DOI:
10.1039/C9RA03640G
(Paper)
RSC Adv., 2019,
9, 19855-19868
Notoamide-type alkaloid induced apoptosis and autophagy via a P38/JNK signaling pathway in hepatocellular carcinoma cells†
Received
14th May 2019
, Accepted 19th June 2019
First published on 25th June 2019
Abstract
Bioassay-guided fractionation of a coral-associated fungus Aspergillus ochraceus LZDX-32-15 resulted in the isolation of eleven notoamide-type alkaloids, including four new congeners, namely notoamides W-Z (1–4). The structures of the new alkaloids were determined by extensive analyses of spectroscopic data (1D and 2D NMR, HRESIMS), while ECD data were used for the configurational assignment. Three alkaloids (6, 10, 11) exerted potent inhibition against a panel of hepatocellular carcinoma (HCC) cell lines with IC50 values ranging from 0.42 to 3.39 μM, that are comparable to the data for paclitaxel. Notoamide G (6) inhibited the viability of HepG2 and Huh-7 cells via both apoptosis and autophagy pathways. Notoamide G activated the expression of caspase-3, caspase-8, and caspase-9, in association with the degradation of the downstream gene PARP in a dose-dependent manner, suggesting that notoamide G induced apoptosis via a mitochondrial pathway and a dead receptor-mediated pathway. In addition, notoamide G increased the autophagic vacuole in both HepG2 and Huh-7 cells in a dose-dependent manner after 24 h through the significant upregulation of the key proteins Beclin1 and LC3B. Further investigation revealed that notoamide G promoted P38 and JNK phosphorylation, whereas the total protein of P-38 and JNK was slightly influenced. Accordingly, the antitumor proliferation of notoamide G in HCC cells was mechanistically mediated by apoptosis and autophagy through a P38/JNK signaling pathway, while notoamide G was considered as a potent lead for further development as an antitumor agent.
1. Introduction
Hepatocellular carcinoma (HCC) is a major contributor to cancer and mortality, and it is a highly fatal disease presenting the third leading cause of death from malignancy worldwide.1–3 The incidence of HCC is highest in Asia and Africa, where the endemically high prevalence of hepatitis B and hepatitis C strongly predisposes the development of chronic liver disease and the subsequent development of HCC.4–6 Infection with HBV and HCV in a cirrhotic patient increases the risk of HCC with an odds ratio of 165 compared to 17 for hepatitis C and 23 for hepatitis B alone.7–10 Unfortunately, the diagnosis of HCC is made too late with advanced disease when patients have become symptomatic and have some degree of liver impairment. At the late stage, there is virtually no effective treatment that would improve survival rate. The proliferation of tumors originated from their ability to avoid programmed cell death (apoptosis), thus compounds with the capability to induce tumor apoptosis have potential for the treatment of cancer. There are two main apoptotic pathways, an intrinsic pathway and an extrinsic pathway, to induce tumor apoptosis.11 The intrinsic pathway is mitochondria-mediated apoptosis which is induced by cytochrome c release and caspase-9 activation. The extrinsic pathway is death receptor-mediated apoptosis which involves dead receptor-mediated interactions and caspase-8 activation. Both pathways activated the execution of caspases, including caspase-3, caspase-6, and caspase-7. These caspases activate various cytoplasmic endonuclease and proteases, which ultimately cause protein cleavage, and DNA breakdown.12–14 A series of Bcl-2 family members (Bax, Bcl-2, and Bcl-xL) can regulate mitochondrial membrane permeabilization and control the release of cytochrome c, and thereby regulating apoptosis. Autophagy is a highly regulated cell death process which is characterized by bulk degradation of cellular proteins and organelles. The autophagy-induced degradation is mediated by the formation of autophagosomes following by lysis of intra-autophagosomal contents (lysosomes).15 Modulating autophagy has recently emerged as a promising therapeutic approach. However, the roles of autophagy in cancer treatment are complicated.16 It was reported that apoptosis and autophagy could interact through the cross talk of respective molecular mechanisms.17 Beclin 1 protein is a central regulator of autophagy in tumor cells, while the anti-apoptotic protein Bcl-2 inhibits autophagy by interaction with Beclin 1.17 LC3 is an ubiquitin-like protein that could facilitate apoptosis by mediating caspase-8 activation. Accumulation of LC3 leads to caspase-8 aggregation and autoactivation, resulting in apoptosis. JNK and p38 MAPK pathways were regarded as important regulators in the control of the balance of autophagy and apoptosis,18 and JNK also contributes to autophagic induction by upregulation LC3 and Beclin 1.19–22 Sorafenib was the first-line systematic treatment agent of advanced HCC. However, sorafenib is limited in patients with a specific genetic background such as mutations in receptor tyrosine kinases.23 Other HCC chemotherapeutic agents include doxorubicin,24 cisplatin,25 mitomycin26 and epirubicin,27 which resulted in some significant side effects and poorer tolerability on patients. Thus, the development of effective treatment which has anti-cancer activity with less adverse effects is still urgent. Natural products have been proved to be the promising source for the discovery of antitumor agents to overcome tumors by chemotherapy.28
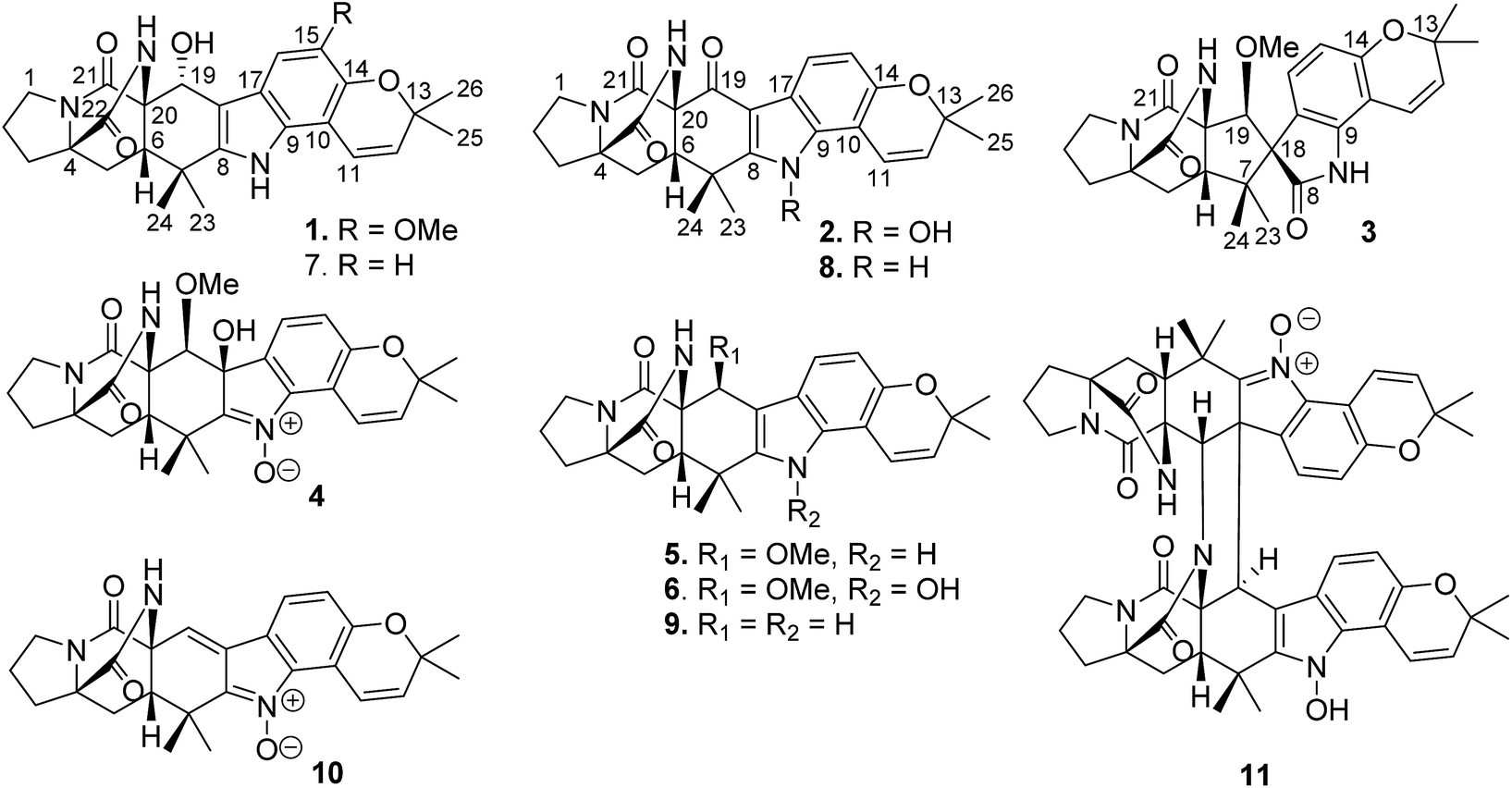
Among naturally occurring alkaloids, notoamide-type congeners are a group of fungal derived natural products that are characterized by the presence of a bicyclo[2.2.2]diazaoctane ring system and densely functionalized indole-derived units.29,30 Some of these alkaloids have been found to show antitumor activity, as exemplified by notoamide C that induced G2/M cell cycle arrest in the HeLa and L1210 cell lines, and stephacidin B showing potent inhibition against a panel of tumor cell lines.31–33 In the course of our search for antitumor compounds from marine microorganisms, a bioassay detection uncovered the EtOAc fraction of the cultured fungus Aspergillus ochraceus LZDX-32-15 to possess significant inhibition against a panel of tumor cell lines. Chromatographic separation of the bioactive fraction resulted in the isolation of 11 notoamide-type alkaloids, including four new congeners (Fig. 1). Herein, the structure elucidation of the new compounds, antitumor effects toward HCC cells, and the mode of action were described.
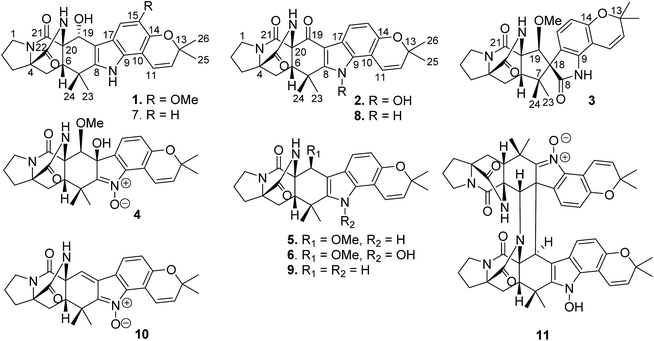 |
| | Fig. 1 Structures of notoamide-type alkaloids 1–11. | |
2. Experimental section
2.1. General procedure
Specific rotations were measured by an Autopol III automatic polarimeter (Rudolph Research Co., Ltd.). IR spectra were recorded on a Thermo Nicolet Nexus 470 FT-IR spectrometer. ECD spectra were measured on a JASCO J-810/J-815 spectropolarimeter. 1H and 13C NMR spectra were recorded on a Bruker Avance-400FT NMR spectrometer. HRESIMS spectra were obtained on a Bruker APEX IV 70 eV FT-MS spectrometer and on a Thermo DFS spectrometer using a matrix of 3-nitrobenzyl alcohol. HF254 silica gel for TLC was obtained from Qingdao Marine Chemistry Co. Ltd. Sephadex LH-20 (18–110 μm) was purchased from Pharmacia. The HPLC fingerprint chromatogram was prepared by HPLC-DAD Waters e2695 Separations module, Waters 2998 Photodiode Array Detector, and Waters Symmetry C18 (5 μm 4.6 × 250 mm). Semi-preparative HPLC was performed on an Alltech 426 pump using UV detector, and the Prevail C18 column (5 μm) was used for separation. Human liver tumor cell lines (HepG2, Sk-hep1, Hep3B, SUN475, SUN449) were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA), and the cell lines SMMC-7721 and Huh-7 were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China).
2.2. Fungal strain and identification
Fungus Aspergillus ochraceus LZDX-32-15 was isolated from a gorgonian coral (LZDX-32), which was collected from the South China Sea, in May 2014. The strain was identified by comparing the morphological character and analysis of the ITS region of the rDNA sequence with those of standard records. The morphological examination was performed by scrutinizing the fungal culture, the mechanism of spore production, and the characteristics of the spores. For inducing sporulation, the fungal strains were separately inoculated onto potato dextrose agar. All experiments and observations were repeated at least twice leading to the identification of the strain LZD-14-1 as Aspergillus ochraceus. The strain LZD-32-15 was deposited at the State Key Laboratory of Natural and Biomimetic Drugs, Peking University.
2.3. Fermentation
Solid afar culture: malt extract (15 g), peptone (3 g), 1000× microelement solution (1 mL), recombination vitamin (vitamin B1), riboflavin, niacin, vitamin B6, calcium pantothenate, inositol, p-aminobenzoic acid (0.5 mg each, biotin 0.25 mg), agar (20 g), pH 7.2. The medium (7 L) was prepared and autoclaved at 15 psi for 30 min. When the temperature dropped to about 50 °C, pour them into 670 plate Petri dishes (9 mm). After the medium become solid, each dish was inoculated with 100 μL of the spore inoculum and incubated at 25 °C for 10 days.
2.4. Extraction and isolation
The fermented material was cut into pieces and was packed into Fernbach flasks (500 mL), then was extracted successively with EtOAc (3 × 500 mL). After evaporation under vacuum, the EtOAc extract (3.46 g) was subjected to a vacuum liquid chromatography (silica gel, 200–300 mesh) with PE/acetone (from 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 0:1, gradient) as an eluent to obtain six fractions (F1 to F6). The MTT assay revealed that F5 fraction showed significant inhibition against human liver tumor cell line SMMC7721 with inhibitory rate of 80% in a dose of 10 μg mL−1. Thus, fraction F5 (1.34 g) was chromatographed on a silica gel (300–400 mesh) column eluting with dichloromethane/acetone (15:1) to collect 3 subfractions (F5-1, F5-2, F5-3). F5-2 (140.4 mg) was purified on a semi-preparative reversed phase (RP) HPLC using MeOH/H2O = 66:44 (2 mL min−1) as a mobile phase to obtain 11 (11.9 mg, Rt 54.8 min, 0.34% yield) and four subfractions including F5-2-1 (42.0 mg), F5-2-2 (30.4 mg), F5-2-3 (32.5 mg), F5-2-4 (33.5 mg). F5-2-1 was purified on a semi-preparative reversed phase (RP) HPLC using MeOH/H2O = 1:1 (2 mL min−1) as a mobile phase to obtain 1 (5.2 mg, Rt 24.3 min, 0.15% yield), 3 (11.2 mg, Rt 26.8 min, 0.32% yield), 6 (23.4 mg, Rt 29.5 min, 0.68% yield). F5-2-2 was purified on a semi-preparative RP-HPLC using MeCN/H2O = 35:65 (2 mL min−1) as a mobile phase to obtain 2 (21.3 mg, Rt 24.8 min, 0.62% yield), 8 (7.8 mg, Rt 26.5 min, 0.23% yield). Compound 4 (10.6 mg, Rt 18.5 min, 0.31% yield) and compound 10 (18.6 mg, Rt 23.6 min, 0.54% yield) were prepared from F5-2-3 by semi-preparative RP-HPLC using MeCN/H2O = 48:52 (2 mL min−1) as a mobile phase. Compound 5 (8.2 mg, Rt 19.4 min, 0.24% yield), 7 (11.4 mg, Rt 23.5 min, 0.33% yield) and 9 (12.6 mg, Rt 26.6 min, 0.36% yield) were prepared from F5-2-4 by semi-preparative RP-HPLC using MeCN/H2O = 42:58 (2 mL min−1) as a mobile phase.
:1 to 0:1, gradient) as an eluent to obtain six fractions (F1 to F6). The MTT assay revealed that F5 fraction showed significant inhibition against human liver tumor cell line SMMC7721 with inhibitory rate of 80% in a dose of 10 μg mL−1. Thus, fraction F5 (1.34 g) was chromatographed on a silica gel (300–400 mesh) column eluting with dichloromethane/acetone (15:1) to collect 3 subfractions (F5-1, F5-2, F5-3). F5-2 (140.4 mg) was purified on a semi-preparative reversed phase (RP) HPLC using MeOH/H2O = 66:44 (2 mL min−1) as a mobile phase to obtain 11 (11.9 mg, Rt 54.8 min, 0.34% yield) and four subfractions including F5-2-1 (42.0 mg), F5-2-2 (30.4 mg), F5-2-3 (32.5 mg), F5-2-4 (33.5 mg). F5-2-1 was purified on a semi-preparative reversed phase (RP) HPLC using MeOH/H2O = 1:1 (2 mL min−1) as a mobile phase to obtain 1 (5.2 mg, Rt 24.3 min, 0.15% yield), 3 (11.2 mg, Rt 26.8 min, 0.32% yield), 6 (23.4 mg, Rt 29.5 min, 0.68% yield). F5-2-2 was purified on a semi-preparative RP-HPLC using MeCN/H2O = 35:65 (2 mL min−1) as a mobile phase to obtain 2 (21.3 mg, Rt 24.8 min, 0.62% yield), 8 (7.8 mg, Rt 26.5 min, 0.23% yield). Compound 4 (10.6 mg, Rt 18.5 min, 0.31% yield) and compound 10 (18.6 mg, Rt 23.6 min, 0.54% yield) were prepared from F5-2-3 by semi-preparative RP-HPLC using MeCN/H2O = 48:52 (2 mL min−1) as a mobile phase. Compound 5 (8.2 mg, Rt 19.4 min, 0.24% yield), 7 (11.4 mg, Rt 23.5 min, 0.33% yield) and 9 (12.6 mg, Rt 26.6 min, 0.36% yield) were prepared from F5-2-4 by semi-preparative RP-HPLC using MeCN/H2O = 42:58 (2 mL min−1) as a mobile phase.
Notoamide W (1). Pale yellow powder, [α]25D +20.0 (c 0.1, MeOH), UV (MeOH) λmax 241, 306 nm; CD (MeOH) λmax (Δε) 225 (+20.04); IR (KBr) νmax 3518, 3402, 3311, 2970, 1713, 1664, 1585, 1472, 1384 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 478.2343 [M + H]+ (calcd for C27H31N3O5, 478.2343).
Table 1 1H NMR data of 1–4 (600 MHz, δ in ppm, J in Hz)
| |
1 |
2 |
3 |
4 |
| 1 |
3.22, m; 3.33, m |
3.34, m; 3.40, m |
3.41, m; 3.42, m |
3.39, m; 3.40, m |
| 2 |
1.85, m; 1.99, m |
1.86, m; 2.03, m |
1.80, m; 2.01, m |
1.83, m; 2.02, m |
| 3 |
1.86, m; 2.54, m |
1.86, m; 2.55, m |
1.83, m; 2.54, m |
1.84, m; 2.53, m |
| 5 |
2.02, d (7.6) |
2.13, dd (5.6, 12.0) |
1.79, dd (9.5, 12.0) |
1.87, dd (7.8, 12.0) |
| 2.17, dd (8.8, 12.0) |
1.94, dd (9.7, 12.0) |
2.04, dd (10.4, 12.0) |
| 6 |
2.65, t (7.6) |
2.85, dd (5.6, 8.8) |
3.81, dd (9.5, 9.7) |
3.52, dd (7.8, 10.4) |
| 11 |
6.94, d (9.8) |
7.25, d (10.0) |
6.60, d, (10.0) |
7.74, d (10.4) |
| 12 |
5.77, d (9.8) |
5.81, d (10.0) |
5.75, d, (10.0) |
5.92, d (10.4) |
| 15 |
|
6.76, d (8.4) |
6.40, d (8.3) |
6.86, d (8.0) |
| 16 |
7.03, s |
7.87, d (8.4) |
6.92, d (8.3) |
7.35, d (8.0) |
| 19 |
5.05, d (8.2) |
|
4.91, s |
4.10, s |
| 23 |
1.30, s |
1.30, s |
0.81, s |
1.29, s |
| 24 |
1.00, s |
1.51, s |
0.66, s |
1.14, s |
| 25 |
1.39, s |
1.41, s |
1.37, s |
1.39, s |
| 26 |
1.40, s |
1.41, s |
1.38, s |
1.41, s |
| NH-indole |
10.46, s |
|
10.76, s |
|
| OH-19 |
5.21, d (8.2) |
|
|
|
| OH-20 |
|
|
|
6.26, s |
| N–OH |
|
11.94, s |
|
|
| CONH |
7.64, s |
8.71, s |
8.40, s |
7.83, s |
| OMe |
3.78, s |
|
3.04, s |
3.30, s |
Table 2 13C NMR data of 1–4 (125 MHz, δ in ppm)
| |
1 |
2 |
3 |
4 |
| 1 |
43.9 |
44.2 |
43.9 |
43.9 |
| 2 |
24.5 |
24.5 |
24.9 |
24.0 |
| 3 |
29.0 |
28.9 |
29.5 |
28.6 |
| 4 |
66.9 |
67.1 |
68.9 |
66.3 |
| 5 |
30.1 |
30.9 |
30.1 |
30.0 |
| 6 |
44.7 |
51.6 |
55.7 |
49.5 |
| 7 |
35.3 |
36.4 |
44.0 |
36.1 |
| 8 |
141.2 |
152.9 |
177.9 |
152.0 |
| 9 |
127.8 |
130.8 |
139.2 |
139.6 |
| 10 |
106.4 |
105.9 |
104.8 |
111.4 |
| 11 |
118.8 |
117.5 |
117.1 |
115.4 |
| 12 |
129.9 |
130.5 |
130.7 |
133.0 |
| 13 |
75.6 |
75.8 |
76.3 |
76.2 |
| 14 |
138.3 |
150.2 |
152.8 |
153.9 |
| 15 |
143.9 |
113.6 |
108.8 |
116.4 |
| 16 |
102.8 |
121.3 |
125.6 |
123.7 |
| 17 |
120.0 |
115.4 |
121.4 |
128.9 |
| 18 |
109.1 |
105.4 |
67.9 |
77.5 |
| 19 |
59.8 |
183.9 |
83.2 |
75.6 |
| 20 |
62.8 |
67.4 |
65.1 |
61.8 |
| 21 |
168.4 |
167.0 |
169.3 |
168.0 |
| 22 |
172.8 |
172.3 |
173.4 |
171.7 |
| 23 |
28.5 |
26.4 |
19.4 |
22.4 |
| 24 |
22.3 |
19.2 |
22.6 |
13.0 |
| 25 |
27.4 |
27.3 |
28.0 |
27.4 |
| 26 |
27.4 |
27.5 |
28.2 |
27.4 |
| OMe |
56.9 |
|
60.1 |
59.5 |
Notoamide X (2). Yellow powder, [α]25D 84.0 (c 0.1, MeOH), UV (MeOH) λmax 243, 310, 346 nm; CD (MeOH) λmax (Δε) 228 (+12.42); IR (KBr) νmax 3444, 2920, 1744, 1715, 1661, 1647, 1454, 1020 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 462.2032 [M + H]+ (calcd for C26H28N3O5, 462.2029).
Notoamide Y (3). Yellow powder, [α]25D −60.0 (c 0.1, MeOH), UV (MeOH) λmax 237, 308 nm; CD (MeOH) λmax (Δε) 224 (+10.33), 241 (−7.01), 254 (+1.08), 269 (−3.16); IR (KBr) νmax 3612, 3419, 2920, 1681, 1661, 1646, 1445, 1022 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 478.2343 [M + H]+ (calcd for C27H31N3O5, 478.2342).
Notoamide Z (4). Yellow powder, [α]25D 28.0 (c 0.1, MeOH), UV (MeOH) λmax 241, 307 nm; ECD (MeOH) λmax (Δε) 235 (+12.58), 276 (+3.81); IR (KBr) νmax 3429, 2918, 1672, 1656, 1661, 1052 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 494.2206 [M + H]+ (calcd forC27H31N3O6, 494.2232).
2.5. Cell culture
The human HCC cell lines HepG2, HuH-7, Hep3b, SK-Hep1, SMMC-7721, SNU475 and SNU449 cells were cultured in Dulbecco's modified Eagle medium (Coring, Coring, NY, USA) and RPMI 1640 medium containing 10% fetal bovine serum (Gibco, Gaithersburg, MD, USA). All cells were cultured in a humidified atmosphere of 5% CO2 incubator at 37 °C. The medium was changed every 2–3 days and subcultured once they reached ∼90% confluence.
2.6. Cell viability assay
Cell viability was evaluated using the CCK8 (Dojindo, Japan) and MTT assays according to the manufacturer's instruction. In brief, cells were plated at a density of 1.0 × 104 cells per well in 96-well plates and allowed to attach overnight. Then the cells were treated with or without compound at the indicated concentration. After 48 h, the culture media was changed with 10% CCK-8 solution and cells were incubated for 1 h at 37 °C in a humidified 5% CO2 atmosphere. By the same manner, MTT solutions containing 1 mg mL−1 and MTT 100 μL were added into each well and incubated for 2 h at 37 °C. Then, the medium was changed with the same volume of DMSO. After incubation, absorbance is read at 450 nm for CCK-8 and 570 nm for MTT by a spectrophotometer and the quantity of formazan product is directly proportional to the number of living cells in culture.
2.7. Cell cycle analysis
For cell cycle analysis, cells were harvested by trypsinization and fixed by treatment in 70% ethanol at 4 °C overnight. The fixed cells were rinsed twice with PBS, and re-suspended in a propidium iodine (PI) solution (50 μg mL−1) containing 0.5 mg mL−1 RNase A (Sigma), and incubated at 37 °C for 30 min. The fluorescence of the PI-labeled cells was then measured using a FACS Calibur system (Nippon Becton Dickinson), and the percentages of cells in G0–G1, S, and G2–M phase were determined using the ModFit program (Nippon Becton Dickinson).
2.8. Western blot analysis
Protein lysates of HCC cells were prepared in RIPA buffer (Millipore, Temecula, CA, USA) containing cocktail protease inhibitors (Roche, IN, USA). Proteins were separated on bis–tris gels (Life Technologies) and transferred to a PVDF membrane (Millipore). The membranes were blocked and probed with primary antibodies of caspase-3, caspase-8, caspase-9, Bax, Bcl2, cytochrome c, PARP, Beclin1, LC3B, P38, p-P38, JNK and p-JNK (Santa Cruz Biotechnologies, Heidelberg, Germany). α-Tubulin was used as host gene control (MBL). The protein–antibody complexes were visualized using the Odyssey Imager (LI-COR Biosciences).
2.9. Analysis of apoptosis
Cell apoptosis was evaluated using the FITC Annexin V Apoptosis Detection Kit (Dojindo, Japan) according to the manufacturer instruction. Cells were plated at a density of 3 × 105 cells per well in 6-well plates and allowed to attach overnight. Then the cells were treated with or without compound at the indicated concentration for 48 h. After drug treatment, the cells were incubated with 200 μL binding buffer and stained with Annexin V-FITC and PI in the dark for 20 min. Then, the cells were assessed using flow cytometry (BD, USA).
2.10. Autophagy assay
Cell autophagy was evaluated using the DAPGreen Autophagy Detection Kit (Dojindo, Japan) according to the manufacturer instruction. In brief, HepG2, Huh-7 were plated at a density of 3 × 105 cells per well in 6-well plates and allowed to attach overnight. The cells were washed with culture medium and then incubated at 37 °C for 30 min with 0.1 μM DAPGreen solution. After the cells were washed twice, the culture medium containing compound 6 (0, 1, 2, 4 μM) was added to the well. After 24 hours of incubation, the cells were assessed using flow cytometry (BD, USA) or fluorescence microscope (Olympus, Japan).
3. Results and discussion
3.1. Structure determination of new compounds
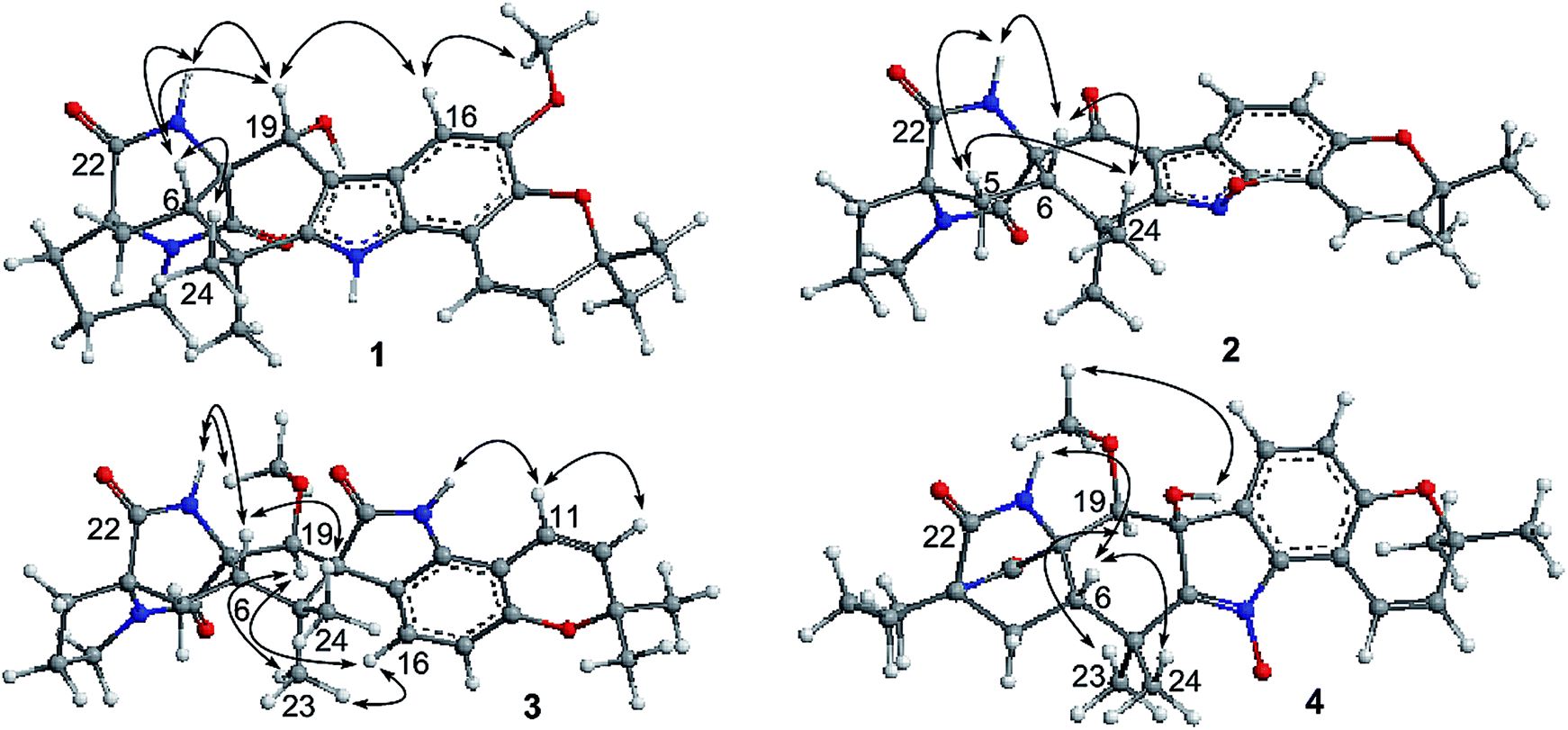
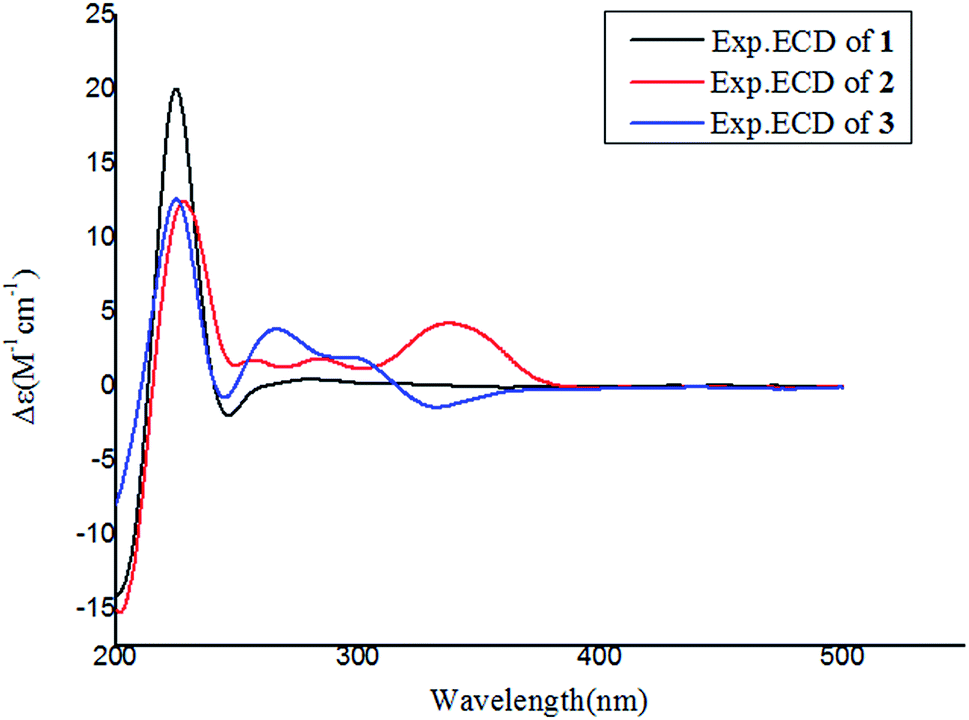
Notoamide W (1) has a molecular formula of C27H31N3O5 as determined by the HRESIMS and NMR data, requiring 14 degrees of unsaturation. The IR absorptions at 3311, 1713, and 1664 cm−1 suggested the presence of hydroxy, carbonyl and phenyl groups. The 1H NMR spectrum exhibited the resonances for four methyl singlet, two vicinal coupling olefinic protons at δH 6.94 (1H, d, J = 9.8 Hz) and 5.77 (1H, d, J = 9.8 Hz), a phenyl proton at δH 7.03 (1H, s), a methoxy singlet at δH 3.78 (3H, s), an oxymethine at δH 5.05 (1H, d, J = 8.2 Hz), three D2O exchangeable protons at δH 5.21 (1H, d, J = 8.2 Hz), 7.64 (1H, s), and 10.46 (1H, s), in addition to a number of alkyl protons. The APT spectrum displayed a total of 27 carbon resonances including 12 sp2 carbons, of which two carbonyl carbons, six aromatic resonances for a phenyl group and four olefinic resonances for two double bonds were recognized. The 2D NMR (COSY, HMQC, and HMBC) data established the planer structure of 1 to be a notoamide-type alkaloid, closely related to notoamide R (7),34 a prenylated indole alkaloid co-isolated from the same fraction. The distinction was attributed to the substitution in the aromatic ring, in which a single proton at δH 7.03 (1H, s, H-16) in the aromatic ring suggested it to be panta-substituted. The HMBC correlation from the methoxy protons (δH 3.78, s) to C-15 (δC 143.9) and H-16 to C-9 (δC 127.8), C-14 (δC 138.3), and C-16, in association with the NOE interaction between MeO and H-16 confirmed the location of a methoxy group at C-15. The relative configuration of 1 was identified by the NOESY data (Fig. 2). The NOE correlation correlations from the amide NH (δH 7.64) to H-19 (δH 5.05) and H-6 (δH 2.65), in addition to the NOE correlations from H-6 to H-19 and H3-24 (δH 1.30) demonstrated the amide group across C-4 and C-20 to be oriented in the same face as H-6 and H-19. The positive Cotton effect (CE) at 225 nm was contributed by the n → π* transition of the amide moiety in the diketopiperazine ring (Fig. 3). Based on the ECD rule for diketopiperazine-bearing analogues,35,36 the CE around 200–250 nm reflected the configuration of the stereogenic centers at the bridgehead of the bicyclo[2.2.2] diazaoctanediketopiperazinecore. Thus, the positive CE of 1 at 225 nm was in agreement with 4S and 20R configurations. Therefore, the absolute configuration of C-6 and C-19 was in accordance with S and R, respectively.
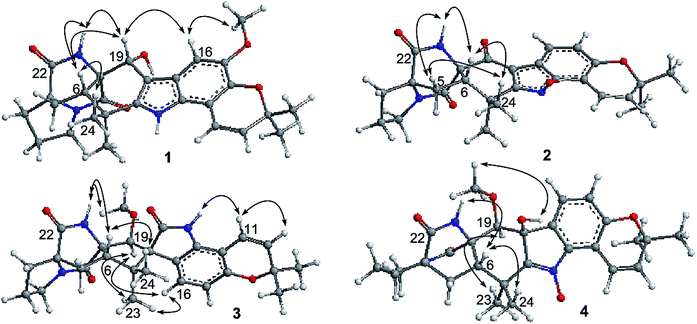 |
| | Fig. 2 Key NOE correlations of 1–4. | |
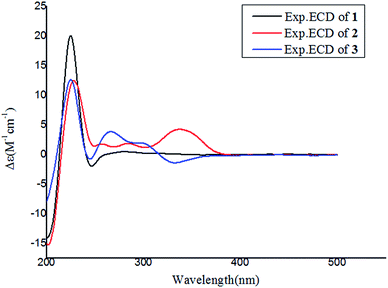 |
| | Fig. 3 Experimental ECD spectra of 1–3 in MeOH. | |
The HRESIMS and NMR data determined the molecular formula of notoamide X (2) as C26H27N3O5, containing an oxygen atom more than that of the co-isolated notoamide I (8).37 Diagnostic 2D NMR data revealed the planer structure of both compounds was close similarity. The difference was attributed to a D2O exchangeable proton at δH 11.94 (s), which showed the NOE interaction with H-11, H3-23, and H3-24. The absence of the HMBC correlation of this proton with the carbons in the indole unit, in association with the molecular composition, suggested that the nitrogen atom in the indole moiety was substituted by a hydroxy group instead of NH group. The NOE correlations from H-6 (δH 2.85) to the amide proton at δH 8.71 (s) and H3-24 (δH 1.51) (Fig. 2) clarified 2 having the same relative configuration as notoamide I. Based on the ECD rule of diketopiperazine core, the positive CE at 228 nm for the n → π* transition of the amide moiety (Fig. 3) assumed 4S and 20R configurations. Accordingly, the absolute configuration of C-6 was assigned to S referring to the NOE data.
Notoamide Y (3) was determined to be a 19-methoxylated analogue of sclerotiamide, a paraherquamide-related indolinone,37 based on the comparative analyses of 1D and 2D NMR correlations in association with the HRESIMS data. The NOE relationships from the amide NH (δH 8.40, s) to H-6 (δH 3.83, t, J = 9.5 Hz) and MeO (Fig. 2) indicated the amide group to be oriented in the same face as H-6 and MeO. In addition, the NOE interactions from H-16 (δH 6.92, d, J = 8.3 Hz) to H-19 (δH 4.91, s) and H3-13 (δH 0.81, s) and between H-19 and H3-13, declared the spiro-face of indolinone, in which the aromatic ring of the indolinone was oriented in the same face toward H-19. The ECD spectrum of 3 showed a positive Cotton effect around 224 nm (Fig. 3), agreeing 4S and 20R configuration. Therefore, 18R and 19S configurations were identified.
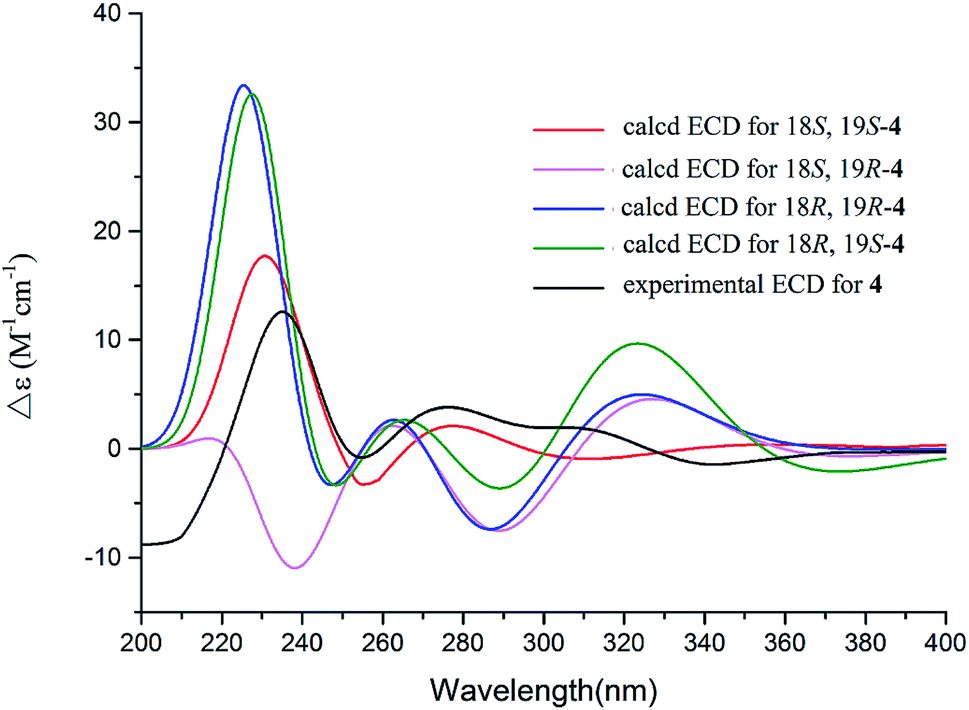
The 1D and 2D NMR data (Tables 1 and 2) in association with the HRESIMS data established the planer structure of notoamide Z (4) to be the same as taichunamide F.38 However, the opposite phase of the specific rotation of 4 ([α]25D +28.0, MeOH) in comparison with the known analogue ([α]25D −3.4, MeOH) suggested 4 to be a stereoisomer of taichunamide F. The NOE interactions from the amide NH (δH 7.83, s) to H-6 (δH 3.52, dd) identified the same face of the amide group toward H-6. The same face of OH-18 (δH 6.26, s) and MeO (δH 3.30, s) was evident from their NOE interaction. In addition, the NOE correlations between H-6/H3-24 (δH 1.29, s) and H-19 (δH 4.10, s)/H3-23 (δH 1.14, s) clarified H-19 in opposite face to H-6. The positive CE at 235 nm reflected 4S and 20R configurations, thus the stereogenic centers at C-18 and C-19 were assigned as S configuration. These assignments were further supported by the calculated ECD data for the model molecules with (18S, 19S)-4, (18R, 19R)-4, (18R, 19S)-4, and (18S, 19R)-4 at the B31YP/6-311++G(d,p) level in the gas phase, using the B3LYP/6-31+G(d) optimized geometries after conformational searches via the MMFF94S force field (Fig. 4),39 in which the ECD data of 20S, 10S-4 matched well with the experimental spectrum.
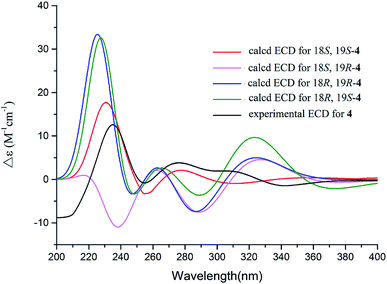 |
| | Fig. 4 Experimental ECD spectra (200–500 nm) of 4 in MeOH and the calculated ECD spectra of 4 at the B3LYP/6-311++G(2d,2p) level. | |
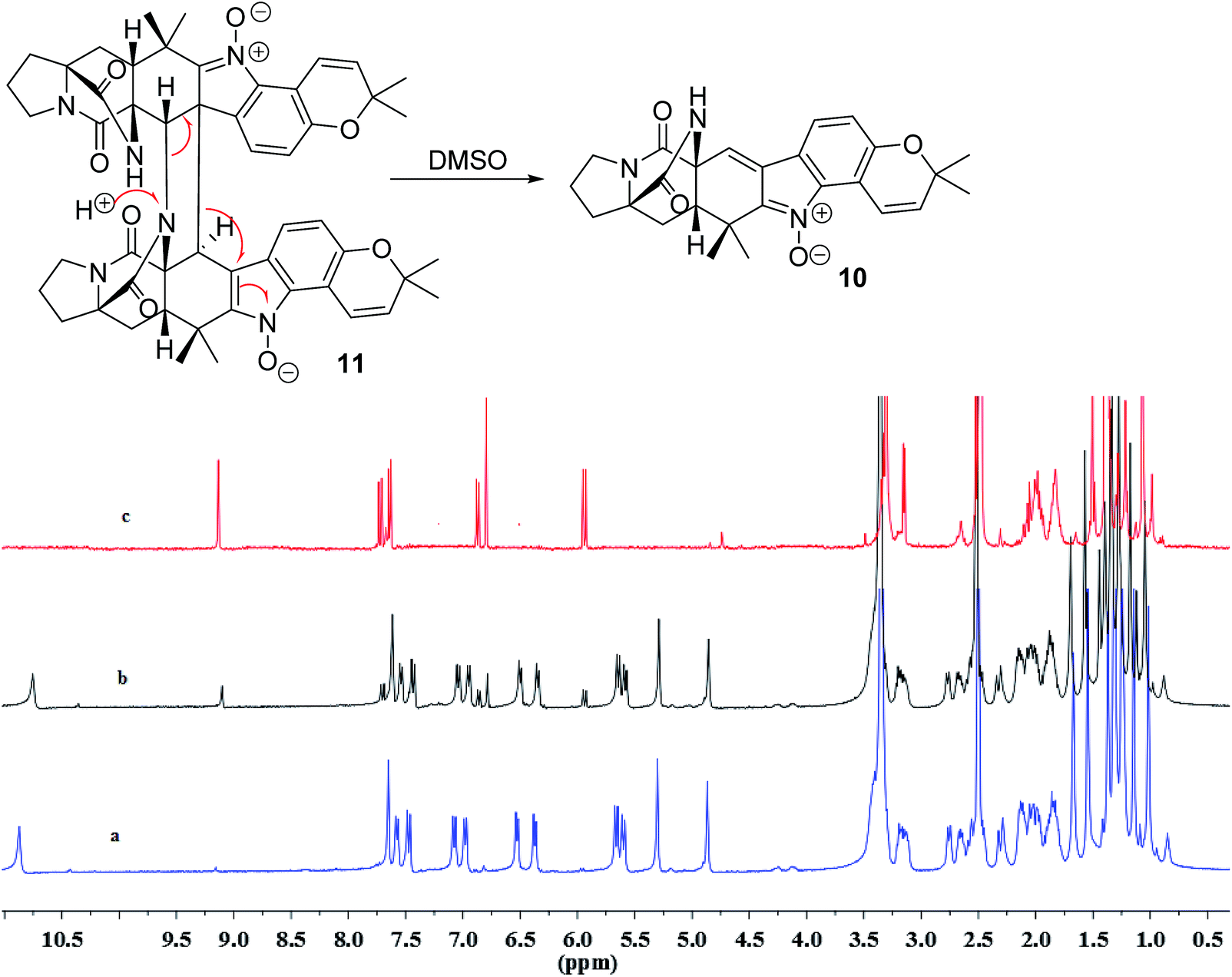
In addition, seven known notoamide-type alkaloids were identical to notoamide F (5),36 notoamide G (6),36 19 epi-notoamide R (7),40 notoamide I (8),36 stephacidin A (9),41 avrainvillamide (10),40 and a dimer of notoamide-type alkaloid stephacidin B (11),42 based on the comparative analyses of NMR and ESIMS data in association with the specific rotation and X-ray diffraction. It is noteworthy that stephacidin B (11) was unstable in DMSO solution, in which part of 11 was spontaneously decomposed into its monomer 10 via an retro-Diels–Alder reaction (Fig. 5).
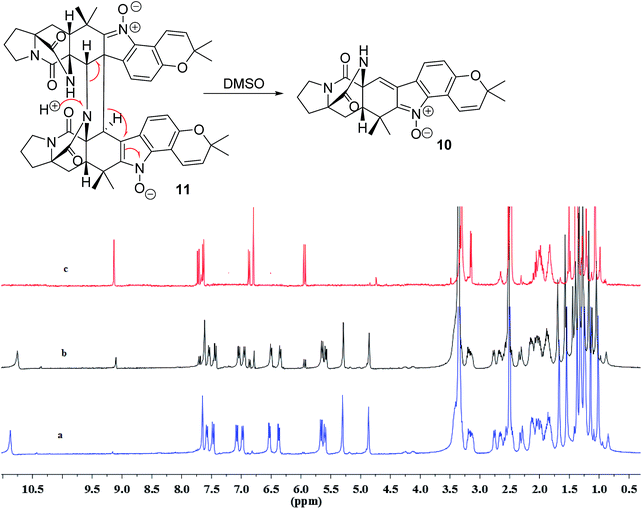 |
| | Fig. 5 Spontaneous conversion of 11 to 10 in DMSO after 24 h. (a) 1H NMR spectrum of 11, (b) 1H NMR spectrum of 11 in DMSO-d6 after 24 h, (c) 1H NMR spectrum of 10. 1H NMR spectra were measured in DMSO-d6. | |
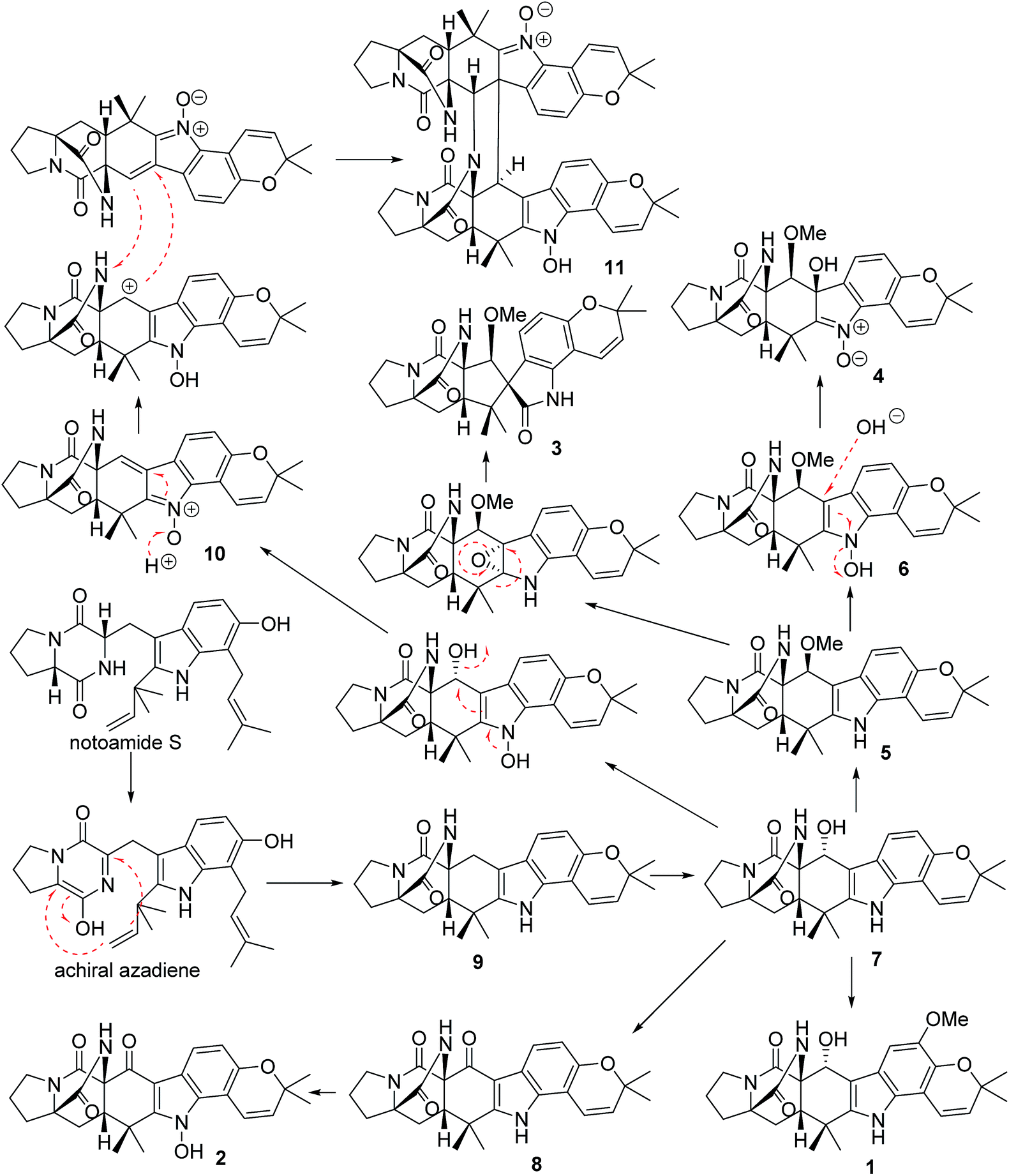
In the biogenetic speculation, notoamide S43 was considered as the precursor to derive the isolated alkaloids. A tautomerization and intramolecular hetero-Diels–Alder (IMDA) reaction occurred to generate the amide group in 9, which was converted to 7 via a hydroxylation. Methylation of 7 derived 1, while oxidation of 7 formed a ketone derivative 8. The latter compound further converted to 2 by a hydroxylation at the nitrogen atom of indole ring. Compound 5 is likely to be derived from 7 via a methylation at C-19 via a nucleophilic attachment instead of methylation at the hydroxyl group due to the variation of the chiral configuration at C-19 between 5 and 7. The conversion of 5 to 6 was followed the similar process as 7 to 2. Nucleophilic addition of 6 by a hydroxyl group and the double bond rearrangement generated 4. Apoxidation of 5 at the indole nucleus and subsequent migration of C-7 to form C-7/C-18 bond resulted in the production of 3. Compound 10 was suggested to be derived from 7 via hydroxylation and an retro-Diels–Alder rearrangement, while the dimerization of 10 to form 11 was achieved by IMDA addition (Scheme 1).
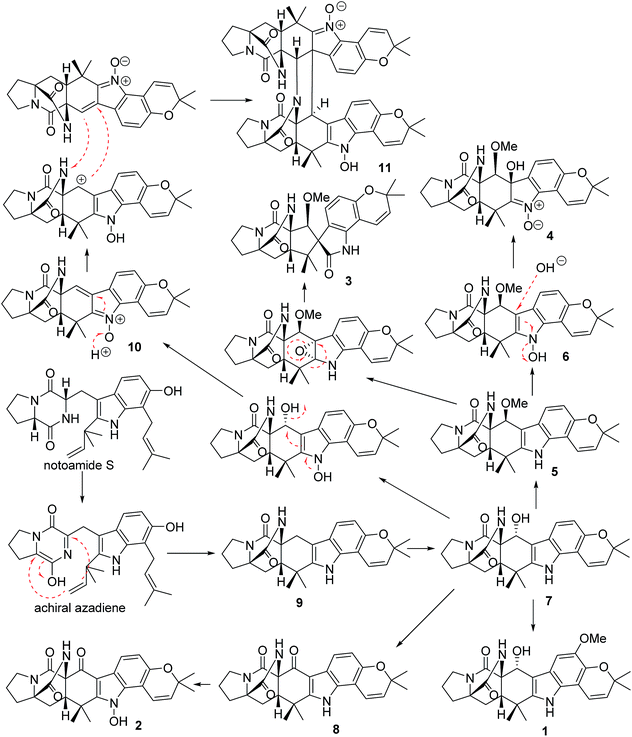 |
| | Scheme 1 Biogenetic speculation of the relationship among 1–11. | |
3.2. Inhibitory effects against the growth of tumor cells
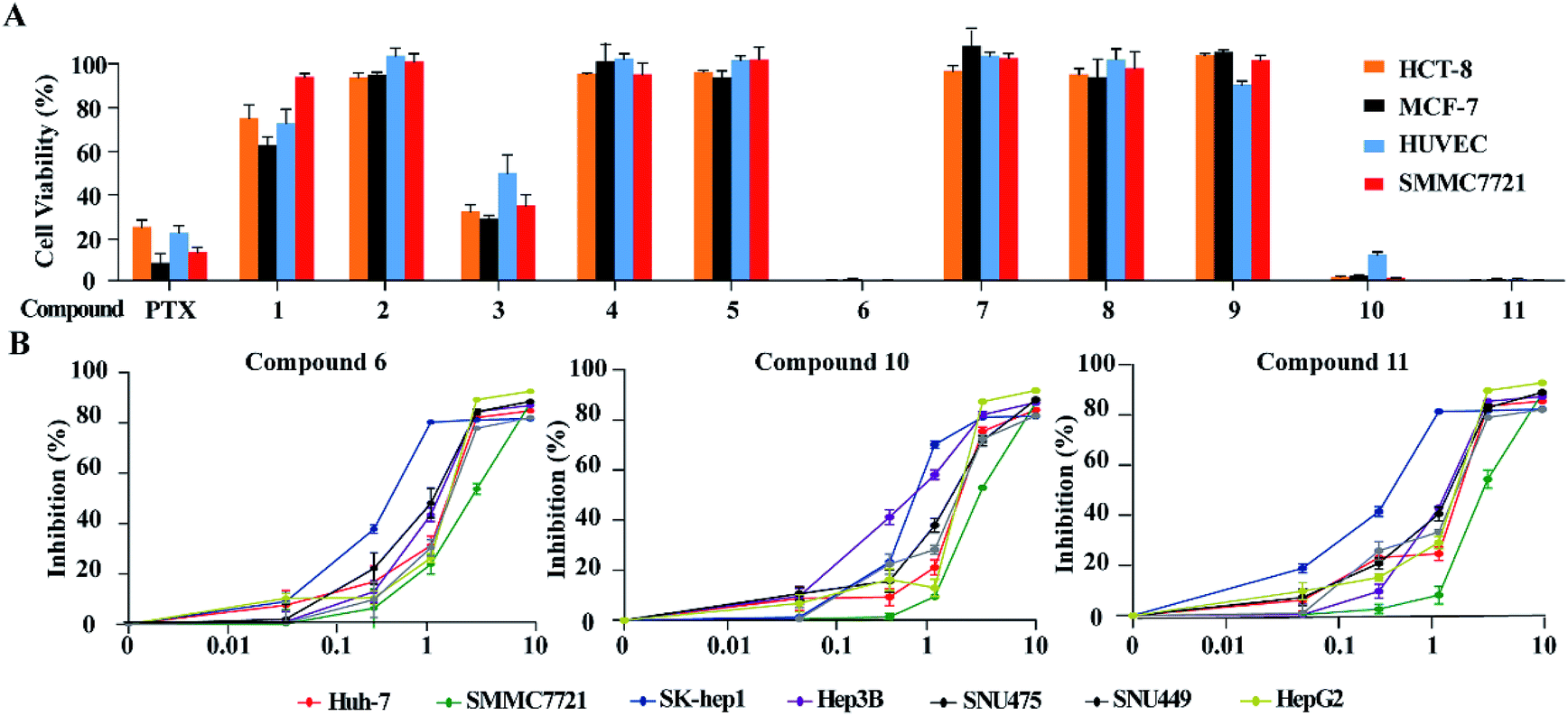
Based on the MTT method, the anti-tumor effects of all alkaloids were screened using human HCC cell line SMMC-772, human colorectal carcinoma cell line HCT-8, human breast cancer cell line MCF-7 and human umbilical vein endothelial cell HUVEC. As shown in Fig. 6A and Table 3, notoamide G (6), avrainvillamide (10), and stephacidin B (11) exhibited significant growth inhibition against the panel of tumor cell lines with the inhibitory rate more than 95% under a dose of 10 μM. Under the same concentration, compounds 2, 4-5, 7–9 exerted no to weak cytotoxic effects toward the selected tumor cell lines, in which around 60% and 20% growth inhibitions were observed after treatment with 3 and 1, respectively. It is noteworthy that these compounds are also cytotoxic toward human umbilical vein endothelial cell HUVEC. Comparison of the activity between compounds 5 and 6 revealed that the hydroxylation at the nitrogen atom of indole core significantly improved the inhibitory effect. The substituent at C-19 was highly sensitive, while additional hydroxylation at C-18 such as 4 significantly reduced the activity. Congeners without substitution at the nitrogen atom of indole showed weak activity, with the exception of compound 3 whose structure was characterized by a spiro-form at the indolone moiety. These results revealed that the capability of congeners to inhibit the tumor cell lines is closely related to the substitution or structural rearrangement.
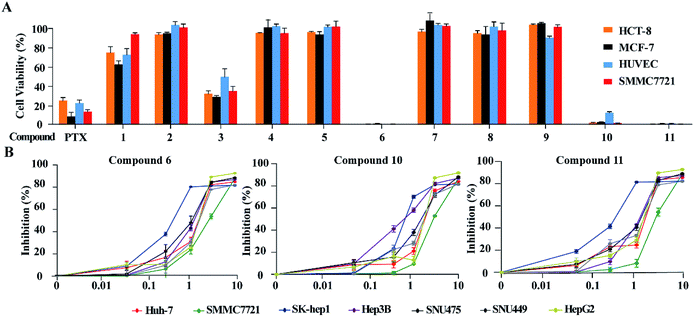 |
| | Fig. 6 Cytotoxic effects of alkaloids on human HCC cells. (A) Cytotoxic effects of 11 alkaloid compounds on HCT-8, MCF-7, SMMC7721 and HUVEC were detected by MTT assays. Cells were treated with 10 μM of each compound for 48 h. MTT solution (1 mg mL−1 MTT, 100 μL) was added into each well and incubated for 2 h at 37 °C. Then, the medium was changed with the same volume of DMSO. Cell viability was measured at absorbance of 570 nm. Paclitaxel (PTX) at concentration of 10 μM was used as positive control. (B) Seven liver cancer cell lines were seeded into 96-well plates at a concentration of 1 × 104 per well and treated with indicated concentrations of compounds 6, 10 and 11, respectively. Cell viability was assessed by CCK8 assays after 48 h of exposure to compounds. Data is represented as mean ± S.D. | |
Table 3 Inhibitory effects of 6, 10, and 11 toward a panel of tumor cell linesa
| No. |
IC50 (μM) |
| SMMC-7721 |
HCT-8 |
MCF-7 |
HUVEC |
| Cells were treated with various concentrations of 1–11 (0.01, 0.10, 1.00, 10.00 μM) for 48 h. After incubation, cell viability was evaluated by CCK-8 assay. Present mean values (±SD) obtained from three separate experiments. PTX was used as a positive control. |
| PTX |
0.074 |
4.03 |
0.086 |
0.513 |
| 6 |
0.244 ± 0.025 |
0.087 ± 0.012 |
0.219 ± 0.082 |
0.279 ± 0.068 |
| 10 |
0.059 ± 0.001 |
0.209 ± 0.015 |
2.443 ± 0.415 |
1.225 ± 0.116 |
| 11 |
0.091 ± 0.003 |
0.194 ± 0.003 |
0.403 ± 0.015 |
0.268 ± 0.083 |
Since compounds 6, 10 and 11 were sensitive toward SMMC7721 cells, the bioassay (CCK8 assay) of these compounds was extended to a panel of HCC cell lines including HepG2, Huh7, Hep3B, SUN475, and SUN449, as well as a liver adenocarcinoma cell line Sk-hep1. The results showed that compounds 6, 10 and 11 significantly reduced the proliferation of these liver cancer cell lines in a dose-dependent manner (Fig. 6B) with IC50 values ranging from 0.42 μM to 3.39 μM (Table 4). Among the liver cancer cells detected, HepG2 and SK-Hep1 are known to contain wild type p53 gene, while other cells are p53 deficient or mutated. In addition, three cell lines including Hep3B, SNU475 and SNU449 were established from HCC patients infected with HBV and contained integrated HBV DNA sequence in genomes. The anticancer-effects against these HCC cells suggested that compounds 6, 10 and 11 could inhibit HCC cell proliferation without the relation to the p53 gene mutation and HBV infection status of HCC cells.
Table 4 Inhibitory effects of 6, 10, and 11 toward a panel of liver cancer cell lines
| |
IC50 (μM) |
| 6 |
10 |
11 |
| Huh-7 |
1.54 ± 0.08 |
1.90 ± 0.18 |
1.40 ± 0.09 |
| HepG2 |
1.31 ± 0.11 |
1.52 ± 0.13 |
1.20 ± 0.10 |
| SMMC-7721 |
2.70 ± 0.12 |
3.39 ± 0.15 |
3.19 ± 0.24 |
| Hep3b |
1.38 ± 0.09 |
0.63 ± 0.05 |
1.29 ± 0.12 |
| SUN475 |
1.10 ± 0.06 |
1.52 ± 0.09 |
1.11 ± 0.07 |
| SUN449 |
1.92 ± 0.12 |
1.90 ± 0.11 |
1.47 ± 0.11 |
| Sk-hep1 |
0.66 ± 0.05 |
0.98 ± 0.04 |
0.42 ± 0.05 |
3.3. Induction of apoptosis in HepG2 and Huh-7
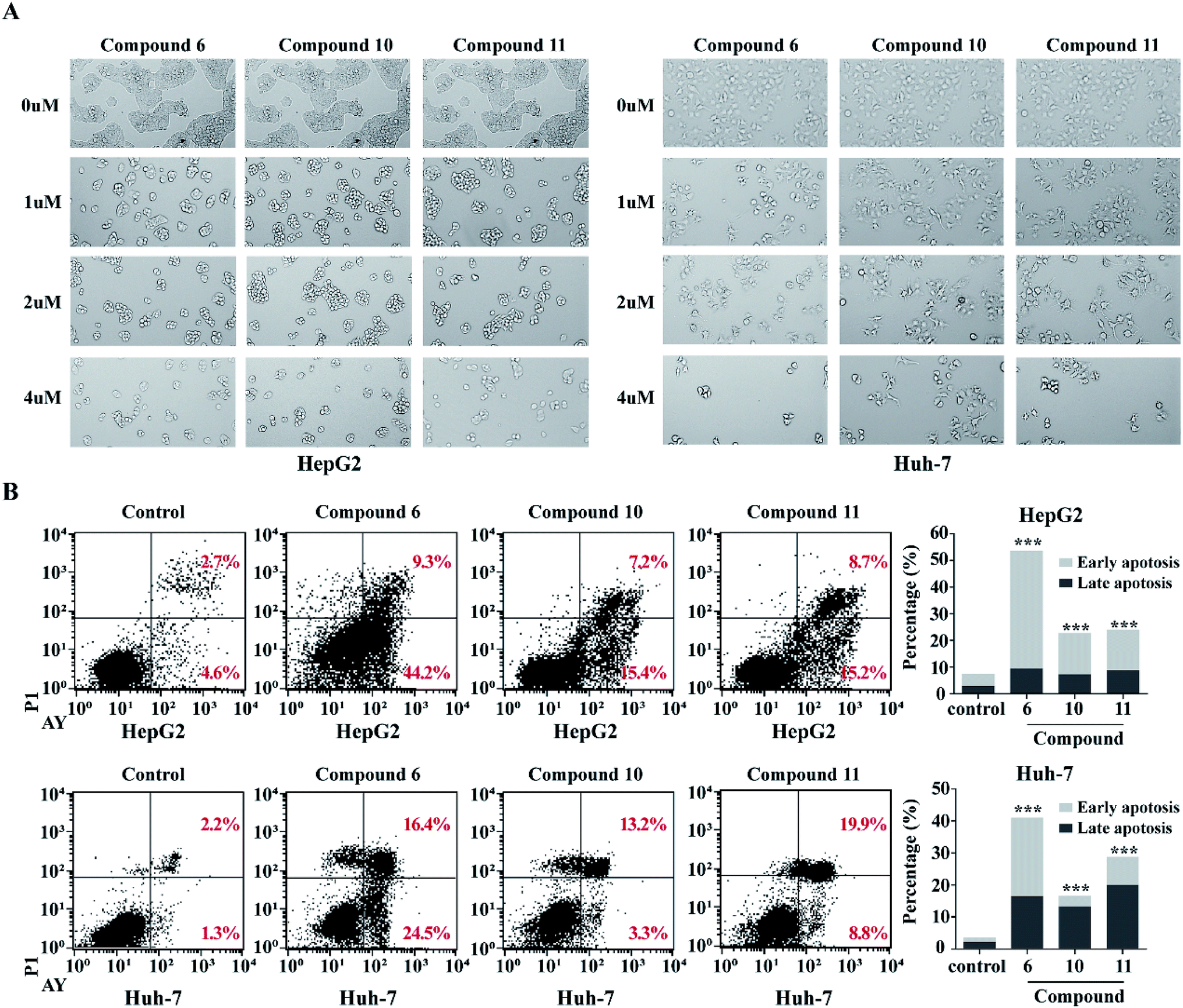
In order to detect whether the proliferation inhibition of HCC cell lines related to the cell apoptosis, two HCC cell lines HepG2 and Huh-7 were selected for further investigation. After treatment of HepG2 and Huh-7 cells with compounds 6, 10 and 11 with different concentrations (1, 2, 4 μM) for 48 h, cell morphologies were observed and captured using a light microscope. The HepG2 and Huh-7 cells in control group grew well, and formed the fusion colony cells. In contrast, the compound-treated tumor cells were changed morphologically to cell shrinkage, pyknosis and dissociated from bottle wall (Fig. 7A). Compared to the control HepG2 and Huh-7 cells, the clone formation in the cells treated with compounds 6, 10 and 11 was obviously declined in a dose-dependent manner. These results suggested that compounds 6, 10 and 11 might induce tumor cell apoptosis. Subsequently, Annexin V-FITC/PI assay was performed to detect apoptosis percentage by flow cytometry. As shown in Fig. 7B, compounds 6, 10 and 11 significantly increased the proportion of early and late stage apoptotic cells in both HepG2 and Huh-7 cell lines, of which compound 6 increased the early stage apoptotic cells of HepG2 from 4.62% to 44.72% and of Huh-7 from 1.29% to 24.46%, and increased the late stage apoptotic cells of HepG2 from 2.74% to 9.24% and of Huh-7 from 2.19% to 16.44%. Compounds 10 and 11 increased early stage apoptotic cells of HepG2 from 4.62% to 15.41% and 15.16%, and late stage apoptotic cells from 2.74% to 7.20% and 8.73%, respectively. However, compounds 10 and 11 slightly increased the early stage apoptotic cells of Huh-7 from 1.28% to 3.30% and 8.83%, but obviously increased the proportion of late stage apoptotic cells from 2.19% to 13.24% and 19.89% respectively. These data clearly indicated that compound 6 was more active than compounds 10 and 11 to induce apoptosis of HCC cells. However, these alkaloids showed slight induction to affect the cell cycle of the selected liver cancer cells.
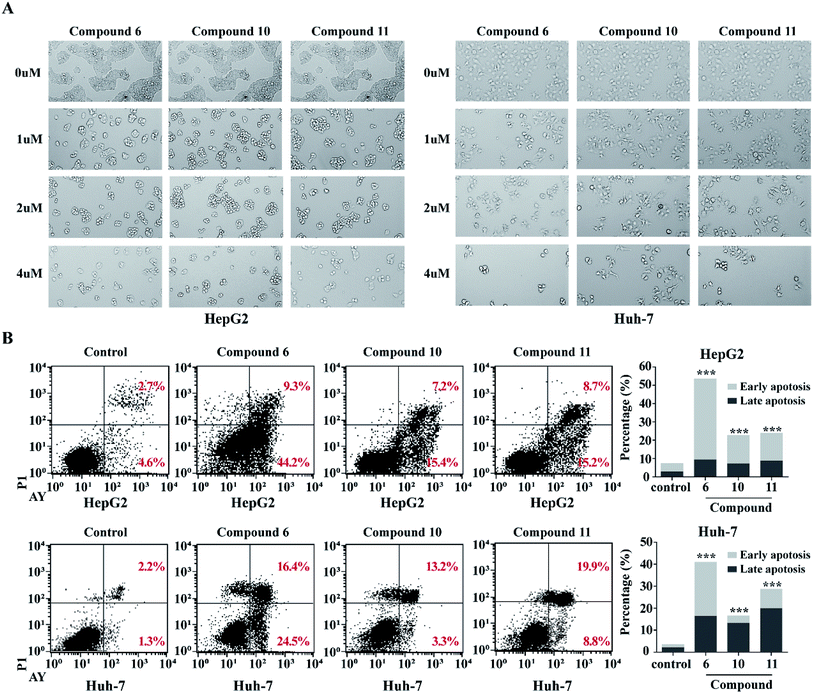 |
| | Fig. 7 Compounds 6, 10 and 11 induced apoptosis in HCC cells. (A) Morphological changes of HepG2 and Huh7 treated with increasing concentrations of (0 μM, 1 μM, 2 μM, 4 μM) of compounds 6, 10 and 11 were shown. (B) Functions of compounds 6, 10 and 11 in cell apoptosis in HepG2 and Huh7 cells. Cells were treated with 4 μM compound for 48 h. Annexin V/PI double staining with flow cytometry analysis was executed for cell apoptosis. DMSO was used as negative control. ***p < 0.001. | |
3.4. Apoptosis-related proteins expression induced by notoamide G (6)
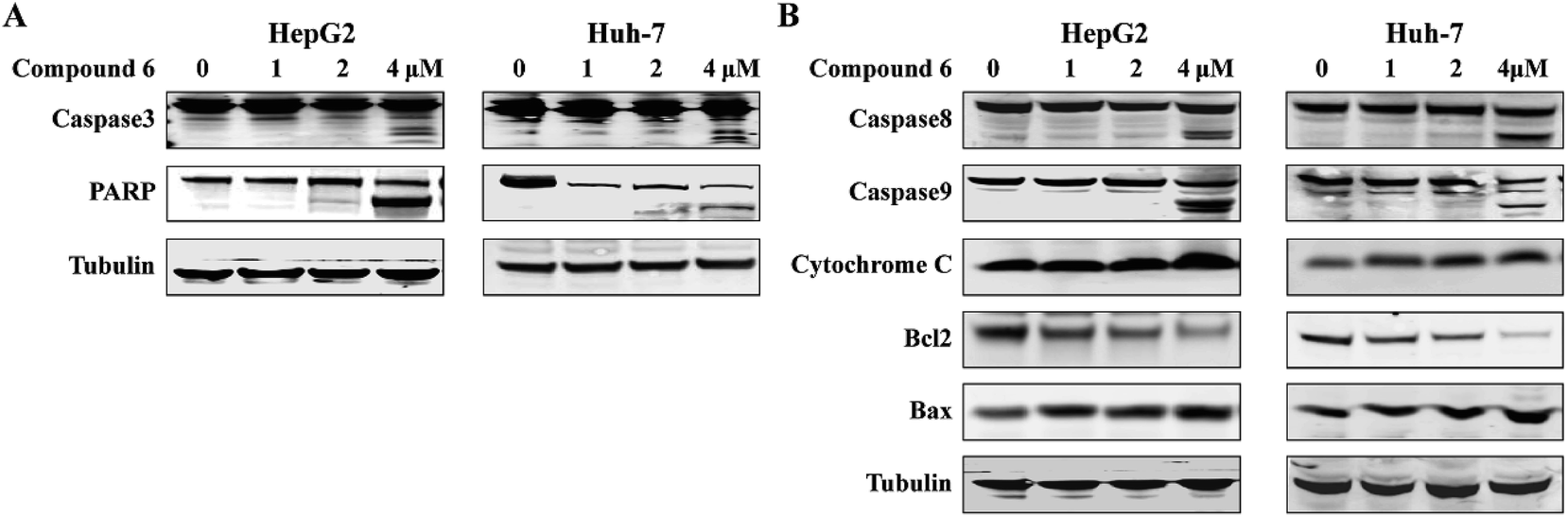
Apoptosis is one of the mechanisms responsible for the physiological deletion of tumor cells which are intrinsically programmed, while the mechanism of apoptosis is complex and involves many pathways. In order to understand the mode of this action, HepG2 and Huh-7 cells were treated respectively with notoamide G in the doses of 0, 1, 2, and 4 μM for 24 h. Western blot analysis showed that caspase-3 was activated in HepG2 and Huh-7 cells that were treated with notoamide G (4 μM) and followed by cleavage of PARP, a typical caspase-3 substrate in a dose-dependent manner (Fig. 8A). Two main pathways to activate caspase-3 included the intrinsic (mitochondrial) and extrinsic (death receptor) pathway of apoptosis. The observation that the up-regulation of cytochrome c and caspase-9 fragment (intrinsic pathway marker), as well as caspase-8 fragment (extrinsic pathway marker) (Fig. 8B) suggested that notoamide G induced apoptosis probably through both the mitochondrial-mediated apoptotic pathway and dead receptor-mediated pathway. In the mitochondrial-regulating apoptosis system, the Bcl-2 family proteins control the permeability of mitochondrial membrane and regulate apoptosis, among which Bcl-2 functions as an anti-apoptotic protein and Bax functions as a pro-apoptotic protein. Further Western blot analysis showed that the expression of the Bcl-2 protein decreased and Bax protein increased in 6-treated HCC cells in a dose-dependent manner (Fig. 8B), suggesting that the abnormal expression of Bcl-2 and Bax might be one important factor contributing to the increase apoptosis observed in HepG2 and Huh-7 cells after treatment by notoamide G.
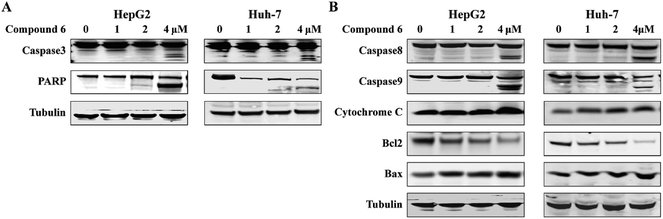 |
| | Fig. 8 Functions of compound 6 on expression of apoptosis-related proteins. HepG2 and Huh7 cells were treated with increasing concentrations of (0 μM, 1 μM, 2 μM, 4 μM) of compound 6. (A) Expression of caspase-3 and PARP fragment (B) and caspase-8 fragment, caspase-9 fragment, cytochrome c, Bcl-2 and Bax were detected by Western blot assays. DMSO was used as negative control. Tubulin was used as a loading control for all whole cell extract samples. | |
3.5. Induction of autophagy in HepG2 and Huh-7 cells by notoamide G
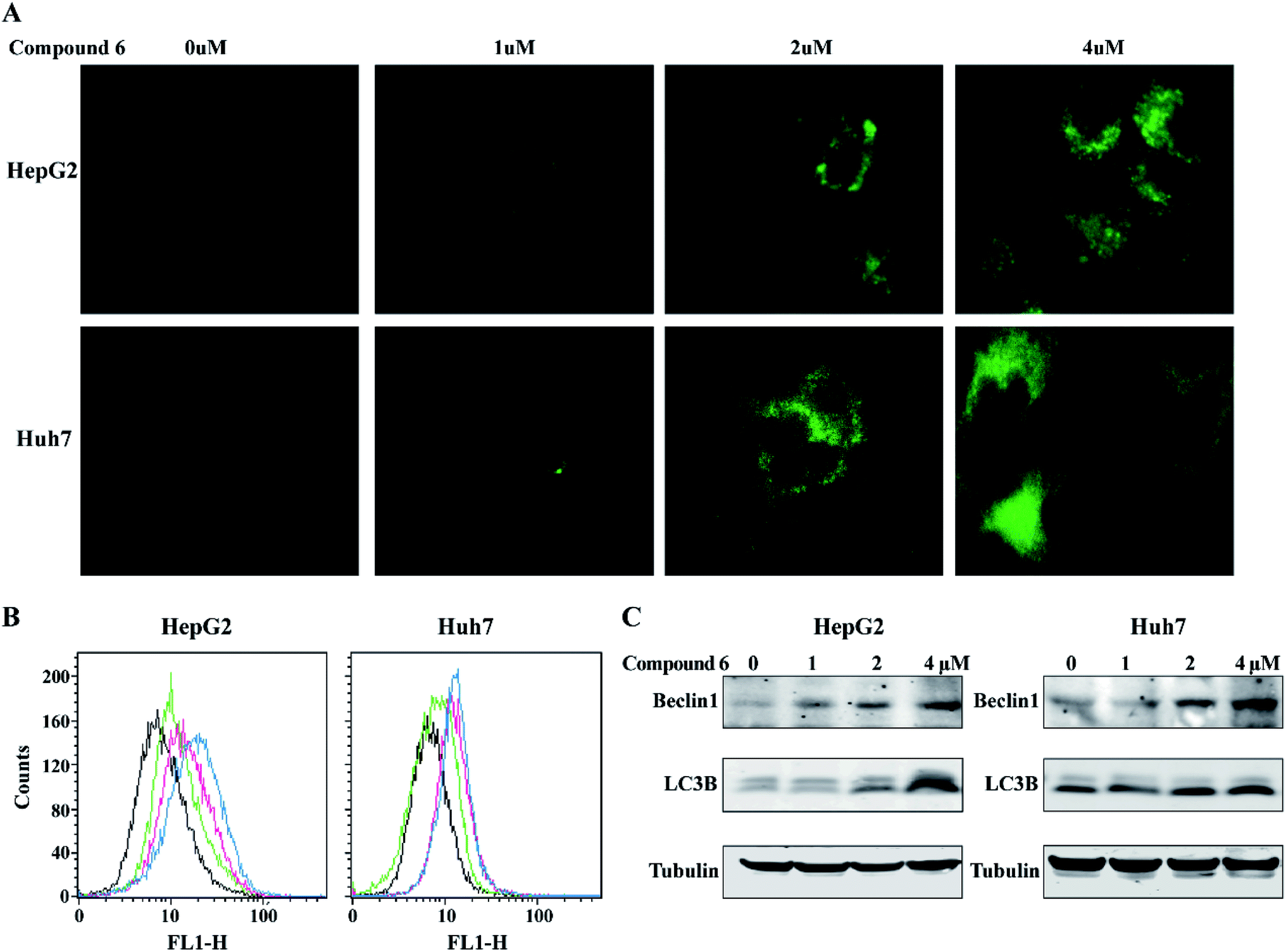
To determine whether the inhibitory effects of notoamide G toward tumor cell lines related to the induction of autophagy, DAPGreen was used to detect the autophagic vacuoles in HepG2 and Huh-7 cells. As shown in Fig. 9A and B, the autophagic vacuoles in both HepG2 and Huh-7 cells were increased in a dose-dependent manner after the treatment of notoamide G for 24 h as detected by flow cytometry and fluorescence microscopy. In the Western blot assays, the expression of two autophagy-related markers, Beclin1 and LC3B (I and II) was upregulated in HepG2 and Huh-7 cells after the treatment of notoamide G for 24 h (Fig. 9C). These findings indicated that both Beclin1 and LC3B paly key role to mediate the autophagy in HepG2 and Huh-7 cell lines which were treated by notoamide G.
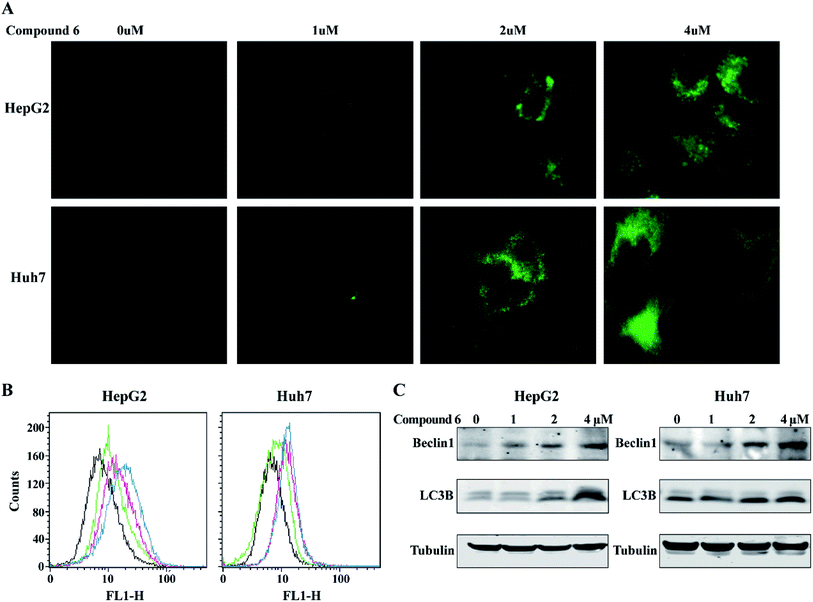 |
| | Fig. 9 Compound 6 induced autophagy in HCC cells. (A and B) HepG2 and Huh7 cells were incubated at 37 °C for 30 minutes with 0.1 μM DAPGreen working solution, and then treated with increasing concentrations of (0, 1, 2, 4 μM) of compound 6. Fluorescence was observed by fluorescence microscope (A) or flow cytometry (B). (C) Expression level of Beclin1 and LC3B (I and II) were detected by Western blot analysis. DMSO was used as negative control. Tubulin was used as a loading control for all whole cell extract samples. | |
3.6. Notoamide G activated P38/JNK signaling pathway
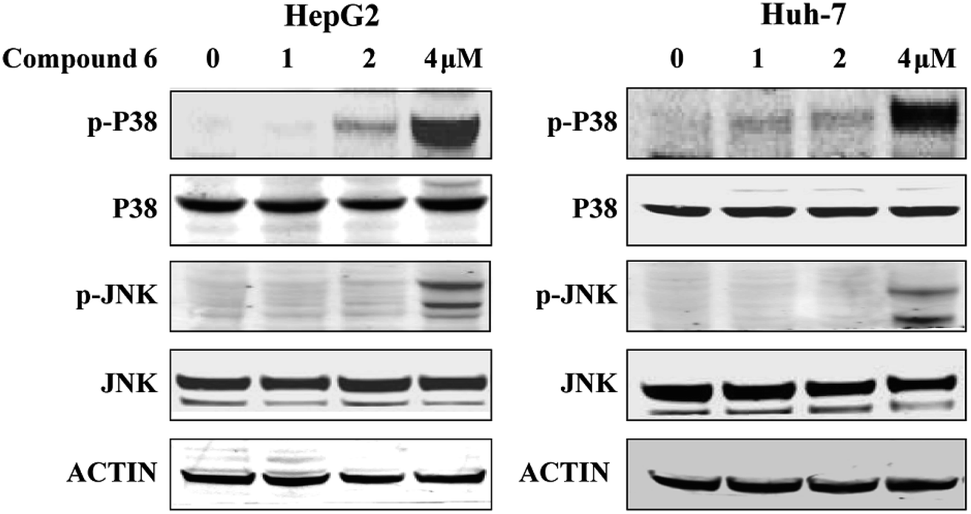
To investigate whether the JNK and P38 MAPK (Mitogen Activated Protein Kinase) pathway was involved in the 6-induced apoptosis and autophagy, the phosphorylation levels of P38 and JNK MAPK in HepG2 and Huh-7 cells treated by notoamide G were evaluated. The results showed that expression of p-P38 and p-JNK MAPK were increased in a dose-dependent manner, whereas the total protein of P38 and JNK were almost no changed (Fig. 10). These findings suggested that notoamide G might activate P38/JNK MAPK signaling pathway for the apoptosis and autophagy in HepG2 and Huh-7 cells.
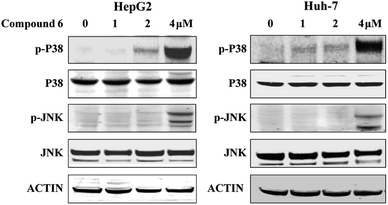 |
| | Fig. 10 Compound 6 induced p38/JNK phosphorylation in HCC cells. HepG2 and Huh7 cells were treated with indicated increasing doses of compound 6 and immunoblotted with indicated antibodies. DMSO was used as negative control. Tubulin was used as a loading control for all whole cell extract samples. | |
4. Discussion
In present protocol for fermentation, the yield of notoamide G (6) and its congeners is lower than 1% in the EtOAc extract. High cost is estimated to accumulate and purify the desired compound for further in vivo test or industry manufacture if the present culture condition was utilized for large scale fermentation. The alternative approach to improve the yields should be considered. Several effects for chemical synthesis of notoamides were successful,44,45 but the multiple steps with low yield implied that the synthesis approach could not meet the requirement of the manufacture in pharmaceutical industry. Strategies for engineering fungal natural product biosynthesis become available to overcome this problem. Meroterpene 6 was structurally characterized by the formation with a tryptophan, a proline and an isoprene unit. Thus, the biosynthetic gene clusters were attributed to nonribosomal peptide synthetase (NRPS) gene cluster and terpene gene cluster. Whole genome sequencing of fungus A. ochraceus LZDX-32-15 resulted in a detailed map of genome, in which the relevant biosynthetic gene clusters were identified by the bioinformatics and comparative transcriptomics data analyses, in association with RT-qPCR analyses. The biogenetic manipulation including global transcriptional perturbation, manipulating specific biosynthetic pathways either in the native host or in a heterologous host, and the specific engineering of enzymes to synthesize target compound in vivo or in vitro, is under progress.
Apoptosis and autophagy are differently involved in the regulation of tumor cell proliferation. Notoamide G induced remarkable apoptosis in HepG2 and Huh7 cells in a dose-dependent manner. Mechanistically, notoamide G significantly activated caspase-8, caspase-9 and cytochrome c, suggesting that the apoptosis induced by notoamide G was related to both intrinsic and extrinsic apoptotic pathway. Consistently, decreasing the expressions of Bcl-2 protein and increasing the expression of Bax protein in 6-treated HCC cells compared with the control cells, suggested that the increased apoptosis observed in hepatocytes was mediated at least in part, through mitochondria-mediated apoptosis. Understanding of the relationship between tumor autophagy and anti-tumor effect will be helpful to explore more effective and predictive therapeutic strategy for tumor treatment. In this work, autophagosome formation was obviously observed in 6-treated HCC cells. In addition, expression of Beclin 1 and LC3B, two molecular markers of autophagy, was significantly upregulated in a dose-dependent manner after tumor cells were treated by notoamide G, suggesting that Beclin 1 and LC3B were the potential targets for notoamide G to induce the tumor autophagy. It has been reported that apoptosis and autophagy could interact through the cross talk of respective molecular mechanisms. Beclin 1 protein is a central regulator of autophagy in tumor cells, while the anti-apoptotic protein Bcl-2 inhibits autophagy by interaction with Beclin 1. The downregulation of Bcl-2 expression and activation of caspase-8 in 6-treated HCC cells, indicating the upregulation of Beclin 1 and LC3B to be closely related to the suppression of Bcl-2 expression. JNK and p38 MAPK pathways were regarded as important regulators in the control of the balance of autophagy and apoptosis.46 In response to stress signals, JNK contributes to autophagic induction by upregulation of LC3 and Beclin 1.47 The significant up-regulation of p-P38 and p-JNK in HepG2 and Huh-7 cells after treated by notoamide G indicated that P38/JNK signaling pathway activation might be one of the major causes for notoamide G to increase the apoptosis and autophagy in hepatic tumor cells.
5. Conclusion
In present work, 11 notoamide-type alkaloids with diverse substitution and different rearrangement were isolated from a coral-associated fungus A. ochraceus LZDX-32-15, of which four were identified as new alkaloids. Bioassay results revealed that compounds 6, 10, and 11 exerted remarkable activities to inhibit a panel of HCC cell lines. However, the remaining alkaloids showed weak to no inhibitory effects toward the selected tumor cell lines. The inhibitory effects against the tumor cell proliferation were mediated by the compound-induced cell apoptosis and autophagy. In summary, Notoamide G (6) could simultaneously induced apoptosis and autophagy that were mediated by the activation of P38/JNK signaling pathway. Notoamide G is a new natural scaffold to inhibit tumor cell viability by both apoptosis and autophagy, suggesting that notoamide G is a novel therapeutic agent for the treatment of HCC.
Author contributions
LH and DL isolated all compounds; TZ and GG did the bioassay; JH and XC detected the mode of action; PP helped to revise manuscript; WL elucidated the structures and edited the article.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by the National Key Research and Development Program of China (2018YFC03109004, 2015CB755906), DY135-B2-05, and the National Natural Science Foundation of China (21861142006, 81630089, 81872793, U1606403, 21502004).
References
- S. Tellapuri, P. D. Sutphin, M. S. Beg, A. G. Singal and S. P. Kalva, Staging systems of hepatocellular carcinoma: a review, Indian J. Gastroenterol., 2018, 37, 481–491 CrossRef PubMed
 .
. - A. Quaglia, Hepatocellular carcinoma: a review of diagnostic challenges for the pathologist, J. Hepatocell. Carcinoma, 2018, 5, 99–108 CrossRef .
- B. Momin, A. J. Millman, D. B. Nielsen, M. Revels and C. B. Steele, Promising practices for the prevention of liver cancer: a review of the literature and cancer plan activities in the National Comprehensive Cancer Control Program, Cancer Causes Control, 2018, 29, 1265–1275 CrossRef .
- P. Rawla, T. Sunkara, P. Muralidharan and J. P. Raj, Update in global trends and aetiology of hepatocellular carcinoma, Contemp. Oncol., 2018, 22, 141–150 Search PubMed .
- A. Baecker, X. Liu, C. L. Vecchia and Z. F. Zhang, Worldwide incidence of hepatocellular carcinoma cases attributable to major risk factors, Eur. J. Cancer Prev., 2018, 27, 205–212 Search PubMed .
- H. Nakayama and T. Takayama, Management before hepatectomy for hepatocellular carcinoma with cirrhosis, World J. Hepatol., 2015, 7, 2292–2302 CrossRef PubMed .
- C. de Martel, D. Maucort-Boulch, M. Plummer and S. Franceschi, World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma, Hepatology, 2015, 62, 1190–1200 CrossRef CAS PubMed .
- M. G. Peters, Hepatitis B virus infection: what is current and new, Top Antivir. Med., 2019, 26, 112–116 Search PubMed .
- V. K. Chaturvedi, A. Singh, S. K. Dubey, H. F. Hetta, J. John and M. P. Singh, Molecular mechanistic insight of hepatitis B virus mediated hepatocellular carcinoma, Microb. Pathog., 2019, 128, 184–194 CrossRef CAS .
- S. Piconese, I. Cammarata and V. Barnaba, Viral hepatitis, inflammation, and cancer: a lesson for autoimmunity, J. Autoimmun., 2018, 95, 58–68 CrossRef CAS .
- S. Elmore, Apoptosis: a review of programmed cell death, Toxicol. Pathol., 2007, 35(4), 495–516 CrossRef CAS PubMed .
- E. A. Slee, C. Adrain and S. J. Martin, Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis, J. Biol. Chem., 2001, 276, 7320–7326 CrossRef CAS PubMed .
- D. J. Burgess, Apoptosis: refined and lethal, Nat. Rev. Cancer, 2013, 13, 79 CrossRef CAS PubMed .
- C. F. A. Warren, M. W. Wong-Brown and N. A. Bowden, BCL-2 family isoforms in apoptosis and cancer, Cell Death Dis., 2019, 10, e177 CrossRef PubMed .
- A. Thorburn, D. H. Thamm and D. L. Gustafson, Autophagy and cancer therapy, Mol. Pharmacol., 2014, 85(6), 830–838 CrossRef PubMed .
- A. El-Khattouti, D. Selimovic, Y. Haikel and M. Hassan, Crosstalk between apoptosis and autophagy: molecular mechanisms and therapeutic strategies in cancer, J. Cell Death, 2013, 6, 37–55 Search PubMed .
- J. A. Pan, E. Ullman, Z. Dou and W. X. Zong, Inhibition of protein degradation induces apoptosis through a microtubule-associated protein 1 light chain 3-mediated activation of caspase-8 at intracellular membranes, Mol. Cell. Biol., 2011, 31, 3158–3170 CrossRef CAS PubMed .
- X. Sui, N. Kong, L. Ye, W. Han, J. Zhou, Q. Zhang, C. He and H. Pan, p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents, Cancer Lett., 2014, 344(2), 174–179 CrossRef CAS PubMed .
- E. Seki, D. A. Brenner and M. A. Karin, Liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches, Gastroenterology, 2012, 143(2), 307–320 CrossRef CAS PubMed .
- Y. Y. Zhou, Y. Li, W. Q. Jiang and L. F. Zhou, MAPK/JNK signalling: a potential autophagy regulation pathway, Biosci. Rep., 2015, 35(3), e00199 Search PubMed .
- H. Pang, L. Cai, Y. Yang, X. Chen, G. Sui and C. Zhao, Knockdown of osteopontin chemosensitizes MDA-MB-231 cells to cyclophosphamide by enhancing apoptosis through activating p38 MAPK pathway, Cancer Biother.Radiopharm., 2011, 26(2), 165–173 CrossRef CAS PubMed .
- X. Lou, Q. Zhou, J. Chiu, J. I. Chao, Y. J. Lee and T. S. Hsu, Regulation of gamma-H2AX and securing contribute to apoptosis by oxaliplatin via a p38 mitogen-activated protein kinase-dependent pathway in human colorectal cancer cells, Toxicol. Lett., 2008, 179, 63–70 CrossRef PubMed .
- J. M. Llovet, S. Ricci, V. Mazzaferro, P. Hilgard, E. Gane, J. F. Blanc, A. C. de Oliveira, A. Santoro, J. L. Raoul, A. Forner, M. Schwartz, C. Porta, S. Zeuzem, L. Bolondi, T. F. Greten, P. R. Galle, J. F. Seitz, I. Borbath, D. Häussinger, T. Giannaris, M. Shan, M. Moscovici, D. Voliotis and J. Bruix, Sorafenib in advanced hepatocellular carcinoma, N. Engl. J. Med., 2008, 359, 378–390 CrossRef CAS PubMed .
- W. Sun and R. Cabrera, Systemic treatment of patients with advanced, unresectable hepatocellular carcinoma: emergence of therapies, J. Gastrointest. Cancer, 2018, 49, 107–115 CrossRef CAS PubMed .
- X. Zhang, J. Li and M. Yan, Targeted hepatocellular carcinoma therapy: transferrin modified, self-assembled polymeric nanomedicine for co-delivery of cisplatin and doxorubicin, Drug Dev. Ind. Pharm., 2016, 42, 1590–1599 CrossRef CAS PubMed .
- C. P. Lin, H. C. Yu, J. S. Cheng, K. H. Lai, G. H. Lo, P. I. Hsu, C. K. Lin, H. H. Chen, C. C. Lo, H. L. Liang and H. H. Tseng, Clinical effects of intra-arterial infusion chemotherapy with cisplatin, mitomycin c, leucovorin and 5-fluorouracil for unresectable advanced hepatocellular carcinoma, J. Chin. Med. Assoc., 2004, 67, 602–610 Search PubMed .
- Z. Bai, Y. Qin, G. Zhu, G. Zhao, G. Deng and J. S. He, Efficacy and safety of epirubicin applied in transcatheter arterial chemoembolization for hepatocellular carcinoma: a meta-analysis, J. Cancer Res. Ther., 2018, 14, 133–138 CrossRef PubMed .
- B. Hu, H. M. An, S. S. Wang, J. J. Chen and L. Xu, Preventive and therapeutic effects of chinese herbal compounds against hepatocellular carcinoma, Molecules, 2016, 21, e142 CrossRef PubMed .
- A. Kai, H. Kato, D. H. Sherman, R. M. Williams and S. Tsukamoto, Isolation of a new indoxyl alkaloid, amoenamide B, from Aspergillus amoenus NRRL 35600: biosynthetic implications and correction of the structure of speramide B, Tetrahedron Lett., 2018, 59, 4236–4240 CrossRef CAS PubMed .
- K. R. Klas, H. Kato, J. C. Frisvad, F. Yu, S. A. Newmister, A. E. Fraley, D. H. Sherman, S. Tsukamoto and R. M. Williams, Structural and stereochemical diversity in prenylated indole alkaloids containing the bicyclo[2.2.2] diazaoctane ring system from marine and terrestrial fungi, Nat. Prod. Rep., 2018, 35, 532–558 RSC .
- S. Tsukamoto, H. Kato, M. Samizo, Y. Nojiri, H. Onuki, H. Hirota and T. Ohta, Notoamides F-K, prenylated indole alkaloids isolated from a marine-derived Aspergillus sp, J. Nat. Prod., 2008, 71, 2064–2067 CrossRef CAS PubMed .
- H. Kato, T. Yoshida, T. Tokue, Y. Nojiri, H. Hirota, T. Ohta, R. M. Williams and S. Tsukamoto, Notoamides A-D: prenylated indole alkaloids isolated from a marine-derived fungus, Aspergillus sp, Angew. Chem., 2007, 46, 2254–2256 CrossRef CAS PubMed .
- Y. Ding, J. R. de Wet, J. Cavalcoli, S. Li, R. J. Greshock, K. A. Miller, J. M. Finefield, J. D. Sunderhaus, T. J. McAfoos, S. Tsukamoto, R. M. Williams and D. H. Sherman, Genome-based characterization of two prenylation steps in the assembly of the stephacidin and notoamide anticancer agents in a marine-derived Aspergillus sp, J. Am. Chem. Soc., 2010, 132, 12733–12740 CrossRef CAS PubMed .
- S. Tsukamoto, H. Umaoka, K. Yoshikawa, T. Ikeda and H. Hirota, Notoamide O, a structurally unprecedented prenylated indole alkaloid, and notoamides P-R from a marine-derived fungus, Aspergillus sp, J. Nat. Prod., 2010, 73, 1438–1440 CrossRef CAS PubMed .
- D. M. H. Jacobus, W. T. Marian, H. N. Jan and C. J. O. Harry, Desulfurization of epidithiodioxopiperazines. a mechanistic and chiroptical study, J. Am. Chem. Soc., 1979, 101, 1159–1162 CrossRef .
- M. W. Robert, K. Ewa, C. Hazel and G. Tomasz, Remarkable, enantiodivergent biogenesis of brevianamide A and B, J. Am. Chem. Soc., 1989, 111, 3064–3065 CrossRef .
- S. Tsukamoto, H. Kato, M. Samizo, Y. Nojiri, H. Onuki, H. Hirota and T. Ohta, Notoamides F-K, prenylated indole alkaloids isolated from a marine-derived Aspergillus sp, J. Nat. Prod., 2008, 71, 2064–2067 CrossRef CAS PubMed ; N. P. Lavey, J. A. Coker, E. A. Ruben and A. S. Duerfeldt, Sclerotiamide: the first non peptide based natural product activator of bacterial caseinolytic protease P, J. Nat. Prod., 2016, 79, 1193–1197 CrossRef PubMed .
- B. X. Zhang, W. F. Zheng, X. Q. Wang, D. Q. Sun and C. Z. Li, Total synthesis of notoamides F, I, and R and sclerotiamide, Angew. Chem., Int. Ed., 2016, 55, 10435–10438 CrossRef CAS PubMed .
- P. J. Stephens and N. Harada, ECD cotton effect approximated by the Gaussian curve and other methods, Chirality, 2010, 22, 229–233 CAS .
- S. Cai, Y. Luan, X. Kong, T. Zhu, Q. Gu and D. Li, Isolation and photoinduced conversion of 6-epi-stephacidins from Aspergillus taichungensis, Org. Lett., 2013, 15, 2168–2171 CrossRef CAS PubMed .
- J. S. Huang, Y. Shu, D. Vyas, C. Fairchild, A. Menendez, K. Krampitz, R. Dalterio, S. E. Klohr and Q. Gao, Stephacidin A and B: two structurally novel, selective inhibitors of the testosterone-dependent prostate LNCaP cells, J. Am. Chem. Soc., 2002, 124, 14556–14557 CrossRef .
- C. Escolano, B. Stephacidin, the avrainvillamide dimer: a formidable synthetic challenge, Angew. Chem., Int. Ed., 2005, 44, 7670–7673 CrossRef CAS PubMed .
- H. Kato, T. Nakahara, K. Sugimoto, K. Matsuo, I. Kagiyama, J. C. Frisvad, D. H. Sherman, R. M. Williams and S. Tsukamoto, Isolation of notoamide S and enantiomeric 6-epi-stephacidin A from the fungus Aspergillus amoenus: biogenetic implications, Org. Lett., 2015, 17(3), 700–703 CrossRef CAS PubMed .
- J. D. Sunderhaus, T. J. McAfoos, J. M. Finefield, H. Kato, S. Li, S. Tsukamoto, D. H. Sherman and R. M. Williams, Synthesis and bioconversions of notoamide T: a biosynthetic precursor to stephacidin A and notoamide B, Org. Lett., 2013, 151, 22–25 CrossRef PubMed .
- G. D. Artman, A. W. Grubbs and R. M. Williams, Concise, asymmetric, stereocontrolled total synthesis of stephacidins A, B and notoamide B, J. Am. Chem. Soc., 2007, 129, 6336–6342 CrossRef CAS PubMed .
- X. Sui, N. Kong, L. Ye, W. Han, J. Zhou, Q. Zhang, C. He and H. Pan, p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents, Cancer Lett., 2014, 344(2), 174–179 CrossRef CAS PubMed .
- Y. Zhou, Y. Li, W. Jiang and L. Zhou, MAPK/JNK signaling: a potential autophagy regulation pathway, Biosci. Rep., 2015, 35(3), e00199 Search PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03640g |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Peter Prokschc,
Xiangmei Chen*b and
Wenhan Lin
a,
Peter Prokschc,
Xiangmei Chen*b and
Wenhan Lin