
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational elucidation of the binding mechanisms of curcumin analogues as bacterial RecA inhibitors†
Zi-Yuan Zhou‡
ab,
Jing Yuan‡a,
Qing Panc,
Xiao-Mei Moa,
Yong-Li Xiea,
Feng Yin b,
Zigang Li*b and
Nai-Kei Wong*a
b,
Zigang Li*b and
Nai-Kei Wong*a
aDepartment of Infectious Diseases, Shenzhen Third People's Hospital, The Second Hospital Affiliated to Southern University of Science and Technology, Shenzhen 518112, China. E-mail: wongnksz@163.com
bDepartment of Chemical Biology, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen 518055, China. E-mail: lizg@pkusz.edu.cn
cShenzhen Key Laboratory of Microbial Genetic Engineering, College of Life Sciences and Oceanology, Shenzhen University, Shenzhen 518055, China
First published on 25th June 2019
Abstract
Antimicrobial resistance (AMR) presents as a serious threat to global public health, which urgently demands action to develop alternative antimicrobial strategies with minimized selective pressure. The bacterial SOS response regulator RecA has emerged as a promising target in the exploration of new classes of antibiotic adjuvants, as RecA has been implicated in bacterial mutagenesis and thus AMR development through its critical roles in error-prone DNA repair. The natural product curcumin has been reported to be an effective RecA inhibitor in several Gram-negative bacteria, but details on the underlying mechanisms are wanting. In order to bridge the gap in how curcumin operates as a RecA inhibitor, we used computational approaches to model interactions between RecA protein and curcumin analogues. We first identified potential binding sites on E. coli RecA protein and classified them into four major binding pockets based on biological literature and computational findings from multiple in silico calculations. In docking analysis, curcumin–thalidomide hybrids were predicted to be superior binders of RecA compared with bis-(arylmethylidene)acetone curcumin analogues, which was further confirmed by MMGBSA calculations. Overall, this work provides mechanistic insights into bacterial RecA protein as a target for curcumin-like compounds and offers a theoretical basis for rational design and development of future antibiotic adjuvants.
Introduction
In the last two decades, antimicrobial resistance (AMR) has spurred the rapid emergence and spread of multidrug-resistant (MDR) bacterial pathogens across the globe, threatening to reset our daily healthcare to pre-antibiotic era conditions. This current crisis has its roots in a dwindling drug development pipeline and concomitant overconsumption of antibiotics, both in food production and in the clinic.1–4 A critical lack of new antibiotics is acutely felt in the contexts of nosocomial infections by Gram negative MDR pathogens such as carbapenem-resistant Enterobacteriaceae (e.g. Klebsiella pneumoniae, Escherichia coli), Pseudomonas aeruginosa, and Acinetobacter baumannii, which contribute to profoundly increased disease burden, costs of medical treatment, risks of surgical or transplant failure and patient mortality.5–9 Concerted efforts are urgently needed to institute new strategies for preserving the efficacy of existing drugs, guiding the rational use of antibiotics, and establishing new modalities of antimicrobial treatment in order to check or at least slow down the development and spread of AMR.10Considerable incentives have been put on developing new compounds that target components of the bacterial adaptive response to selective pressure induced by antibiotics themselves. Notably, the bacterial RecA protein plays pivotal roles in several key processes leading to the acquisition of antibiotic resistance, namely, SOS response, horizontal gene transfer,11 and mutagenesis.12,13 RecA is a 38 kDa DNA recombinase best known for its regulatory function in bacterial DNA repair.14 As a DNA-dependent ATPase, it requires ATP to bind ssDNA (single-stranded DNA) in order to form a nucleoprotein filament.13 Bacterial DNA recombination is key to DNA repair and genome maintenance. In this process, RecA catalyzes the central steps of DNA recombination, aligning, pairing and DNA strand exchange reaction. In addition, RecA itself critically controls the initiation of the SOS response, by triggering autocatalytic cleavage of the transcriptional repressor LexA.14,15 In the contexts of AMR development under sublethal concentrations of antibiotics, RecA hyperactivity in SOS response results in runaway expression of inducible low-fidelity DNA repair systems (e.g. the error-prone polymerase V16,17) that drive consequent mutations,18 paving the way for genomic instability and mutagenesis-driven resistance acquisition.19 Taken as a whole, RecA seems a promising target for blocking the evolution or acquisition of bacterial antibiotic resistance.
One of the alternative approaches to conventional antibiotic regimens is to reduce antibiotic dosage while enhancing its performance via the complementary use of antibiotic potentiators or adjuvants.20–22 Previous studies on pharmacological strategies for RecA inhibition have focused on RecA binding via the ATP binding pocket for small molecule inhibitors like suramin and congo red.23 The natural product curcumin has been recognized as a potential therapeutic agent or adjuvant24 for the treatment of an array of diseases including cancer25 and Alzheimer's disease.26,27 Curcumin has shown potential of an antibacterial agent, though relatively little is known about the molecular mechanisms at work. The compound exhibits broad-range antimicrobial activity against Gram negative bacteria including Escherichia coli, Pseudomonas aeruginosa, and Salmonella typhimurium.28,29 Curcumin also reportedly inhibited the growth of clinical isolates of Helicobacter pylori in vitro and eradicated the pathogen in a mouse model.30 However, several factors have hampered progress in research on curcumin analogues as therapeutically viable antibiotic adjuvants, which include a lack of structural information for predicting the drug binding site(s) of RecA protein, and poor pharmacokinetic/pharmacodynamic (PK/PD) properties of known compounds.24
Despite its ability to inhibit antibiotic-induced SOS response,28,29,31 curcumin reportedly did not interfere with the ATP hydrolysis required for RecA activity.23 The molecular underpinnings of how curcumin binds and inhibits RecA remain obscure, as there have been no attempts on theoretical or experimental investigation on the subject thus far. Predictions on curcumin–RecA interactions by computational approaches seem opportune in this light. It is increasingly being appreciated that rational drug design can be greatly assisted by the successful application of docking software and other allied computational approaches such as MMGBSA calculation, and MD simulation.32–35 Since the discovery and elaborate characterization of RecA protein more than a decade ago, a sizable body of works have been devoted to the development of RecA inhibitors into as antibiotic adjuvants.18,36,37 However, the absence of any RecA-based docking studies restricted the exploration of the curcumin scaffold in rational drug design.
To address the gap in our knowledge on how curcumin operates as a RecA inhibitor, we here use computational approaches including docking analysis to elucidate the possible mechanisms underlying binding between RecA protein and curcumin analogues. Our work utilized known X-ray structures of bacterial RecA proteins to make predictions on potential RecA–curcumin binding sites based on literature resources. We further predicted the key amino acid residues involved by multiple algorithms, including 3D-structure based algorithms PASS,38 ConCavity,39 POCASA,40 GHECOM,41,42 DoGSiteScore43 SiteHound.44 The binding regions were then classified into four binding pockets (pockets A, B, C and D) based on their locations on RecA and physiological functions (Fig. 1). Two groups of curcumin analogues, bis-(arylmethylidene)acetone analogues and curcumin–thalidomide hybrids, were analysed for their predicted binding affinities with respect to each of these binding pockets. Structural insights gained herein should provide a theoretical foundation for future experimental exploration including rational design of curcumin skeleton based compounds as antibiotic adjuvants to help combat RecA-mediated mutagenesis and AMR development.
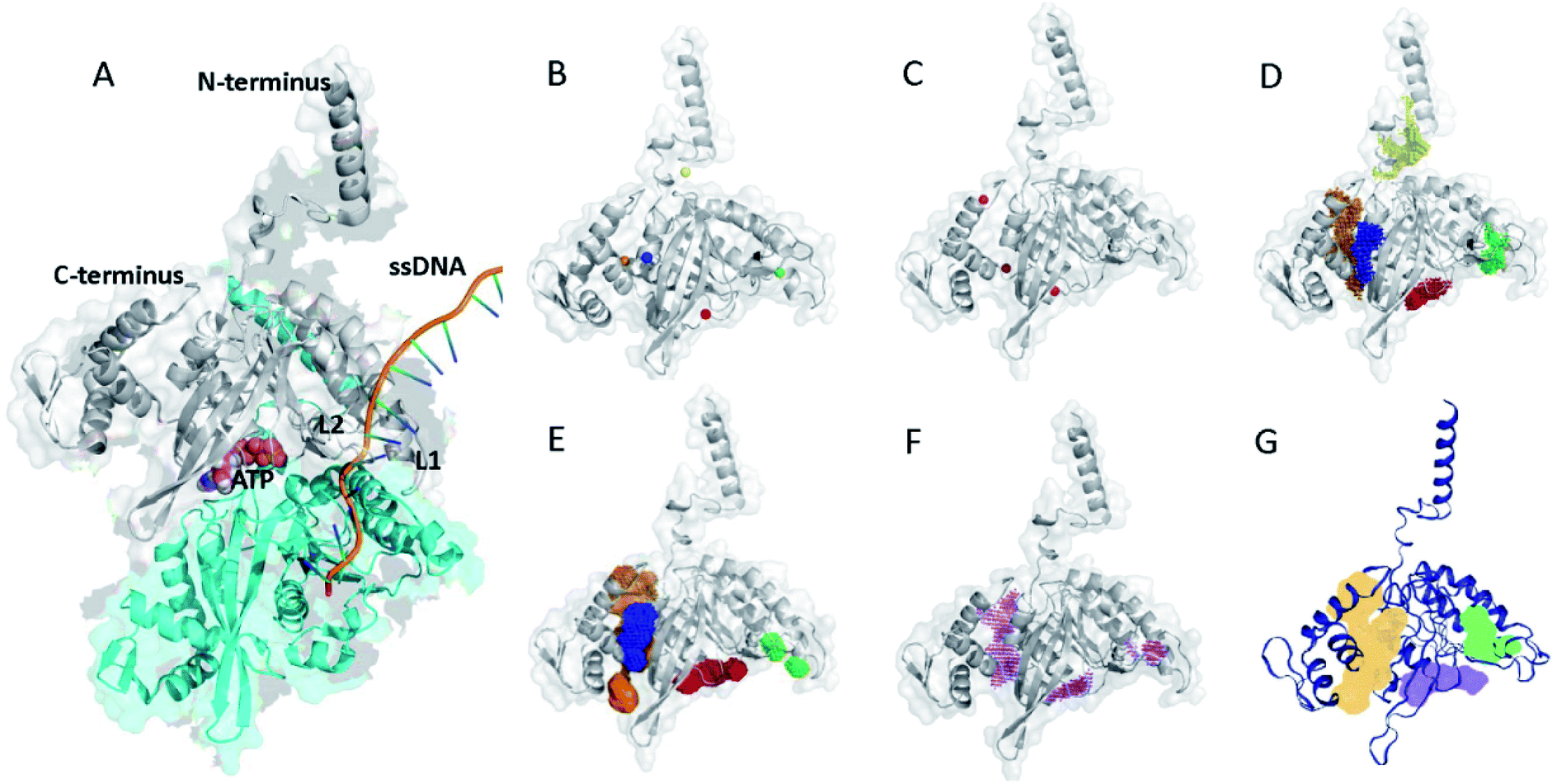 | ||
| Fig. 1 (A) X-ray structure of E. coli RecA with ATP and ssDNA. The structure was adopted with modifications from a reported crystal structure (PDB ID: 3CMT).13 (B) Potential binding sites of RecA inhibition predicted by POCASA (coloured spheres).40 (C) Potential binding sites of RecA inhibition predicted by PASS (red spheres).38 Prediction of potentially druggable target pockets by RecA inhibitors based on the 3D structure of E. coli RecA by using different algorithms, including (D) POCASA, (E) GHECOM, (F) ConCavity and (G) DoGSiteScore. Coloured patches represent spatial span of predicted binding pockets. | ||
Results and discussion
Predictions on binding sites and key amino acid residues for bacterial RecA protein
Bacterial RecA protein has been recognized as a target of pharmaceutical importance.16 However, no 3D binding information between the binding pockets of RecA and curcumin analogues is available to this date. In order to make predictions on the binding sites for drug–protein interactions, we first analyzed the 3D structure of RecA. RecA X-ray crystal structures of multiple bacterial species, including structures from Mycobacterium tuberculosis,45 Mycobacterium smegmatis46 and Escherichia coli13 are available in the PDB (Protein Data Bank) database. These RecA proteins all share very high similarity in 3D structures. The E. coli RecA (EcRecA) protein has frequently been used as a prototype of bacterial RecA in computation.14 The RecA filament can adopt two conformations, namely: the extended conformation with DNA and ATP (or non-hydrolysable ATP analog) represents the active state, while the compressed conformation formed in the absence of DNA represents the inactive state of RecA.13,47 Most of the reported RecA structures resemble the inactive state,13 in which the L1 and L2 regions essential for ssDNA binding are unresolved in the X-ray structure.48 Perturbing the active state of RecA or trapping RecA in its inactive state could block its physiological functions.47–49 Thus, the X-ray structure of E. coli RecA protein with PDB ID of 3CMT,13 which represents the active state, was applied throughout this study in the hope of gaining more realistic glimpses into RecA function.Electrostatic potential of the protein surface (upper left of Fig. 2) offers useful clues for inferring the function of RecA protein. The loop regions L1 and L2 are marked on Fig. 2. The positively charged residues Lys248 (ref. 52) and Lys250 (blue region above L2),53,54 together with Arg243 and Lys245 (blue region of pocket C, close to L2)55 were reported to be important for the binding of RecA to dsDNA in the homologous pairing process.56 The algorithm eF-surf (electrostatic surface of functional-site) was used to calculate the molecular surface of the protein, in manners consistent with the eF-site database.50 eF-seek is a web server for the discovery of similar binding sites for uploaded protein structures.51 Each of the points above the line in the density plot (upper right of Fig. 2) corresponds to a “significant” match between the binding sites in RecA and the pockets in the eF-seek database.51 On the other hand, the points in the lower left region indicate the non-similar binding sites. Since the number of dots could be too large (about 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500) to be fully displayed, a density plot is shown instead of a scatter plot below the threshold line.
500) to be fully displayed, a density plot is shown instead of a scatter plot below the threshold line.
 | ||
| Fig. 2 Similarity search for RecA binding sites based on analysis of electrostatic potential and molecular topology of RecA. Upper left: eF-surf calculated electrostatic potential (from −0.1 V to 0.1 V) for the molecular surface of RecA protein.50 Predicted binding pockets and loop regions (L1 and L2) are displayed. Upper right: 2D density plot for the similarity search of candidate RecA binding pockets and all other binding sites. Calculations were conducted by using the eF-seek web server. Scale for the density plot is as presented on the right. Curves on the plot represent the threshold line established with the Matthews' correlation coefficient (MCC) as an evaluative measure.51 The definitions of the Z-score, coverage and score are provided in the notes of table in this figure. | ||
Table in Fig. 2 shows the predicted binding residues ranked by their scores. Each row in this table corresponds to a dot in the density plot (upper right of Fig. 2). Exhaustive calculations could be very time-intensive and subsequently generate hundreds of rows ranked by their score. For clarity, we selected four of these rows for discussion. Entry 1 to entry 4 represent pocket A to pocket D, respectively. The first row of this table refers to the dots highlighted with cyan in the Fig. 2 density plot, which represents the ATPase region for RecA protein. Some of the predicted key residues are also pivotal for ATPase activities, such as Glu68, Glu96, Tyr103, Gln194, etc.52,55 The predicted ligand for entry 1 in the table of Fig. 2 is ADP, in agreement with reports on RecA structures.52,55 Again, the eF-seek web server was used for our calculations.51 The clique search algorithm of this server was used for analysis of representative binding sites in the eF-site database,50 which contains about 17500 binding sites by 2007 and has been updated about twice per year since then.50 Currently, the database contains binding pockets of bacterial RecA protein, including the X-ray structure (PDB ID: 3CMT) that we applied in this study. For each query, eF-seek carried out a comparison against all binding sites in this representative ligand database.50 The similarity between the predicted binding sites on protein and each binding site in the database is evaluated by two measures, specifically, the Z-score and coverage.51 A predicted binding site with a larger Z-score and a higher coverage value would share more features similar to that of the query.
We performed parallel analysis with the algorithm SiteHound,44 whose results are as shown in Fig. 3. The “cluster data” table displays the top 6 ranking interacting energy clusters (or putative binding sites). Potential interfacial residues of RecA identified by prediction of protein interfaces are listed in the Table in Fig. 3, which describes the ranking, key residues and volume for each cluster. The sum of the energy values of all the points that belong to the same cluster or total interaction energy (TIE), which is an indication of the binding strength between RecA corresponding pockets and clusters, is used to rank the cluster. The volume of the pockets (clusters) in Å3 reflects degree of correlation with the ranking of TIE. The colouring of the clusters in Fig. 3 corresponds to their ranking number in the Table in Fig. 3. The Cartesian coordinates (x, y, z) for each centre indicate the location of each cluster. This information is useful for generating a docking box centred on a putative binding site. For example, residues Glu68, Ser69, Ser70, Gly71, Lys72, Thr73, Thr74, Leu77, Asp94, Glu96, Leu99, Asp100, Tyr103, Ala104, Asp144 of the cluster 1 in Fig. 3 is located at the ligand binding sites with the lowest energy (−1097.5 kcal mol−1) and the biggest volume (87 Å3) best predicted by SiteHound. The cluster 1 (red) and cluster 4 (light blue) are adjacent to each other. Collectively, these two clusters indicate the ATPase core (pocket A) of RecA. The key residues Glu68, Glu96, Tyr103 predicted in cluster 1 and 4 were reported to be significant for RecA ATPase activity.52 Similarly, the cluster 2 and cluster 3 are also adjacent with each other. Together, they mark the ssDNA binding region (pocket D) with Ser172, Arg176, Met197, Ile199, Thr210, Gly211, Gly212 and Asn213 as key residues. Some of these key residues are underlined in the table in Fig. 3, which means that they are also predicted by the eF-seek as key residues, as listed in table in Fig. 3.
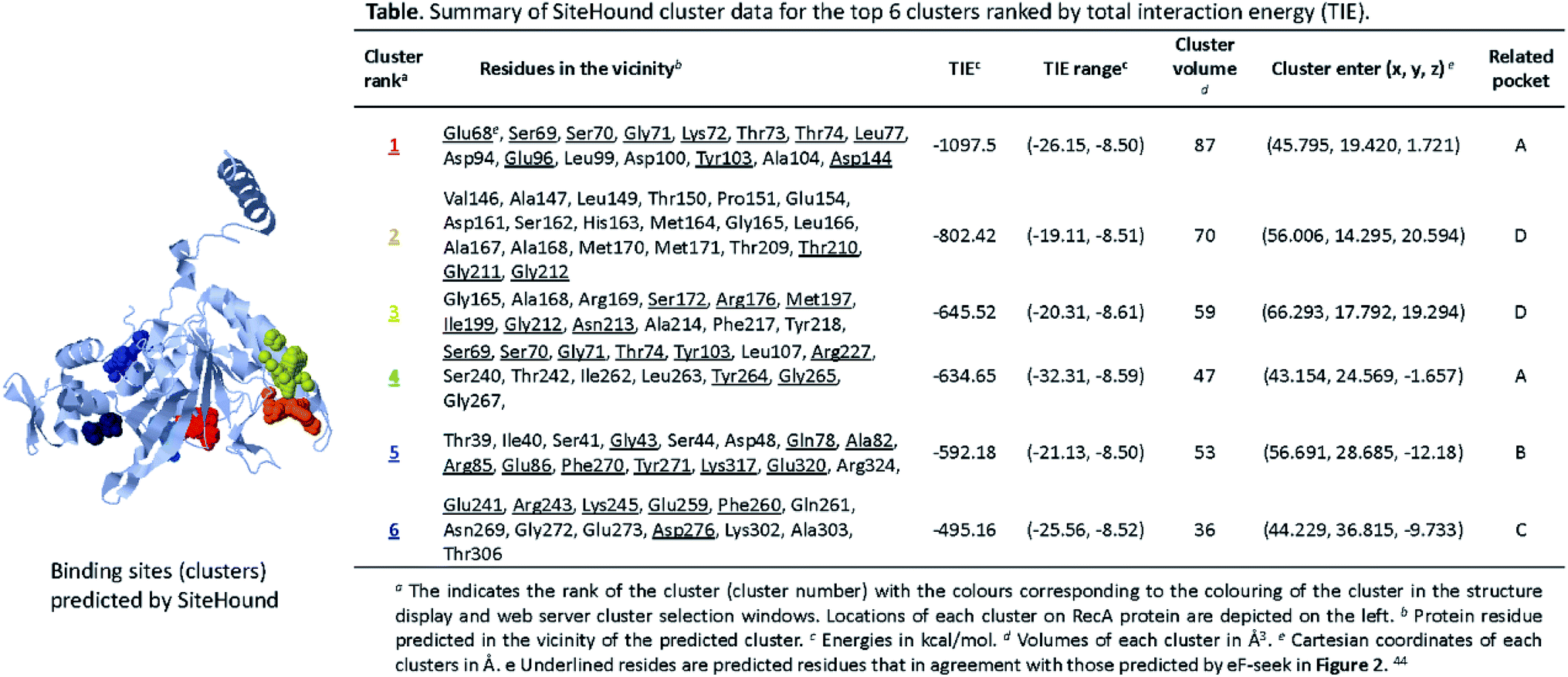 | ||
| Fig. 3 Ligand binding sites predicted by SiteHound.44 The structure displayed in the above presents a 3D view of predicted clusters (coloured spheres) in relation to the RecA protein structure. Calculations for the clusters were performed on the basis of TIE (total interacting energy) by using the Jmol Java applet. The clusters are depicted in as spheres to indicate approximate geometry. Detailed cluster data of the corresponding clusters (specified by coloured) are as shown in the above table. | ||
Structure optimization for curcumin and tetrahydrocurcumin
As an initial step to model RecA–curcumin analogue interactions, we set out to determine the most stable tautomer of curcumin for binding mechanism calculations. Tautomers of selected curcumin analogues were investigated via quantum mechanics calculations by the density functional theory (DFT) with conductor-like polarizable continuum model (CPCM)57,58 and solute electron density (SMD)59 using water as a solvent molecule in this work. Gibbs free energies of three possible tautomers of curcumin and two possible tautomers of tetrahydrocurcumin were estimated (Fig. 4). A single point energy calculation was conducted using the B3LYP/6-31G(d) optimized geometries with B3LYP/6-31++G(d,p) basis set. For optimized estimation of solvation effects, we followed the convention of comparing available experimental data and calculated values for adjustment purposes.60 For example, the SMD training set contain more than two thousand experimental solvation free energies.61 Accordingly, SMD, which is based on the universal solvation model, was used in the single point energy calculations in this study.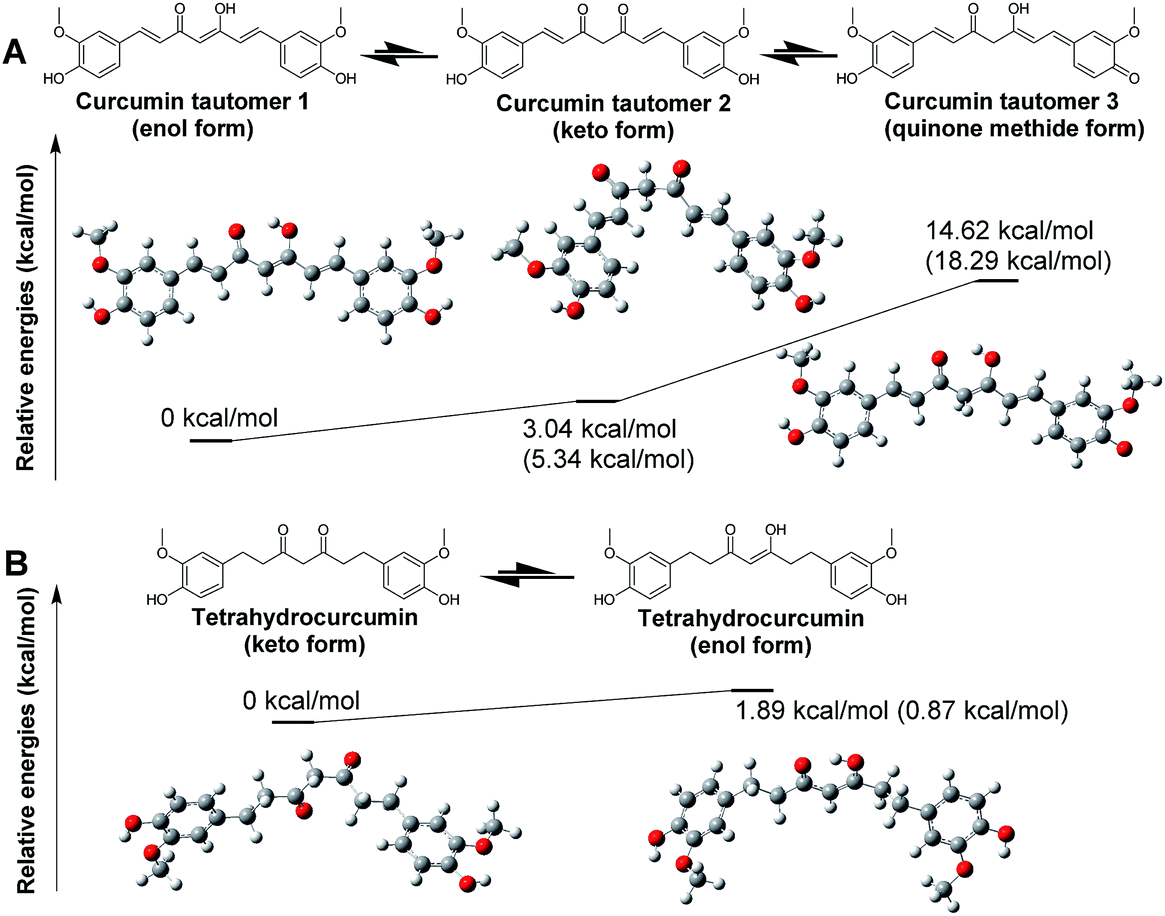 | ||
| Fig. 4 Optimized geometries and relative free energies of (A) curcumin (B) tetrahydrocurcumin. The relative Gibbs free energies in water are presented in kcal mol−1. Values outside the blankets are results for structure optimization and frequency analysis under DFT B3LYP/6-31G(d) using the CPCM solvation model in water. Values presented in brackets are calculated by energy and frequency analysis by the DFT B3LYP/6-31G++(d,p) method using SMD solvation model in water. | ||
As anticipated, the enol form of curcumin tautomer 1 (enol form) is more stable than its keto counterpart tautomer 2 (keto form) in water (Fig. 4A). This result suggests that the curcumin molecule likely exists predominantly in enol form in water. This is also in agreement with the DFT computational estimation and experimental results reported by Tsonko et al.62 Based on the similarity of the scaffolds, it is reasonable to speculate that the other curcumin analogues in Schemes 1 and 2 would similarly adopt the enol form. The energy difference between the enol and keto forms is small, which means that the keto form of curcumin could nonetheless also exist during interactions with RecA.
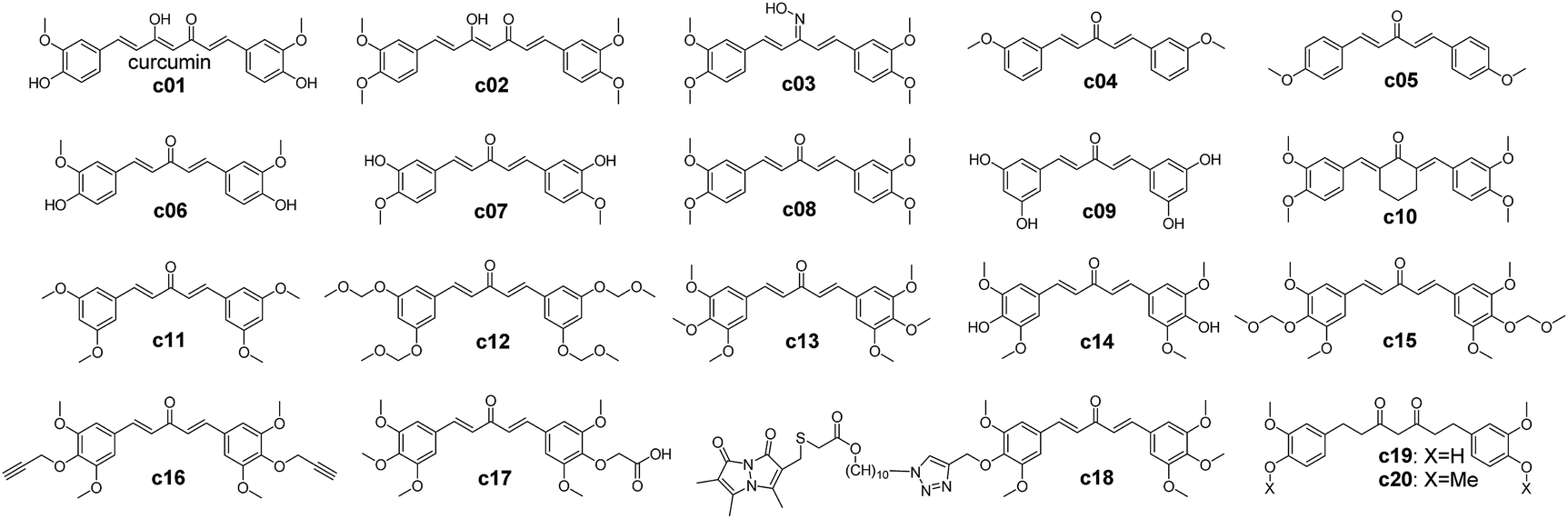 | ||
| Scheme 1 Chemical structures of curcumin analogues.65,66 | ||
 | ||
| Scheme 2 Molecular structures of the curcumin–thalidomide analogues. c21, c37 to c40 are thalidomide and thalidomide analogues, c23 to c27 are the compound 3 to 7 from ref. 73. c22 and c28 to c36 are the compounds designed in this study based on the predicted binding modes and feasibility of synthetic routes. | ||
In contrast to curcumin, tetrahydrocurcumin is predicted to assume the keto form in water based on DFT optimization. Tetrahydrocurcumin (Fig. 4B) is a commercially available analogue of curcumin, and could be used as a starting material for organic synthesis. It has a smaller delocalized conjugation system than curcumin, which imparts instability on the enol form of tetrahydrocurcumin. Analysis on potential energy profiles in Fig. 4 suggests that docking and subsequent MMGBSA calculations could be meaningfully pursued with the enol tautomers of compounds with curcumin skeleton and keto tautomers of compounds with tetrahydrocurcumin skeleton.
Binding mechanism studies of curcumin analogues as RecA specific inhibitors
In order to furnish details on the binding between RecA and curcumin analogues, the analogues in Scheme 1 were docked with each of the predicted pockets in RecA. Of note, pocket D of bacterial RecA has been reported to interact with nucleotide triplet phosphate groups.13,52 The backbone and side chain amide group of the key residue Asn213 was speculated to form hydrogen bonds with the 5′ of the phosphate group, thereby participating in the interaction between RecA ssDNA binding region and ssDNA.13 Similarly, Met197 from the loop region (L2) also assists in this process.13 The backbone amide groups of Gly211 and Gly212 from L2 of RecA form hydrogen bonds with the second phosphate group, while the Ser172 and Arg176 form hydrogen bonds with the third phosphate group.13 van der Waals contacts of the third phosphate group with the aliphatic groups at Lys198, Ile199 and Thr208 help to stabilize the binding of ssDNA with RecA.13 Among these residues in pocket D, Ile199 is crucial, for its mutation was reported to impair the DNA recombination and repair function of RecA.63,64 Inhibitors that could block this region by forming polar interactions with these residues are thus anticipated to interfere with the physiological function of bacterial RecA (Fig. 2 and 3).To further investigate the effect of structural modifications on binding energies, a set of reported curcumin analogues was selected to calculate the compounds' binding energies with different binding sites of RecA protein (Scheme 1 and Table 1). Two kinds of docking software were used in this study for the docking study of the curcumin analogues. The binding energies estimation results by Autodock Vina predict that this selected set of compounds preferentially bind pocket B rather than pocket D, but this observation was not supported by Ledock estimation with the same docking parameters. Molecules with a larger scaffold (c01, c02, c18, c19) seem to have higher predicted binding affinities toward pocket A and pocket B than the rest of compounds (c03–c17), though the differences are moderate. Introduction of a large group by click reaction did not result in significant higher predicted binding energies (c18, which was previously reported as GO-Y080).65 Yamakoshi and co-workers also reported that the bis-(arylmethylidene)acetone skeleton of c03–c17 could give rise to cytotoxicity effects of these compounds.65 In addition, the 3-oxo-1,4-pentadine structure (c13–c18) is essential for cytotoxicity.65 Among all these curcumin analogues, the 3-, 4-, 5-substituted compounds such as c13 and c15 showed the highest cytotoxicity.65 As c03–c17 failed to show higher predicted binding energies than curcumin or tetrahydrocurcumin, further computational analysis and rational design on lead compounds may take this finding into consideration.
| Entry | Binding energies estimatedc (kcal mol−1) | |||
|---|---|---|---|---|
| Pocket A | Pocket B | Pocket C | Pocket D | |
| a Curcumin in enol form.b Curcumin in keto form.c Binding energies outside the brackets were estimated by Autodock Vina,67 binding energies in the brackets were estimated by Ledock (http://www.lephar.com/). | ||||
| c01a | −7.8 (−3.94) | −8.6 (−3.24) | −6.5 (−3.06) | −6.0 (−3.61) |
| c01b | −7.2 (−3.90) | −8.0 (−3.80) | −7.6 (−3.28) | −5.7 (−3.91) |
| c02 | −7.3 (−3.28) | −7.9 (−2.91) | −6.5 (−2.66) | −5.7 (−3.02) |
| c03 | −6.7 (−2.89) | −7.2 (−3.04) | −6.5 (−2.71) | −5.9 (−3.02) |
| c04 | −6.7 (−3.23) | −7.0 (−3.03) | −6.8 (−3.19) | −5.2 (−2.91) |
| c05 | −6.5 (−3.51) | −7.4 (−3.12) | −6.9 (−3.20) | −5.3 (−2.88) |
| c06 | −7.2 (−3.86) | −7.3 (−3.46) | −7.0 (−4.09) | −5.6 (−4.01) |
| c07 | −6.9 (−4.14) | −7.8 (−3.79) | −7.1 (−3.86) | −5.7 (−3.90) |
| c08 | −6.6 (−3.18) | −7.3 (−2.88) | −6.5 (−2.95) | −5.4 (−3.15) |
| c09 | −7.0 (−4.30) | −7.6 (−4.69) | −7.0 (−4.75) | −5.9 (−4.33) |
| c10 | −7.0 (−3.53) | −7.6 (−3.64) | −7.0 (−3.20) | −6.2 (−3.23) |
| c11 | −6.7 (−3.11) | −7.2 (−2.63) | −6.7 (−3.12) | −5.3 (−2.99) |
| c12 | −6.5 (−3.35) | −7.3 (−2.81) | −5.6 (−2.82) | −5.7 (−3.34) |
| c13 | −6.9 (−3.30) | −7.1 (−3.00) | −6.5 (−2.51) | −5.4 (−3.06) |
| c14 | −6.7 (−3.48) | −7.3 (−3.59) | −6.6 (−3.48) | −5.6 (−3.86) |
| c15 | −6.5 (−3.25) | −7.0 (−2.31) | −5.8 (−2.68) | −5.3 (−3.21) |
| c16 | −6.6 (−4.11) | −7.0 (−3.48) | −6.1 (−3.50) | −5.3 (−3.75) |
| c17 | −6.9 (−2.83) | −7.3 (−2.52) | −6.2 (−2.37) | −5.3 (−2.91) |
| c18 | −7.4 (−4.23) | −6.6 (−5.14) | −5.5 (−4.34) | −6.2 (−4.88) |
| c19 | −7.3 (−3.96) | −7.9 (−3.37) | −7.3 (−3.44) | −6.4 (−4.73) |
| c20 | −7.3 (−3.28) | −7.9 (−2.91) | −6.5 (−2.66) | −5.7 (3.02) |
Interestingly, c19 (tetrahydrocurcumin) have the highest Vina binding energy for pocket D. This is probably due to the C–C single bonds in tetrahydrocurcumin molecule (c19) allowing it to be more flexible than its curcumin counterpart (c01) and other curcumin-like molecules (c02–c18) in Table 1. However, in MMGBSA calculations, the estimated binding energies did not suggest any advantages of tetrahydrocurcumin over its curcumin counterpart. The possible binding modes for both keto and enol forms of curcumin (c01) RecA and tetrahydrocurcumin (c19) with the ssDNA binding region (pocket D) were depicted in Fig. 5. The hydroxyl group of these compounds form hydrogen bonds with the amide group of Asn213. Other key polar interactions include hydrogen bonds of these molecules with the backbone of Met197, Ala167 and/or Ala168, and with the side chain of Thr208. Given the hydrophobic features of curcumin and tetrahydrocurcumin scaffolds, it seems logical to speculate that hydrophobic interactions predominate in the binding of curcumin with bacterial RecA. Both the keto and enol forms of curcumin were predicted to make van der Waals contacts with Gly212 (Fig. 5). The enol-form and keto-form of curcumin showed similar binding potencies with the pocket D region of RecA (Table 2).
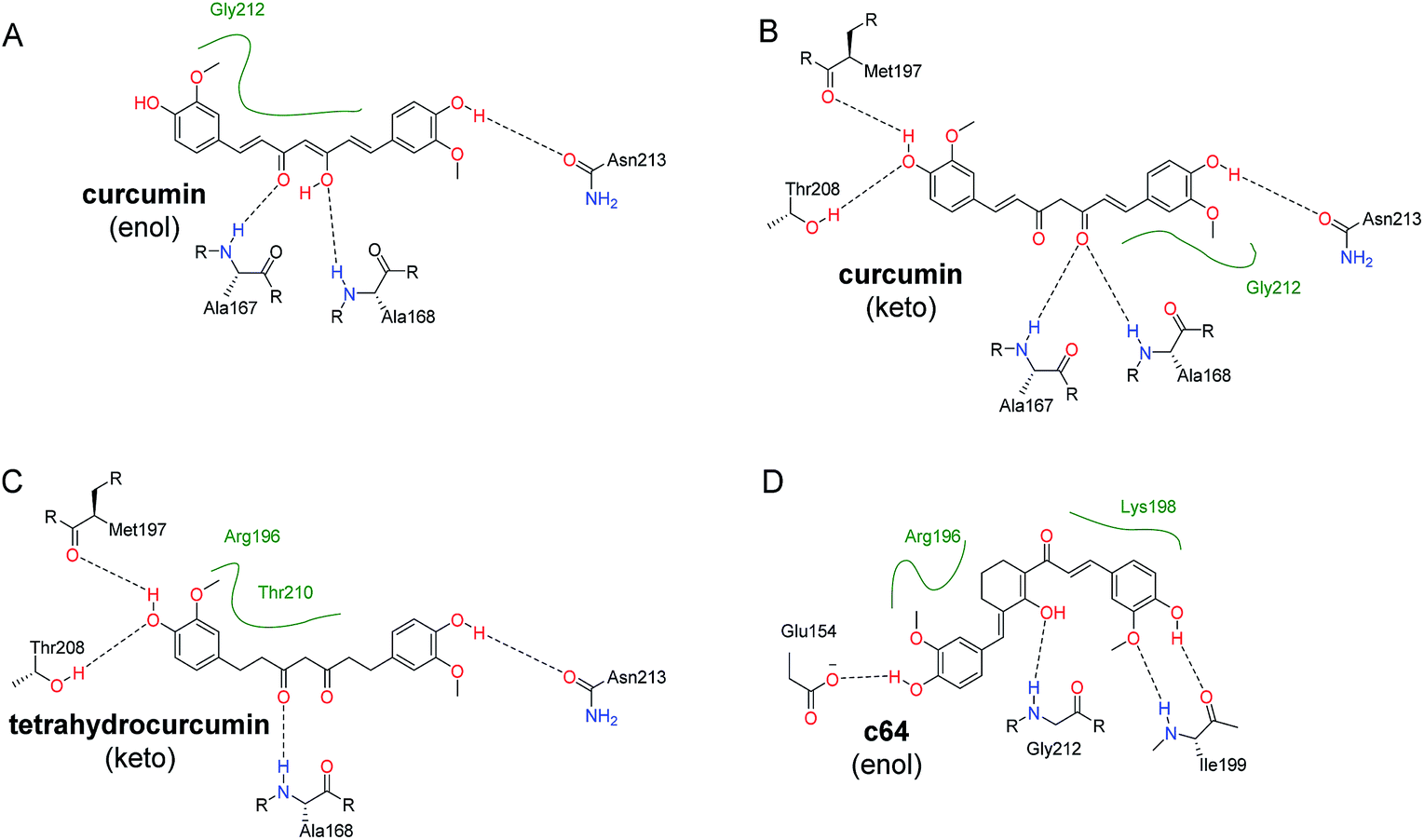 | ||
| Fig. 5 Predicted binding modes of representative compounds with the ssDNA binding region (pocket D). 2D figures were generated by Poseview68,69 and refined with Chemdraw. Hydrophobic interactions are displayed as green contact curves. (A) Curcumin (c01) in enol form; (B) curcumin (c01) in keto form; (C) tetrahydrocurcumin (c19) in keto form. (D) c64 in enol form (as depicted in Scheme S1†). | ||
| Energy termsa | RecA-inhibitor complexes | |||
|---|---|---|---|---|
| RecA + curcumin (enol) | RecA + curcumin (keto) | RecA + c19 | RecA + c64 | |
| a Definition of the energy terms: ΔGvdW = van der Waals contribution from MM. ΔGele = EEL, electrostatic energy as calculated by the MM force field. ΔGsolv,p = EPB/EGB, which is the electrostatic contribution to the solvation free energy calculated by MMPBSA or MMGBSA respectively. ΔGsolv,np = Esurf (or Ecavity/Enpolar) for MMGBSA (MMPBSA) calculation. Nonpolar contribution to the solvation free energy. ΔGMM = ΔGele + ΔGvdW. ΔGsolv = ΔGsolv,p + ΔGsolv,np. ΔGbind = ΔGMM + ΔGsolv, final estimated binding free energy calculated from the terms above. (kcal mol−1), which is the binding free energies in the absence of entropic contribution. This is described as eqn (6) in the ESI. | ||||
| ΔGvdW | −36.38 | −33.33 | −35.29 | −38.15 |
| ΔGele | −22.62 | −14.27 | −19.85 | −20.11 |
| ΔGsolv,p | 39.36 | 27.41 | 35.09 | 38.62 |
| ΔGsolv,np | −4.94 | −4.06 | −3.67 | −4.88 |
| ΔGMM | −59.00 | −47.60 | −55.14 | −58.27 |
| ΔGsolv | 34.42 | 23.36 | 31.42 | 33.74 |
| ΔGbind | −24.59 | −24.24 | −23.72 | −24.51 |
To further validate the binding energies, an MM-GBSA approach was adopted in re-calculation of the binding energies of several key analogues. The MM-GBSA approach was chosen for its ease and efficiency in ranking binding affinities of receptor and ligands compared with other algorithms such as MM-PSBA.33,34 In general, the binding energies calculated by the MM-GBSA approach tend to give excellent correlation with binding energies generated by experimental approaches.35
To further investigate the binding potency of the key molecules, we optimized the RecA–c01 (enol or keto form) and RecA–c19 with Amber16, and then refined the structures with a short MD simulation for the MMGBSA calculation. Detailed MMGBSA results are listed in Table 2. No significant difference was observed for RecA–curcumin (enol form, ΔGbind = −24.59 kcal mol−1) complex and RecA–curcumin (keto form, ΔGbind = −24.24 kcal mol−1) complex. Similarly, the ΔGbind of RecA–tetrahydrocurcumin and RecA–c64 (−23.72 and −24.51 kcal mol−1) did not show significant differences compared with that of the RecA–curcumin complexes. Compared with values for the RecA–curcumin enol/keto complexes, differences in the calculated binding free energies between these complexes were not significant. These energetic results suggest that ssDNA binding region of RecA (pocket D) does not have significant preference for the enol or keto tautomer of curcumin. Overall, we envision that simple modifications of the curcumin scaffold with different substitution groups (e.g. methyl, methoxyl, hydroxyl groups) would not markedly improve the predicted binding energies.
Binding mechanism studies on curcumin–thalidomide conjugates as RecA specific inhibitors
Combinatorial antimicrobial therapy has been a mainstay of clinical treatment of bacterial infections.70 However, while antimicrobial combinations of existing agents have almost been exclusively emphasized in clinical practice and research, insufficient progress has been seen in the development of novel antibiotic adjuvants.71 In order to keep AMR at bay, a more comprehensive approach is desired for the development of antibiotic adjuvants encompassing both antibiotics and other bioactive compounds.71 One possible strategy to economize on the costs of drug discovery is to generate hybrid molecules that inherit the pharmacological advantages of parent compounds. In this study, we attempted to rationally design a panel of hybrid molecules containing a curcumin scaffold and thalidomides, followed by predictions of their binding energies.Curcumin scaffold based structural modifications in Scheme 1 do not seem to allow lower predicted energies than curcumin itself, which means that no analogues in Scheme 1 are predicted to be significantly more potent than curcumin itself (see Table 1). Furthermore, the curcumin analogues (c03–c17) with bis-(arylmethylidene)acetone skeleton may give rise to greater cytotoxicity. Since curcumin is a hydrophobic compound with calculated logP = 3.62 (https://chemaxon.com) and very low water solubility of 0.00575 mg mL−1 (https://www.drugbank.ca), introducing hydrophilic groups while keeping the scaffold of the curcumin unchanged could be a promising direction in rational drug design of curcumin-like compounds as RecA inhibitors. Thalidomide is an FDA approved drugs with higher water solubility (2.55 mg mL−1, https://www.drugbank.ca) and lower calculated logP of 0.16 (https://chemaxon.com) than curcumin. Incorporation of the thalidomide moiety as a hydrophilic group into the curcumin scaffold may help circumvent constraints during structural modification of curcumin to achieve higher binding affinity with RecA. Although thalidomide was once withdrawn due to severe teratogenicity, its safety for adults was confirmed. It has recently been reintroduced by FDA as a therapeutic agent for cancer such as human multiple myeloma and for some inflammatory diseases.72 Introduction of a thalidomide moiety into the curcumin skeleton has been attempted previously. In fact, hybrid compounds of thalidomide and curcumin (c23–c27 in Scheme 2) have been successfully designed and synthesized by Liu et al. for treating human multiple myeloma.73
Compared with the case of curcumin analogues, significantly higher binding scores (Table 1) were observed for the thalidomide–curcumin hybrids (Table 3). This applies to both of the scores generated by Autodock Vina and Ledock. Majority of these hybrid compounds are predicted to be more potent bacterial RecA inhibitors than the curcumin analogues. Interestingly, although curcumin was reportedly unable to interfere with RecA ATPase activity, which implicitly means that curcumin is not a major binder of RecA pocket A, the possibility for thalidomide-curcumin hybrids to interact with RecA pocket A cannot be ruled out.
| Entry | Estimated binding energiesa (kcal mol−1) | |||
|---|---|---|---|---|
| Pocket A | Pocket B | Pocket C | Pocket D | |
| a Binding energies outside the brackets were estimated by Autodock Vina67 and binding energies within the brackets were estimated by Ledock (http://www.lephar.com/). | ||||
| c21 | −7.7 (−2.97) | −7.7 (−3.09) | −6.4 (−2.85) | −5.6 (−3.02) |
| c22 | −11.5 (−5.29) | −10.5 (−6.20) | −8.6 (−5.04) | −8.3 (−4.53) |
| c23 | −10.2 (−5.35) | −10.1 (−4.43) | −8.5 (−4.22) | −7.6 (−4.83) |
| c24 | −9.3 (−3.98) | −9.7 (−4.36) | −7.9 (−3.83) | −8.5 (−4.61) |
| c25 | −10.8 (−4.68) | −9.3 (−4.73) | −8.8 (−5.76) | −7.4 (−5.38) |
| c26 | −8.9 (−4.19) | −9.4 (−3.97) | −8.0 (−4.15) | −7.4 (−4.37) |
| c27 | −9.8 (−4.76) | −9.4 (−4.49) | −8.8 (−5.18) | −7.0 (−4.74) |
| c28 | −11.5 (−4.94) | −10.7 (−4.80) | −9.8 (−4.39) | −8.8 (−5.57) |
| c29 | −10.1 (−5.55) | −9.9 (−4.50) | −8.3 (−4.17) | −7.5 (−5.02) |
| c30 | −10.0 (−5.52) | −9.4 (−6.00) | −8.1 (−6.16) | −7.5 (−6.42) |
| c31 | −9.8 (−6.52) | −9.1 (−6.42) | −8.0 (−6.18) | −8.7 (−5.00) |
| c32 | −9.7 (−3.93) | −11.1 (−5.69) | −7.6 (−4.11) | −8.9 (−4.89) |
| c33 | −8.5 (−4.40) | −9.3 (−4.38) | −7.2 (−4.33) | −9.1 (−5.60) |
| c34 | −8.8 (−4.67) | −9.6 (−5.36) | −7.7 (−4.95) | −6.7 (−5.33) |
| c35 | −10.0 (−5.13) | −10.9 (−6.14) | −8.4 (−5.20) | −8.7 (−5.48) |
| c36 | −9.4 (−4.24) | −9.1 (−5.15) | −7.3 (−4.51) | −6.5 (−4.27) |
| c37 | −9.8 (−6.52) | −9.1 (−6.42) | −8.0 (−6.18) | −5.5 (−5.00) |
| c38 | −7.3 (−2.49) | −7.9 (−2.19) | −8.1 (−2.12) | −6.1 (−2.39) |
| c39 | −7.8 (−3.32) | −7.8 (−3.37) | −6.5 (−3.17) | −5.9 (−3.27) |
| c40 | −7.7 (−3.21) | −7.8 (−2.96) | −7.2 (−2.95) | −5.8 (−3.14) |
The c22 and c24 are speculated to adopt the enol form as they feature a curcumin scaffold, while c32 is predicted to adopt the keto form for its tetrahydrocurcumin scaffold (Fig. 6). The binding energies (ΔGbind) of both the enol and keto forms were recalculated by MMGBSA. The free energy gap between the enol and keto forms for tetrahydrocurcumin is quite small (Fig. 4B). As shown in Table 4, the ΔGbind value of the c32 (keto form)–RecA complex (ΔGbind = −38.46 kcal mol−1) is less negative compared with that of c32 (enol form)–RecA complex (ΔGbind = −43.32 kcal mol−1), which suggests that the enol form of c32 likely binds more firmly the pocket D of RecA than does the keto form of c32. Thus, the binding process of c32 with RecA could trigger c32 to adopt an enol form.
 | ||
| Fig. 6 Predicted binding modes of RecA pocket D with (A) c22; (B) c24; (C) c32 in enol form; and (D) c32 in keto form. 2D figures were generated by Poseview68,69 and refined with ChemDraw. Hydrophobic interactions are displayed as green contact curves. | ||
| Energy termsa | RecA-inhibitor complexes | ||
|---|---|---|---|
| RecA + c22 | RecA + c24 | RecA + c32 (enol)/c32 (keto) | |
| a Definitions of the energy terms are the same as that of Table 2. | |||
| ΔGvdW | −76.32 | −43.11 | −82.68/−68.72 |
| ΔGele | −16.75 | −7.29 | −31.24/−1.49 |
| ΔGsolv,p | 53.46 | 28.51 | 79.13/38.87 |
| ΔGsolv,np | −7.37 | −5.83 | −8.54/−7.11 |
| ΔGMM | −93.07 | −50.41 | −113.92/−70.21 |
| ΔGsolv | 46.09 | 22.67 | 70.60/31.75 |
| ΔGbind | −46.98 | −27.73 | −43.32/−38.46 |
To elaborate on our proposition that curcumin–thalidomide hybrids are more potent than other curcumin-like compounds, the binding energies of 20 curcumin building blocks (compounds c41–c60, in Scheme S1†) and 25 additional curcumin analogues (compounds c61–c85, in Scheme S2†) were further predicted (see structural details in the ESI, Pages S13–S15†). As shown in Table S3,† the additional curcumin analogues in Scheme S2† consistently showed significantly larger predicted binding energies (e.g. energies range from −5.6 to −6.4 for pocket D) than that of building blocks in Scheme S1† (e.g. energies range from −4.3 to −5.2 for pocket D). Therefore, the additional curcumin analogues in Scheme S2† are predicted to be more potent RecA inhibitors than the building blocks in the Scheme S1.† However, none of the compounds from these two groups are predicted to be more potent than the thalidomide–curcumin hybrids (e.g. energies range from −6.5 to −9.1 for pocket D), as shown in Table 3.
The above results indicate that the curcumin–thalidomide hybrids in Scheme 2 could shed light on new directions in the rational design of novel curcumin analogues as bacterial RecA inhibitors for blocking the bacterial SOS pathway. Sources of the 85 compounds investigated in this study are as listed in Table 5.
| Entry | Compounds | Classification | Applications | Ref. |
|---|---|---|---|---|
| 1 | c01–c20 | Curcumin and curcumin analogues | Inhibitors of bacterial RecA protein; cancer treatment | 65 and 66 |
| 2 | c23–c27 | Curcumin–thalidomide conjugates | Treatment of multiple myeloma | 73 |
| 3 | c22, c28–c37 | Rational drug design of this study | Predicted RecA protein inhibitors | None |
| 4 | c21, c38–c40 | Thalidomide and its analogues | “Building blocks” for rational design | None |
| 5 | c41–c55 | Reported curcumin “building blocks” | Creating antimicrobial food-contact surfaces | 74 |
| 6 | c56–c85 | Additional curcumin analogues | Inhibitors of bacterial sialidase | 75 and 76 |
Conclusion
In summary, we have advanced an account on the possible binding mechanisms underlying RecA inhibition by curcumin and its analogues. The binding pockets and related key amino acid residues on the bacterial RecA protein were predicted by multiple algorithms. We first utilized a known X-ray structure of bacterial RecA to predict the binding sites based on computational studies and biological literature. The RecA binding sites were classified into four different binding pockets. Pocket A is the ATP binding region. Pocket B could be an important modulator of RecA protein conformation, with yet unclarified physiological functions. Pocket C defines the dsDNA binding region, while pocket D constitutes the ssDNA binding region. These four binding pockets provide a fuller picture of drug–protein interactions for the rational design of RecA inhibitors as antibiotic adjuvants. On the basis of the predicted binding pockets, two groups of curcumin analogues were analysed for their binding affinities with RecA. First, bis-(arylmethylidene)-acetone skeleton compounds were investigated for their binding affinities with each of the RecA binding sites. Second, when a thalidomide group was introduced into the curcumin scaffold in calculations, we found a significant increase in the predicted binding affinities. The curcumin–thalidomide hybrids could constitute an opportunity for drug design of curcumin-skeleton based antibiotic adjuvants as RecA inhibitors. In this light, a group of hybrids of (tetrahydro)-curcumin–thalidomide was designed in this study taking into account the above-described binding mechanisms and feasibility of synthetic routes (c22, c28–c37). The compounds were further predicted for their binding affinities with RecA. These curcumin–thalidomide hybrids were estimated to bind RecA more efficiently than existing compounds. The theoretical predictions reported in this work offer insights into how RecA inhibition may be achieved through interactions with curcumin and its analogues. The knowledge thus gained could prove useful to future drug design and development of antibiotic adjuvants targeting bacterial RecA protein.Experimental
DFT optimization of the structures
All optimizations and Gibbs free energy calculations in Fig. 2 were performed with the Gaussian 09 program77 by using the density functional theory, which has been credited as a powerful tool for clarifying detailed reaction mechanisms and predicting stereoselectivity and chemoselectivity in both organic and biological reactions.78 All geometries of the molecules were fully optimized in water with CPCM solvent model at the B3LYP/6-31G(d) level of DFT theory with Gaussian 09 software package.79Molecular docking of RecA and inhibitors
In order to explore the binding mode of RecA with its inhibitors, molecular docking was performed by using the Ledock and AutoDock Vina programs.67 For running Autodock Vina, the 3D structure files for all curcumin analogues and the RecA protein were applied in pdbqt format, which is a format similar to the pdb format. The major difference is that pdbqt contains information about polar hydrogen, while the pdb usually does not contain information about hydrogen atoms.80 In this work, the pdbqt files for the curcumin analogues were prepared with the open sourced chemical toolbox, Open Babel.81 The search area was specified by size and coordinates in the 3D space (grid box for AutoDock). Vina only searches for the possible docking modes in this specified search area of the receptor. In this study, we have four different grid boxes, one for each of the pockets A, B, C and D. The geometry parameters of each pocket are as listed in the ESI.†MM-GBSA calculations
The MM-GBSA calculation procedure used here is similar to what we previously used for identifying small molecule inhibitors of a known target.32 The compounds in Tables 2 and 4 were further optimized using a multiple-step procedure, which includes the format conversion of small molecules using Open Babel, 2000 steps of energy-minimization, 100 ps of molecular dynamic simulation, 4000 steps of energy-minimization, and then molecular mechanics/generalized born and surface area (MM-GBSA) binding energy calculation using AMBER 16 software package. This procedure is similar to the known binding estimation after refinement (BEAR) protocol. More details about the calculation are listed in the ESI (Page S16†). The MM-PBSA model is theoretically more rigorous and computationally heavier than MM-GBSA, but it does not always a better correlation with experimental binding free energy when compared the MM-PBSA and MM-GBSA methods in calculation terms of solvation.33,34 In general, the binding energies calculated by MM-GBSA approach provide excellent correlation with experimental binding energies. In this regard, the rescoring of docking complexes using MM-GBSA has emerged as a computationally important approach in structure-based drug design.35 To ensure the accuracy of MMGBSA calculations, BEAR (binding estimation after refinement),82 a classical protocol that have successfully applied in our previous studies, was used for the MMGBSA in this work.32Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by high-performance computing platform of Peking University. The Special Support Funds of Shenzhen for Introduced High-Level Medical Team grants (SZSM201512005) for Prof. Lanjuan Li and Dr Jing Yuan, and the China Postdoctoral Science Foundation (2018M641097) are acknowledged. We also thank Prof. Xinhao Zhang for his useful suggestions during the course of manuscript preparation.References
- C.-A. D. Burnham, J. Leeds, P. Nordmann, J. O'Grady and J. Patel, Nat. Rev. Microbiol., 2017, 15, 697–703 CrossRef CAS PubMed
.
- L. J. V. Piddock, Nat. Rev. Microbiol., 2017, 15, 639–640 CrossRef CAS PubMed
- S. J. Howard, S. Hopwood and S. C. Davies, Sci. Transl. Med., 2014, 6, 1–2 CrossRef PubMed
- K. U. Jansen, C. Knirsch and A. S. Anderson, Nat. Med., 2018, 24, 10–20 CrossRef CAS PubMed
- J. A. Otter and G. L. French, Lancet Infect. Dis., 2010, 10, 227–239 CrossRef PubMed
- D. H. Kett, E. Cano, A. A. Quartin, J. E. Mangino, M. J. Zervos, P. Peyrani, C. M. Cely, K. D. Ford, E. G. Scerpella and J. A. Ramirez, Lancet Infect. Dis., 2011, 11, 181–189 CrossRef PubMed
- M. E. Falagas, A. C. Kastoris, A. M. Kapaskelis and D. E. Karageorgopoulos, Lancet Infect. Dis., 2010, 10, 43–50 CrossRef CAS PubMed
- D. E. Karageorgopoulos and M. E. Falagas, Lancet Infect. Dis., 2008, 8, 751–762 CrossRef PubMed
- J. Li, R. L. Nation, J. D. Turnidge, R. W. Milne, K. Coulthard, C. R. Rayner and D. L. Paterson, Lancet Infect. Dis., 2006, 6, 589–601 CrossRef CAS PubMed
- E. A. Ashley, J. Recht, A. Chua, D. Dance, M. Dhorda, N. V. Thomas, N. Ranganathan, P. Turner, P. J. Guerin, N. J. White and N. P. Day, J. Antimicrob. Chemother., 2018, 73, 1737–1749 CrossRef CAS PubMed
- A. Babić, A. B. Lindner, M. Vulić, E. J. Stewart and M. Radman, Science, 2008, 319(5869), 1533–1536 CrossRef PubMed
- A. Yakimov, G. Pobegalov, I. Bakhlanova, M. Khodorkovskii, M. Petukhov and D. Baitin, Nucleic Acids Res., 2017, 45, 9788–9796 CrossRef CAS PubMed
- Z. Chen, H. Yang and N. P. Pavletich, Nature, 2008, 453, 489–494 CrossRef CAS PubMed
- M. M. Cox, Crit. Rev. Biochem. Mol. Biol., 2007, 42, 41–63 CrossRef CAS PubMed
- M. M. Cox, Nat. Rev. Mol. Cell Biol., 2007, 8, 127–138 CrossRef CAS PubMed
- K. Schlacher, P. Pham, M. M. Cox and M. F. Goodman, Chem. Rev., 2006, 106, 406–419 CrossRef CAS PubMed
- K. Schlacher, K. Leslie, C. Wyman, R. Woodgate, M. M. Cox and M. F. Goodman, Mol. Cell, 2005, 17, 561–572 CrossRef CAS PubMed
- P. Bellio, L. Di Pietro, A. Mancini, M. Piovano, M. Nicoletti, F. Brisdelli, D. Tondi, L. Cendron, N. Franceschini, G. Amicosante, M. Perilli and G. Celenza, Phytomedicine, 2017, 29, 11–18 CrossRef CAS PubMed
- M. A. Kohanski, D. J. Dwyer, B. Hayete, C. A. Lawrence and J. J. Collins, Cell, 2007, 130, 797–810 CrossRef CAS PubMed
- C. Gonzalez-Bello, Bioorg. Med. Chem. Lett., 2017, 27, 4221–4228 CrossRef CAS PubMed
- J. Bueno, J. Microb. Biochem. Technol., 2016, 8, 169–176 CAS
- M. A. Farha and D. D. Brown, Nat. Biotechnol., 2013, 31, 120–122 CrossRef CAS PubMed
- T. J. Wigle and S. F. Singleton, Bioorg. Med. Chem. Lett., 2007, 17, 3249–3253 CrossRef CAS PubMed
- K. M. Nelson, J. L. Dahlin, J. Bisson, J. Graham, G. F. Pauli and M. A. Walters, J. Med. Chem., 2017, 60, 1620–1637 CrossRef CAS PubMed
- G. Elias, P. J. Jacob, E. Hareeshbabu, V. B. Mathew, B. Krishnan and K. Krishnakumar, Int. J. Pharm. Sci. Res., 2015, 6, 2671–2680 CAS
- S. R. Rainey-Smith, B. M. Brown, H. R. Sohrabi, T. Shah, K. G. Goozee, V. B. Gupta and R. N. Martins, Br. J. Nutr., 2016, 115, 2106–2113 CrossRef CAS PubMed
- K. H. M. Cox, A. Pipingas and A. B. Scholey, J. Psychopharmacol., 2015, 29, 642–651 CrossRef CAS PubMed
- P. Bellio, F. Brisdelli, M. Perilli, A. Sabatini, C. Bottoni, B. Segatore, D. Setacci, G. Amicosante and G. Celenza, Phytomedicine, 2014, 21, 430–434 CrossRef CAS PubMed
- S. A. Marathe, R. Kumar, P. Ajitkumar, V. Nagaraja and D. Chakravortty, J. Antimicrob. Chemother., 2013, 68, 139–152 CrossRef CAS PubMed
- R. De, P. Kundu, S. Swarnakar, T. Ramamurthy, A. Chowdhury, G. B. Nair and A. K. Mukhopadhyay, Antimicrob. Agents Chemother., 2009, 53, 1592–1597 CrossRef CAS PubMed
- Y. Oda, Mutat. Res. Lett., 1995, 348, 67–73 CrossRef CAS
- Z. Zhou, Y. Yuan, S. Zhou, K. Ding, F. Zheng and C.-G. Zhan, Bioorg. Med. Chem. Lett., 2017, 27, 3739–3743 CrossRef CAS PubMed
- T.-J. Hou, J.-M. Wang, Y.-Y. Li and W. Wang, J. Comput. Chem., 2011, 32, 866–877 CrossRef CAS PubMed
- S.-Y. Huang, S. Z. Grinter and X. Zou, Phys. Chem. Chem. Phys., 2010, 12, 12899–12908 RSC
- C. R. W. Guimaraes, Methods Mol. Biol., 2012, 819, 255–268 CrossRef CAS PubMed
- Md K. Alam, A. Alhhazmi, John F. DeCoteau, Y. Luo and C. R. Geyer, RecA Inhibitors Potentiate Antibiotic Activity and Block Evolution of Antibiotic Resistance, Cell Chem. Biol., 2016, 23, 381–391 CrossRef PubMed
- J. Z. Sexton, T. J. Wigle, Q. He, M. A. Hughes, G. R. Smith, S. F. Singleton, A. L. Williams and L.-A. Yeh, Curr. Chem. Genomics, 2010, 4, 34–42 CrossRef CAS PubMed
- G. P. Brady Jr and P. F. W. Stouten, J. Comput.-Aided Mol. Des., 2000, 14, 383–401 CrossRef CAS
- J. A. Capra, R. A. Laskowski, J. M. Thornton, M. Singh and T. A. Funkhouser, PLoS Comput. Biol., 2009, 5, e1000585 CrossRef PubMed
- J. Yu, Y. Zhou, I. Tanaka and M. Yao, Bioinformatics, 2010, 26, 46–52 CrossRef CAS PubMed
- T. Kawabata, Proteins, 2009, 78, 1195–1211 CrossRef PubMed
- T. Kawabata and N. Go, Proteins, 2007, 68, 516–529 CrossRef CAS PubMed
- A. Volkamer, D. Kuhn, T. Grombacher, F. Rippmann and M. Rarey, J. Chem. Inf. Model., 2012, 52, 360–372 CrossRef CAS PubMed
- M. Hernandez, D. Ghersi and R. Sanchez, Nucleic Acids Res., 2009, 37, 413–416 CrossRef PubMed
- S. Datta, N. Ganesh, N. R. Chandra, K. Muniyappa and M. Vijayan, Proteins, 2003, 50, 474–485 CrossRef CAS PubMed
- M.-C. Unciuleac, S. Shuman and P. C. Smith, J. Bacteriol., 2016, 198, 1521–1533 CrossRef CAS PubMed
- M. S. VanLoock, X. Yu, S. Yang, A. L. Lai, C. Low, M. J. Campbell and E. H. Egelman, Structure, 2003, 11, 187–196 CrossRef CAS PubMed
- D. A. McGrew and K. L. Knight, Crit. Rev. Biochem. Mol. Biol., 2003, 38, 385–432 CrossRef CAS PubMed
- J. C. Drees, S. L. Lusetti and M. M. Cox, J. Biol. Chem., 2004, 279, 52991–52997 CrossRef CAS PubMed
- K. Kinoshita, Y. Murakami and H. Nakamura, Nucleic Acids Res., 2007, 35, 398–402 CrossRef PubMed
- K. Kinoshita and H. Nakamura, Protein Sci., 2005, 14, 711–718 CrossRef CAS PubMed
- R. M. Story and T. A. Steitz, Nature, 1992, 355, 374–376 CrossRef CAS PubMed
- X. Xing and C. E. Bell, Biochemistry, 2004, 43, 16142–16152 CrossRef CAS PubMed
- T. Shinohara, S. Ikawa, W. Iwasaki, T. Hiraki, T. Hikima, T. Mikawa, N. Arai, N. Kamiya and T. Shibata, Nucleic Acids Res., 2015, 43, 973–986 CrossRef CAS PubMed
- R. M. Story, I. T. Weber and T. A. Steitz, Nature, 1992, 355, 318–325 CrossRef CAS PubMed
- H. Kurumizaka, S. Ikawa, A. Sarai and T. Shibata, Arch. Biochem. Biophys., 1999, 365, 83–91 CrossRef CAS PubMed
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed
- C. K. Kim, B.-H. Park, H. W. Lee and C. K. Kim, Org. Biomol. Chem., 2013, 11, 1407–1413 RSC
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- A. C. Chamberlin, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2008, 112, 8651–8655 CrossRef CAS PubMed
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
- M. Kolev Tsonko, A. Velcheva Evelina, A. Stamboliyska Bistra and M. Spiteller, Int. J. Quantum Chem., 2005, 102, 1069–1079 CrossRef
- K. Hörtnagel, O. N. Voloshin, H. H. Kinal, N. Ma, C. Schaffer-Judge and R. D. Camerini-Otero, J. Mol. Biol., 1999, 286, 1097–1106 CrossRef PubMed
- F. Larminat, C. Cazaux, M. Germanier and M. Defais, J. Bacteriol., 1992, 174, 6264–6269 CrossRef CAS PubMed
- H. Yamakoshi, H. Ohori, C. Kudo, A. Sato, N. Kanoh, C. Ishioka, H. Shibata and Y. Iwabuchi, Bioorg. Med. Chem., 2010, 18, 1083–1092 CrossRef CAS PubMed
- H. Ohori, H. Yamakoshi, M. Tomizawa, M. Shibuya, Y. Kakudo, A. Takahashi, S. Takahashi, S. Kato, T. Suzuki, C. Ishioka, Y. Iwabuchi and H. Shibata, Mol. Cancer Ther., 2006, 5, 2563–2571 CrossRef CAS PubMed
- O. Trott, A. J. Olson and J. AutoDock Vina, Comput. Chem., 2010, 31, 455–461 CAS
- K. Stierand, P. C. Maaß and M. Rarey, Bioinformatics, 2006, 22, 1710–1716 CrossRef CAS PubMed
- P. C. Fricker, M. Gastreich and M. Rarey, J. Chem. Inf. Comput. Sci., 2004, 44, 1065–1078 CrossRef CAS PubMed
- L. Kalan and G. D. Wright, Expert Rev. Mol. Med., 2011, 13, 1–17 Search PubMed
- R. Domalaon, T. Idowu, F. Schweizer, G. G. Zhanel and F. Schweizer, Clin. Microbiol. Rev., 2018, 31(2), 1–45 CrossRef PubMed
- M. E. Franks, G. R. Macpherson and W. D. Figg, Lancet, 2004, 363, 1802–1811 CrossRef CAS
- K. Liu, D. Zhang, J. Chojnacki, Y. Du, H. Fu, S. Grant and S. Zhang, Org. Biomol. Chem., 2013, 11, 4757–4763 RSC
- N. Dogra, R. Choudhary, P. Kohli, J. D. Haddock, S. Makwana, B. Horev, Y. Vinokur, S. Droby and V. Rodov, J. Agric. Food Chem., 2015, 63, 2557–2565 CrossRef CAS PubMed
- W. Li, R. Zhang, W. Li, Y. He, D. Zhou, W. Li and G. Zheng, Aging, 2019, 11, 771–782 Search PubMed
- B. R. Kim, J.-Y. Park, H. J. Jeong, H.-J. Kwon, S.-J. Park, I.-C. Lee, Y. B. Ryu and W. S. Lee, J. Enzyme Inhib. Med. Chem., 2018, 33, 1256–1265 CrossRef CAS PubMed
- , M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, R. A., Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- Y. Li, F. Li, Y. Zhu, X. Li, Z. Zhou, C. Liu, W. Zhang and M. Tang, Struct. Chem., 2016, 27, 1165–1173 CrossRef CAS
- B. Zhou, J. Hu, F. Xu, Z. Chen, L. Bai, E. Fernandez-Salas, M. Lin, L. Liu, C.-Y. Yang, Y. Zhao, D. McEachern, S. Przybranowski, B. Wen, D. Sun and S. Wang, J. Med. Chem., 2018, 61, 462–481 CrossRef CAS PubMed
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed
- G. Degliesposti, C. Portioli, M. D. Parenti and G. Rastelli, J. Biomol. Screening, 2010, 16, 129–133 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00064j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |