
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGinsenoside Rk1 inhibits cell proliferation and promotes apoptosis in lung squamous cell carcinoma by calcium signaling pathway
Xining Anabc,
Rongzhan Fuabc,
Pei Maabc,
Xiaoxuan Ma *abc and
Daidi Fan*abc
*abc and
Daidi Fan*abc
aShaanxi Key Laboratory of Degradable Biomedical Materials, School of Chemical Engineering, Northwest University, 229 North Taibai Road, Xi'an, Shaanxi 710069, China. E-mail: xiaoxuanma@nwu.edu.cn; fandaidi@nwu.edu.cn; Fax: +86-29-8830-5118; Tel: +86-29-8830-5118
bShaanxi R&D Center of Biomaterials and Fermentation Engineering, School of Chemical Engineering, Northwest University, 229 North Taibai Road, Xi'an, Shaanxi 710069, China
cBiotech. & Biomed. Research Institute, Northwest University, 229 North Taibai Road, Xi'an, Shaanxi 710069, China
First published on 13th August 2019
Abstract
Ginsenoside Rk1 (Rk1) is a rare saponin extracted from Sun Ginseng (SG) and has been shown to have an anti-tumor effect; however, the potential role of its in lung squamous cell carcinoma remains elusive. In this study, we investigated the anti-proliferative activity and involved mechanism of Rk1 against lung squamous cell carcinoma in vitro and in vivo. First, MTT assay, cell colony formation assay and cell cycle assay showed that Rk1 effectively inhibited cell proliferation and colony formation, and induced cell arrest at G1 phase. Following AV/PI staining, JC-10 staining, Western blot and immunohistochemistry indicated that Rk1 induced caspase-dependent apoptosis. In addition, Rk1 induced ER stress, causing the release of Ca2+, resulting in intracellular calcium and mitochondrial calcium overload. Intracellular calcium overload activated the calpain–caspase-12 and calpain–caspase-7–PARP pathways, while mitochondrial calcium overload caused mitochondrial membrane potential reduced, and the release of cytochrome c. BAPTA-AM (Ca2+ scavengers) and calpeptin (calpain inhibitors) significantly attenuated Rk1-induced apoptosis. Moreover, Rk1 significantly inhibited the growth of SK-MES-1 xenograft tumors with low toxic side effects. In summary, this study for the first time demonstrated that Rk1 had significant antitumor effects against lung squamous cell carcinoma and great potential to serve as a novel anticancer agent.
1. Introduction
Lung cancer is a type of malignant tumor with the fastest growth in morbidity and mortality and threatens human life.1 Non-small cell lung cancer (NSCLC) accounts for about 80–85% of all lung cancer cases, and is mainly divided into lung squamous cell carcinoma, adenocarcinoma, and large cell carcinoma.2 Among these three main types, lung squamous cell carcinoma accounts for 40% to 50% of NSCLC, and has a close relationship with smoking.3,4 Patients with advanced non-small cell lung cancer have extremely low 5 year survival rate of approximately 15%.5 Chemotherapy is the most commonly used treatments for patients with lung squamous cell carcinoma, however it causes a series of toxic side effects, such as bone marrow suppression, liver damage, immunosuppression, nephrotoxicity and neurotoxicity, which will seriously affect the quality of life of patients.6 Therefore, it is essential to study new types of anti-tumor drug with low side effects against lung squamous cell carcinoma.Ginseng, the dried root of Panax ginseng C. A. Mey., has been used as a traditional medicine for thousands of years in the Eastern countries and is now recognized as one of the world's most admitted herbal medicines.7 It serves as treatments for a variety of diseases, such as central nervous system disorders, fatigue, cardiovascular system damage, liver and kidney function damage, metabolic disorders.8 Ginsenoside is the main active ingredient in ginseng,9 which has a wide range of pharmacological activities,10 including immunity enhancing,11 anti-tumor,12 anti-inflammatory,13 and hemolysis preventing effects.14 The ginsenoside can be converted into a rare ginsenoside by degrading a partial glycosyl group of ginsenoside or changing its side chain structure.15 Studies have shown that the medicinal activity of rare ginsenoside is more significant than that of ginsenoside.16
Ginsenoside Rk1 is a rare ginsenoside extracted from Sun Ginseng (SG),17 and its structure is shown in Fig. 1A. Rk1 has been reported to have anti-tumor effects against liver cancer and melanoma. Young-Joo Kim et al. reported that ginsenoside Rk1 induces apoptosis in human hepatoma cells by inhibiting telomerase activity.18 Ji Seong Kim et al. researched the anti-tumor effect of ginsenoside Rk1 on melanoma cell SK-MEL-2.19 However, few studies have explored the antitumor activity of Rk1 on lung cancer, especially lung squamous cell carcinoma. And the anti-lung squamous cell carcinoma action and functional mechanism of ginsenoside Rk1 remain unclear.
 | ||
| Fig. 1 Rk1 significantly suppressed lung squamous cell carcinoma growth in vitro. (A) The chemical structure of the ginsenoside Rk1. (B) The cell viability of SK-MES-1 and H226 cells was measured via the MTT assay after Rk1 treatment. (C) Giemsa-stained colonies were observed under an inverted microscope. Data are presented as the means ± SD of triplicate experiments, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control. | ||
In this study, we demonstrated for the first time the antitumor-activity of Rk1 against lung squamous cell carcinoma in vivo and in vitro. Its underlying mechanism of anticancer effects was further elucidated, involving endogenous apoptosis of calcium signaling.
2. Materials and methods
2.1 Chemical reagents
Rk1 (purity > 98%) was purchased from Puruifa Technology Development Co., Ltd (Chengdu, China). Gefitinib was purchased from AstraZeneca (Cheshire, UK). Alpha minimum Eagle's medium (α-MEM) and Roswell Park Memorial Institute (RPMI)-1640 cell culture media and penicillin/streptomycin were obtained from HyClone (LA, USA). Fetal bovine serum (FBS) was purchased from GIBCO (NY, USA). Giemsa staining solution was purchased from Xi'an Heart Biological Technology (Xi'an, China). Hoechst 33342 fluorescent dye and JC-10 mitochondrial membrane potential detection kit were obtained from Beyotime Institute of Biotechnology (Shanghai, China). Fluo-3/AM fluorescent dye and Rhod-2/AM fluorescent dye were purchased from Med Chem Express (NJ, USA). Z-VAD-FMK, 2-APB, BAPTA-AM and Calpeptin were purchased from Med Chem Express (NJ, USA). The primary antibodies against Bcl-2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000; cat. no. ab32124), and Bax (1:1000–1:10000; cat. no. ab32503) were obtained from Abcam (Cambridge, MA, USA). The primary antibodies against, p21 (1:1000; cat. no. 2947), CDK4 (1:1000; cat. no. 12790), CyclinD1 (1:1000; cat. no. 2978), p53 (1:1000; cat. no. 2527), Cytochrome c (1:1000; cat. no. 11940), cleaved caspase 3 (1:1000; cat. no. 9664), cleaved caspase 9 (1:1000; cat. no. 20750), calpain (1:1000; cat. no. 2556), caspase 12 (1:1000; cat. no. 2202), cleaved caspase 7 (1:1000; cat. no. 8438), cleaved PARP (1:1000; cat. no. 5625), GRP78 (1:1000; cat. no. 3177), CHOP (1:1000; cat. no. 5554), IRE1α (1:1000; cat. no. 3294), ATF6 (1:1000; cat. no. 65880) and β-actin (1:1000; cat. no. 4970) were obtained from Cell Signaling Technology (Danvers, MA, USA). Polyethylene glycol (PEG-400) was purchased from Longyuan Chemical (Zhengzhou, China).
:1000; cat. no. ab32124), and Bax (1:1000–1:10000; cat. no. ab32503) were obtained from Abcam (Cambridge, MA, USA). The primary antibodies against, p21 (1:1000; cat. no. 2947), CDK4 (1:1000; cat. no. 12790), CyclinD1 (1:1000; cat. no. 2978), p53 (1:1000; cat. no. 2527), Cytochrome c (1:1000; cat. no. 11940), cleaved caspase 3 (1:1000; cat. no. 9664), cleaved caspase 9 (1:1000; cat. no. 20750), calpain (1:1000; cat. no. 2556), caspase 12 (1:1000; cat. no. 2202), cleaved caspase 7 (1:1000; cat. no. 8438), cleaved PARP (1:1000; cat. no. 5625), GRP78 (1:1000; cat. no. 3177), CHOP (1:1000; cat. no. 5554), IRE1α (1:1000; cat. no. 3294), ATF6 (1:1000; cat. no. 65880) and β-actin (1:1000; cat. no. 4970) were obtained from Cell Signaling Technology (Danvers, MA, USA). Polyethylene glycol (PEG-400) was purchased from Longyuan Chemical (Zhengzhou, China).
2.2 Cell culture
Human lung squamous carcinoma cells (SK-MES-1 and H226) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). These cells were cultured in α-MEM and RPMI-1640 medium respectively supplemented with 10% FBS and 1% penicillin/streptomycin (Thermo Fisher Scientific, Inc.) in a 5% carbon dioxide atmosphere incubator at 37 °C.2.3 Cell viability assay
Cell viability was measured by MTT assay according to the manufacturer's protocols. SK-MES-1 and H226 cells were seeded into 96-well plates at a density of 1 × 104 cells per well, and cultured for 24 h. Cells were then treated with different concentrations of Rk1 (0, 50, 100, 150 and 200 μM) for 24 or 48 h. Subsequently, the supernatant was discarded, and a mixture of 50 μL MTT (5 mg mL−1 MTT in PBS solution) and 100 μL of the medium was incubated for 2 to 4 h. Finally, 150 μL of DMSO was added to each well and shaken on a shaker for 10 min until the formazan crystals were dissolved. Finally, at the wavelength of 490 nm, the absorbance was read with a microplate reader (Power Wave XS2, Bio-tek Instruments Inc., USA). All experiments were performed in triplicate.2.4 Colony formation assay
SK-MES-1 and H226 cells were seeded in 6-well plates (1000 cells per well), and the culture medium was changed every four days for about 14 days. When each colony exceeded 50 cells, cells were treated with different concentrations of Rk1 (50, 100 and 150 μM) solutions for 24 h. Subsequently, the medium was removed, the cells were washed twice with phosphate buffered saline (PBS), and the cells were fixed with 4% paraformaldehyde for 10 min, and then colonies were stained with Giemsa staining solution for 15 min according to the manufacturer's protocols. Finally, the number of colony cells was observed with a fluorescence microscope (Nikon, Tokyo, Japan). All experiments were performed in triplicate.2.5 Detection of cell cycle by flow cytometry
The effect of Rk1 on cell cycle distribution was examined by flow cytometry (Becton Dickson, CA) after propidium iodide (PI) staining. Briefly, SK-MES-1 and H226 cells were seeded into 6-well plates (2 × 105 cells per mL) and cultured for 24 hours. Cells were treated with different concentrations of Rk1 (0, 50, 100, 150 μM) for 24 h. Subsequently, the cells were digested, centrifuged, and collected, and the cells were fixed with ice-cold 75% ethanol, and placed in a refrigerator at 4 °C overnight. The ethanol was removed by washing with PBS, and the cells were co-treated with a mixture containing 0.5 mg mL−1 RNase A, and stained with 50 mg mL−1 PI. Finally, flow cytometry was used to measure the PI fluorescence of each nuclear. All experiments were performed in triplicate.2.6 Hoechst 33342 staining assay
Apoptotic status was observed by Hoechst 33342 staining. Briefly, SK-MES-1 and H226 cells were seeded into 96-well plates (1 × 104 per well), and cells were treated with different concentrations of Rk1 (0, 50, 100 and 150 μM) for 48 h. After discarding the supernatant, the cells were washed twice with PBS, and then 10 μg mL−1 of Hoechst 33342 was added to each well, and cultured at 37 °C for 15 min. Finally, cell staining was observed using a fluorescence microscope (Nikon, Tokyo, Japan) under UV irradiation. All experiments were performed in triplicate.2.7 Quantification of apoptosis using Annexin V/PI staining assay
SK-MES-1 and H226 cells (2 × 105 cells per mL) were plated in a 6-well plate and cells were treated with different concentrations of Rk1 (0, 50, 100 and 150 μM) for 24 h. The cells were then collected and suspended in binding buffer. After staining with a mixture of 5 μL Annexin V and 5 μL PI solution, the cells were incubated for 30 min in the dark at 37 °C. Finally, the stained cells were examined by flow cytometry. All experiments were performed in triplicate.2.8 Measurement of intracellular calcium levels
The [Ca2+]i level in the SK-MES-1 and H226 cells was determined by Fluo-3/AM fluorescence staining according to the manufacturer's protocols. SK-MES-1 and H226 cells were plated at a density of 2 × 105 cells per well in a 6-well plate and cultured for 24 h. Then incubated with vehicle (α-MEM or RPMI-1640) or Rk1 (50, 100, and 150 μM) for 24 h, cells were washed with PBS and incubated with 5 μM Fluo-3/AM for 30 min at 37 °C in the dark. The cells were then washed with PBS two times, incubated with PBS for 10 min at 37 °C. Finally, the stained cells were examined by flow cytometry. All experiments were performed in triplicate.2.9 Measurement of calcium levels in the mitochondria
The [Ca2+]m level in the SK-MES-1 and H226 cells was determined by Rhod-2/AM fluorescence staining according to the manufacturer's protocols. SK-MES-1 and H226 cells were plated at a density of 2 × 105 cells per well in a 6-well plate and cultured for 24 h. Then incubated with vehicle (α-MEM or RPMI-1640) or Rk1 (50, 100, and 150 μM) for 24 h, cells were washed with PBS and incubated with 10 μM Rhod-2/AM for 30 min at 37 °C in the dark. The cells were then washed with PBS two times, incubated with PBS for 10 min at 37 °C. Finally, the stained cells were examined by flow cytometry. All experiments were performed in triplicate.2.10 Measurement of the mitochondrial membrane potential (MMP)
MMP was measured by JC-10 assay kit (Beyotime, Jiangsu, China) according to the manufacturer's protocols. SK-MES-1 and H226 cells (2 × 105 cells per mL) were placed in 6-well plates, and cells were treated with different concentrations of Rk1 (0, 50, 100, and 150 μM) for 24 h, and then the cells were collected and resuspended. The cell suspension was incubated with JC-10 for 30 min at 37 °C and washed twice with PBS. When the mitochondrial membrane potential is high, JC-10 is an aggregate, which produces red fluorescence (excitation 490 nm, emission 570 nm); when the mitochondrial membrane potential decreases, JC-10 is a monomer, which produces green fluorescence (excitation 525 nm, emission 590 nm). Flow cytometry was used to detect the relative proportion of red-green fluorescence to measure the proportion of mitochondrial depolarization. All experiments were performed in triplicate.2.11 Western blot assay
The cells were lysed on ice for 30 min with a buffer containing RIPA and phenylmethylsulfonyl fluoride (PMSF). The lysate was then centrifuged at 12000 g for 20 min at 4 °C, and the protein concentration in the resulting supernatant was measured using a BCA Protein Assay Kit (Thermo Scientific, Fremont, CA, USA). 20 μg of protein were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. Next, the PVDF membrane was blocked with 5% skim milk for 2 h, then incubated with the desired primary antibody overnight, and then incubated with the secondary antibody for 1 h at room temperature. The immunocomplexes were finally analyzed using an ECL system (PerkinElmer, Waltham, MA, USA). All experiments were performed in triplicate.
2.12 Human lung squamous cell carcinoma xenograft mouse model
Four-week-old female nude mice (12 ± 2 g) were purchased from Hunan SJA Experimental Animal Co., Ltd. (Hunan, China). All experiments were performed in compliance with the Animal Ethics Procedures and Guidelines of the People's Republic of China and approved by the Animal Ethics Committee of Northwest University, Shaanxi, China (NWU-AWC-20180620M). The mice were housed under sterile conditions. After one week of adaptation of the mice, SK-MES-1 cells (5–6 × 106 cells each) were inoculated into the left axilla of the mice. Two weeks after the inoculation, tumors with large rice grains grew, indicating that the tumor was successfully inoculated. When the tumor volume reached 150–200 mm3, the mice were randomly divided into 4 groups (n = 5) according to tumor volume and body weight: control group (0.9% saline solution), RK1 treatment group (10 and 20 mg kg−1 d−1) and positive control Gefitinib treatment group (20 mg kg−1 d−1). The body weight and tumor volume of the mice were recorded every three days, and the tumor volume was calculated using the following equation: tumor volume = (length × width2) × 1/2. Twenty-one days after the administration, the mice were sacrificed, and the xenograft tumor and its major organs were taken out.2.13 Histopathology and immunohistochemistry
Fresh tissue specimens were immersed in a 10% formalin solution, then dehydrated, impregnated with wax, dewaxed and sectioned, and the section thickness was 4 μm. Tumors and organs (heart, lung, spleen, liver and kidney) were stained with hematoxylin and eosin (H&E). In addition, tumor specimens were immunostained with cleaved caspase 3. The sections were observed under an optical microscope (Leica, Germany).2.14 Statistical analysis
Data are shown as the means ± standard deviation (SD). EC50 values were analyzed by GraphPad software. Statistical differences between group means were assessed by one-way ANOVA and two-tailed t-tests using SPSS 19.0 software (IBM, Armonk, NY, USA). A p value of <0.05 was considered statistically significant.3. Results
3.1 Rk1 inhibited proliferation of lung squamous carcinoma cells in vitro
The anti-proliferative effects of Rk1 on lung squamous cell carcinoma cell lines SK-MES-1 and H226 were detected by MTT assay. The results indicate that Rk1 had cytotoxic effect and significantly reduced the survival rate of lung squamous cell carcinoma cells. After 48 h of treatment with 50, 100, 150 μM Rk1 (Fig. 1B), for SK-MES-1 cells, the cell death rates were 34.52 ± 2.37%, 59.78 ± 1.98% and 84.25 ± 2.12%, respectively. And the EC50 value is 82.235 ± 1.32 μM. For H226 cells, the cell death rates were 29.68 ± 2.49%, 55.88 ± 2.11% and 77.03 ± 1.96%. And the EC50 value is 85.401 ± 1.16 μM. Thus, in lung squamous cell carcinoma cells, cell viability was significantly reduced by Rk1 treatment in a concentration- and time-dependent manner.3.2 Rk1 inhibited the colony formation of lung squamous cell carcinoma
In the colony formation assay, the effect of Rk1 on the ability of single cells to form colony clusters was investigated in SK-MES-1 and H226 cells. The results showed that the number of colonies of SK-MES-1 and H226 cells was significantly reduced by Rk1 treatment in a concentration-dependent manner (Fig. 1C). In SK-MES-1 cells, the 150 μM Rk1 treatment reduced the number of colonies by about 80% compared to that of the control. Similar results were observed in H226 cells, and the number of colonies was significantly reduced by more than 75% at 150 μM Rk1 treated compared to the control group. Therefore, Rk1 was able to inhibit the formation of colonies of lung squamous cell carcinoma.3.3 Rk1 induced cell cycle arrest at G1 phase
To demonstrate the effect of Rk1 on the proliferation of lung squamous cell carcinoma cells was related to cell cycle distribution, flow cytometry was performed to examine the effect of RK1 on cell cycle distribution of SK-MES-1 and H226 cells (Fig. 2A). The results showed that the percentage of cells in the G1 phase of the two cells was increased after 24 h of Rk1 treatment, while the percentage of cells in the S phase and the G2 phase was gradually decreased. In SK-MES-1 cells, treatment with 150 μM Rk1 increased the percentage of cells in the G1 phase by 14.50 ± 0.23% and decreased the percentage of cells in the S phase by 20.20 ± 0.18% compared with the control. Similarly, treatment with 150 μM Rk1 induced a significant increase (22.03 ± 0.31%) in H226 cells in G1 phase and an obvious reduction (18.24 ± 0.26%) in cells in S phase compared with the control. The expression of cell cycle related proteins was further analyzed by Western blotting (Fig. 2B). The results showed that the protein expression levels of cyclin D1 and CDK4 were down-regulated, while the protein expression levels of p53 and p21WAF1/CIP1 were up-regulated in a dose-dependent manner. Therefore, Rk1 inhibited cell proliferation by regulating related proteins in G1 phase. | ||
| Fig. 2 Rk1 induced G1 cell cycle arrest in lung squamous cell carcinoma cell lines. (A) The cell cycle distribution was analyzed via flow cytometry. (B) Cells were treated with the indicated concentrations of Rk1 for 24 h, and the expression levels of G1 transition-related proteins were evaluated via immunoblot analysis. Values are presented as the means ± SD, n = 3; *p < 0.05, **p < 0.01 compared with the control. | ||
3.4 Rk1 induced apoptosis of lung squamous carcinoma cells
Hoechst 33342 staining and Annexin V/PI staining were performed to study the Rk1-induced SK-MES-1 and H226 cells apoptosis. Results of Hoechst 33342 staining showed that the number of cells was significantly reduced, the number of bright blue cells was elevated, and the cell membrane shrinkage with Rk1-treated in a dose-dependent manner (Fig. 3A). According to flow cytometry analysis, the early apoptotic rate of 150 μM Rk1-treated SK-MES-1 cells was increased by 41.54 ± 0.45% compared with the control for 24 h. Similar to these results, the early apoptotic rate of H226 cells was increased by 48.87 ± 0.52% (Fig. 3B). These results demonstrated that Rk1 was capable of inducing apoptosis in lung squamous cell carcinoma cells. | ||
| Fig. 3 Rk1 induced apoptosis in SK-MES-1 and H226 cells. (A) Hoechst 33342 staining was used to evaluate cell morphology. Luminous cells are indicative of apoptotic cells. Scale bars = 200 μm. (B) Apoptosis was analyzed via flow cytometry after Annexin V/PI staining. (C) Cells were treated with the indicated concentrations of Rk1 for 24 h, and the levels of apoptosis-related proteins were determined by Western blot. (D) Immunohistochemical staining was performed to analyze the expression level of cleaved caspase 3 in tumor tissues. Scale bars = 200 μm (E) SK-MES-1 cells were pretreated with 50 μM Z-VAD-fmk for 2 h and then treated with 100 μM Rk1 for 48 h. Cell inhibition was determined via MTT assay. Values are presented as the means ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control. | ||
To investigate a mechanism related to Rk1-induced apoptosis in lung squamous carcinoma cells, Western blotting was used to detect the expression of apoptosis-related proteins (Fig. 3C). The results showed that the expression of cleaved caspase-9, -3, Bax and cytosolic cytochrome c were upregulated, and Bcl-2 was downregulated by Rk1 in a concentration-dependent manner, indicating that the intrinsic pathway was involved in Rk1-induced apoptosis. Meanwhile, immunohistochemical analysis of tumor tissues showed that the average area of caspase 3 staining positive after Rk1 treatment was larger than that of the control (Fig. 3D). To further investigate the role of caspase in Rk1-induced apoptosis, SK-MES-1 and H226 cells were pretreated with Z-VAD-fmk (widespread caspase inhibitor) for 2 h and then treated with Rk1 for 24 h (Fig. 3E). As shown, Z-VAD-fmk significantly inhibited RK1-induced death of lung squamous cell carcinoma cells, which confirming that Rk1-induced apoptosis was caspase-dependent.
3.5 Rk1 induced ER stress to release calcium ions into the cytosol
ER stress is related to the induction of apoptosis as well as autophagy.20 To assess whether Rk1 induced ER stress, Western blotting was performed to detect proteins associated with ER stress (Fig. 4A). The results indicated that the protein levels of GRP78, ATF-6, and IRE1α were increased under Rk1 treatment. However, an increase in the protein levels of CHOP was not observed in either of the cell lines. These results indicate that Rk1 was capable of inducing ER stress.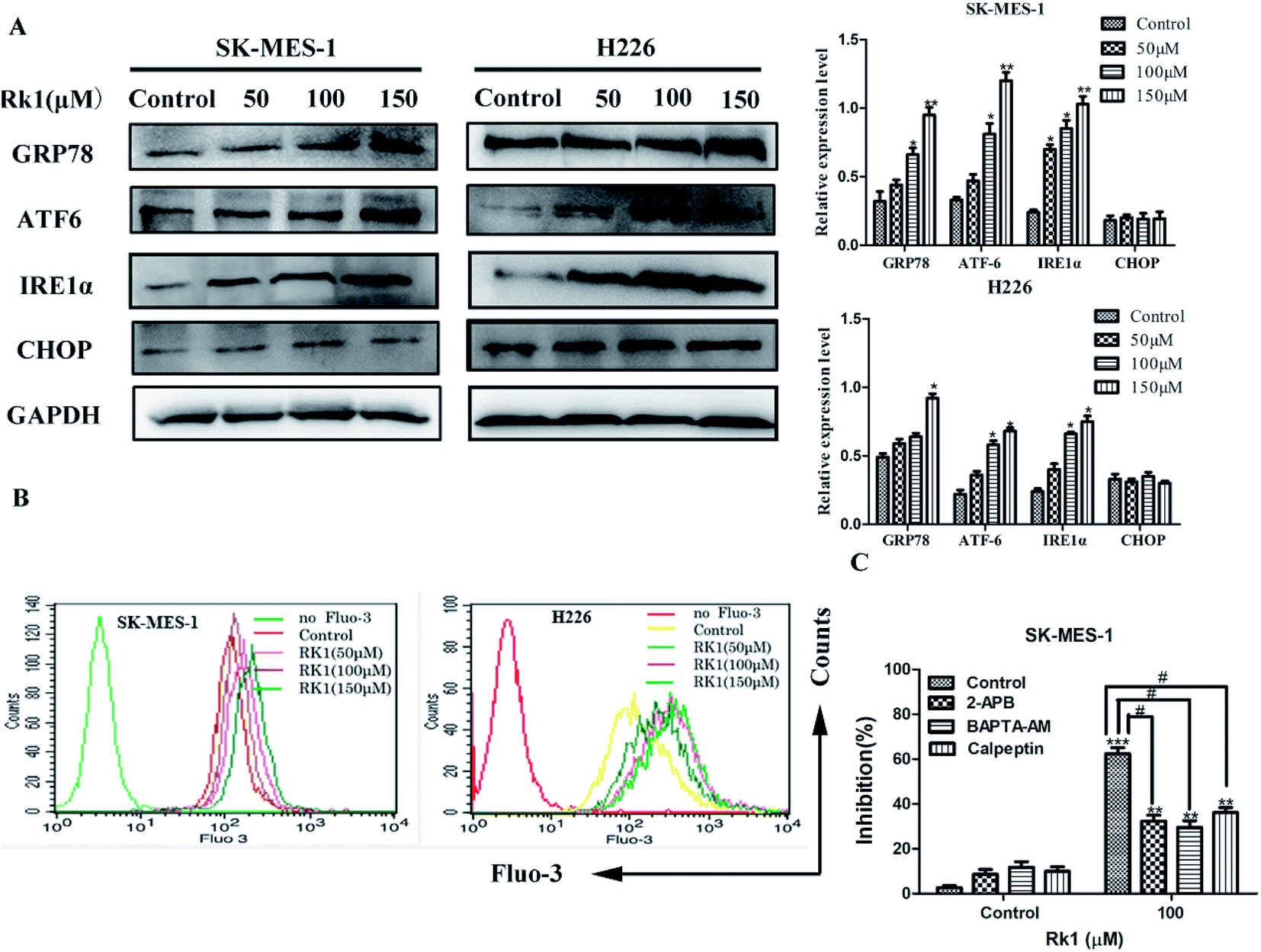 | ||
| Fig. 4 Rk1 induced endoplasmic reticulum stress to release calcium ions in SK-MES-1 and H226 cells. (A) Cells were treated with the indicated concentrations of Rk1 for 24 h. The levels of GRP78, ATF-6, IRE1α and CHOP were determined via Western blot. (B) Intracellular calcium levels were analyzed via flow cytometry after Fluo-3/AM staining. (C) SK-MES-1 cells were pretreated with 100 μM 2-APB (IP3Rs channel inhibitor), 40 μM BAPTA-AM (intracellular Ca2+ chelating agent) or 50 μM calpeptin (calpain inhibitor) for 2 h and then treated with 100 μM Rk1 for 24 h. Cell inhibition was determined via MTT. Values are presented as the means ± SD, n = 3; *p < 0.05, **p < 0.01 compared with the control, #p < 0.05, ##p < 0.01, ###p < 0.001 in contrast to the Rk1-treated group. | ||
During ER stress, calcium ions are released from the ER lumen, thereby increasing cytosolic calcium overload and subsequently causing apoptosis.21 First, SK-MES-1 and H226 cells were stained with Fluo-3/AM to detect changes of calcium levels in the cytoplasm (Fig. 4B). According to flow cytometry results, calcium levels in the cytoplasm of Rk1-treated SK-MES-1 and H226 cells were significantly increased in a dose-dependent manner. Pretreatment with BAPTA-AM (intracellular Ca2+ chelating agent) for 2 h, followed by Rk1 treatment for 48 h, MTT results showed a significant increase in the survival rate of SK-MES-1 cells, indicating that calcium ions were involved in the apoptosis induced by Rk1 (Fig. 4C). Since ER is the main calcium reservoir in the cell, the ER regulates calcium release and uptake through calcium-related membrane channels in the lumen of the ER.22 To determine whether Rk1-induced changes in calcium in SK-MES-1 and H226 cells were associated with ER calcium channels, inositol-1,4,5-triphosphate receptor (IP3Rs) on the ER calcium channel was blocked with 2-aminoethoxydiphenyl borate (2-APB) (Fig. 4C). As shown, 2-APB treatment significantly reduced Rk1-induced cell death. These data indicated that Rk1 induces ER stress, releasing Ca2+ into the cytoplasm through the IP3Rs channel, resulting in Ca2+ overload in the cytoplasm causing apoptosis.
3.6 Rk1 increased intracellular calcium levels to activate calpain-induced apoptosis
It is reported that excess calcium ions in the cells can bind to Ca2+-dependent enzymes, such as calpain, calcineurin activation, which activates downstream proteins to trigger apoptosis.23 The effect of Rk1 on calpain activation was examined by Western blotting. The Fig. 5A showed that Rk1 could increase the expression of calpain levels in a concentration-dependent manner. The calcium chelator BAPTA/AM blocked the Rk1-induced increase of calpain protein levels (Fig. 4C), indicating that Rk1-induced calpain activation was calcium-dependent. In addition, following the calpain inhibitor (calpeptin) pretreatment, Rk1 treatment for 48 h showed a significant increase in cell viability, confirming that Rk1-induced apoptosis is dependent on calpain. These results indicated that Rk1 induced an increase in intracellular calcium levels, resulting in calpain activation and subsequent apoptosis.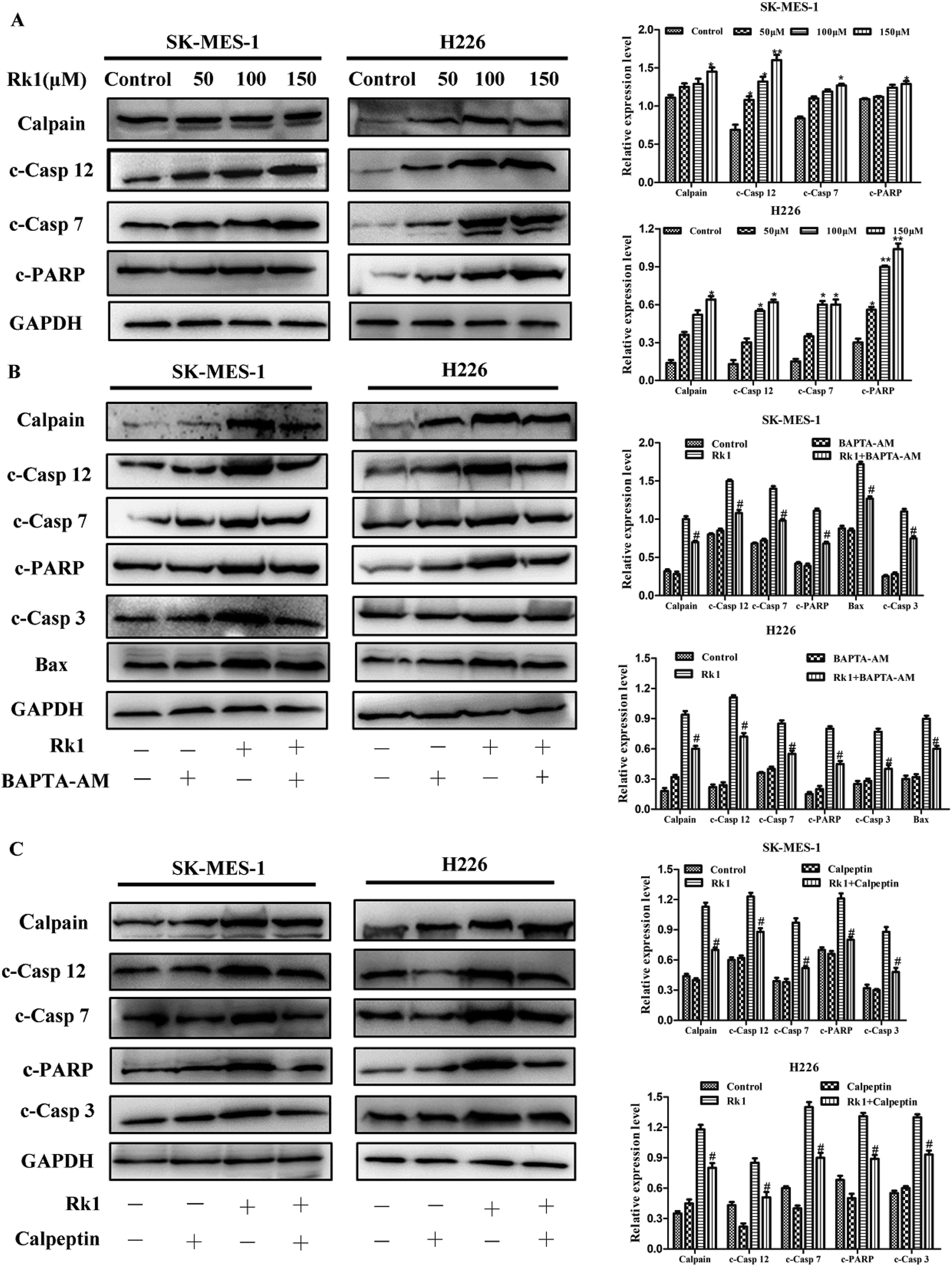 | ||
| Fig. 5 Rk1 increased intracellular calcium levels to activate calpain-induced apoptosis in SK-MES-1 and H226 cells. (A) Cells were treated with the indicated concentrations of Rk1 for 24 h. The levels of calpain, cleaved caspase 12, 7 and cleaved PARP were determined via Western blotting. (B) BAPTA-AM pretreated cells using the above methods, and calpain, cleaved caspase 12, 7, 3, cleaved PARP and Bax were analyzed by Western blotting. (C) Calpeptin pretreated cells using the above methods, and calpain, cleaved caspase -12, -7, -3 and cleaved PARP were analyzed by Western blotting. Values are presented as the means ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control, #p < 0.05, ##p < 0.01, ###p < 0.001 in contrast to the Rk1-treated group. | ||
Then the activation of calpain leads to the activation of caspases-12 and -7. Western blot results showed that Rk1 significantly increased caspase-12, -7, -3 and PARP cleavage levels in SK-MES-1 and H226 cells (Fig. 5A). These results indicated that Rk1 causes caspase-12, -7, -3 activation and PARP cleavage. To determine whether calcium and calpain are upstream of caspase 12–caspase 3 and caspase 7–PARP, SK-MES-1 and H226 cells were pretreated with BAPTA/AM or calpeptin for Western blot analysis (Fig. 5B and C). We found that the protein expression levels of cleavage caspase-12, -3, -7 and PARP were decreased compared with Rk1 treatment. These results indicated that the caspase 12–caspase 3 and caspase 7–PARP cascades are downstream cascades of calcium and calpain. Taken together, our data showed that Rk1 induced apoptosis in SK-MES-1 and H226 cells mainly through calcium-mediated calpain/caspase-12/caspase-3 and calcium-mediated calpain/caspase-7/PARP pathway.
3.7 Rk1 induced mitochondria-mediated apoptosis via mitochondrial Ca2+ overload
Continues increase of the calcium ion in the cytoplasm will induce the increase of calcium uptake in mitochondria and cause mitochondrial damage to initiate apoptosis.24 Firstly, changes in calcium levels in mitochondrial matrices were analyzed using Rhod-2/AM dyes and it was found that calcium levels in mitochondria were increased induced by Rk1 in a concentration-dependent manner (Fig. 6A). Then, we assessed whether Rk1 affected the MMP in lung squamous cell carcinoma cells by using JC-10 fluorescent dye (Fig. 6B). Flow cytometry results showed that the red/green fluorescence intensity ratio in SK-MES-1 and H226 cells under 150 μM Rk1 treatment was significantly lower (42.23% and 43.95%) than that of the control, indicating mitochondrial depolarization. | ||
| Fig. 6 Rk1 induced mitochondria-mediated apoptosis via mitochondrial Ca2+ overload in SK-MES-1 and H226 cells. (A) Mitochondrial calcium levels were analyzed via flow cytometry after Rhod-2/AM staining. (B) MMP was assessed with the fluorescent mitochondrial probe JC-10, and the red/green fluorescence intensity was analyzed via flow cytometry. (C) SK-MES-1 cells were pretreated with 40 μM BAPTA-AM (intracellular Ca2+ chelating agent) for 2 h and then treated with 100 μM Rk1 for 24 h. MMP was assessed with the fluorescent mitochondrial probe JC-10, and the red/green fluorescence intensity was analyzed via fluorescence microscopy. Scale bars = 200 μm. Values are presented as the means ± SD, n = 3. | ||
To verify whether mitochondrial depolarization was related to Ca2+ overload in the mitochondria, SK-MES-1 cells were pretreated with the calcium ion chelating agent BAPTA-AM and stained with JC-10 fluorescent dye (Fig. 6C). Fluorescence microscopy showed that red fluorescence increased and green fluorescence decreased in cells pretreated with BAPTA-AM compared to that of the 150 μM Rk1 treated group. These results indicated that Ca2+ overload in the mitochondria caused a decrease in MMP, resulting in mitochondrial damage.
Western blot analysis showed that the protein expression levels of cleavage of Bax and caspase-3 were both decreased by BAPTA/AM pretreatment compared with that of only Rk1 treatment group. Thus, Rk1 induces an increase in intracellular calcium ions, which in turn increases mitochondrial uptake of calcium, leading to mitochondrial damage-induced apoptosis.
3.8 Rk3 inhibited the growth of SK-MES-1 in vivo
A lung squamous cell carcinoma xenograft model was established by nude mice inoculated with SK-MES-1 cells. After 21 days of treatment, tumor growth was significantly inhibited in the Rk1 treated group compared to control group (Fig. 7A). The tumor inhibition rate (56.37%) at 20 mg kg−1 Rk1 treatment was close to that of the 20 mg kg−1 Gefitinib treatment group (61.95%). Interestingly, there was a significant difference in body weight between the Rk1 treated group and the Gefitinib treated group (Fig. 7B and C). The body weight of the 10 and 20 mg kg−1 Rk1 treatment groups gradually increased and approached that of the control group. Whereas the body weight of the Gefitinib treatment group was significantly reduced by 26.2 ± 2.5%. These results indicated that Rk1 had almost no toxic side effects compared with Gefitinib. | ||
| Fig. 7 Rk1 significantly suppressed lung squamous cell carcinoma growth in vivo. SK-MES-1 lung squamous cell carcinoma xenografts were treated with solvent or Rk1 (10 and 20 mg kg−1 d−1) or with Gefitinib (20 mg kg−1 d−1). (A) Representative image of SK-MES-1 xenograft tumors from the control group, Rk1-treated groups and Gefitinib-treated group. (B) Tumor volume and (C) body weight were measured every three days. (D) H&E staining of tumor specimens and major organs. Scale bars = 200 μm. | ||
H&E staining of tumor tissues and main organs was observed under light microscopy (Fig. 7D). Compared with the control, the number of tumor cells in the Rk1 treatment group was significantly reduced, the morphology of tumor cells was changed, and large areas of vacuoles in the tumor tissues were observed. Histological analysis of kidney, lung, spleen and heart showed no significant damage. These results confirmed that Rk1 has low toxicity.
4. Discussion
Ginsenoside Rk1 is one of the effective active ingredients in ginseng, and studies have shown that it has anti-tumor effect.25 However, the anti-tumor activity and the underlying mechanism against lung squamous cell carcinoma have been rarely reported. In this study, the anti-proliferation and anti-cancer mechanism of Rk1 against lung squamous cell carcinoma was investigated in vitro and in vivo, respectively.In vitro, MTT assay showed that proliferation of lung squamous carcinoma cells was significantly inhibited by Rk1 in a concentration- and time-dependent manner. After incubation with 150 μM Rk1 for 48 h, the inhibition rate of SK-MES-1 cells reached 84.25 ± 2.12%, and the inhibition rate of H226 cells reached 77.03 ± 1.96%. In addition, Rk1 suppressed the colony-forming ability of SK-MES-1 and H226 cells. In vivo, 10 mg kg−1 and 20 mg kg−1 Rk1 significantly inhibited tumor growth from the xenograft nude mouse model, with tumor inhibition rates of 24.15% and 56.37%, respectively. At the same time, the inhibition rate of Rk1 at 20 mg kg−1 was similar to that of the same dose of Gefitinib (61.95%). H&E staining showed that Rk1 significantly inhibited tumor cell growth and induced morphological changes compared to the control group. Both the control group and the Rk1 group had an increase in body weight without significant difference. Contrarily, the body weight of mice treated with Gefitinib was significantly reduced. Therefore, Rk1 exhibit anti-lung squamous cell carcinoma activity with low toxicity effects.
The abnormal activity of cell cycle proteins and the uncontrolled proliferation of tumors are the major features of cancer.26 Consequently, regulation of cell cycle proteins-induced cell cycle arrest is considered to be an attractive target for the treatment of cancer.27 Flow cytometry analysis showed that Rk1 caused G1 phase arrest in lung squamous cell carcinoma. The regulation of the cell cycle mainly depends on the periodic expression, accumulation and degradation of three proteins: cyclin-dependent kinases (CDKs), cyclins (Cyclins), and cyclin kinase inhibitors (CKIs).28 Among them, p53, p21, CDK4, and cyclinD1 proteins are important regulators of cyclins. The p53 and p21 proteins play an important role in the regulation of G1/S detection sites in the cell cycle; CDK4 and cyclinD1 proteins are key regulators that control the cell cycle from G1 to S phase.29 Our results showed that the protein levels of cyclin D1 and CDK4 were down-regulated and the protein levels of p21 and p53 were up-regulated. Here, Rk1 inhibition of SK-MES-1 and H226 cells proliferation was associated with G1 phase arrest.
Certain physiological and pathological conditions, such as hypoxia, oxidative stress and calcium ion imbalance, caused ER stress, the ER homeostasis imbalance.30 ER stress activates the unfolded protein response (UPR), which regulates the expression of ER molecular chaperone GRP78/BiP, the ER stress sensor protein IRE1α, XBP-1S, and ATF-6.31 In the present study, we found that Rk1 enhanced the expression of GRP78, IRE1α and ATF6 protein levels, which indicated that ginsenoside Rk1 could induced the occurrence of ER stress. According to previous reported, severe and sustained ER stress induces apoptosis.32 The major apoptotic pathway of ER stress is mediated by the CHOP/GADD153 gene.33 However, research showed that CHOP protein expression level did not be affected by Rk1 treatment. Therefore, ginsenoside Rk1 induced apoptosis of lung squamous cell carcinoma cells is not mediated through the CHOP pathway associated with ER stress.
The ER, a calcium reservoir, stores calcium ions in cells and is also involved in protein processing.34 It has been reported that under ER stress conditions, calcium is released into the cytoplasm through calcium-related membrane channels in the lumen of the ER, inducing calcium overload.21 Ca2+ plays a very important role in maintaining normal cell function, and acts as a second messenger in cells, regulating various biological functions.35 When intracellular Ca2+ is overloaded, it leads to the activation of pro-apoptotic factors and causes apoptosis.36,37 In the present study, we showed that intracellular calcium levels were elevated in SK-MES-1and H226 cells, mainly due to the release of Ca2+ from ER. Ca2+ release channel is mainly regulated by the inositol 1,4,5-triphosphate receptor (IP3R) on the ER membrane.38 SK-MES-1 and H226 cells were pretreated with IP3R receptor inhibitor 2-APB and found that the calcium ion release channel was blocked, intracellular calcium ions were decreased and resulted the cell death rate was reduced. In addition, the Ca2+ chelating agent BAPTA-AM was used to examine the role of [Ca2+]i in Rk1-induced apoptosis. After BAPTA-AM pretreatment, the cell survival rate was increased. These results indicated that Rk1 induced ER stress, which caused calcium ions in the endoplasmic reticulum to be released into the cytoplasm through the IP3R channel, disrupting intracellular calcium homeostasis, thereby promoting dysfunction and apoptosis of SK-MES-1 and H226 cells.
It has previously been reported that the release of Ca2+ activates calpain, which in turn activates apoptosis-related proteins. In this study, we found that the protein expression level of calpain was significantly increased by Rk1 treatment in a concentration-dependent manner. Previous studies have demonstrated that calpain activates downstream caspase-12 and caspase-7 proteins expressions, which in turn triggers apoptosis.39 The results of elevated levels of caspase-12, caspase-7, PARP, and caspase-3 protein by Rk1 treatment also confirmed this. At the same time, calpain, caspase-12, -7, PARP and caspase-3 protein expression levels were also reduced after BAPTA-AM or calpeptin (calpain inhibitor) pretreatment. These results suggested that Rk1 induced an increase in calcium ions and activated calpain, resulting in the activation of caspase-12 or caspase-7 and cleavage of PARP.
On the other hand, an increase in intracellular free calcium ions is taken up by mitochondria, causing calcium overload in the mitochondria, leading to mitochondrial dysfunction and apoptosis.40 In present study, mitochondrial calcium ions increased and MMP decreased induced by Rk1 in a concentration-dependent manner. When the MMP is lowered, the pro-apoptosis-related protein cytochrome c in the membrane gap, is released into the cytosol.41,42 Western blot analysis showed that the level of Cyto c protein gradually increased. We also found that pro-apoptotic factor Bax protein levels increased and anti-apoptotic factor Bcl-2 protein levels decreased. Pretreatment of cells with BAPTA-AM revealed an increase in mitochondrial membrane potential and a down-regulation of Bax protein expression. That is, Rk1 induced the release of calcium ions, caused calcium overload in the mitochondria, mitochondrial damage and triggered apoptosis.
5. Conclusions
Ginsenoside Rk1 had high anticancer activity against SK-MES-1 and H226 cells, and caused G1 arrest. We also demonstrated that Rk1 induced ER stress, allowing calcium ions to be released from ER into the cytoplasm through the IP3R channel, inducing apoptosis via calcium signaling. The major involved apoptotic mechanisms illustrated in Fig. 8 were the calcium-mediated calpain–caspase-12 pathway, the calcium-mediated calpain–caspase-7–PARP pathway, and the calcium-mediated mitochondria-dependent pathway. Therefore, Rk1 was expected to be a potent candidate of novel anticancer drugs for the treatment of lung squamous cell carcinoma.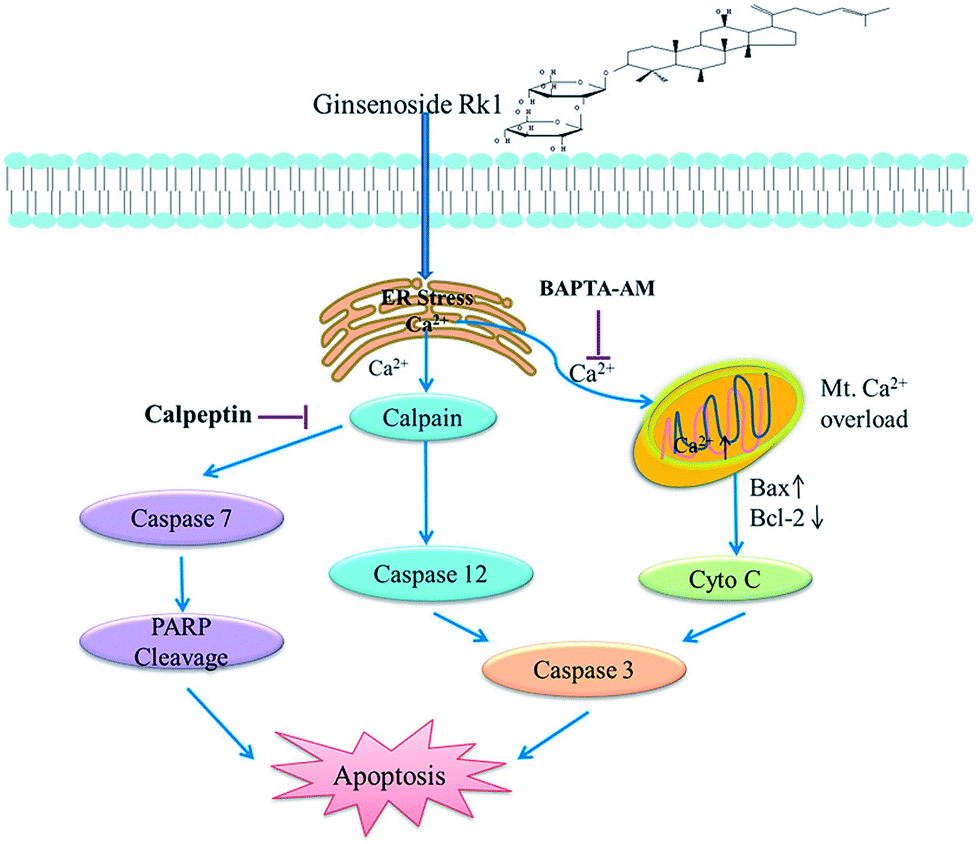 | ||
| Fig. 8 Schematic representation of the hypothesized molecular mechanism underlying the anti-cancer activity of Rk1. The calcium signaling pathway was involved in Rk1-induced apoptosis. | ||
Ethics approval and consent to participate
All experiments were performed in compliance with the Animal Ethics Procedures and Guidelines of the People's Republic of China and approved by the Animal Ethics Committee of Northwest University, Shaanxi, China (NWU-AWC-20180620M).Funding
This work was supported by the National Natural Science Foundation of China (21576223, 21878246, 21808184); Shaanxi Key Laboratory of Degradable Biomedical Materials Program (15JS106, 2016SZSj-35).Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
Not applicable.References
- A. Aggarwal, G. Lewison and S. Idir, et al., The State of Lung Cancer Research: A Global Analysis, J. Thorac. Oncol., 2016, 11, 1040–1050 CrossRef PubMed.
- L. Yan, Y. Yao, L. H. Wang, M. L. Wang and X. H. Fu, Detection of CK19, LUNX, and KS1/4 mRNA expression in the peripheral blood for diagnosis of micrometastases in patients with non-small cell lung cancer and their clinical implications, Genet. Mol. Res., 2015, 14, 15090–15095 CrossRef CAS PubMed.
- T. C. G. A. R. Network, Comprehensive genomic characterization of squamous cell lung cancers, Nature, 2012, 517, 576 CrossRef PubMed.
- G. V. Scagliotti, S. Novello, S. Rapetti and M. Papotti, Current State-of-the-Art Therapy for Advanced Squamous Cell Lung Cancer, American Society of Clinical Oncology Educational Book, 2013, vol. 354 Search PubMed.
- H. R. Sanders and M. Albitar, Somatic mutations of signaling genes in non-small-cell lung cancer, Cancer Genet. Cytogenet., 2010, 203, 7–15 CrossRef CAS PubMed.
- M. McCulloch, C. See and X. Shu, et al., Astragalus-Based Chinese Herbs and Platinum-Based Chemotherapy for Advanced Non–Small-Cell Lung Cancer: Meta-Analysis of Randomized Trials, J. Clin. Oncol., 2006, 24, 419 CrossRef CAS PubMed.
- K. S. Kang, H. Jungyeob, K. Young-Joo, P. J. Hill, C. Eun-Ju and Y. Noriko, Heat-processed Panax ginseng and diabetic renal damage: active components and action mechanism, J. Ginseng Res., 2013, 37, 379–388 CrossRef CAS PubMed.
- Y. Kimura, M. Sumiyoshi and M. Sakanaka, Effects of Ginsenoside Rb1 on Skin Changes, J. Biomed. Biotechnol., 2012, 2012, 946242 Search PubMed.
- C. N. Gillis, Panax ginseng pharmacology: a nitric oxide link?, Biochem. Pharmacol., 1997, 54, 1–8 CrossRef CAS PubMed.
- X. Song, J. Chen, K. Sakwiwatkul, R. Li and S. Hu, Enhancement of immune responses to influenza vaccine (H3N2) by ginsenoside Re, Int. Immunopharmacol., 2010, 10, 351–356 CrossRef CAS PubMed.
- L. L. Xu, T. Han and J. Z. Wu, et al., Comparative research of chemical constituents, antifungal and antitumor properties of ether extracts of Panax ginseng and its endophytic fungus, Phytomedicine, 2009, 16, 609–616 CrossRef CAS PubMed.
- K. T. Choi, Botanical characteristics, pharmacological effects and medicinal components of Korean Panax ginseng C A Meyer, Acta Pharmacol. Sin., 2008, 29, 1109–1118 CrossRef CAS PubMed.
- G. X. Li and Z. Q. Liu, The protective effects of ginsenosides on human erythrocytes against hemin-induced hemolysis, Food Chem. Toxicol., 2008, 46, 886–892 CrossRef CAS PubMed.
- S. Y. Nah, Ginseng ginsenoside pharmacology in the nervous system: involvement in the regulation of ion channels and receptors, Front. Physiol., 2014, 5, 98 Search PubMed.
- Y. J. Kim, D. Zhang and D. C. Yang, Biosynthesis and biotechnological production of ginsenosides, Biotechnol. Adv., 2015, 33, 717–735 CrossRef CAS PubMed.
- T. Chen, B. Li, Q. Ye, Z. Qiu and Q. Peng, Functional mechanism of Ginsenosides on tumor growth and metastasis, Saudi J. Biol. Sci., 2018, 25, 917–922 CrossRef CAS PubMed.
- I. H. Park, N. Y. Kim and S. B. Han, et al., Three new dammarane glycosides from heat processed ginseng, Arch. Pharmacal Res., 2002, 25, 428–432 CrossRef CAS.
- Y. J. Kim, J. H. Park, H. C. Kwon, N. K. Su, H. Y. Kim and H. O. Yang, Inhibition of telomerase activity and induction of apoptosis by ginsenoside Rk1 in HepG2 cells, Cancer Res., 2007, 67, 1206–1216 Search PubMed.
- S. K. Ji, E. J. Joo and J. Chun, et al., Induction of apoptosis by ginsenoside Rk1 in SK-MEL-2-human melanoma, Arch. Pharmacal Res., 2012, 35, 717–722 CrossRef PubMed.
- A. D. Kim, K. A. Kang and Z. Rui, et al., Ginseng saponin metabolite induces apoptosis in MCF-7 breast cancer cells through the modulation of AMP-activated protein kinase, Environ. Toxicol. Pharmacol., 2010, 30, 134–140 CrossRef CAS PubMed.
- M. Hammadi, A. Oulidi and G. Florian, et al., Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78, FASEB J., 2013, 27, 1600–1609 CrossRef CAS PubMed.
- H. Llewelyn Roderick and M. D. Bootman, Redoxing calcium from the ER, Cell, 2005, 120, 4–5 CrossRef PubMed.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Regulation of cell death: the calcium-apoptosis link, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef CAS PubMed.
- M. R. Duchen, Mitochondria and calcium: from cell signalling to cell death, J. Physiol., 2010, 529, 57–68 CrossRef PubMed.
- Y. U. Qian, K. W. Zeng, M. A. Xiao-Li, Y. Jiang, T. U. Peng-Fei and X. M. Wang, Ginsenoside Rk1 suppresses pro-inflammatory responses in lipopolysaccharide-stimulated RAW264.7 cells by inhibiting the Jak2/Stat3 pathway, Chin. J. Nat. Med., 2017, 15, 751 Search PubMed.
- G. I. Evan and K. H. Vousden, Proliferation, cell cycle and apoptosis in cancer, Nature, 2001, 411, 342–348 CrossRef CAS PubMed.
- T. Otto and P. Sicinski, Cell cycle proteins as promising targets in cancer therapy, Nat. Rev. Cancer, 2017, 17, 93–115 CrossRef CAS PubMed.
- E. A. Nigg, Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle, Bioessays, 2010, 17, 471–480 CrossRef PubMed.
- Y. Li, C. W. Jenkins, M. A. Nichols and Y. Xiong, Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21, Oncogene, 1994, 9, 2261–2268 CAS.
- M. Schröder and R. J. Kaufman, ER stress and the unfolded protein response, Mutat. Res., 2005, 569, 29–63 CrossRef PubMed.
- B. Kim, H. S. Kim and E. J. Jung, et al., Curcumin induces ER stress-mediated apoptosis through selective generation of reactive oxygen species in cervical cancer cells, Mol. Carcinog., 2016, 55, 918–928 CrossRef CAS PubMed.
- D. H. Shin, D. G. Leem and J. S. Shin, et al., Compound K induced apoptosis via endoplasmic reticulum Ca 2+ release through ryanodine receptor in human lung cancer cells, J. Ginseng Res., 2018, 42, 165–174 CrossRef PubMed.
- O. Nobumichi, Y. Satoshi, H. Takayuki, O. Kikuo and H. Hidetoshi, TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death, EMBO J., 2014, 24, 1243–1255 Search PubMed.
- J. Meldolesi and T. Pozzan, The endoplasmic reticulum Ca 2+ store: a view from the lumen, Trends Biochem. Sci., 1998, 23, 10 CrossRef CAS PubMed.
- G. Hajnóczky, E. Davies and M. Madesh, Calcium signaling and apoptosis, Biochem. Biophys. Res. Commun., 2003, 304, 445–454 CrossRef.
- D. J. Mcconkey, The role of calcium in the regulation of apoptosis, J. Leukocyte Biol., 1996, 59, 775 CrossRef CAS PubMed.
- C. Cui, R. Merritt, L. Fu and Z. Pan, Targeting calcium signaling in cancer therapy, Acta Pharm. Sin. B, 2017, 7, 3–17 CrossRef PubMed.
- M. G. Jeschke, G. G. Gauglitz and J. Song, et al., Calcium and ER stress mediate hepatic apoptosis after burn injury, J. Cell. Mol. Med., 2010, 13, 1857–1865 CrossRef.
- R. Rosario, P. Paolo and F. Davide, et al., Calcium and apoptosis: facts and hypotheses, Oncogene, 2003, 22, 8619–8627 CrossRef PubMed.
- S. Gyorgy and R. Rosario, Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood?, FEBS Lett., 2004, 567, 111–115 CrossRef PubMed.
- M. A. Campos, Mitochondrial apoptosis, An. R. Acad. Nac. Med., 2004, 121, 627 Search PubMed.
- N. Joza, S. A. Susin and E. Daugas, et al., Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death, Nature, 2001, 410, 549–554 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |