; proksch@uni-duesseldorf.de; Fax: +49 211 81 11923; Tel: +49 211 81 14175 Tel: +49 211 81 14163
bDepartment of Pharmacy, Faculty of Mathematic and Natural Sciences, Udayana University, 80361 Bali, Indonesia
cDepartment of Organic Chemistry, University of Debrecen, P.O.B. 400, 4002 Debrecen, Hungary
dInstitute for Physiological Chemistry, University Medical Center of the Johannes Gutenberg University Mainz, Duesbergweg 6, 55128 Mainz, Germany
eState Key Laboratory of Natural and Biomimetic Drugs, Peking University, 100191 Beijing, China
First published on 13th August 2019
Abstract
An endophytic fungus Bulgaria inquinans (isolate MSp3-1), isolated from mistletoe (Viscum album), was subjected to fermentation on solid Czapek medium. Chromatographic workup of the crude EtOAc extract yielded five new natural products (1–5). Subsequent application of the “One Strain, MAny Compounds” (OSMAC) strategy on this strain by the addition of a mixture of salts (MgSO4, NaNO3 and NaCl) to solid Czapek medium induced the accumulation of nine additional new secondary metabolites (6–13, 16), with most of them (8, 10–12) not detectable in cultures lacking the salt mixture. The structures of the new compounds were established on the basis of the 1D/2D NMR and HRESIMS data. The TDDFT-ECD method was applied to determine the absolute configurations of the new compounds 1, 4 and 6 as well as of the previously reported bulgarialactone B (14), for which the absolute configuration was unknown so far. The modified Mosher's method was performed to assign the absolute configurations of 12 and 13. TDDFT-ECD analysis also allowed determining the absolute configuration of (+)-epicocconone, which had an enantiomeric absolute configuration in the tricyclic moiety compared to that of bulgarialactone B (14). All the isolated metabolites were evaluated for their cytotoxic activity. Compound 2 was found to possess strong cytotoxic activity against the murine lymphoma cell line L5178Y with an IC50 value of 1.8 μM, while the remaining metabolites were shown to be inactive.
Introduction
Fungi are well-known producers of novel drug leads, as exemplified by the fascinating discovery of beta-lactam antibiotics, cyclosporin A, caspofungin, lovastatin and fingolimod.1–3 In particular, endophytic fungi, which live asymptomatically within plant tissues, have been recognized for their capability to produce therapeutically interesting natural products.4 Remarkable examples include the antimycotic natural products cryptocandin5 and cryptocin,6 the insecticidal compound nodulisporic acid A,7 the mitochondrial toxin phomoxanthone A8,9 as well as the immunosuppressant diterpene pyrones subglutinols A and B,10 among others. Interestingly, isolation of the important anticancer agents, paclitaxel from Taxomyces andreanae, an endophyte of Taxus brevifolia,11 and camptothecin produced by Entrophospora infrequens, a fungus associated with Nothapodytes foetida,12 as well as the identification of lycopodine-type alkaloids recently detected in an UV-irradiated strain of Paraboeremia, a fungal endophyte of Lycopodium serratum var. longipetiolatum,13 highlights the special importance of endophytes as a reservoir of metabolites previously known only from the host plant.14,15Gene clusters involved in the biosynthesis of fungal secondary metabolites often remain silent under standard laboratory culture conditions, leading to a frequent rediscovery of known metabolites.16,17 To overcome this problem, several strategies for enhancing the biosynthetic potential of fungi can be applied. One of them is the OSMAC (One Strain, MAny Compounds) approach,18 a powerful experimental method used to enhance the chemical diversity of microorganisms using the selective modification of the fermentation parameters, such as the media type and composition, the physical parameters (pH value, temperature, aeration conditions), the addition of enzyme inducers/inhibitors and chemical elicitors.16–19 Even a difference in water quality in preparing the culture media, such as exchanging tap water for distilled water, was reported to influence the pattern of the main metabolites of Paraphaeosphaeria quadriseptata, due to the presence of traces of metal ions (Cu2+, Cd2+ and Cr3+) in tap water.20 Successful application of the OSMAC approach on the fungal endophyte, Dothideomycete sp. CRI7 by changing the medium type (PDB vs. Czapek malt medium) as well as by using different nutrient sources (potato for PDB medium and malt extract for Czapek malt medium) resulted in distinct secondary metabolite production of this fungus.21,22 Interestingly, a series of studies have reported the isolation of halogenated natural products from fungal cultures grown on media to which different halide salts had been added, thus highlighting the fungal capability to utilize different halogen sources when present in the media.23–26 In addition, supplementing media with trace elements, e.g. the addition of CuCl2 to cultures of Pestalotiopsis sp. Z233 or the addition of ZnSO4 to cultures of Aspergillus clavatus, induced the production of new sesquiterpenes possessing tyrosinase inhibitory activity,27 and the production of a new metabolite, clavatustide C,28 respectively.
As part of our ongoing studies aimed at influencing the biosynthetic capacity of endophytic fungi utilizing the OSMAC approach, we investigated the metabolic profiles of Bulgaria inquinans (isolate MSp3-1), an ascomycete fungus isolated from sprouts of common mistletoe (Viscum album). Previous chemical investigations of this fungus resulted in the isolation of azaphilone pigments, namely bulgarialactones A–D,29,30 of which bulgarialactones A and B exhibited antimicrobial, cytotoxic and nematocidal activities.29 Moreover, quinones containing a benzofluoranthenequinone nucleus, bulgarhodin and bulgarein31 as well as cytotoxic anthraquinone dimers, bulgareones A and B,32 were isolated from this fungus.
In the present study, we report the isolation and structure elucidation of 14 new natural products, including five new natural products (1–5) derived from B. inquinans cultured on solid Czapek medium and nine new compounds (6–13, 16) isolated from this fungus when grown on solid Czapek medium with the addition of a salt mixture (MgSO4, NaNO3 and NaCl). Furthermore, determination of the absolute configurations of the new compounds and of the known derivative, bulgarialactone B (14), is described herein. The cytotoxicity assay results of the isolated natural products are briefly discussed.
Results and discussion
Chemical investigation of B. inquinans grown on solid Czapek medium resulted in the isolation of five new natural products, namely butyrolactones (1–3, 5) and a new metabolite bearing a diol moiety (4) together with known compounds, such as the azaphilone pigment bulgarialactone B (14)29 as well as phenylbutyrolactone IIa (15)33,34 and xenofuranone B (17)35 (Fig. 1). After this fermentation, we studied the chemical profiles of the fungus when grown in the presence of different salts that had been added to solid Czapek medium. B. inquinans was cultured on solid Czapek medium following the addition of either NaCl, NaBr, NaI, NaNO3, or (NH4)2SO4 (3.5 g of each) or following the addition of salt mixtures: (a) MgSO4·7H2O, NaNO3 and NaCl (2.5 g each), (b) FeSO4·7H2O, NaNO3 and NaCl (2.5 g each) or (c) ZnSO4·7H2O, NaNO3 and NaCl (2.5 g each), as described in the Experimental section. B. inquinans failed to grow on media containing either (NH4)2SO4, or mixtures (b) and (c). No changes in the chromatographic profiles were observed following the addition of either NaCl, NaBr, NaI or NaNO3 to solid Czapek medium when compared to chromatograms of the fungus grown on the medium without salts. However, the presence of a mixture of MgSO4, NaNO3 and NaCl in the medium resulted in a significant change in the metabolite profile of B. inquinans, as indicated by HPLC-DAD analysis (Fig. S1, ESI†). Chromatographic workup of this fungal extract led to the isolation of nine new secondary metabolites (6–13, 16), including a butyrolactone derivative 6, two unusual 1,3-oxazine containing natural products (7 and 8), five new α-pyrones (9–13) and (−)-(S)-flavipesin B (16), together with the known compound bulgarialactone D (18)30 (Fig. 1). Compounds 9, 13 and 18 were subsequently also detected (albeit in minor amounts) in HPLC chromatograms of the fungus following cultivation on solid Czapek medium without the addition of salts. The OSMAC approach nevertheless enhanced their production, enabling isolation and structural characterization of these compounds. Moreover, the accumulation of compounds 8 and 10–12 was only induced in the presence of the salt mixture, whereas these latter compounds were not detected in fungal cultures lacking salts.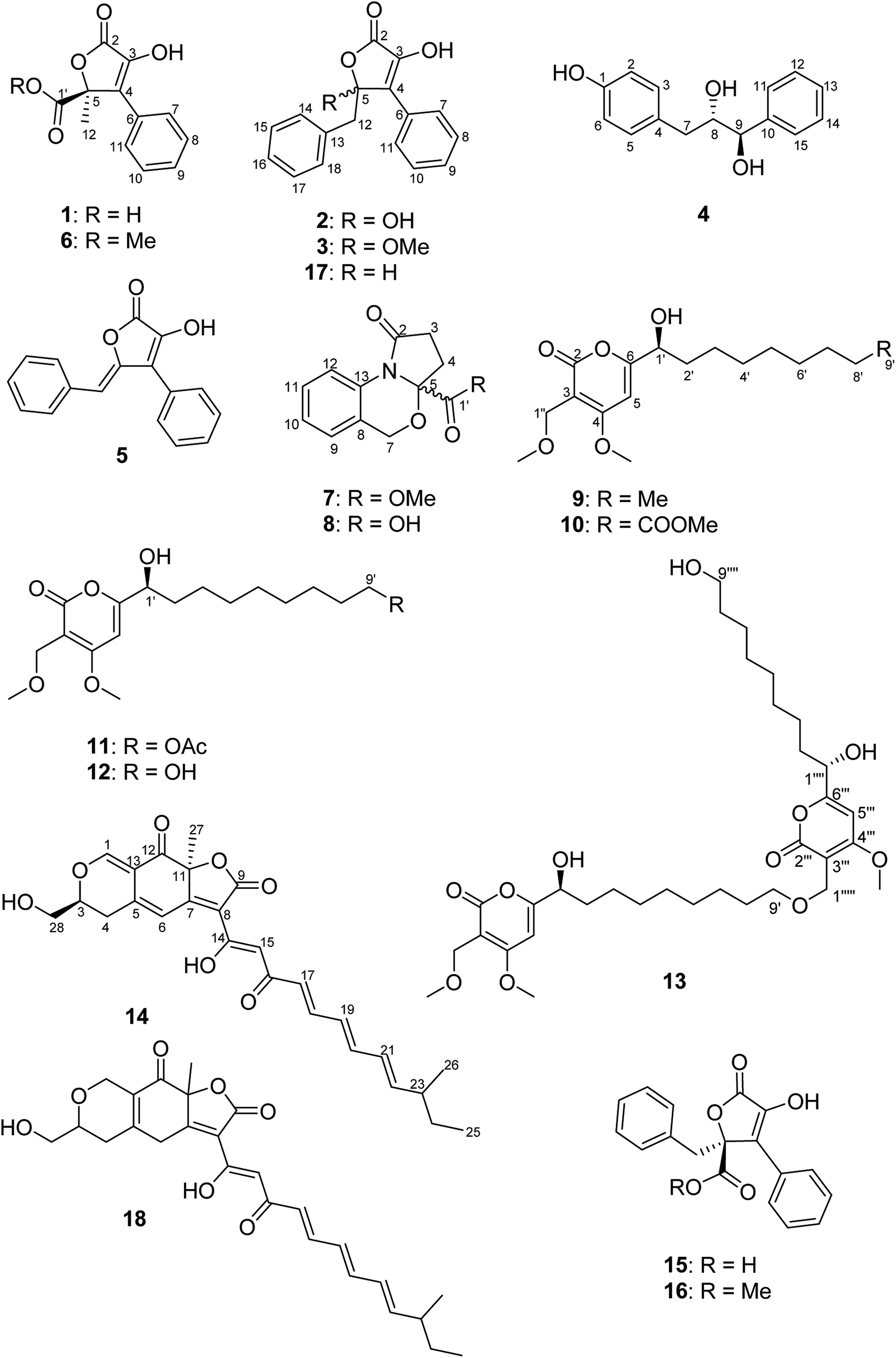 | ||
| Fig. 1 Structures of compounds 1–18 isolated from B. inquinans. | ||
Compound 1 was obtained as a yellow solid. Its UV spectrum displayed a maximum absorption at 288 nm, which was characteristic for butyrolactone-type metabolites.36 The HRESIMS spectrum exhibited a prominent pseudomolecular ion peak at m/z 253.0600 [M + H]+, attributed to the molecular formula C12H10O5, indicating 8 degrees of unsaturation. The 1H NMR data of 1 (Table 1) revealed signals of one methyl at δH 1.74 (H3-12) and five aromatic protons resonating at δH 7.74 (H-7/H-11), 7.41 (H-8/H-10) and 7.34 (H-9), implying the presence of a mono-substituted benzene ring. Moreover, the 13C NMR data of 1 (Table 1) displayed signals of a carbonyl carbon at δC 170.3 (C-2), two sp2 carbons at δC 140.6 (C-3) and 131.2 (C-4), and one oxygenated sp3 carbon at δC 85.2 (C-5), attributed to a conjugated five-membered lactone ring. The HMBC correlations observed from H-7/H-11 to C-4 and from H3-12 to C-4, C-5 and C-1′ (Fig. 2) provided the connections of the aromatic ring and of the methyl group to the butyrolactone ring as well as the attachment of the carboxylic acid group to C-5.
| Position | 1 | 2 | 3 | 5 | 6 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| a Recorded at 600 MHz (1H) and 150 MHz (13C). | ||||||||||
| 2 | 170.3, C | 168.9, C | 168.1, C | 166.6, C | 169.9, C | |||||
| 3 | 140.6, C | 141.4, C | 143.0, C | 141.4, C | 140.8, C | |||||
| 4 | 131.2, C | 127.3, C | 123.9, C | 126.5, C | 130.6, C | |||||
| 5 | 85.2, C | 107.4, C | 110.4, C | 148.1, C | 84.9, C | |||||
| 6 | 131.4, C | 132.3, C | 131.8, C | 130.5, C | 131.2, C | |||||
| 7/11 | 128.7, CH | 7.74, br d (7.5) | 129.4, CH | 8.00, br d (7.5) | 128.8, CH | 7.94, br d (7.4) | 130.4, CH | 7.57, br d (7.5) | 128.6, CH | 7.66, br d (7.3) |
| 8/10 | 129.6, CH | 7.41, br t (7.5) | 129.5, CH | 7.46, br t (7.5) | 129.8, CH | 7.48, br d (7.4) | 129.8, CH | 7.51, br t (7.5) | 129.8, CH | 7.42, br t (7.3) |
| 9 | 129.7, CH | 7.34, tt (7.5, 1.2) | 129.4, CH | 7.37, tt (7.5, 1.6) | 129.8, CH | 7.39, tt (7.4, 1.1) | 128.9, CH | 7.45, tt (7.5, 1.3) | 129.8, CH | 7.35, tt (7.3, 1.2) |
| 12 | 22.1, CH3 | 1.76, s | 44.9, CH2 | 3.39, d (13.8); 3.34, d (13.8) | 44.6, CH2 | 3.37, d (13.8); 3.34, d (13.8) | 109.6, CH | 5.97, s | 22.2, CH3 | 1.79, s |
| 13 | 135.7, C | 135.2, C | 135.3, C | |||||||
| 14/18 | 131.4, CH | 6.85, br d (6.9) | 131.5, CH | 6.85, br d (6.9) | 130.9, CH | 7.68, br d (7.4) | ||||
| 15/17 | 128.8, CH | 7.10, br t (6.9) | 128.8, CH | 7.10, br t (6.9) | 129.6, CH | 7.35, br t (7.4) | ||||
| 16 | 127.9, CH | 7.12, tt (6.9, 1.6) | 128.0, CH | 7.12, tt (6.9, 1.5) | 128.8, CH | 7.25, tt (7.4, 1.1) | ||||
| 5-OMe | 50.9, CH3 | 3.24, s | ||||||||
| 1′ | 172.4, C | 171.4, C | ||||||||
| 1′-OMe | 53.9, CH3 | 3.76, s | ||||||||
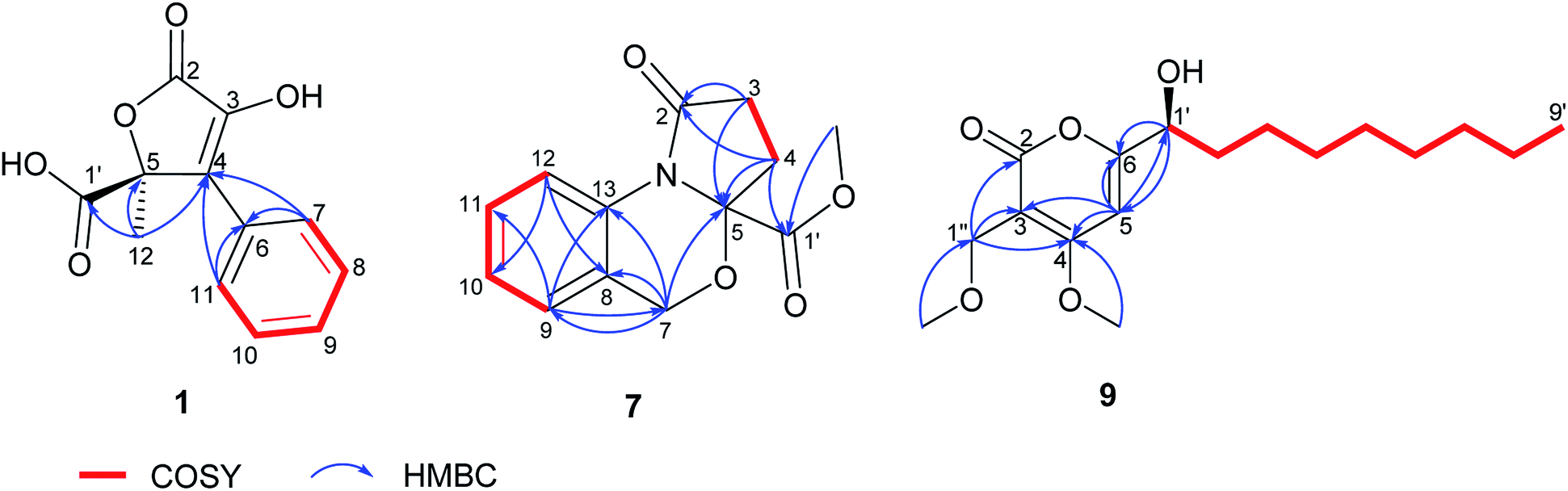 | ||
| Fig. 2 COSY and selected HMBC correlations of 1, 7 and 9. | ||
In order to elucidate the absolute configuration of 1, the TDDFT-ECD protocol was performed on the arbitrarily chosen (S) stereoisomer. B3LYP/6-31+G(d,p) and CAM-B3LYP/TZVP PCM/MeCN reoptimization of the initial 13 MMF conformers resulted in three and eight low-energy conformers over a 1% Boltzmann-population, respectively. ECD spectra computed at various levels for both sets of conformers effectively reproduced the experimental ECD spectrum of 1 (Fig. 3), allowing the elucidation of the absolute configuration as (S). Accordingly, the structure of 1 was established as a new natural product, for which the name bulgariline A is proposed (Fig. 1).
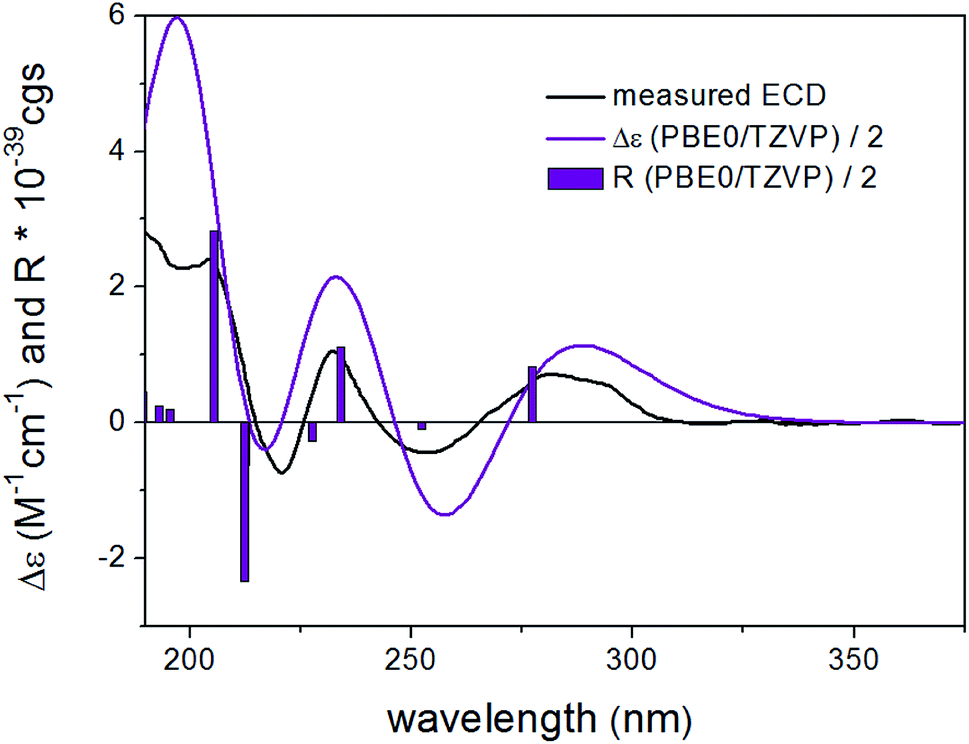 | ||
| Fig. 3 Experimental ECD spectrum (black) of 1 in MeCN compared with the Boltzmann-weighted PBE0/TZVP PCM/MeCN ECD spectrum (purple) of (S)-1 computed for the eight low-energy CAM-B3LYP/TZVP PCM/MeCN conformers. The bars represent the rotational strength of the lowest-energy conformer. | ||
The molecular formulae of 2 and 3 were assigned as C17H14O4 and C18H16O4, respectively, based on their prominent pseudomolecular ion peaks in the HRESIMS spectra. The 1H and 13C NMR data of 2 (Table 1) were almost identical to those of phenylbutyrolactone IIa (15), except for the replacement of the carboxylic acid functionality with a hydroxyl group, as indicated by the absence of a carbonyl signal in the 13C NMR spectrum, as well as a 28 amu difference in the molecular weight of 2 compared with that of phenylbutyrolactone IIa (15). The NMR data of 3 (Table 1) were in good agreement with those of 2, apart from the presence of a methoxy signal at δH 3.24 (δC 50.9). Accordingly, a methoxy substituent was assigned to C-5 instead of a hydroxyl group as in 2, based on the evident HMBC correlation from 5-OMe to C-5. Thus, 2 and 3 were elucidated as new butyrolactone derivatives and were named as bulgarilines B and C, respectively. The zero value of their specific rotations indicated that 2 and 3 were obtained as racemic mixtures.
The molecular formula of 4 was determined as C15H16O3 based on its HRESIMS data, implying 8 degrees of unsaturation. Investigation of its 1H NMR data (Table 2) revealed typical signals of phenyl and p-hydroxy phenyl moieties in the structure of 4. In addition, consecutive COSY correlations were observed between a set of methylene protons resonating at δH 2.51/2.84 (H2-7) and two O-methine protons at δH 3.87 and 4.53 (H-8 and H-9), which permitted the assignment of a 1,2-propanediol moiety. The HMBC correlations from H2-7 to C-3, C-4, C-5, C-8 and C-9 and in turn from H-9 to C-10, C-11 and C-15 established the connectivity of the partial structures. The relative configuration of 4 was deduced to be erythro, based on the large coupling constant value between the two vicinal methines at positions 8 and 9 (5.8 Hz), while a smaller value (J = 2.5 Hz) is suggested for threo, as previously reported for related vicinal diols.37,38 Hence, the structure of 4 was elucidated and the trivial name bulgariol is suggested for this compound.
| Position | 4a | |
|---|---|---|
| δC, type | δH (J in Hz) | |
| a Recorded at 600 MHz (1H) and 150 MHz (13C). | ||
| 1 | 156.6, C | |
| 2/6 | 115.9, CH | 6.68, br d (8.5) |
| 3/5 | 131.4, CH | 7.02, br d (8.5) |
| 4 | 131.6, C | |
| 7 | 38.9, CH2 | 2.51, dd (14.2, 9.2) |
| 2.84, dd (14.2, 3.1) | ||
| 8 | 77.8, CH | 3.87, ddd (9.2, 5.8, 3.1) |
| 9 | 78.2, CH | 4.53, d (5.8) |
| 10 | 143.5, C | |
| 11/15 | 128.4, CH | 7.41, br d (7.0) |
| 12/14 | 129.0, CH | 7.33, br t (7.0) |
| 13 | 128.3, CH | 7.26, tt (7.0, 1.5) |
DFT reoptimization of the initial 41 MMFF conformers of (8R,9S)-4 resulted in 15 and 22 low-energy conformers over a 1% Boltzmann-population. Despite the flexibility of the molecule and the substantially different ECD spectra of the individual low-energy conformers, the Boltzmann-averaged ECD spectra computed at various levels for both sets of conformers gave moderate to good mirror-image agreement with the experimental ECD spectrum (Fig. 4). Furthermore, the sign of the highest-wavelength ECD transition was the same for all the conformers over a 1.2% Boltzmann-population, allowing elucidation of the absolute configuration as (8S,9R).
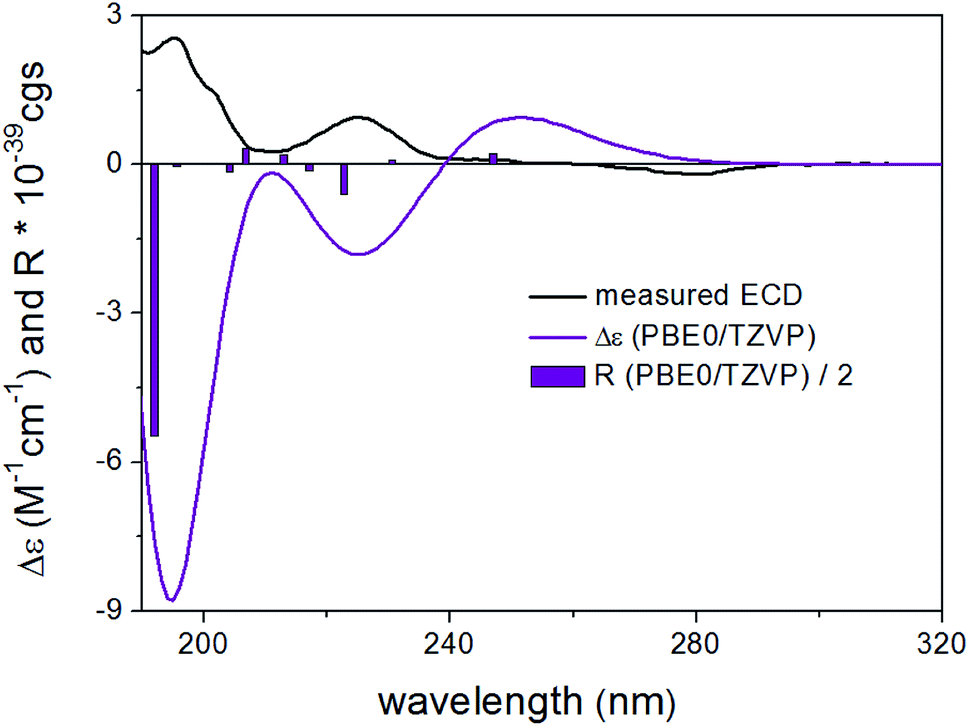 | ||
| Fig. 4 Experimental ECD spectrum (black) of 4 in MeCN compared with the Boltzmann-weighted PBE0/TZVP PCM/MeCN ECD spectrum (purple) of (8R,9S)-4 computed for the 22 low-energy CAM-B3LYP/TZVP PCM/MeCN conformers. The bars represent the rotational strength of the lowest-energy conformer. | ||
Compound 5 had the molecular formula C17H12O3, requiring 12 degrees of unsaturation. The 1H and 13C NMR data of 5 (Table 1) were similar to those of 2 and 3. However, an additional olefinic signal appeared at δH 5.97 (H-12), which provided HMBC correlations to C-4, C-5, C-13, C-14/C-18, along with a long-range correlation to C-2. Thus, the position of the double bond was deduced at Δ5(12), contributing to one additional degree of unsaturation of 5 compared to 2 and 3. The geometry of the exocyclic double bond C-5/C-12 in 5 was determined based on the NOESY spectrum. The NOE correlations observed between H-12 with H-7/H-11 implied the close proximity of H-12 to the aromatic ring, thus confirming the Z configuration of the double bond.39,40 Compound 5, (5Z)-3-hydroxy-4-phenyl-5-(phenylmethylene)-2(5H)-furanone, was reported previously only as a synthetic product.41 In the present study, we report 5 for the first time as a natural product and provide its complete NMR data (Table 1).
The molecular ion of 6 was 14 amu larger than that of 1, as indicated by the HRESIMS spectrum, corresponding to the molecular formula C13H12O5. The 1H and 13C NMR data of 6 (Table 1) were in perfect agreement with those of 1, except for the additional methoxy group signal at δH 3.76/δC 53.9, which showed the respective HMBC correlation to C-1′, hinting at a substitution of the carboxylic acid group with a carbomethoxy attached to C-5 in the structure of 6. Finally, 6 was elucidated as a methyl ester of bulgariline A to which the trivial name bulgariline D is given.
The absolute configuration of 6 was concluded to be the same as that of 1 on the basis of biogenetic considerations, similar positive values of their specific rotation, [α]20D +16 for 1 and [α]25D +26 for 6, as well as the identical ECD spectra of 1 and 6. Furthermore, TDDFT-ECD calculations performed on the arbitrarily chosen (R) enantiomer gave mirror-image agreement (Fig. S106, ESI†), verifying the (S) absolute configuration of 6. It is interesting to compare the ECD spectra of 1 and 6 with those of the related compounds chaetobutenolide C, WF-3681 and WF-3681 methyl ester described by some of us in 2017 and where the carboxylic acid/ester group is attached through two carbons to the central unit.36 The positive transition at ca. 260–280 nm, which is stronger in two recent cases, and the negative one around 220 nm, belonging to the homochiral derivatives, becomes more complex in 1 and 6, but they are still in accordance with the two characteristic bands. Moreover, 6 was proven to be a true natural product, by incubating 1.0 mg of 1 in 1.0 mL MeOH containing 0.1% formic acid at room temperature for 1 week, which did not result in methylation, as indicated by HPLC analysis.
Nitrogen-containing metabolites (7 and 8) were obtained as yellow solids. The HRESIMS spectra exhibited prominent pseudomolecular ion peaks at m/z 248.0919 [M + H]+ and m/z 234.0760 [M + H]+, consistent with the molecular formula C13H13NO4 and C12H11NO4, respectively, both corresponding to 8 degrees of unsaturation. Inspection of the 1H NMR spectrum of 7 (Table 3) revealed the resonances of four aromatic protons, three sets of methylenes and one methoxy group. The COSY correlations of the aromatic protons from H-9 until H-12 suggested a 1,2-disubstituted aromatic ring for 7, as corroborated by HMBC correlations from H-9 to C-11 and C-13 as well as from H-12 to C-8 and C-10. The last spin system observed in the COSY spectrum between δH 2.60/2.67 (H2-3) and 2.30/2.54 (H2-4) afforded HMBC correlations from H2-3 to C-2 and C-5 and from H2-4 to C-5 and C-1′, respectively. Additionally, the HMBC correlation from 1′-OMe to C-1′ established the substructure of an oxoproline residue bearing a 5-carbomethoxy group (Fig. 2). Further HMBC correlations from the methylene at δH 4.96/5.10 (H2-7) to C-5, C-8, C-9 and C-13 established the linkage between these two substructures, which accounted for the last degree of unsaturation in 7. The 1H and 13C NMR data of 8 were in a good agreement with those of 7, apart from the absence of an OMe signal, which is in accordance with the 14 amu difference in the molecular weight of 8 compared to 7, thus indicating a carboxylic acid group attached at C-5, instead of a carbomethoxy function. Accordingly, 7 and 8 possess unusual heterocyclic structures containing a 1,3-oxazine nucleus, which rarely occurs in natural products. A few examples of natural 1,3-oxazines include the antimycobacterial oxazinin A, isolated from the Eurotiomycetes strain 110162,42 as well as salinazinones A and B from the bacterial strain Streptomyces sp. KMF-004.43 The latter two compounds have been also reported from the marine-derived bacterium Streptomyces spinoverrucosus as spinoxazines A and B.44 However, compared to the aforementioned natural products, the structures of 7 and 8 lack a keto function in the 1,3-oxazine core, which is mostly attributed to synthetic compounds.45,46 The trivial names bulgarixines A (7) and B (8) were assigned to these compounds. Considering the possibility that 7 might arise as an artifact of its non-methoxylated analogue (8) during the isolation process, an experiment was carried out by incubating 0.5 mg of 8 in 0.5 mL MeOH containing 0.1% formic acid for 1 week at room temperature. HPLC analysis showed that no methylated product was present in the chromatogram, thus indicating that 7 is a natural product. The baseline ECD curves of both 7 and 8 indicated that both of them were isolated as a racemate.
| Position | 7a | 8a | ||
|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, typeb | δH (J in Hz) | |
| a Recorded at 600 MHz (1H) and 150 MHz (13C).b Chemical shifts extracted from HSQC and HMBC spectra. | ||||
| 2 | 174.3, C | 174.3, C | ||
| 3 | 30.6, CH2 | 2.60, ddd (17.2, 10.1, 2.3) | 30.5, CH2 | 2.59, ddd (17.2, 10.1, 2.2) |
| 2.67, dt (17.2, 9.6) | 2.70, dt (17.2, 9.7) | |||
| 4 | 31.4, CH2 | 2.30, dt (13.5, 10.1) | 31.3, CH2 | 2.28, dt (13.5, 10.1) |
| 2.54, ddd (13.5, 9.6, 2.3) | 2.54, ddd (13.5, 9.7, 2.2) | |||
| 5 | 92.6, C | 92.6, C | ||
| 7 | 66.1, CH2 | 4.96, d (15.8); | 65.7, CH2 | 4.93, d (15.7) |
| 5.10, d (15.8) | 5.19, d (15.7) | |||
| 8 | 124.4, C | 124.6, C | ||
| 9 | 125.5, CH | 7.10, br d (8.0) | 125.2, CH | 7.10, br d (8.0) |
| 10 | 125.9, CH | 7.14, td (8.0, 1.2) | 125.3, CH | 7.13, td (8.0, 1.2) |
| 11 | 128.5, CH | 7.27, br t (8.0) | 128.0, CH | 7.26, br t (8.0) |
| 12 | 120.4, CH | 8.27, br d (8.0) | 120.3, CH | 8.29, br d (8.0) |
| 13 | 134.0, C | 133.8, C | ||
| 1′ | 171.4, C | 172.6, C | ||
| 1′-OMe | 53.7, CH3 | 3.73, s | ||
Compound 9 was afforded as a brown solid. Its molecular formula was determined as C17H28O5 on the basis of its HRESIMS spectrum, requiring 4 degrees of unsaturation. Its UV spectrum displayed a maximum absorption at around 299 nm, suggesting a pyrone nucleus.47,48 Detailed analysis of the 1H and 13C NMR data of 9 (Table 4) indicated signals of an isolated olefinic proton at δH 6.65 (H-5), an oxygenated methine at δH 4.40 (H-1′) and an isolated methylene at δH 4.31 (H2-1′′), in addition to seven aliphatic methylenes observed at δH 1.31–1.82 (H2-2′–H2-8′), two methoxy groups at δH 4.00 and 3.32, one methyl group at δH 0.90 (H3-9′), along with signals for a carbonyl at δC 166.7 (C-2) and sp2 carbons at δC 101.8 (C-3), 171.7 (C-4) and 171.4 (C-5). COSY correlations observed from H-1′ to H3-9′ allowed the assignment of a 1-nonanol side-chain (Fig. 2). The HMBC correlations from H-5 to C-3, C-4 and C-6, as well as from H2-1′′ to C-2, C-3 and C-4, confirmed the presence of a conjugated α-pyrone nucleus in the structure of 9. Additionally, the position of two methoxy groups (4-OMe and 1′′-OMe) as substituents of the α-pyrone ring was deduced based on their respective HMBC correlations. Finally, the presence of HMBC correlations from H-5 to C-1′ and in turn from H-1′ to C-5 and C-6, indicated the connection of the 1-nonanol side-chain with the α-pyrone nucleus at C-6 (Fig. 2). The structure of 9 is closely related to dothideopyrone B, which was isolated from the endophyte Dothideomycete sp. LRUB20,49 and with dothideopyrone F, a recently reported α-pyrone from the endolichenic fungus Dothideomycetes sp. EL003334.47 However, 9 features two extra methylene protons in the saturated aliphatic chain in comparison with dothideopyrone B,49 and an additional OMe at C-1′′ in comparison with dothideopyrone F.47 Therefore, compound 9 was elucidated as a new α-pyrone derivative, for which the trivial name bulgariapyrone A is suggested.
| Position | 9a | 10a | 11a | 12a | 13b,d | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC, typec | δH (J in Hz) | δC, type | δH (J in Hz) | δC, typec | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| a Recorded at 600 MHz (1H) and 150 MHz (13C).b Recorded at 600 MHz (1H) and 125 MHz (13C).c Chemical shifts extracted from the HSQC and HMBC spectra.d Signals for another monomeric unit are identical, except for δC 166.8 (C, C-2′′′), 102.3 (C, C-3′′′), 171.5 (C, C-4′′′), 94.4 (CH, C-5′′′), 171.1 (C, C-6′′′), 26.9 (CH2, C-7′′′′), as well as signals at δC 33.6 (CH2, C-8′′′′)/δH 1.52 (2H, m, H-8′′′′), δC 63.0 (CH2, C-9′′′′)/δH 3.53 (2H, t, J = 6.7 Hz, H-9′′′′) and δC 62.3 (CH2, C-1′′′′′)/δH 4.34 (2H, s, H-1′′′′′).e Signals can be interchangeable.f ov stands for overlapped signals. | ||||||||||
| 2 | 166.7, C | 166.8, C | 166.7, C | 166.8, C | 166.7, C | |||||
| 3 | 101.8, C | 101.8, C | 101.8, C | 101.8, C | 101.9, C | |||||
| 4 | 171.7, C | 171.7, C | 171.7, C | 171.7, C | 171.7, C | |||||
| 5 | 94.4, CH | 6.65, s | 94.4, CH | 6.65, s | 94.4, CH | 6.65, s | 94.4, CH | 6.65, s | 94.5, CH | 6.65, br s |
| 6 | 171.4, C | 171.3, C | 171.3, C | 171.3, C | 171.3, C | |||||
| 1′ | 71.5, CH | 4.40,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) dd (7.9, 4.5) dd (7.9, 4.5) |
71.5, CH | 4.40, m | 71.5, CH | 4.40, dd (7.9, 4.5) | 71.5, CH | 4.40, dd (8.0, 4.5) | 71.5, CH | 4.40, br dd (7.9, 4.4) |
| 2′ | 36.3, CH2 | 1.68,m; 1.82, m |
36.2, CH2 | 1.68, m; 1.83, m | 36.2, CH2 | 1.69, m; 1.82, m | 36.2, CH2 | 1.68, m; 1.82, m | 36.3, CH2 | 1.68, m; 1.82, m |
| 3′ | 26.2, CH2 | 1.42, m | 26.1, CH2 | 1.43, m | 26.1, CH2 | 1.44, m | 26.1, CH2 | 1.44, m | 26.1, CH2 | 1.44, m; 1.41 m |
| 4′ | 30.4,e CH2 | 1.27–1.37, ovf | 30.2,e CH2 | 1.31–1.38, ov | 30.2,e CH2 | 1.31–1.39, ov | 30.4,e CH2 | 1.31–1.39, ov | 30.4,e CH2 | 1.30–1.37, ov |
| 5′ | 30.5,e CH2 | 1.27–1.37, ov | 30.1,e CH2 | 1.31–1.38, ov | 30.3,e CH2 | 1.31–1.39, ov | 30.5,e CH2 | 1.31–1.39, ov | 30.5,e CH2 | 1.30–1.37, ov |
| 6′ | 30.6,e CH2 | 1.27–1.37, ov | 30.2,e CH2 | 1.31–1.38, ov | 30.4,e CH2 | 1.31–1.39, ov | 30.6,e CH2 | 1.31–1.39, ov | 30.6,e CH2 | 1.30–1.37, ov |
| 7′ | 33.0, CH2 | 1.27–1.37, ov | 26.0, CH2 | 1.61, p (7.4) | 27.0, CH2 | 1.36, ov | 26.9, CH2 | 1.34, ov | 27.1, CH2 | 1.34, ov |
| 8′ | 23.7, CH2 | 1.31, ov | 34.8, CH2 | 2.31, t (7.4) | 29.7, CH2 | 1.62, p (6.7) | 33.6, CH2 | 1.52, p (6.7) | 30.6, CH2 | 1.54, m |
| 9′ | 14.4, CH3 | 0.90,t (7.0) |
176.0, C | 65.7, CH2 | 4.05, t (6.7) | 63.0, CH2 | 3.53, t (6.7) | 71.6, CH2 | 3.46, t (6.6) | |
| 11′ | 173.1, C | |||||||||
| 12′ | 20.8, CH3 | 2.02, s | ||||||||
| 1′′ | 64.1, CH2 | 4.31, s | 64.1, CH2 | 4.30, s | 64.1, CH2 | 4.31, s | 64.1, CH2 | 4.30, s | 64.1, CH2 | 4.30, s |
| 4-OMe | 57.7, CH3 | 4.00, s | 57.7, CH3 | 4.00, s | 57.7, CH3 | 4.00, s | 57.7, CH3 | 4.00, s | 57.7, CH3 | 4.00, s |
| 9′-OMe | 51.9, CH3 | 3.65, s | ||||||||
| 1′′-OMe | 58.3, CH3 | 3.32, s | 58.3, CH3 | 3.32, s | 58.3, CH3 | 3.32, s | 58.3, CH3 | 3.32, s | 58.3, CH3 | 3.32, s |
Compounds 10–12 exhibited similar UV spectra as 9, indicative of pyrone derivatives (see ESI†). Their molecular formulae were determined as C18H28O7, C19H30O7 and C17H28O6, respectively, as indicated by their HRESIMS spectra. The 1H and 13C NMR data of 10–12 (Table 4) were similar to those of 9 and indicated that 10–12 retained the same α-pyrone core structure as 9, differing from the latter only in the nature of their aliphatic side-chains. The molecular weight of 10 was 44 amu larger than that of 9, which together with an additional methoxy signal at δH 3.65/δC 51.9 and an additional carbonyl resonating at δC 176.0 (C-9′), implied that the aliphatic chain of 10 is terminated by a carbomethoxy group instead of a methyl group. This deduction was corroborated by the observed HMBC correlation from H2-7′/H2-8′ to C-9′, in addition to a further degree of unsaturation of 10 in comparison with 9. The 1H NMR data of 11 (Table 4) displayed an additional oxygenated methylene at δH 4.05 (H2-9′) and a signal of a downfield shifted methyl group at δH 2.02 (H3-12′), which along with the observed HMBC correlations from H2-9′ to C-7′, C-8′ and C-11′, as well as from H3-12′ to C-11, hinted at the presence of an acetoxy group at the terminus of the aliphatic side-chain in 11. The molecular weight of compound 12 was 16 amu larger than that of 9. Moreover, the 1H NMR data of 12 (Table 4) displayed an extra oxygenated methylene (δH 3.53) split into a triplet, which revealed a COSY correlation to δH 1.52 (H-8′), suggesting the presence of a hydroxyl group at C-9′. Accordingly, compounds 10–12 were established as new α-pyrone analogues, for which the trivial names bulgariapyrones B–D are proposed.
In order to assign the absolute configurations of 9–12, the modified Mosher's reaction was carried out, and 12 was chosen as a model compound for this reaction. Both primary and secondary alcohol groups in 12 were converted to either (S)- or (R)-MTPA esters. Based on the calculated values of Δδ(S)–(R) of MTPA esters, the absolute configuration at C-1′ was established as (S), which is in agreement with the literature data of previously described α-pyrone derivatives related to 12 (Fig. 5).47 Similar specific optical rotation values of 9–11 suggested that these α-pyrones share the same (S) absolute configuration at C-1′.
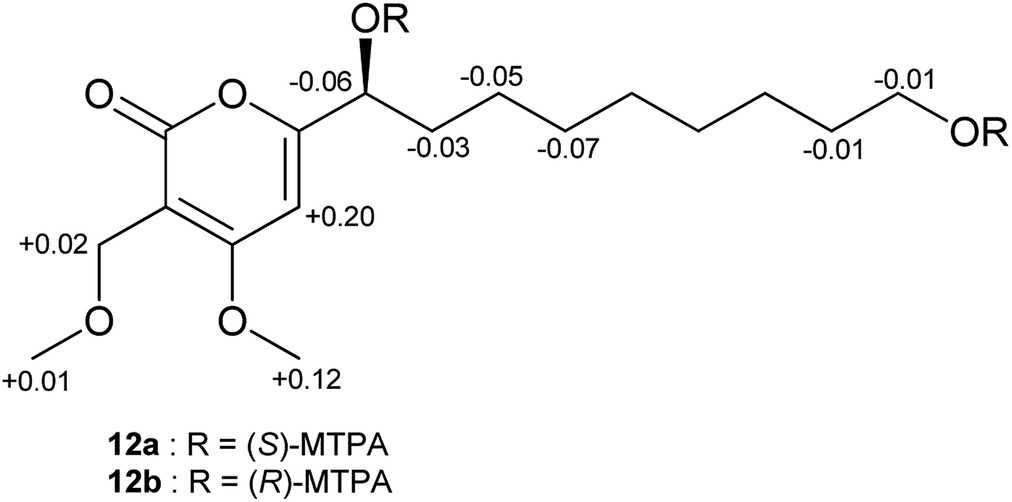 | ||
| Fig. 5 Δδ(S)–(R) values in ppm for the MTPA esters of 12. | ||
The molecular formula of 13 was determined as C33H52O11, based on the prominent pseudomolecular ion peak at m/z 625.3587 [M + H]+ in the HRESIMS spectrum, corresponding to 8 degrees of unsaturation. Analysis of the molecular formula and of the 1D/2D NMR data of 13 (Table 4, ESI†) suggested it contained two substructures similar to bulgariapyrone D (12). However, the NMR data of 13 revealed additional signals of an isolated methylene at δH 4.34/δC 62.3 (H2-1′′′′′) and one set of methylene as a triplet at δH 3.46/δC 71.6 (H2-9′), suggesting an asymmetry of the molecule. The HMBC correlations observed from H2-1′′′′′ to C-2′′′, C-3′′′, C-4′′′ and C-9′, and in turn from H2-9′ to C-7′, C-8′ and C-1′′′′′, together with the NOESY correlation H2-9′/H2-1′′′′′ allowed connecting these two monomeric units via an ether bridge between C-1′′′′′ and C-9′, suggesting that 13 could be formed through a condensation reaction between two molecules of 12. Thus, the planar structure of 13 was elucidated as shown and the name bulgariapyrone E is proposed. Notably, only two related naturally occurring symmetrical α-pyrone dimers have been reported so far, multiforisin D, previously isolated from Gelasinospora multiforis,50 and dothideopyrone D, a metabolite from the endophytic fungus Dothideomycete sp. LRUB20,49 which emphasize the rare nature of these dimeric compounds. Moreover, compound 13 seems to be a product of a “head to tail” condensation, where the side chain of one monomeric unit is connected through an ether bond to the C-1′′′′′ of the α-pyrone nucleus of the other monomeric unit, which creates an asymmetry in the structure of 13 and makes it structurally different from known symmetrical C2-type bis-pyrone dimers. As the condensation of primary alcohols might take place under acidic conditions, an experiment was carried out by incubating 0.2 mg of 12 in 0.2 mL MeOH containing different concentrations of formic acid (0.1%, 1%, 5% and 10%), at room temperature for 1 week, followed by HPLC analysis. The HPLC chromatograms showed no formation of compound 13, which indicated that 13 is a true natural product, and not an artifact arising during the isolation procedure.
Following the same Mosher's reaction protocol for compound 13 as for 12, the (S)- or (R)-MTPA ester products were obtained (see the Experimental section). The MALDI-MS spectra of the reaction products indicated that all the primary and secondary alcohol groups in 13 reacted with the reagent (m/z 1295 [M + Na]+). Thus, the absolute configuration of 13 was unequivocally assigned as (S) for both stereocenters at C-1′ and C-1′′′′, which is in accordance with biosynthetic considerations and the reported configuration of the structurally related homodimer dothideopyrone D.49
Compound 14 was obtained as a dark red solid. Its molecular formula was determined as C26H28O7, on the basis of a prominent pseudomolecular ion peak at m/z 453.1910 [M + H]+ in the HRESIMS spectrum, requiring 13 degrees of unsaturation. Detailed analysis of the 1D and 2D NMR spectra showed that 14 is a known compound, bulgarialactone B,29 first isolated from the same fungus more than two decades ago. However, in this first paper, the absolute configuration was not determined for bulgarialactone B and the relative configuration was elucidated on the basis of the weak NOESY correlation between H-3 and H3-27. Further reports on bulgarialactone B focused on the Hsp90 inhibitory activity and were not engaged in the elucidation of the absolute configuration.30,51 The SciFinder database indicates the absolute configuration for the closely related compound epicocconone, which lacks a chirality centre in the unsaturated side-chain and exhibits potent fluorescent properties, but the original paper52 reported only the relative configuration of the two chirality centres as elucidated by the comparison of the HF/6-31G(d,p) optimized low-energy geometries of the high-temperature molecular dynamics trajectories with the experimental NMR data. Subsequent papers did not address the elucidation of the absolute configuration of epicocconone.
Bulgarialactone B (14) had positive Cotton effects (CEs) at 437 and 269 nm and negative ones at 315 and 232 nm. In order to determine the absolute configuration of bulgarialactone B, the solution TDDFT-ECD method was applied on the arbitrarily chosen (3S,11S,23R) and (3S,11S,23S) stereoisomers.53,54 Although the core part was expected to govern mostly the ECD spectrum, the C-23 chirality centre located in the allylic position of the conjugated π system of the side-chain was also supposed to have at least a minor contribution, which would have allowed distinguishing the C-23 epimers. A MMFF (Merck Molecular Force Field) conformational search of the two epimers resulted in 549 and 541 conformer clusters in a 21 kJ mol−1 energy window, respectively, indicating high conformational flexibility. These geometries were reoptimized at the CAM-B3LYP/TZVP PCM/MeCN level. Despite the large number of low-energy conformers, the Boltzmann-averaged ECD spectra of both epimers computed at four different levels (B3LYP/TZVP, BH&HLYP/TZVP, CAM-B3LYP/TZVP and PBE0/TZVP, all with PCM for MeCN) gave consistently moderate to good agreement with the experimental ECD spectrum (Fig. 6 and 7). Interestingly, the difference between the computed ECD spectra of the epimers was rather small, which suggested that the influence of C-23 on the overall ECD is marginal. Accordingly, the absolute configuration of the core part could be elucidated as (3S,11S), while the C-23 chirality centre remained unassigned. By analyzing the individual ECD spectra of the low-energy computed conformers, it turned out that the features of the ECD spectra were influenced by the helicity of the dihydropyrane ring and the orientation of the conjugating side-chain, the latter of which was also described by Syzgantseva et al. to influence the UV characteristics.55
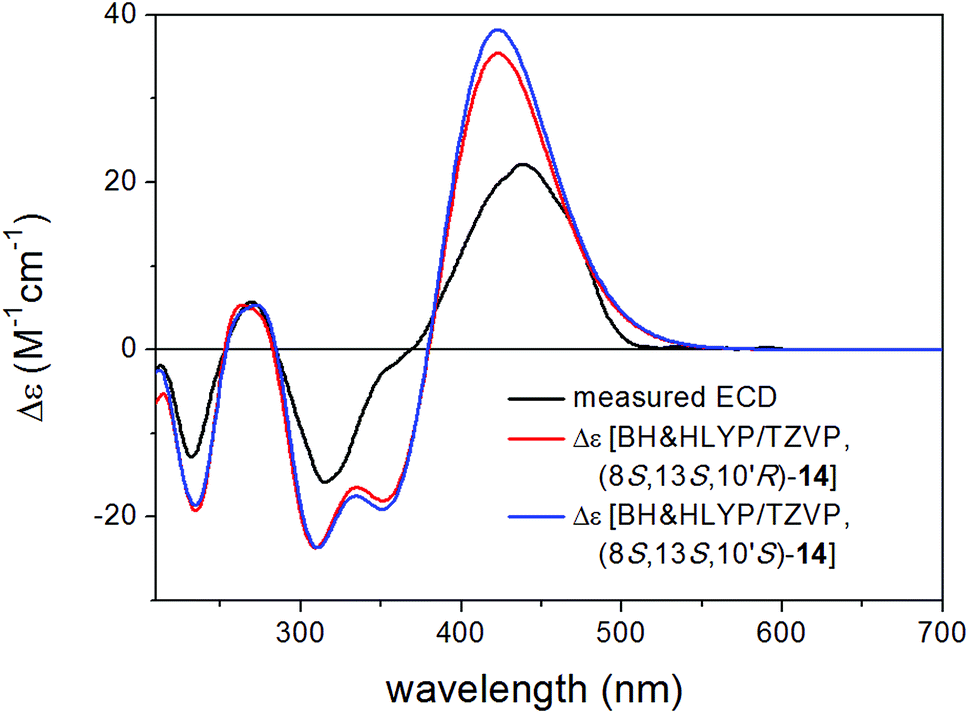 | ||
| Fig. 6 Experimental ECD spectrum of 14 in MeCN compared with the Boltzmann-weighted BH&HLYP/TZVP PCM/MeCN spectra of (3S,11S,23R)-14 and (3S,11S,23S)-14 computed for the low-energy (≥1%) CAM-B3LYP/TZVP PCM/MeCN conformers (26 and 26 conformers, respectively). | ||
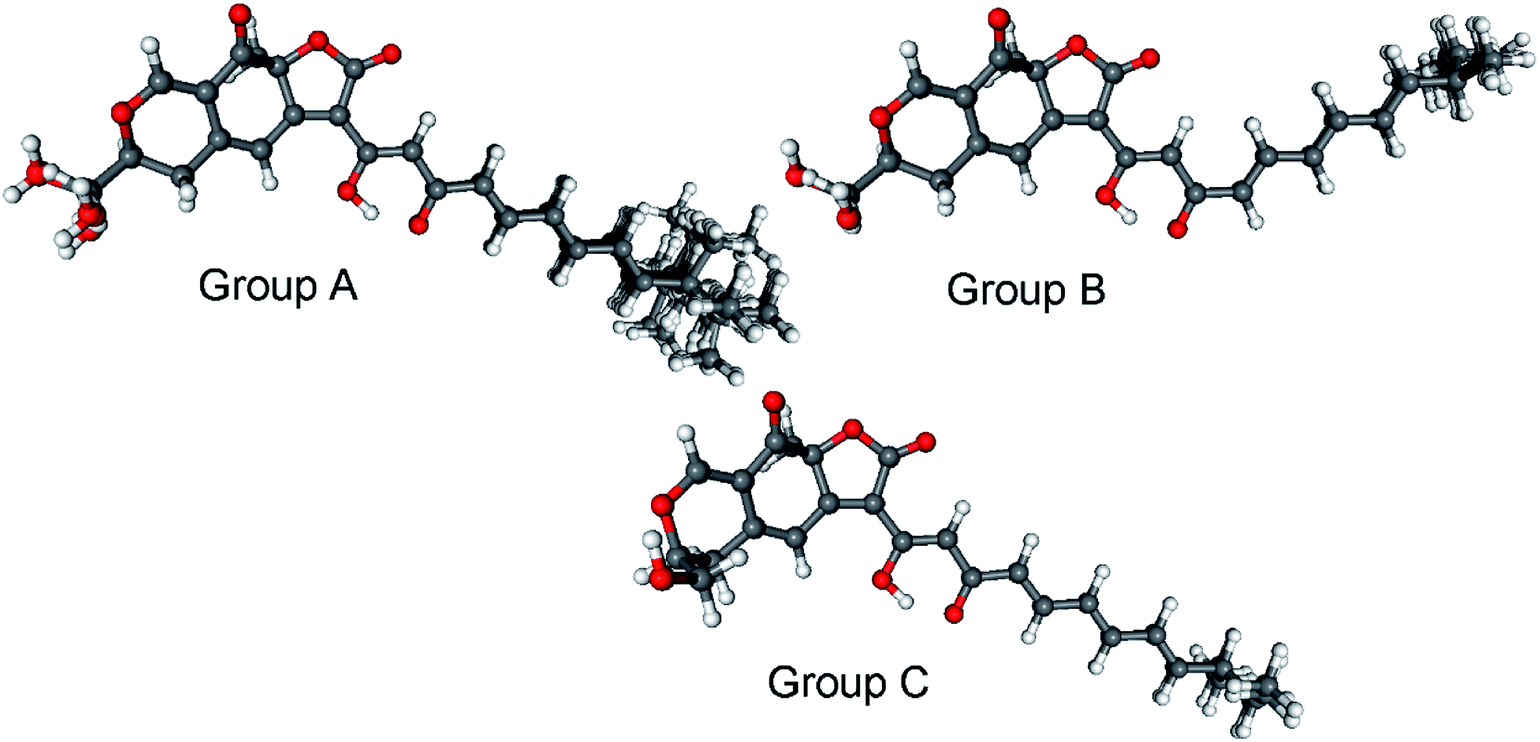 | ||
| Fig. 7 Classification of the 26 low-energy (≥1%) CAM-B3LYP/TZVP PCM/MeCN conformers of (3S,11S,23R)-14 into conformer groups. Group A (70.3%) contains conformers A, B, C, D, E, F, H, I, J, L, M, N, Q, R, S, T, U, V, W, Y, Z; group B (5.6%) contains conformers G, K, X; group C (3.2%) contains conformers O and P. | ||
The experimental ECD spectra of 14 and that of (+)-epicocconone reported by Bell and Karuso52 have a mirror image relationship, suggesting that (+)-epicocconone showing an intense negative CE above 400 nm has an (3R,11R) absolute configuration in Epicoccum nigrum,52 which is an example of the chiral switching of the tricyclic skeleton.56 It is also clear that the absolute configuration presented in the SciFinder database for epicocconone57 is incorrect and it should specify only the relative configuration.
The planar structure of compound 16 was shown to be identical to the previously described flavipesin B, isolated from the fungal endophyte Aspergillus flavipes AIL8,58 which has been previously reported as a synthetic compound.59,60 In a recent study, it was isolated from an engineered strain of A. nidulans.61 However, 16 possesses an opposite sign of the specific optical rotation, [α]25D −72 (MeOH), compared to that of the reported value for flavipesin B: [α]25D +133 (acetone).58 Therefore, in this study, we assigned 16 as (−)-(S)-flavipesin B.
The structures of the remaining known compounds isolated from B. inquinans were established as phenylbutyrolactone IIa (15),33,34,59 xenofuranone B (17)35,67 and bulgarialactone D (18),30 based on their spectroscopic data as well as on a comparison with the literature.
All the isolated compounds were evaluated for cytotoxicity towards the murine lymphoma cell line L5178Y. However, only 2 exhibited pronounced activity against the L5178Y cell line, with an IC50 value of 1.8 μM, which was stronger than that of the positive control kahalalide F (IC50 4.3 μM). The presence of a hydroxy group attached to C-5 in the 2-furanone ring in the structure of 2 was apparently important for its cytotoxic activity as no activity was found for compounds 3, phenylbutyrolactone IIa (15), 16 and xenofuranone B (17). Furthermore, a closely related derivative, 4-O-demethylisobutyrolactone II, which bears an additional hydroxy group on both aromatic rings at the positions 9 and 16 (tyrosine-derived residues) compared to 2, was shown to be inactive against the L5178Y cell line,62 thus suggesting that the phenyl substituents were required for its cytotoxicity. The remaining compounds showed no significant cytotoxic properties. Interestingly, related α-pyrones were previously reported to be nontoxic or exhibited only moderate cytotoxicity against several cancer cell lines.49 A number of α-pyrone analogues, however, revealed promising biological activities as antibacterial agents,63 HIV-1 protease inhibitors,64 tyrosinase inhibitors65 or inhibitors of nitric oxide production,47 making these metabolites attractive scaffolds for synthetic chemical studies. In our study, bulgarialactone B (1) revealed no cytotoxicity against the tested L5178Y cell line, albeit in a previous study it was reported to possess antitumor activity against an ascitic ovarian carcinoma xenograft.30
Butyrolactone analogues similar to the derivatives isolated in this study were previously described from Aspergillus species,58,62,66–68 Here we report B. inquinans as a source of this type of metabolites for the first time. Moreover, the results obtained upon application of the OSMAC approach employing a mixture of salts (MgSO4, NaNO3 and NaCl) highlighted the usefulness of this strategy not only for diversifying secondary metabolites produced by this strain, but also to afford rare natural product scaffolds, as exemplified by the isolation of 1,3-oxazine derivatives (7 and 8) and of an unusual α-pyrone dimer 13. Another set of OSMAC experiments was performed to investigate which salt mainly contributes to the changes in the metabolic profile of B. inquinans, by adding separately MgSO4·7H2O (2.5 and 3.5 g), and a mixture of NaNO3 and NaCl (2.5 g each) to Czapek medium with and without MgSO4. The fungus failed to grow in the presence of the mixture of NaNO3 and NaCl, when MgSO4 was completely excluded from Czapek medium, suggesting that Mg2+ ions are apparently critical for fungal growth. However, the addition of MgSO4 alone did not result in any changes in the HPLC chromatogram in comparison with the chromatogram of the fungus grown on solid Czapek medium, suggesting that the fungus was only metabolically affected by adding the mixture of these three salts simultaneously. Interestingly, OSMAC studies with the marine-derived fungus, Ascotricha sp. ZJ-M-5, involving MgCl2 in Czapek Dox broth medium, previously reported that Mg2+ ions influenced the secondary metabolites profile of this fungus.69 With respect to the findings in this study, the compounds obtained from the OSMAC experiments in the presence of Mg2+ were mostly methoxylated derivatives. Thus, it may be speculated that Mg2+ ions under certain conditions might trigger O-alkylation reactions.
Experimental section
General procedures
A Jasco P-2000 polarimeter was used to measure the optical rotations. 1H (600 and 300 MHz), 13C (150, 125 and 75 MHz) and 2D NMR spectra were recorded on Bruker AVANCE DMX 600, 500 and 300 NMR spectrometers. The chemical shifts (δ) were referenced to the residual solvent peaks at δH 3.31 (MeOH-d4) ppm for 1H or δC 49.0 (MeOH-d4) ppm for 13C. Mass spectra (ESI) were measured with a Finnigan LCQ Deca mass spectrometer. HRESIMS spectra were recorded with an UHR-QTOF maXis 4G (Bruker Daltonics) mass spectrometer. HPLC analysis was performed with a Dionex UltiMate 3000 with an UltiMate 3000 pump coupled to a photodiode array detector (DAD 3000 RS). Detection wavelengths were set at 235, 254, 280 and 340 nm. The column was prefilled with Eurospher 100–10 C18, 125 × 4 mm (Knauer, Germany). The following gradient was used for routine analysis (MeOH: 0.1% HCOOH in H2O): 0 min (10% MeOH); 5 min (10% MeOH); 35 min (100% MeOH); 45 min (100% MeOH). Semipreparative HPLC was performed with a Merck Hitachi Chromaster HPLC system (UV detector 5410; pump 5110; column Eurospher 100–10 C18, 300 × 8 mm, Knauer, Germany; flow rate 5 mL min−1). Column chromatography was performed using Silica 60 M (0.040–0.063 mm; Macherey-Nagel, Germany), Silica gel 90 C18-reversed phase and Sephadex LH-20. TLC plates pre-coated with silica gel 60 F254 (Macherey-Nagel, Germany) were used for analysis, detection was under UV 254 and 366 nm. ECD spectra were recorded on a JASCO J-810 spectropolarimeter.Fungal material and fermentation
The fungus Bulgaria inquinans (isolate MSp3-1) was isolated from a healthy sprout of mistletoe (Viscum album),70 collected in January 2017 at Jülich, Germany. Fungal identification was carried out according to a standard molecular biology protocol,70 followed by a BlastN search in the NCBI database. The sequence was submitted to the GenBank (accession no. MK246763). The fungal strain is kept in one of the author's laboratory (P. P.).The fungus was cultivated on solid Czapek medium, which was prepared by autoclaving 200 mL of liquid Czapek medium with the addition of 5.0 g of bacto agar in a 1 L Erlenmeyer flask. The composition of liquid Czapek medium was 10.0 g dextrose, 20.0 g mannitol, 20.0 g maltose, 3.0 g yeast extract, 1.0 g corn steep liquor, 0.5 g tryptophan, 0.5 g K2HPO4·3H2O, 0.3 g MgSO4·7H2O and 1 L of distilled water (pH value of the medium adjusted between 7.2–7.8). The fermentation was performed in 15 flasks at room temperature, under static conditions for 27 days.
OSMAC experiments were carried out by growing the fungus on solid Czapek medium containing either 3.5 g NaCl, 3.5 g NaBr, 3.5 g NaI, 3.5 g NaNO3, 3.5 g (NH4)2SO4 or mixtures of (a) MgSO4·7H2O, NaNO3 and NaCl (2.5 g of each), (b) FeSO4·7H2O, NaNO3 and NaCl (2.5 g of each), or (c) ZnSO4·7H2O, NaNO3 and NaCl (2.5 g of each), added to each 1 L flask followed by extraction when the flasks were completely overgrown by the fungus. Based on the chromatographic profiles obtained from these experiments, a large-scale fermentation of B. inquinans was carried out by adding a mixture of MgSO4·7H2O, NaNO3 and NaCl (2.5 g of each) to solid Czapek medium. The fungus was grown under static conditions for 33 days followed by extraction.
Extraction and isolation
The fungal culture grown on solid Czapek medium was extracted with 500 mL EtOAc added to each flask followed by concentration in vacuo to afford the crude extract (5.1 g). The extract was then loaded on silica gel 60 (VLC) and eluted successively with n-hexane–EtOAc followed by CH2Cl2–MeOH to obtain 13 fractions (V1–V13). Fractions V3, V4 and V5 eluted with n-hexane–EtOAc (6:4), (4:6) and (2:8), respectively, were subjected to further separation based on their HPLC chromatograms. Fraction V5 (1.7 g) was separated over Sephadex LH-20 and eluted with MeOH to afford nine subfractions (V5.1–V5.9). Purification of the V5.3 subfraction (65.9 mg) was achieved by semipreparative HPLC using ACN–H2O (from 65% to 100% ACN, 20 min), to yield bulgarialactone B (14, 27.0 mg). Fraction V4 (507.9 mg) was submitted to Sephadex LH-20, employing CH2Cl2–MeOH (1:1) as the mobile phase to obtain 12 subfractions (V4.1–V4.12). Semipreparative HPLC was used to purify the subfraction V4.10 (270.0 mg), using MeOH-0.1% HCOOH in H2O (from 30% to 100% MeOH, 25 min), to afford the new compounds 1 (20.0 mg) and 4 (2.2 mg) along with phenylbutyrolactone IIa (15, 124.0 mg). In a similar manner, the separation of fraction V3 (150 mg) on Sephadex LH-20 resulted in 10 subfractions (V3.1–V3.10). Purification by semipreparative HPLC of the V3.6 subfraction (26.0 mg), employing MeOH-0.1% HCOOH in H2O as the eluent (from 65% to 100% MeOH, 20 min), yielded the new natural products 2 (2.8 mg), 3 (1.5 mg) and 5 (1.9 mg) together with xenofuranone B (17, 1.2 mg).
The fungal culture obtained from the large-scale fermentation on the Czapek medium with the salt mixture was extracted with 500 mL EtOAc added to each flask. Following the previously described procedure, the crude extract (7.7 g) obtained after removal of the solvent was chromatographed on Silica gel 60 (VLC) to afford 13 fractions (MV1–MV13). Fractions MV3, MV4, MV6 and MV9 eluted with n-hexane–EtOAc (6:4), (4:6), 100% EtOAc and DCM–MeOH(1:9), respectively, were selected for further isolation work-up, guided by their HPLC results. The separation of fraction MV9 (1.5 g) was carried out on a Silica gel 90 C18-reversed phase column by a step gradient elution employing mixtures of H2O–MeOH to yield 10 subfractions (MV9.1–MV9.10). The MV9.7 subfraction (95.1 mg) was submitted to Sephadex LH-20 using CH2Cl2–MeOH (1:1) as the eluent to yield seven subfractions (MV9.7.1–MV9.7.7). Purification of the MV9.7.2 subfraction (24.5 mg) was achieved by semipreparative HPLC using MeOH-0.1% HCOOH in H2O (from 65% to 100% MeOH, 20 min) to afford 13 (9.3 mg). The new compound 12 (30.4 mg) was afforded by the purification of MV9.7.5 (64.7 mg) with semipreparative HPLC using MeOH-0.1% HCOOH in H2O (from 40% to 100% MeOH, 20 min). The new compound 8 (1.5 mg) was obtained by the separation of the MV9.1 subfraction (40.0 mg) on Sephadex LH-20, and final purification was achieved by semipreparative HPLC using MeOH-0.1% HCOOH in H2O (from 25% to 100% MeOH, 20 min). Fraction MV6 (452.5 mg) was applied on Sephadex LH-20 employing MeOH as the eluent to yield nine subfractions (MV6.1–MV6.9). Purification of the MV6.6 subfraction (21.0 mg) was carried out by semipreparative HPLC using ACN–H2O (from 60% to 100% ACN, 25 min) to afford bulgarialactone D (18, 4.0 mg). The new compounds 9 (2.6 mg), 10 (9.1 mg) and 11 (2.6 mg) were obtained from purification of MV6.3 subfraction (59.3 mg) by semipreparative HPLC using MeOH-0.1% HCOOH in H2O (from 55% to 100% MeOH, 20 min). Furthermore, fraction MV4 (622.4 mg) was chromatographed on Sephadex LH-20, eluting with CH2Cl2–MeOH (1:1), to give eight subfractions (MV4.1–MV4.8). The MV4.6 subfraction (54.4 mg) was then purified by semipreparative HPLC using MeOH-0.1% HCOOH in H2O (from 40% to 100% MeOH, 25 min), which yielded 3 (14.6 mg) and 6 (3.2 mg) along with 16 (6.5 mg). Application of the latter procedure for the purification of fraction MV3 (164.9 mg) by semipreparative HPLC following separation on Sephadex LH-20 yielded compounds 5 (2.8 mg) and 7 (2.0 mg). The total amounts of 3 and 5, both from the fungal culture grown on solid Czapek medium and from the OSMAC experiment, were 16.1 and 4.7 mg, respectively.
Bulgariline A (1): yellow solid; [α]20D +16 (c 0.20, MeOH); UV (MeOH, PDA): λmax 288, 218 nm; ECD (MeCN, λ [nm] (Δε), c 0.529 mM): 294sh (+0.56), 282 (+0.71), 254 (−0.44), 232 (+1.05), 221 (−0.74), 205sh (+2.42); 1H and 13C NMR data, see Table 1; HRESIMS m/z 235.0600 [M + H]+ (calcd for C12H11O5, 235.0601).
Bulgariline B (2): yellow solid; [α]20D 0 (c 0.20, MeOH); UV (MeOH, PDA): λmax 291, 204 nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 283.0962 [M + H]+ (calcd for C17H15O4, 283.0965).
Bulgariline C (3): yellow solid; [α]20D 0 (c 0.20, MeOH); UV (MeOH, PDA): λmax 293, 201 nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 297.1123 [M + H]+ (calcd for C18H17O4, 297.1121).
Bulgariol (4): yellow, solid; [α]20D −25 (c 0.20, MeOH); UV (MeOH, PDA): λmax 277 nm; ECD (MeCN, λ [nm] (Δε), c 0.409 mM): 280 (−0.19), 245sh (+0.10), 225 (+0.95), 201sh (+1.52), 195 (+2.55); 1H and 13C NMR data, see Table 2; HRESIMS m/z 267.0989 [M + Na]+ (calcd for C15H16NaO3, 267.0992).
(5Z)-3-Hydroxy-4-phenyl-5-(phenylmethylene)-2(5H)-furanone (5): yellow solid; UV (MeOH, PDA): λmax 336, 263, 245 nm; 1H and 13C NMR data, see Table 1; HRESIMS m/z 265.0861 [M + H]+ (calcd for C17H13O3, 265.0859).
Bulgariline D (6): yellow solid; [α]25D +26 (c 0.20, MeOH); UV (MeOH, PDA): λmax 287, 217 nm; ECD (MeCN, λ [nm] (Δε), c 0.201 mM): 296sh (+2.70), 281 (+3.94), 253sh (−0.95), 249 (−1.18), 231 (+2.99), 221 (−3.22), 218sh (−2.90), 204sh (+7.03); 1H and 13C NMR data, see Table 1; HRESIMS m/z 249.0757 [M + H]+ (calcd for C13H13O5, 249.0757).
Bulgarixine A (7): yellow solid; [α]25D 0 (c 0.11, MeOH); UV (MeOH, PDA): λmax 243, 206 nm; 1H and 13C NMR data, see Table 3; HRESIMS m/z 248.0919 [M + H]+ (calcd for C13H14NO4, 248.0917).
Bulgarixine B (8): yellow solid; [α]25D 0 (c 0.20, MeOH); UV (MeOH, PDA): λmax 244, 206 nm; 1H and 13C NMR data, see Table 3; HRESIMS m/z 234.0760 [M + H]+ (calcd for C12H12NO4, 234.0761).
Bulgariapyrone A (9): brown solid; [α]25D −48 (c 0.20, MeOH); UV (MeOH, PDA): λmax 300, 206 nm; 1H and 13C NMR data, see Table 4; HRESIMS m/z 313.2013 [M + H]+ (calcd for C17H29O5, 313.2010).
Bulgariapyrone B (10): brown solid; [α]25D −72 (c 0.40, MeOH); UV (MeOH, PDA): λmax 299, 211 nm; 1H and 13C NMR data, see Table 4; HRESIMS m/z 357.1919 [M + H]+ (calcd for C18H29O7, 357.1908).
Bulgariapyrone C (11): brown solid; [α]25D −70 (c 0.20, MeOH); UV (MeOH, PDA): λmax 300, 206 nm; 1H and 13C NMR data, see Table 4; HRESIMS m/z 371.2070 [M + H]+ (calcd for C19H31O7, 371.2064).
Bulgariapyrone D (12): brown solid; [α]25D −90 (c 0.20, MeOH); UV (MeOH, PDA): λmax 298, 212 nm; 1H and 13C NMR data, see Table 4; HRESIMS m/z 329.1960 [M + H]+ (calcd for C17H29O6, 329.1959).
Bulgariapyrone E (13): brown solid; [α]25D −32 (c 0.20, MeOH); UV (MeOH, PDA): λmax 299, 208 nm; 1H and 13C NMR data, see Table 4; HRESIMS m/z 625.3587 [M + H]+ (calcd for C33H53O11, 625.3582).
Bulgarialactone B (14): dark red solid; [α]20D +344 (c 0.10, CHCl3); UV (MeOH, PDA): λmax 441, 322 nm; ECD (MeCN, λ [nm] (Δε), c 0.147 mM): 470sh (+14.51), 437 (+22.20), 417sh (+18.22), 352sh (−2.59), 323sh (−14.77), 315 (−15.84), 269 (+5.71), 232 (−12.87); HRESIMS m/z 453.1910 [M + H]+ (calcd for C26H29O7, 453.1908).
(−)-(S)-Flavipesin B (16): yellow solid; [α]25D −72 (c 0.20, MeOH); UV (MeOH, PDA): λmax 290, 204 nm; 1H and 13C NMR data, see ESI;† HRESIMS m/z 325.1073 [M + H]+ (calcd for C19H17O5, 325.1071).
Mosher ester analysis of 12 and 13
Both (S)- and (R)-MTPA esters of 12 were prepared in NMR tubes by the addition of either (R)-MTPA-Cl (10 μL, 53.44 μmol) or (S)-MTPA-Cl (10 μL, 53.44 μmol) to a solution of 12 (1.0 mg, 3.05 μmol) and pyridine-d5 (10 μL, 130.75 μmol) in 100 μL CDCl3, according to a protocol described earlier.71 Each reaction mixture was maintained for 3 h at room temperature, and 500 μL CDCl3 was added afterwards. In a similar manner, (S)- and (R)-MTPA esters of 13 were prepared. Ester products were confirmed by LC-ESIMS at m/z 761 [M + H]+ for 12 and by MALDI-MS at m/z 1295 [M + Na]+ for 13.(S)-MTPA ester of 12 (12a): 1H NMR (CDCl3, 600 MHz) δH 6.19 (1H, s, H-5), 5.63 (1H, dd, J = 7.5, 5.3 Hz, H-1′), 4.30 (1H, m, Hb-9′), 4.29 (2H, s, H2-1′′), 4.26 (1H, m, Ha-9′), 3.80 (3H, s, 4-OMe), 3.52 (3H, s, OMe), 3.48 (3H, s, OMe), 3.36 (3H, s, 1′′-OMe), 1.88 (2H, m, H2-2′), 1.64 (2H, p, J = 6.8 Hz, H2-8′), 1.19 (2H, m, H2-3′), 1.25 (2H, m, H2-7′), 1.13–1.25 (6H, m, H2-4′–H2-6′).
(R)-MTPA ester of 12 (12b): 1H NMR (CDCl3, 600 MHz) δH 5.99 (1H, s, H-5), 5.69 (1H, dd, J = 7.3, 5.0 Hz, H-1′), 4.31 (1H, m, Hb-9′), 4.27 (2H, s, H2-1′′), 4.27 (1H, m, Ha-9′), 3.68 (3H, s, 4-OMe), 3.54 (3H, s, OMe), 3.52 (3H, s, OMe), 3.35 (3H, s, 1′′-OMe), 1.91 (2H, m, H2-2′), 1.65 (2H, p, J = 6.8 Hz, H2-8′), 1.26 (2H, m, H2-3′) 1.25 (2H, m, H2-7′) 1.17–1.30 (6H, m, H2-4′–H2-6′).
(S)-MTPA ester of 13 (13a): 1H NMR (CDCl3, 600 MHz) δH 6.20 (1H, s, H-5/H-5′′′), 6.18 (1H, s, H-5′′′/H-5), 5.63 (2H, m, H-1′, H-1′′′′), 4.31 (2H, s, H2-1′′′′′), 4.31 (1H, m, Hb-9′′′′) 4.30 (2H, s, H2-1′′), 4.27 (1H, m, Ha-9′′′′) 3.82 (3H, s, 4-OMe/4′′′-OMe), 3.80 (3H, s, 4′′′-OMe/4-OMe), 3.53 (3H, s, OMe), 3.49 (6H, s, OMe), 3.47 (2H, t, J = 6.8 Hz, H2-9′), 3.37 (3H, s, 1′′-OMe), 1.90 (4H, m, H2-2′, H2-2′′′′), 1.65 (2H, p, J = 6.7 Hz, H2-8′/H2-8′′′′), 1.54 (2H, p, J = 6.7 Hz, H2-8′′′′/H2-8′), 1.27 (4H, m, H2-7′, H2-7′′′′), 1.21 (4H, m, H2-3′, H2-3′′′′), 1.16–1.25 (12H, m, H2-4′–H2-6′, H2-4′′′′–H2-6′′′′).
(R)-MTPA ester of 13 (13b): 1H NMR (CDCl3, 600 MHz) δH 5.99 (2H, s, H-5, H-5′′′), 5.68 (2H, m, H-1′, H-1′′′′), 4.31 (1H, m, Hb-9′′′′), 4.30 (2H, s, H2-1′′), 4.27 (1H, m, Ha-9′′′′), 4.28 (2H, s, H2-1′′′′′), 3.68 (3H, s, 4-OMe/4′′′-OMe), 3.67 (3H, s, 4′′′-OMe/4-OMe), 3.54 (6H, s, OMe), 3.53 (3H, s, OMe), 3.45 (2H, t, J = 6.8 Hz, H2-9′), 3.36 (3H, s, 1′′-OMe), 1.91 (4H, m, H2-2′, H2-2′′′′), 1.66 (2H, p, J = 6.7 Hz, H2-8′/H2-8′′′′), 1.54 (2H, p, J = 6.7 Hz, H2-8′′′′/H2-8′), 1.29 (4H, m, H2-3′, H2-3′′′′), 1.27 (4H, m, H2-7′, H2-7′′′′), 1.16–1.25 (12H, m, H2-4′–H2-6′, H2-4′′′′–H2-6′′′′).
Cytotoxicity assay
Cytotoxicity was assayed against the murine lymphoma cell line L5178Y, using the MTT method.62 Kahalalide F was used as a positive control and a medium containing 0.1% DMSO was included as a negative control.Computational section
Mixed torsional/low-mode conformational searches were carried out by using the Macromodel 10.8.011 software72 with the Merck Molecular Morce Field (MMFF) with an implicit solvent model for CHCl3 applying a 21 kJ mol−1 energy window. Geometry optimizations [B3LYP/6-31+G(d,p) in vacuo and CAM-B3LYP/TZVP73 with the PCM solvent model for MeCN] and TDDFT [B3LYP/TZVP, BH&HLYP/TZVP, CAM-B3LYP/TZVP and PBE0/TZVP with the same or no solvent model as in the preceding optimization step] calculations were performed with Gaussian 09.74 The ECD spectra were generated as the sum of the Gaussians with 3000 and 4200 cm−1 half-height widths using dipole-velocity-computed rotational strength values.75 Boltzmann distributions were estimated from the B3LYP and the CAM-B3LYP energies. The MOLEKEL software package was used for visualization of the results.76Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. P. A. wishes to thank DAAD Program Biodiversity and Health for the doctoral scholarship. P. P. wants to thank the Manchot Foundation for support. We further want to thank Dr Margareta Proksch for help during mistletoe collection. The research of the Hungarian authors was supported by the EU and cofinanced by the European Regional Development Fund under the project GINOP-2.3.2-15-2016-00008. We also thank the National Research, Development and Innovation Office (NKFI K120181 and PD121020). The Governmental Information-Technology Development Agency (KIFÜ) is acknowledged for CPU time.Notes and references
- A. L. Demain, J. Ind. Microbiol. Biotechnol., 2014, 41, 185–201 CrossRef CAS PubMed.
- C. R. Strader, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2011, 74, 900–907 CrossRef CAS PubMed.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- G. A. Strobel, Crit. Rev. Biotechnol., 2002, 22, 315–333 CrossRef CAS PubMed.
- G. A. Strobel, R. V. Miller, C. Martinez-Miller, M. M. Condron, D. B. Teplow and W. Hess, Microbiology, 1999, 145, 1919–1926 CrossRef CAS PubMed.
- J. Li, G. Strobel, J. Harper, E. Lobkovsky and J. Clardy, Org. Lett., 2000, 2, 767–770 CrossRef CAS PubMed.
- J. G. Ondeyka, G. L. Helms, O. D. Hensens, M. A. Goetz, D. L. Zink, A. Tsipouras, W. L. Shoop, L. Slayton, A. W. Dombrowski, J. D. Polishook, D. A. Ostlind, N. N. Tsou, R. G. Ball and S. B. Singh, J. Am. Chem. Soc., 1997, 119, 8809–8816 CrossRef CAS.
- D. Rönsberg, A. Debbab, A. Mándi, V. Vasylyeva, P. Böhler, B. Stork, L. Engelke, A. Hamacher, R. Sawadogo, M. Diederich, V. Wray, W. Lin, M. U. Kassack, C. Janiak, S. Scheu, S. Wesselborg, T. Kurtán, A. H. Aly and P. Proksch, J. Org. Chem., 2013, 78, 12409–12425 CrossRef PubMed.
- P. Böhler, F. Stuhldreier, R. Anand, A. K. Kondadi, D. Schlütermann, N. Berleth, J. Deitersen, N. Wallot-Hieke, W. Wu, M. Frank, H. Niemann, E. Wesbuer, A. Barbian, T. Luyten, J. B. Parys, S. Weidtkamp-Peters, A. Borchardt, A. S. Reichert, A. Peña-Blanco, A. J. García-Sáez, S. Itskanov, A. M. van der Bliek, P. Proksch, S. Wesselborg and B. Stork, Cell Death Dis., 2018, 9, 286 CrossRef PubMed.
- J. C. Lee, E. Lobkovsky, N. B. Pliam, G. Strobel and J. Clardy, J. Org. Chem., 1995, 60, 7076–7077 CrossRef CAS.
- A. Stierle, G. Strobel and D. Stierle, Science, 1993, 260, 214–216 CrossRef CAS PubMed.
- S. C. Puri, V. Verma, T. Amna, G. N. Qazi and M. Spiteller, J. Nat. Prod., 2005, 68, 1717–1719 CrossRef CAS PubMed.
- K. Ishiuchi, D. Hirose, T. Suzuki, W. Nakayama, W.-P. Jiang, O. Monthakantirat, J.-B. Wu, S. Kitanaka and T. Makino, J. Nat. Prod., 2018, 81, 1143–1147 CrossRef CAS PubMed.
- A. H. Aly, A. Debbab and P. Proksch, Appl. Microbiol. Biotechnol., 2011, 90, 1829–1845 CrossRef CAS PubMed.
- A. H. Aly, A. Debbab and P. Proksch, Pharmazie, 2013, 68, 499–505 CAS.
- K. Scherlach and C. Hertweck, Org. Biomol. Chem., 2009, 7, 1753–1760 RSC.
- G. Daletos, W. Ebrahim, E. Ancheeva, M. El-Neketi, W. Lin and P. Proksch, in Chemical Biology of Natural Products; Microbial Co-culture and OSMAC Approach as Strategies to Induce Cryptic Fungal Biogenetic Gene Clusters, ed. P. Grothaus, G. M. Cragg and D. J. Newman, CRC Press, Boca Raton, FL, USA, 2017, pp. 233–284 Search PubMed.
- H. B. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS.
- S. Kusari, C. Hertweck and M. Spiteller, Chem. Biol., 2012, 19, 792–798 CrossRef CAS PubMed.
- P. A. Paranagama, E. M. K. Wijeratne and A. A. L. Gunatilaka, J. Nat. Prod., 2007, 70, 1939–1945 CrossRef CAS PubMed.
- S. P. D. Senadeera, S. Wiyakrutta, C. Mahidol, S. Ruchirawat and P. Kittakoop, Org. Biomol. Chem., 2012, 10, 7220–7226 RSC.
- R. T. Hewage, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Phytochemistry, 2014, 108, 87–94 CrossRef CAS PubMed.
- L. Hammerschmidt, A. H. Aly, M. Abdel-Aziz, W. E. G. Müller, W. Lin, G. Daletos and P. Proksch, Bioorg. Med. Chem., 2015, 23, 712–719 CrossRef CAS PubMed.
- H. Wang, H. Dai, C. Heering, C. Janiak, W. Lin, R. S. Orfali, W. E. G. Müller, Z. Liu and P. Proksch, RSC Adv., 2016, 6, 81685–81693 RSC.
- H. Yamazaki, H. Rotinsulu, R. Narita, R. Takahashi and M. Namikoshi, J. Nat. Prod., 2015, 78, 2319–2321 CrossRef CAS PubMed.
- S. Sureram, C. Kesornpun, C. Mahidol, S. Ruchirawat and P. Kittakoop, RSC Adv., 2013, 3, 1781–1788 RSC.
- B. Wu, X. Wu, M. Sun and M. Li, Mar. Drugs, 2013, 11, 2713–2721 CrossRef PubMed.
- P. Ye, L. Shen, W. Jiang, Y. Ye, C.-T. A. Chen, X. Wu, K. Wang and B. Wu, Mar. Drugs, 2014, 12, 3203–3217 CrossRef PubMed.
- M. Stadler, H. Anke, K. Dekermendjian, R. Reiss, O. Sterner and R. Witt, Nat. Prod. Lett., 1995, 7, 7–14 CrossRef CAS.
- L. Musso, S. Dallavalle, L. Merlini, A. Bava, G. Nasini, S. Penco, G. Giannini, C. Giommarelli, A. De Cesare, V. Zuco, L. Vesci, C. Pisano, F. Dal Piaz, N. De Tommasi and F. Zunino, Bioorg. Med. Chem., 2010, 18, 6031–6043 CrossRef CAS PubMed.
- R. L. Edwards and H. J. Lockett, J. Chem. Soc., Perkin Trans. 1, 1976, 2149–2155 RSC.
- N. Li, J. Xu, X. Li and P. Zhang, Fitoterapia, 2013, 84, 85–88 CrossRef CAS PubMed.
- J. W. van Dijk, C.-J. Guo and C. C. C. Wang, Org. Lett., 2016, 18, 6236–6239 CrossRef CAS PubMed.
- E. Hühner, K. Öqvist and S.-M. Li, Org. Lett., 2019, 21, 498–502 CrossRef PubMed.
- A. O. Brachmann, S. Forst, G. M. Furgani, A. Fodor and H. B. Bode, J. Nat. Prod., 2006, 69, 1830–1832 CrossRef CAS PubMed.
- E. Ancheeva, L. Küppers, S. H. Akone, W. Ebrahim, Z. Liu, A. Mándi, T. Kurtán, W. Lin, R. Orfali, N. Rehberg, R. Kalscheuer, G. Daletos and P. Proksch, Eur. J. Org. Chem., 2017, 2017, 3256–3264 CrossRef CAS.
- N. Tabata, T. Sunazuka, H. Tomoda, T. Nagamitsu, Y. Iwai and S. Omura, J. Antibiot., 1993, 46, 762–769 CrossRef CAS PubMed.
- Z. Lin, L. Marett, R. W. Hughen, M. Flores, I. Forteza, M. A. Ammon, G. P. Concepcion, S. Espino, B. M. Olivera, G. Rosenberg, M. G. Haygood, A. R. Light and E. W. Schmidt, Bioorg. Med. Chem. Lett., 2013, 23, 4867–4869 CrossRef CAS PubMed.
- A. N. Pearce, E. W. Chia, M. V. Berridge, E. W. Maas, M. J. Page, V. L. Webb, J. L. Harper and B. R. Copp, J. Nat. Prod., 2007, 70, 111–113 CrossRef CAS PubMed.
- J. Sikorska, S. Parker-Nance, M. T. Davies-Coleman, O. B. Vining, A. E. Sikora and K. L. McPhail, J. Nat. Prod., 2012, 75, 1824–1827 CrossRef CAS PubMed.
- Scifinder®, Chemical Abstracts Service, Columbus, OH, 2019, RN 73764-04-2 (accessed March 23, 2019).
- Z. Lin, M. Koch, M. H. Abdel Aziz, R. Galindo-Murillo, M. D. Tianero, T. E. Cheatham, L. R. Barrows, C. A. Reilly and E. W. Schmidt, Org. Lett., 2014, 16, 4774–4777 CrossRef CAS PubMed.
- M. C. Kim, J. H. Lee, B. Shin, L. Subedi, J. W. Cha, J.-S. Park, D.-C. Oh, S. Y. Kim and H. C. Kwon, Org. Lett., 2015, 17, 5024–5027 CrossRef CAS PubMed.
- P. Fu, S. La and J. B. MacMillan, J. Nat. Prod., 2015, 79, 455–462 CrossRef PubMed.
- A. N. Cayley, K. A. Gallagher, C. Menard-Moyon, J. P. Schmidt, L. J. Diorazio and R. J. Taylor, Synthesis, 2008, 2008, 3846–3856 CrossRef.
- Y. Zhou, Y. Zhai, X. Ji, G. Liu, E. Feng, D. Ye, L. Zhao, H. Jiang and H. Liu, Adv. Synth. Catal., 2010, 352, 373–378 CrossRef CAS.
- G. S. Kim, W. Ko, J. W. Kim, M.-H. Jeong, S.-K. Ko, J.-S. Hur, H. Oh, J.-H. Jang and J. S. Ahn, J. Nat. Prod., 2018, 81, 1084–1088 CrossRef CAS PubMed.
- D. R. McMullin, T. K. Nsiama and J. D. Miller, J. Nat. Prod., 2014, 77, 206–212 CrossRef CAS PubMed.
- P. Chomcheon, S. Wiyakrutta, N. Sriubolmas, N. Ngamrojanavanich, C. Mahidol, S. Ruchirawat and P. Kittakoop, Phytochemistry, 2009, 70, 121–127 CrossRef CAS PubMed.
- H. Fujimoto, Y. Satoh, M. Nakayama, T. Takayama and M. Yamazaki, Chem. Pharm. Bull., 1995, 43, 547–552 CrossRef CAS PubMed.
- Y. I. Oh, N. A. Kim, Y. H. Kim, T. H. Lee and Y. S. Lee, Saengyak Hakhoechi, 2013, 44, 209–219 CAS.
- P. J. L. Bell and P. Karuso, J. Am. Chem. Soc., 2003, 125, 9304–9305 CrossRef CAS PubMed.
- S. Superchi, P. Scafato, M. Gorecki and G. Pescitelli, Curr. Med. Chem., 2018, 25, 287–320 CrossRef CAS PubMed.
- A. Mandi, I. W. Mudianta, T. Kurtan and M. J. Garson, J. Nat. Prod., 2015, 78, 2051–2056 CrossRef CAS PubMed.
- O. A. Syzgantseva, V. Tognetti, L. Joubert, A. Boulangé, P. A. Peixoto, S. Leleu and X. Franck, J. Phys. Chem. A, 2012, 116, 8634–8643 CrossRef CAS PubMed.
- W. Ebrahim, A. H. Aly, A. Mándi, F. Totzke, M. H. Kubbutat, V. Wray, W. H. Lin, H. Dai, P. Proksch, T. Kurtán and A. Debbab, Eur. J. Org. Chem., 2012, 2012, 3476–3484 CrossRef CAS.
- Scifinder®, Chemical Abstracts Service, Columbus, OH, 2019, RN 371163-96-1 (accessed March 23, 2019).
- Z.-Q. Bai, X. Lin, Y. Wang, J. Wang, X. Zhou, B. Yang, J. Liu, X. Yang, Y. Wang and Y. Liu, Fitoterapia, 2014, 95, 194–202 CrossRef CAS PubMed.
- H. Morishima, K. Fujita, M. Nakano, S. Atsumi, M. Ookubo, M. Kitagawa, H. Matsumoto, A. Okuyama and T. Okabe, Japanese Kokai Tokkyo Koho JP, 1994, 6100445.
- K. Nitta, N. Fujita, T. Yoshimura, K. Arai and Y. Yamamoto, Chem. Pharm. Bull., 1983, 31, 1528–1533 CrossRef CAS.
- J. W. van Dijk and C. C. C. Wang, Org. Lett., 2018, 20, 5082–5085 CrossRef CAS PubMed.
- H. Chen, G. Daletos, M. S. Abdel-Aziz, D. Thomy, H. Dai, H. Brötz-Oesterhelt, W. Lin and P. Proksch, Phytochem. Lett., 2015, 12, 35–41 CrossRef CAS.
- M. P. Singh, F. Kong, J. E. Janso, D. A. Arias, P. A. Suarez, V. S. Bernan, P. J. Petersen, W. J. Weiss, G. Carter and M. Greenstein, J. Antibiot., 2003, 56, 1033–1044 CrossRef CAS PubMed.
- C.-L. Sun, R.-F. Pang, H. Zhang and M. Yang, Bioorg. Med. Chem. Lett., 2005, 15, 3257–3262 CrossRef CAS PubMed.
- D. Zhang, X. Li, J.-S. Kang, H.-D. Choi and B.-W. Son, Bull. Korean Chem. Soc., 2007, 28, 887–888 CrossRef CAS.
- R. R. Parvatkar, C. D'Souza, A. Tripathi and C. G. Naik, Phytochemistry, 2009, 70, 128–132 CrossRef CAS PubMed.
- K. Zhou, L. Zhu, X. Wang, T. Zhang, Y. Wang, W. Dong, B. Ji, H. Yang, G. Du, Q. Hu and M. Zhou, Chem. Nat. Compd., 2016, 52, 591–594 CrossRef CAS.
- X. Pang, J.-Y. Zhao, X.-M. Fang, T. Zhang, D.-W. Zhang, H.-Y. Liu, J. Su, S. Cen and L.-Y. Yu, J. Nat. Prod., 2017, 80, 2595–2601 CrossRef CAS PubMed.
- W.-J. Wang, D.-Y. Li, Y.-C. Li, H.-M. Hua, E.-L. Ma and Z.-L. Li, J. Nat. Prod., 2014, 77, 1367–1371 CrossRef CAS PubMed.
- J. Kjer, A. Debbab, A. H. Aly and P. Proksch, Nat. Protoc., 2010, 5, 479 CrossRef CAS PubMed.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451 CrossRef CAS PubMed.
- Macromodel, Schrödinger, LLC, 2015, http://www.schrodinger.com/MacroModel.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
- U. Varetto, MOLEKEL, v. 5.4, Swiss National Supercomputing Centre, Manno, Switzerland, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC chromatograms, UV, HRESIMS, NMR spectra of 1–13, 16, structure and population of the low-energy conformers of (S)-1, classification of the 22 low-energy conformers of (8R,9S)-4, experimental ECD spectrum of 6 compared with the Boltzmann-weighted PBE0/TZVP PCM/MeCN ECD spectrum of (R)-6, and classification of the 26 low-energy conformers of (3S,11S,23S)-14 into conformer groups. See DOI: 10.1039/c9ra03678d |
| ‡ Present address: G. Daletos, Department of Chemical Engineering, Massachusetts Institute of Technology, 25 Ames Street, Cambridge, Massachusetts 02139, United States. |
| This journal is © The Royal Society of Chemistry 2019 |