
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyurethane ionomers based on amino ethers of ortho-phosphoric acid
I. M. Davletbaeva a,
O. O. Sazonov*a,
A. R. Fazlyeva,
R. S. Davletbaevb,
S. V. Efimovc and
V. V. Klochkovc
a,
O. O. Sazonov*a,
A. R. Fazlyeva,
R. S. Davletbaevb,
S. V. Efimovc and
V. V. Klochkovc
aKazan National Research Technological University, 68 Karl Marx Str., Kazan, Republic of Tatarstan 420015, Russian Federation. E-mail: davletbaeva09@mail.ru; sazonov.oleg2010@gmail.com
bKazan National Research Technical University named after A. N. Tupolev-KAI, 10 Karl Marx Str., Kazan, Republic of Tatarstan 420111, Russian Federation
cKazan Federal University, 18 Kremlyovskaya Str., Kazan, Republic of Tatarstan 420008, Russian Federation
First published on 12th June 2019
Abstract
The etherification of ortho-phosphoric acid with triethanolamine and polyoxypropylene glycol is studied. The reaction process is accompanied by the formation of hyperbranched amino ethers of ortho-phosphoric acid terminated by hydroxyl groups. A specific feature of the chemical structure of the compounds obtained is the existence of ion pairs in their structure separated in space. The reaction of the etherification of ortho-phosphoric acid with glycols becomes possible through the use of tertiary amines. The amino ethers of ortho-phosphoric acid are investigated as a polyol component for the synthesis of polyurethanes with high adhesion characteristics and strength properties. The experimental results presented allow us to relate polyurethanes obtained on the basis of ortho-phosphoric acid amino ethers to polymers of ionomeric nature.
1 Introduction
Ionomers are polymeric materials having a small number of ionic side groups in hydrophobic main chains. Ionic fragments are included in these polymers in the form of cation, anion or zwitterion particles in various concentrations. Depending on the type of polymer, ionic groups can be distributed along the main chain periodically, randomly or as end groups.1–10 The concentration of ionic groups usually varies in the range of 1–15 mol% and they can be completely or partially neutralized. Intensified interaction between ionic pairs and their clustering increases the density of the nodes of their spatial polymer grid. This feature leads to a high probability of self-organization with formation of various types of nano-and microstructures containing ionic clusters and ionconductive channels.11–29 The resulting polymer network also causes strong changes in the mechanical properties of ionomers compared to the original non-ionic polymers. To a certain critical degree, this effect may lead to an increase in abrasion resistance, optical transparency, antistatic properties, interaction with various types of fillers, heat sealability and adhesive properties to various types of materials, such as polymers, ceramics, and metals.Polyurethanes are a class of highly versatile polymers and their range of use can be further broadened by the introduction of ionic groups into their structures. Therefore, growing attention has been paid to the synthesis of polyurethane ionomers.30–44
Polyols bearing ionic or ionizable functions are particularly useful as reactive components for the synthesis of self emulsifying functionalized soft polyurethane segments.45,46 On the basis of triethanolamine, boric acid and polyoxyethyleneglycols, hyperbranched ethers of boric acid used as the basic compounds for the preparation of polyurethanes of ionomeric nature were obtained.47,48 There exist also significant opportunities to study the properties of new polyurethane ionomers containing mixed soft segments involving ionic groups.49
Despite a variety of studies of polyurethane ionomers, however, there are relatively few publications related to the synthesis of polyurethane ionomers containing phosphoric acid units.50–54
Promising polyol components for the production of polyurethane ionomers are ortho-phosphoric ethers. There exist many ways to synthesize them.55–57 The most widely used phosphorylating agent for alcohols is phosphorus oxychloride, a characteristic feature of which is high toxicity. The direct use of affordable and low-toxic H3PO4 (OPA) for the production of ortho-phosphoric acid ethers is limited due to its low reactivity with respect to hydroxyl-containing compounds. As a rule, the etherification reaction takes place in this case at high temperatures. The condensation of OPA and hydroxyl-containing compounds at relatively low temperatures was first carried out by means of catalytic exposure to a given reaction system of equimolar OPA amounts of trialkylamines.58–60 In those works the reaction processes leading to obtaining OPA monophosphate were studied. The etherification was carried out in an organic solvent medium. To achieve high conversions, catalysts were additionally introduced, among which there were imidazoles and derivatives of rhenium oxides (VII). At the same time, the complete etherification of OPA was not considered there.
Our paper demonstrates an evidently simple strategy to synthesis of amino ethers of ortho-phosphoric acid (AEPA) by etherification of OPA with triethanolamine and polyoxypropylene glycol. Due to the branched topological structure containing terminal hydroxyl groups, the incorporation of the polyether component in the AEPA composition and the presence of PO− anions with adjustable content in them, these compounds turned out to be an advanced polyol basis for the synthesis of new polyurethane ionomers. The described new strategy of synthesis may allow industrial production of such polymers in the future.
2 Experimental
2.1 Materials
Polypropylene glycol (PPG), (Wanol 2310) was purchased from Wanhua Chemical (Beijing, China). Triethanolamine (TEOA), triethylamine (TEA), toluene were obtained from Ltd. “Component-reaktiv” (Moscow, Russia). 85% aqueous solution of ortho-phosphoric acid was purchased from Ltd “MCD-Chemicals” (Moscow, Russia). Polyisocyanate “Wannate PM-200” (PIC) was purchased from Kumho Mitsui Chemicals, Inc. (China).2.2 Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[H3PO4]:[PPG] = 1:3:6, 1:6:6, 1:9:6 (AEPA-3, AEPA-6 and AEPA-9, respectively). The calculated amount of OPA and PPG was placed in a round-bottom flask, mixed for two minutes, then TEOA was added to the reaction system. Within two hours, the reaction mass was stirred at T = 80 °C and a residual pressure of 0.7 kPa. The synthesized liquid AEPA-PPG was collected into a stoppered flask. The amount of residual water did not exceed 0.3 wt%.
:[H3PO4]:[PPG] = 1:3:6, 1:6:6, 1:9:6 (AEPA-3, AEPA-6 and AEPA-9, respectively). The calculated amount of OPA and PPG was placed in a round-bottom flask, mixed for two minutes, then TEOA was added to the reaction system. Within two hours, the reaction mass was stirred at T = 80 °C and a residual pressure of 0.7 kPa. The synthesized liquid AEPA-PPG was collected into a stoppered flask. The amount of residual water did not exceed 0.3 wt%.The etherification of OPA with TEA and PPG was carried out similarly to the synthesis of AEPA. Compounds were obtained at molar ratios [TEA]:[H3PO4]:[PPG] = 1:3:6 (EPA-3), 1:3:7 (EPA-3*), 1:6:6 (EPA-6) and 1:9:6 (EPA-9). The reaction was quenched after the desired amount of hydroxylation toward the target product. Reaction progress was monitored by titration to determine a hydroxyl groups concentration.
2.3 Light-scattering of AEPA solution
Dynamic light scattering experiments were carried out on Zetasizer Nano ZS (Malvern, Great Britain). This instrument has 4 mV He–Ne laser, which works on the 632.8 nm wavelength. Measurements were carried out at the 173° detection angle. The experiments were carried out at 25 °C in the disposable plastic cuvettes of 1 cm path length.2.4 Fourier transform infrared spectroscopy analysis (FTIR)
The FTIR spectra of the products were recorded on an InfraLUM FT 08 Fourier transform spectrometer (Lumex, St. Petersburg, Russia) using the attenuated total reflection technique. The spectral resolution was 2 cm−1, and the number of scans was 60.2.5 Measurements of critical micelle concentration (CMC)
The droplet counting method was used to determine the surface tension (σ). The basis of the calculations is the law, where the weight of the drop that comes off the pipette is proportional to the surface tension of the fluid and the radius of the pipette (R): m = 2πRσ/g, where: g is acceleration of gravity; m is the drop mass of the test liquid.2.6 NMR spectroscopy
1H and 31P NMR spectra were obtained on a BrukerAvance II 500 spectrometer (500.13 MHz for 1H and 202.46 MHz for 31P) using an inverse TBI probehead (1H/31P-BB-2D) or a direct BBO probehead (BB-1H-2D). The samples contained 120 μL of the investigated polymer mix and 480 μL of toluene-d8. The spectra were recorded at 25 °C; separate sets of temperature measurements were carried out in the range from 6 °C to 32 °C. The proton chemical shift scale is referenced with respect to the quintet of the methyl group in toluene at 2.09 ppm; for 31P, the external standard signal of 85% H3PO4 (δP = 0) poured in a capillary and placed into the polymer solution was used (it allowed us to assign the more high-field signal to phosphoric acid).2.7 Water concentration measurement
Water concentration was measured on a Mettler Toledo V20 volumetric titrator according to Karl Fischer.2.8 Viscosity measurements
The dynamic viscosity of samples was determined at the temperature 296 K, at atmospheric pressure and using a SVM 3000 Stabinger Viscometer (Anton Paar, Austria) with an error of 0.00005 g cm−3.2.9 Tensile stress–strain measurements
Tensile stress–strain measurements were obtained from the film samples of size 40 mm × 15 mm with Universal Testing Machine Inspekt mini (Hegewald & PeschkeMeß- und Prüftechnik GmbH, Nossen, Germany) at 293 ± 2 K, 1 kN. The crosshead speed was set at 50 mm min−1 and the test continued until sample failure. Minima of five tests were analyzed for each sample and the average values were reported.2.10 The coating adhesion on the breakaway
The value of adhesion of the coating to tearing was measured using a PSO-10MG4S (Ltd. Stroibpibor, Chelyabinsk, Russia) device designed to control the adhesion strength of protective and paint coatings with the base using the method of normal tearing of steel disks.2.11 Thermomechanical analysis (TMA)
The thermomechanical curves of polymer samples were obtained using TMA 402 F (Netzsch, Selb, Germany) thermomechanical analyzer in the compression mode. The sample thickness was 2 mm, and the rate of heating was 3 °C min−1 from −50 °C to 350 °C in the static mode. The load was 2 N.2.12 Mechanical loss tangent measurements (MLT)
The MLT curves of polymer samples were taken using the dynamic mechanical analyzer DMA 242 (Netzsch, Selb, Germany) in the mode of oscillating load. Force and stress–stain correspondence were calibrated using a standard mass. The thickness of the sample was 2 mm. Viscoelastic properties were measured under nitrogen. The samples were heated from −50 °C to 350 °C at the rate of 3 °C min−1 and frequency of 1 Hz. The mechanical loss tangent was defined as the ratio of the viscosity modulus G′′ to the elasticity modulus G′.3 Results and discussion
3.1 Synthesis and characterization of amino ethers of ortho-phosphoric acid
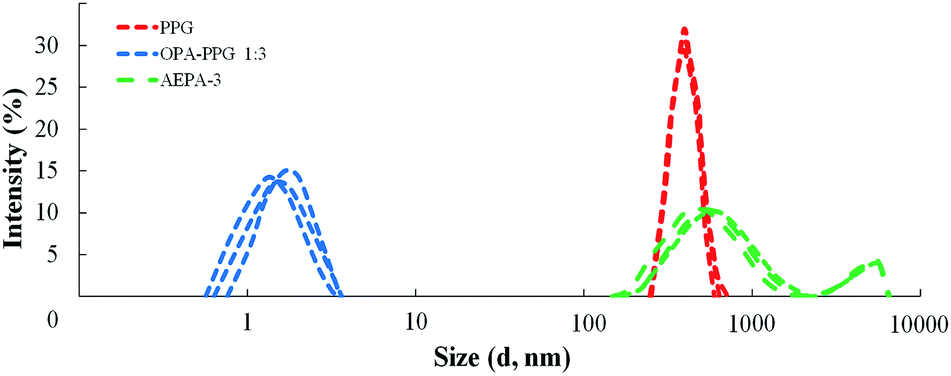
The interaction between TEOA, OPA and PPG was studied at such a stoichiometric ratio, at which under the conditions of formation of total phosphates all three hydroxyl groups of triethanolamine and one hydroxyl group of PPG could potentially be consumed for the etherification reaction. At the same time, the formation of branched amino ethers of ortho-phosphoric acid (AEPA-3) was expected, where the tertiary amine is the main branch center and the phosphates play the function of the subsequent branch centers.To analyze the size of the AEPA-3 obtained, the initial PPG was first investigated. The average hydrodynamic diameter of the PPG particles in toluene was 396 nm (Fig. 1). A relatively wide particle size distribution indicates the formation of PPG associates in toluene.
 | ||
| Fig. 1 Particle size distribution in the environment of toluene, determined by dynamic light scattering. | ||
In order to determine the role of tertiary amines in the etherification reaction of OPA and PGG, the interaction of 1 mole of OPA and 3 moles of PPG at 80 °C in the absence of a tertiary amine was investigated. For the OPA–PPG system (Fig. 1), the average hydrodynamic particle diameter is 1.7 nm. At the same time, particles with an average size of 396 nm are not revealed. The results obtained can be explained by the fact that OPA, due to the formation of hydrogen bonds involving R–OH and hydroxyl groups in the PPG, destroys the associative OH and hydroxyl groups in the PPG, destroys the associative interactions responsible for the formation of large PPG aggregates, and converts the oligoetherdiol to the monomeric form.
For the AEPA-3 solution, the average hydrodynamic particle diameter is 542 nm with a wide interval of their distribution (from 110 nm to 1100 nm) (Fig. 1). In addition, larger particles are formed with sizes up to 4500 nm. The most likely reason for the formation of such large-sized particles and a wide distribution with relatively small sizes of AEPA-3 may be associated with micelle formation processes. The diagram does not observe particles with small sizes. The fact that the PPG has entered into interaction with the OPA, is evidenced by the fact that the sizes and distribution of particles here significantly exceed the size distribution region for PPG. The absence of free OPA was judged by the absence of particle distribution in the region of small sizes.
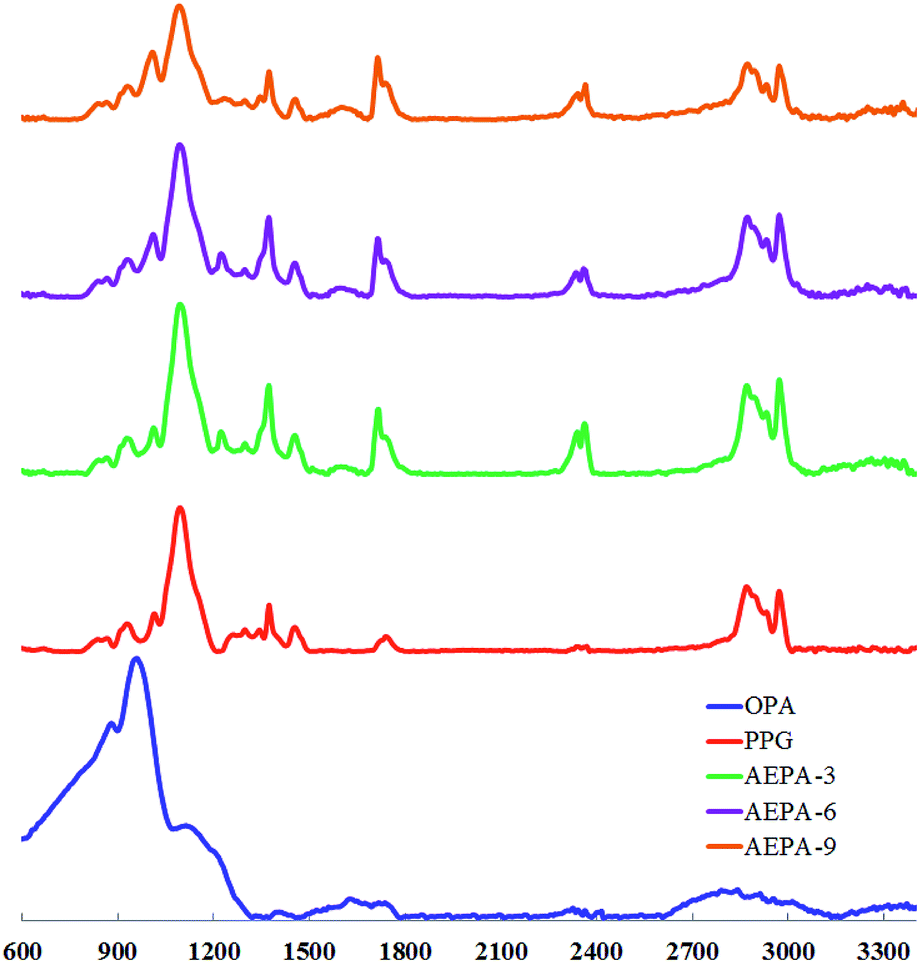
The formation of P–O–C bonds in the composition of AEPA-3 is evidenced by the presence of a band at 980 cm−1 in the FTIR spectra corresponding to the stretching vibrations of the P–O bond in P–O–C (Fig. 2). The intensity of this band is increasing in the series AEPA-3, AEPA-6 and AEPA-9. The high-intensity bands due to the stretching vibrations of the P–O bond in the structure of the P–OH groups of OPA at 875 cm−1 and 950 cm−1 practically disappear.
 | ||
| Fig. 2 FTIR spectra. | ||
On the FTIR spectra, bands appear in the region of 2370 cm−1 and 2430 cm−1, which correspond to the N–H bond in the composition of the tertiary ammonium. In the spectra of AEPA-3 and AEPA-6, new absorption bands appear in the region of 1720 cm−1 and 1745 cm−1, which can reflect the formation of phosphate of the PO− anion.
To establish the completeness of the reaction by acid groups, their content in AEPA-3 was determined by titration. As a result, 21 mg of KOH was consumed to neutralize free P–OH groups in 1 g of AEPA-3. The consumed amount of KOH corresponds to the equivalent of one P–OH group per each reacted molecule of OPA. The obtained data allow to judge about the existence in the composition of AEPA-3 of secondary acid phosphates of polyoxypropylene glycol.
As a result, almost half of the used PPG in the composition of the product of the interaction of 1 mole of TEOA, 3 moles of OPA and 6 moles of PPG remains in its original, unreacted state. This circumstance explains the wide particle size distribution for AEPA-3 (Fig. 1).
Thus, the analysis of the results obtained suggests that the presence of triethanolamine is a condition for the etherification reaction of one P–OH group of ortho-phosphoric acid with glycols at T = 75–80 °C without the need for additional use of cocatalysts.
In order to increase the content of acid phosphates in AEPA, the products of interaction of TEOA, OPA and PPG were obtained, during the synthesis of which the molar ratio of TEOA to PPG remained constant, and the molar excess of OPA relative to TEOA increased (AEPA-6). In these cases, it was assumed that both hydroxyl groups located at the ends of the PPG can enter into the etherification reaction. 50 mg of KOH were consumed to neutralize free P–OH groups in 1 g of AEPA-6. The amount of KOH consumed corresponds to the equivalent of one P–OH group per each reacted molecule of OPA, that is, the formation of secondary acid phosphates of PPG.
The solution of AEPA-6 in toluene is opalescent due to the formation of micellar particles. When measuring their size using the method of light scattering, the sizes of these particles exceed 5000 nm. In this regard, the measurements were performed in acetone. For AEPA-6 (Fig. 2), the average hydrodynamic particle diameter in acetone is 1030 nm, and the size distribution itself turned out to be relatively narrow – from 850 to 1100 nm. The formation of particles of relatively large sizes can be explained by the fact that for a given molar ratio of the reactants, the PPG reacts along both terminal hydroxyl groups. The observed narrow particle size distribution may be due to the homogeneity of the composition of the interaction products in the reaction system under consideration and the fact that the amount of PPG used completely entered the AEPA-6 (Scheme 1b and Fig. 3).
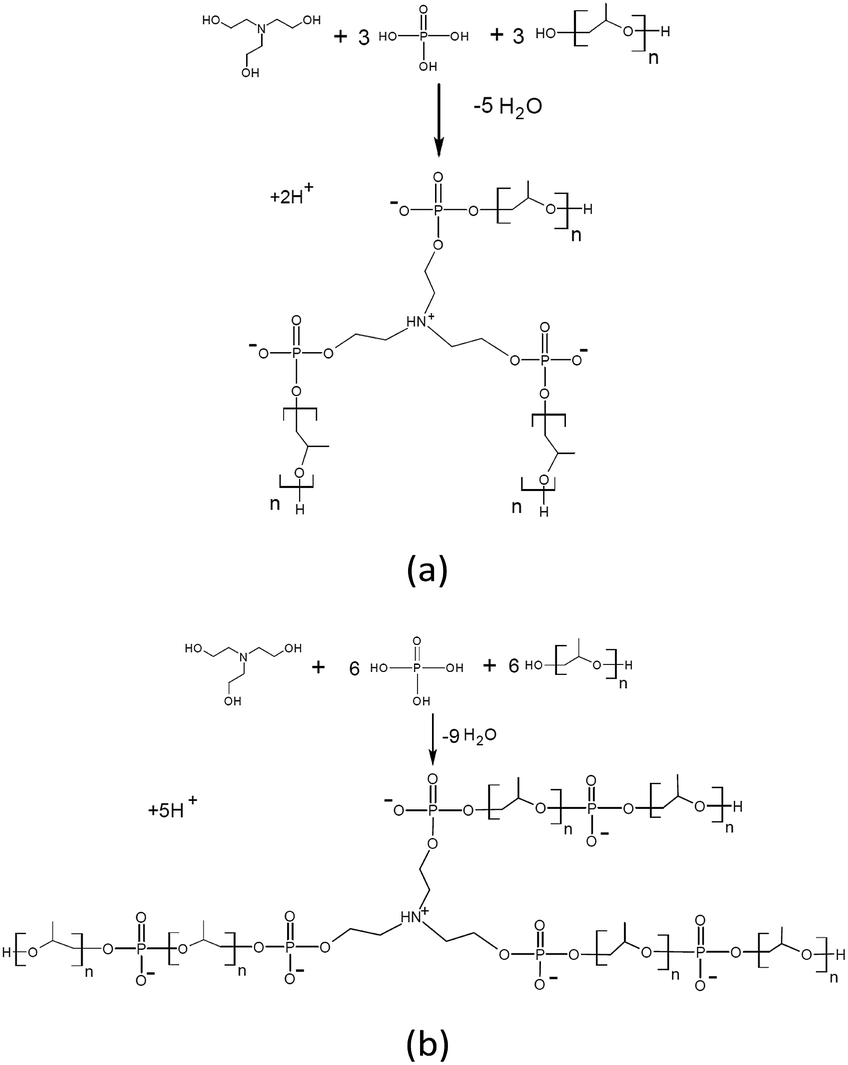 | ||
| Scheme 1 Formation of AEPA-3 (a); AEPA-6 (b). | ||
 | ||
| Fig. 3 Particle size distribution for AEPA-6 in the environment of acetone, determined by dynamic light scattering. | ||
Thus, as a result of the interaction between TEA, OPA and PPG, secondary acid phosphates are predominantly formed. In this regard, it should be noted that OPA is a tribasic acid, which is characterized by a large difference in the dissociation constants of each P–OH group. So, ortho-phosphoric acid is strong in the first stage (KI = 7.52 × 10−3), moderate in the second stage (KII = 6.31 × 10−8) and very weak in the third stage (KIII = 1.26 × 10−12).61 Such a high dissociation constant of the third P–OH group does not allow it to enter into the etherification reaction. Remaining as part of the acidic secondary and primary phosphates of PPG, the third P–OH group, as judged by the data of FTIR-spectroscopy, dissociates, existing in AEPA in the form of the PO− anion.
In order to confirm the formation of spatially separated ion pairs in the AEPA structure, the concentration dependences of the surface tension in aqueous solutions were measured for these compounds (Fig. 4). Since polyoxypropylene glycol exhibits the properties of surfactants, surface-active properties were also measured for PPG.
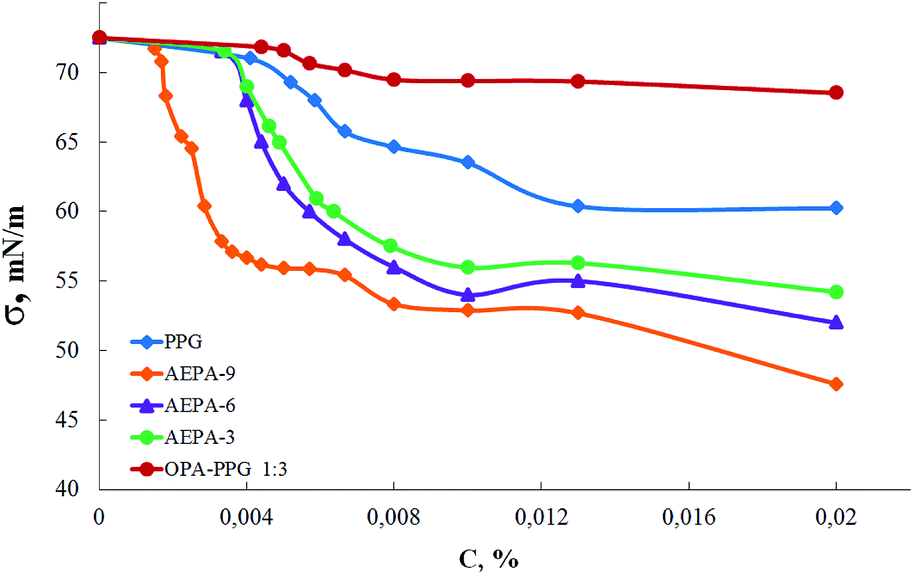 | ||
| Fig. 4 Surface tension isotherms. | ||
To confirm the proposed variants of the AEPA structure, 1H NMR and 31P NMR spectroscopy were used. In connection with the established structure of AEPA, the main issue is related to the existence in the resulting compounds of CH2–OH and OH groups with associated protons, PO− and P–O–C groups.
For this purpose, 1H NMR spectra of AEPA-3 and AEPA-9 were obtained at different temperatures (Fig. 5a). The existence of –OH + H+ ↔ –OH2+ exchange interactions causes a blurred signal of 5.6–5.4 ppm at +6 °C, which with increasing temperature to +14 °C turns into a wide signal in the 5.4 ppm area, and at +20 °C and +30 °C turns into a singlet at δ = 4.8 ppm.
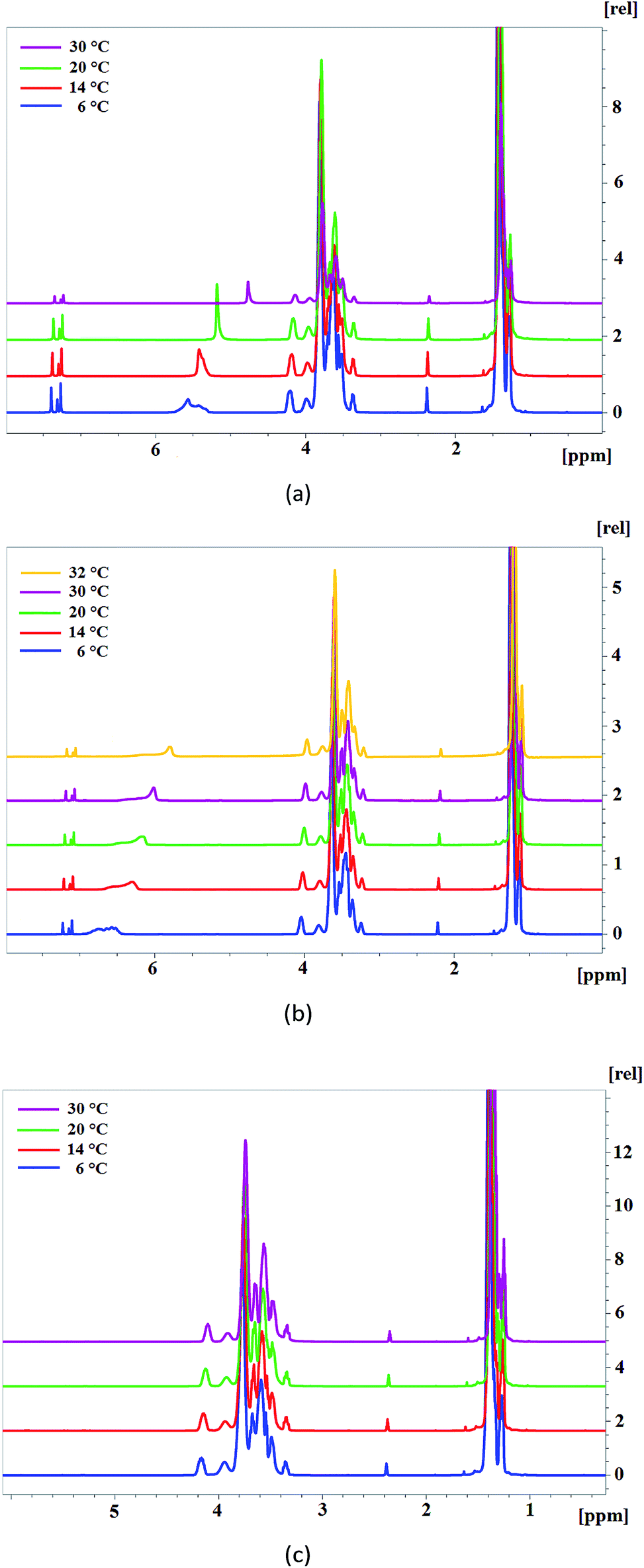 | ||
| Fig. 5 1H NMR spectra of AEPA-9 (a), AEPA-6 (b), PPG (c) at different temperatures. | ||
For AEPA-6, a similar shift of signals with increasing temperature is observed in the range 6.5–6.0 ppm (Fig. 5b).
For PPG under conditions of a low content of OH groups and the absence of their exchange interactions with protons, the corresponding signals on the 1H NMR spectra do not manifest themselves in the entire temperature range (Fig. 5c).
On the 1H NMR spectrum of TEOA, an intense signal is observed at δ = 5.4 ppm, which corresponds to a proton in the structure of hydroxyl groups. The absence of this signal in the structure of AEPA is a consequence of the involvement of all three hydroxyl groups of TEOA in the interaction with OPA.
Since AEPA-3 contains the least amount of free protons, a very weak signal, corresponding to –OH + H+ ↔ –OH2+ exchange interactions, appears at δ = 4.5 ppm (Fig. 6). In all 1H NMR spectra of the AEPA, the signal at δ = 5.0 ppm, characteristic of OPA, does not appear.
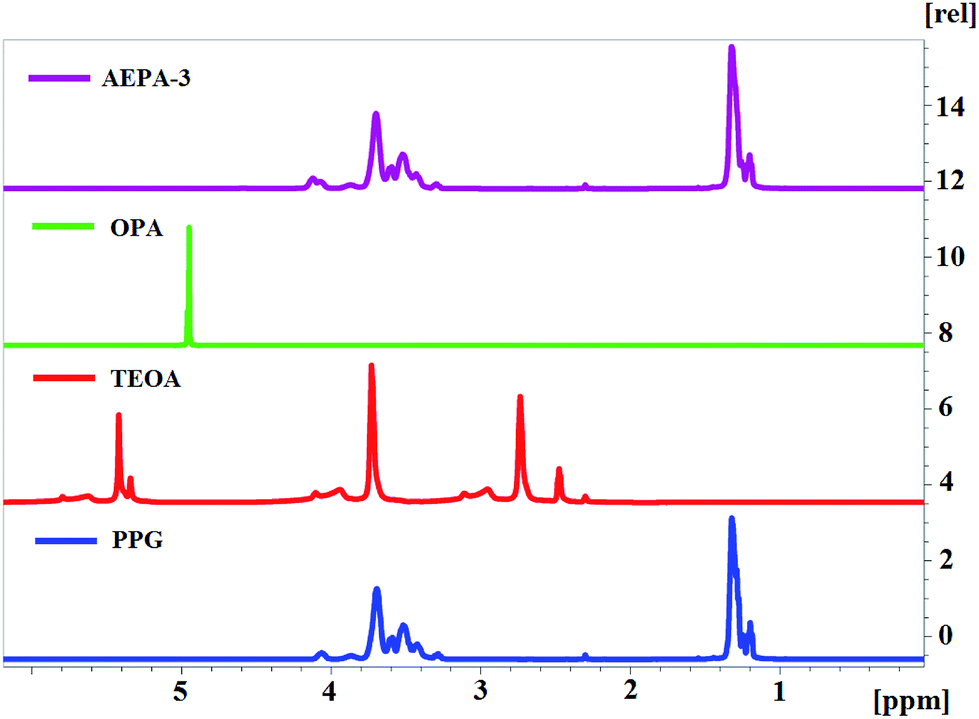 | ||
| Fig. 6 1H NMR spectra. | ||
The 31P NMR spectra of AEPA (Fig. 7) can be divided into two regions. At δ = 1.3–0.9 ppm, the first region corresponds to PO− anions, the position and manifestation of which undergoes noticeable changes during the transition from AEPA-3 to AEPA-6. The second region at δ = 3.0–3.1 ppm corresponds to the phosphorus bound by the ether linkage. There is a regular increase in signal intensity at δ = 3.0–3.1 ppm from AEPA-3 to AEPA-9.
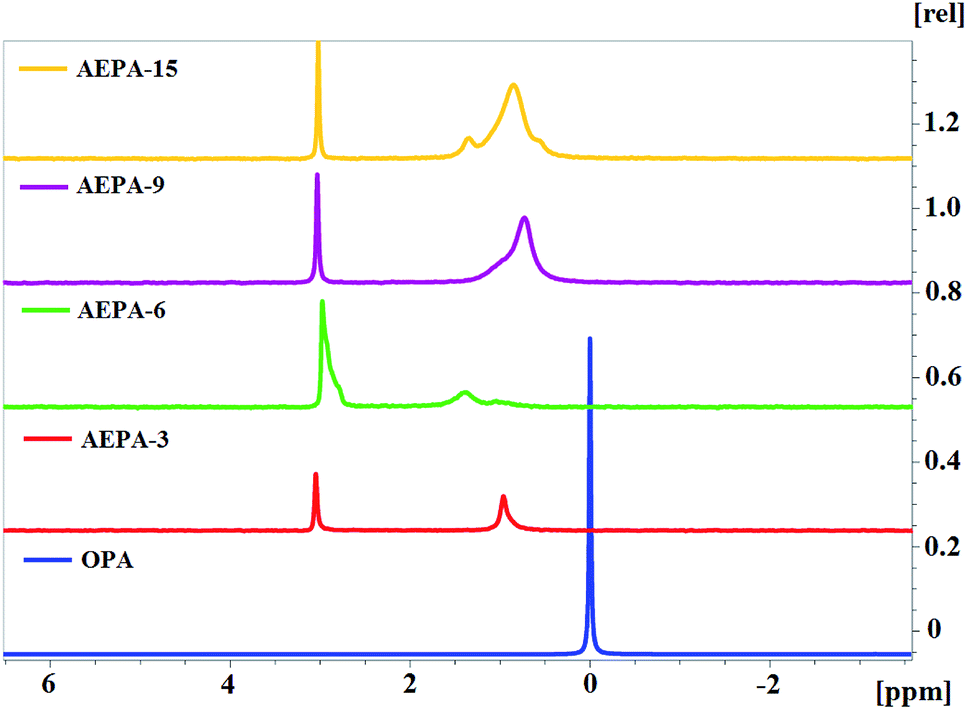 | ||
| Fig. 7 31P NMR spectra. | ||
The 31P NMR spectrum was also measured for AEPA-9, AEPA-15 and OPA (Fig. 7). Synthesis of AEPA-9 and AEPA-15 assumed the initial excess of the content of OPA from its theoretically possible entry into the etherification reaction. Despite the large excess of OPA in the composition of AEPA-15, on its 31P NMR spectrum there is no narrow signal at δ = 0 ppm, which characterizes OPA in the initial state.
The results obtained allow us to conclude that, due to the presence of PO− anions in the AEPA, these branched compounds are involved in the formation of clusters. As a result of a cooperative nature, unreacted OPA molecules also enter into these interactions. This is also indicated by the expansion and complication of the spectra from AEPA-9 to AEPA-15 in the range δ = 0.4–1.6 ppm.
To study the effect of trifunctional TEOA on the topological structure of AEPA, and the effect of tertiary amines on the etherification reactions of OPA, triethanolamine was replaced with triethylamine in order to obtain ortho-phosphoric acid ethers (EPA).
For EPA-6, two signals are observed on the 31P NMR spectrum, similar to AEPA-6 (Fig. 7). For EPA-3*, obtained at a molar ratio [TEA]:[H3PO4]:[PPG] = 1:3:7 (Scheme 2), only one signal is observed in the 31P NMR spectrum at δ = 1.5 ppm. For EPA-3, obtained at the molar ratio [TEA]:[H3PO4]:[PPG] = 1:3:6, only one signal is also observed in the region δ = 3.2–2.8 ppm.
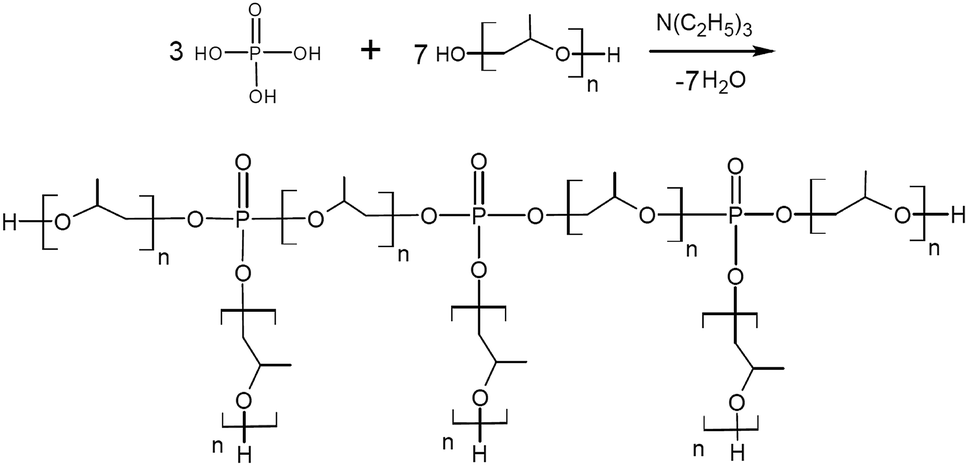 | ||
| Scheme 2 Formation of EPA-3*. | ||
On the basis of the spectra obtained, it can be judged that all phosphorus atoms in EPA-3* and EPA-3 are in the form of complete OPA ethers and therefore cannot participate in the clustering processes. In this case, EPA should exhibit a topological structure with a large number of branches in comparison with EPA (Fig. 8).
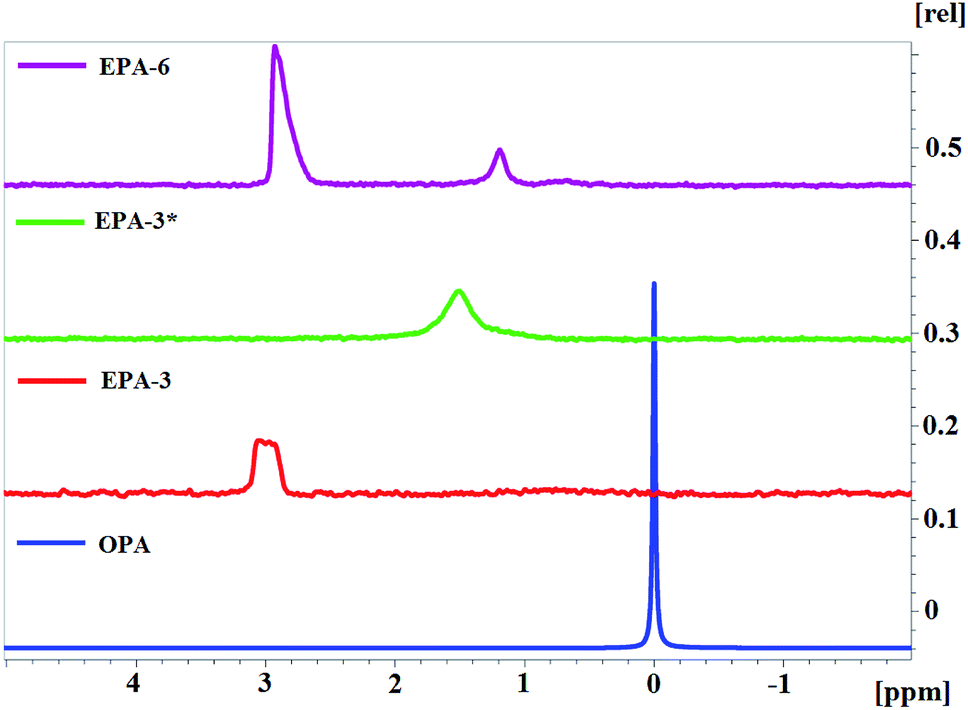 | ||
| Fig. 8 31P NMR spectra. | ||
According to the literature,62,63 hyperbranched polymers and oligomers are characterized by low viscosity compared with linear analogues. Moreover, their viscosity is much lower than viscosity of linear analogues both in solution and in the molten state. The higher the degree of branching, the lower the viscosity of the branched polymer. In this regard, measurements of the dynamic viscosity of the OPA–PPG, AEPA and EPA system, obtained with a different molar excess of OPA relative to the PPG, were carried out (Fig. 9). For the OPA–PPG system (without using tertiary amines), the viscosity increases significantly even at a relatively low content of ortho-phosphoric acid; with a further increase in the molar fraction of OPA, an increase in its viscosity becomes non-additive.
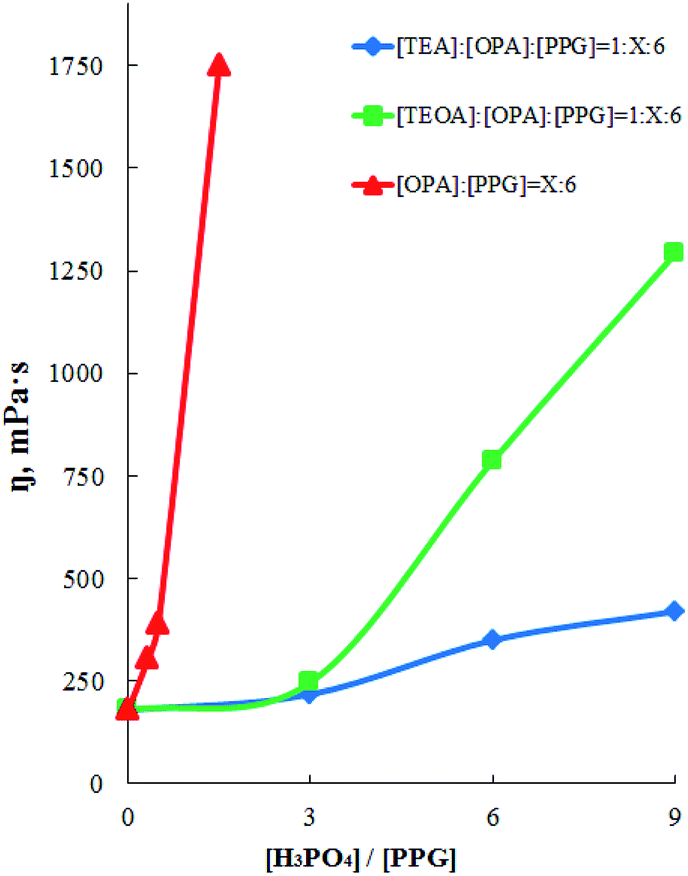 | ||
| Fig. 9 Dependence of dynamic viscosity on molar excess (X) of H3PO4. T = 20 °C. | ||
For AEPA, viscosity increases with an increase in the molar excess of OPA relative to PPG up to 3 only insignificantly. With a further increase in the molar excess of OPA, the contribution to the AEPA structure of unreacted P–OH groups of ortho-phosphoric acid increases. The increase in the content of the acid ethers of OPA is the cause of the formation of viscous associates. As a result, an increase in the relative content of OPA leads to an intensive increase in the dynamic viscosity values, due to the fact that AEPA-6 and AEPA-9 contain unreacted P–OH groups, whose content increases from AEPA-3 to AEPA-9.
For EPA, viscosity with an increase in molar fraction of OPA increases with a smaller intensity, indicating a greater degree of branching of EPA compared to AEPA and that AEPA, unlike EPA, exhibits ionomeric nature (Scheme 2).
Thus, when replacing TEA with TEOA, the hydroxyl groups of triethanolamine are fully involved in etherification with OPA, but the catalytic activity of the tertiary amine weakens as a result of a decrease in its availability in the branched structure of AEPA.
The obtained AEPA were used as a polyol component of ionomeric nature for the synthesis of polyurethanes (AEPA-PU). The isocyanate component served as a polyisocyanate based on 4,4′-diphenylmethane diisocyanate.
According to the table and Fig. 10a, polyurethanes exhibit high adhesive characteristics and strength properties. For comparison purposes, stress–strain relations for polyurethanes are given, in which the AEPA was replaced with PPG (PPG-PU) and the product of the interaction of PPG was replaced with OPA (PPG-OPA-PU). The observed significant differences in the mechanical behaviour of AEPA-PU and PPG-PU are a consequence of the ionomeric nature of AEPA-PU. The most optimal components from the point of view of a combination of adhesive characteristics, modulus of elasticity and strength are AEPA-3-PU and AEPA-6-PU. An increase in the fraction of OPA in the composition of AEPA leads to an increase in the content of PO− anions, but at the same time the number of urethane groups in the composition of AEPA-PU and the predicted density of the nodes of its spatial polymer network decrease. The high content of PO− anions in AEPA-6-PU eliminates the reduction of crosslinking sites in comparison with AEPA-3-PU. As a result, the mechanical properties of AEPA-6-PU turned out to be higher in comparison with AEPA-3-PU. A further increase in the proportion of OPA in the composition of AEPA-9 leads to an increase in the content of associatively bound molecules of unreacted OPA. As a result, adhesion, elastic modulus, strength decreased, and plastic deformation values of AEPA-9-PU increased.
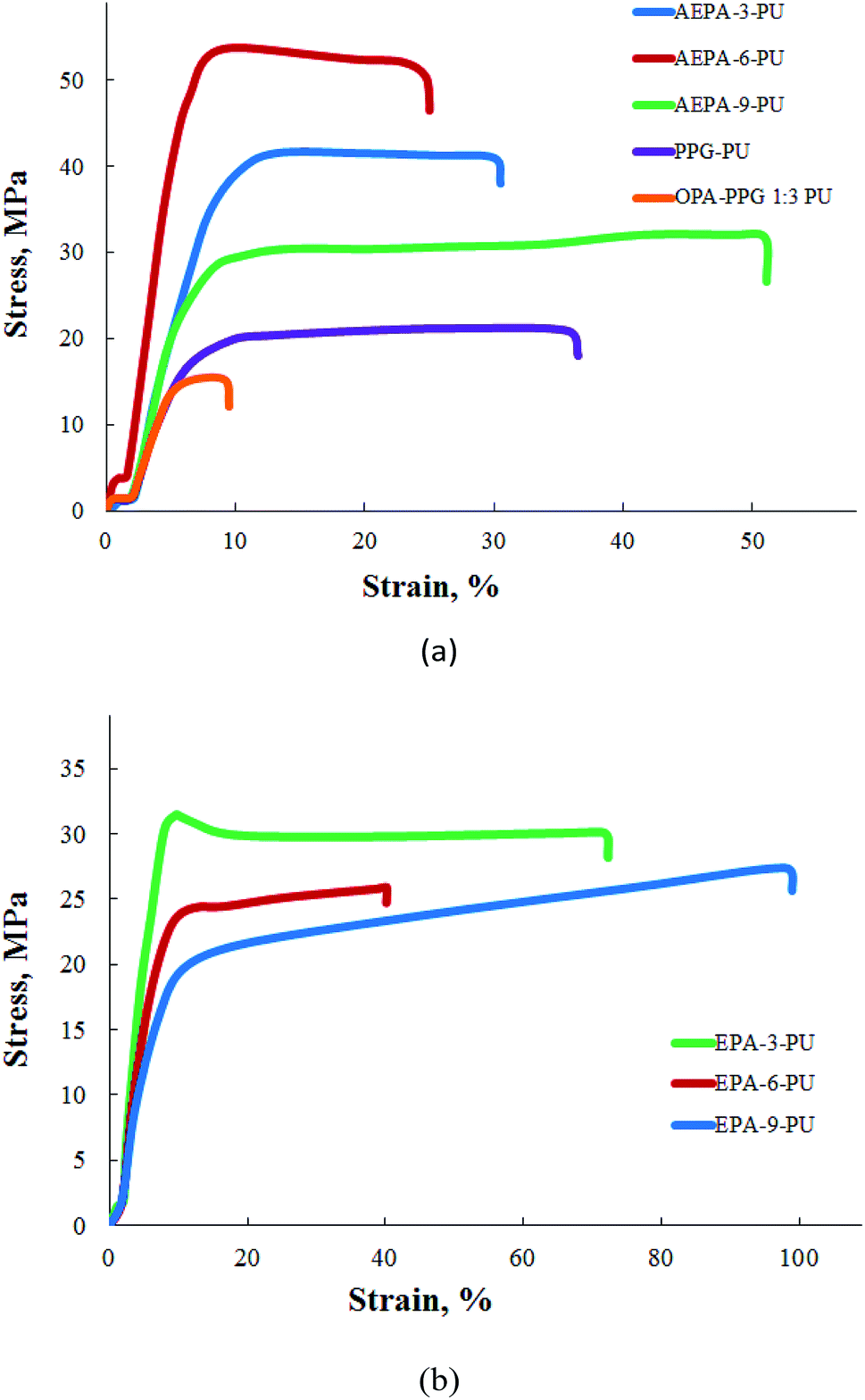 | ||
| Fig. 10 Tensile tests of polymers. | ||
The mechanical behavior of polyurethanes synthesized using EPA (EPA-PU) was also investigated (Table 1 and Fig. 10b). The decrease in both strength and adhesive characteristics for polyurethanes obtained on the basis of EPA is a consequence of the fact that, unlike AEPA, these compounds lack PO− anions.
| Polyol component | Adhesive strength to steel, MPa | Elastic modulus, MPa | Ultimate tensile strength, MPa |
|---|---|---|---|
| AEPA-3 | 4.2 | 1050 | 40 |
| AEPA-6 | 6.0 | 1100 | 55 |
| AEPA-9 | 3.5 | 850 | 30 |
| EPA-3 | 1.0 | 380 | 30 |
| EPA-6 | 1.2 | 310 | 25 |
| EPA-9 | 0.5 | 275 | 25 |
| PPG | 5.0 | 205 | 20 |
| [H3PO4]:[PPG] = 1:3 |
0.4 | 145 | 16 |
Polyurethanes obtained on the basis of AEPA exhibit high heat resistance. The fact that the highest contribution to the heat resistance causes the ionomer component is indicated by the TMA and DMA analysis curves (Fig. 11). So, if deformation processes for AEPA-3-PU begin with a temperature of 140 °C, then for AEPA-6-PU, which is characterized by a higher content of PO− anions, the beginning of deformation corresponds to a temperature of 210 °C. The thermo-destructive flow of AEPA-6-PU starts at 250 °C with a maximum response to the curves of the tangent of the angle of mechanical losses at 290 °C for both AEPA-3-PU and AEPA-6-PU.
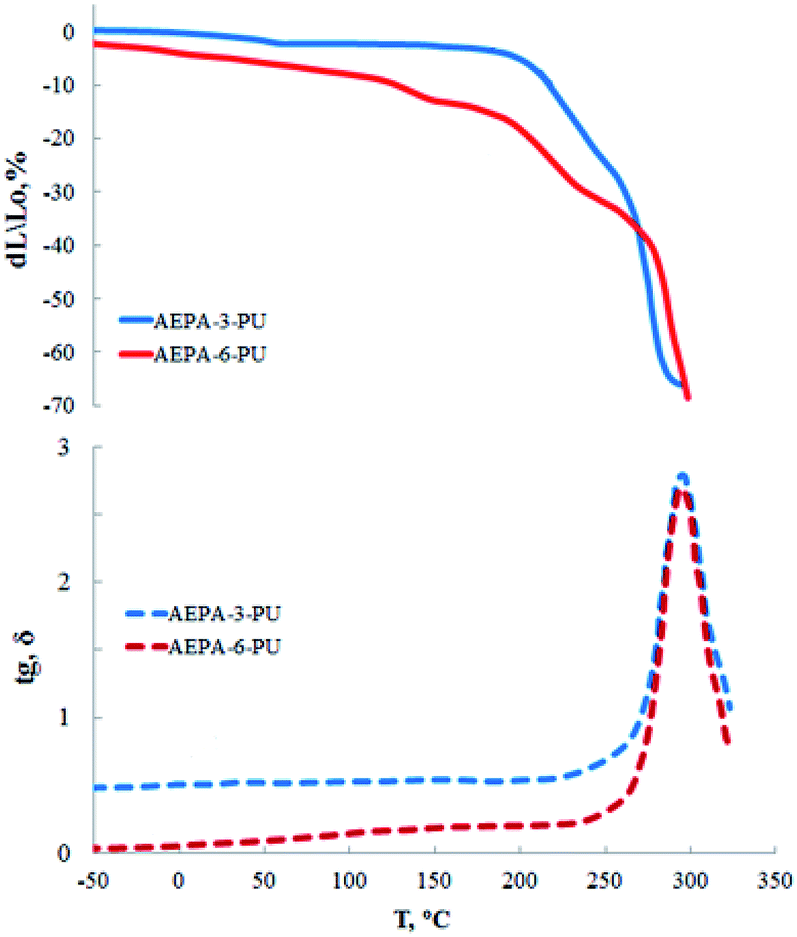 | ||
| Fig. 11 TMA curves and temperature dependence of the mechanical loss tangent. | ||
4 Conclusions
The etherification of ortho-phosphoric acid with polyoxypropylene glycol was studied in the presence of tertiary amines. It was shown that when triethylamine is used, complete OPA etherification occurs, followed by the formation of branched OPA ethers terminated by hydroxyl groups.When replacing TEA with TEOA, the hydroxyl groups of triethanolamine are fully involved in etherification with OPA, but the catalytic activity of the tertiary amine weakens due to a decrease in its availability in the branched AEPA structure. As a result, the most acidic P–OH groups remain unreacted in the amino ethers of ortho-phosphoric acid.
Due to the presence of PO− anions in the AEPA, these branched compounds are involved in the formation of clusters. As a result of a cooperative nature, unreacted OPA molecules also enter into these interactions.
Polyurethane ionomers were synthesized on the basis of AEPA. It was shown that AEPA-PU exhibit significantly higher adhesive strength compared with EPA-PU. The decrease in both strength and adhesive characteristics for polyurethanes obtained on the basis of EPA is a consequence of the fact that, unlike AEPA, these compounds lack PO− anions. With an increase in the content of PO− anions in the composition of AEPA, a regular increase in the heat resistance of AEPA-PU occurs.
The described method of synthesis as well as the results of the present study can be used to produce industrially such polymers in the future.
Conflicts of interest
There are no conflicts to declare.Abbreviation
| OPA | ortho-Phosphoric acid |
| TEOA | Triethanolamine |
| TEA | Triethylamine |
| PPG | Polypropylene glycol |
| PIC | Polyisocyanate “Wannate PM-200” |
| AEPA | Amino ethers of ortho-phosphoric acid |
| AEPA-3 | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEOA]:[H3PO4]:[PPG] = 1:3:6 |
| AEPA-6 | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEOA]:[H3PO4]:[PPG] = 1:6:6 |
| AEPA-9 | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEOA]:[H3PO4]:[PPG] = 1:9:6 |
| EPA | Ethers of ortho-phosphoric acid |
| EPA-3 | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEA]:[H3PO4]:[PPG] = 1:3:6 |
| EPA-3* | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEA]:[H3PO4]:[PPG] = 1:3:7 |
| EPA-6 | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEA]:[H3PO4]:[PPG] = 1:6:6 |
| EPA-9 | Amino ethers of ortho-phosphoric acid synthesized on the basis of [TEA]:[H3PO4]:[PPG] = 1:9:6 |
| PU | Polyurethane |
| AEPA-PU | Polyurethanes synthesized on the basis of AEPA |
| AEPA-3-PU | Polyurethanes synthesized on the basis of AEPA-3 |
| AEPA-6-PU | Polyurethanes synthesized on the basis of AEPA-6 |
| AEPA-9-PU | Polyurethanes synthesized on the basis of AEPA-9 |
| EPA-PU | Polyurethanes synthesized on the basis of EPA |
| EPA-3-PU | Polyurethanes synthesized on the basis of EPA-3 |
| EPA-6-PU | Polyurethanes synthesized on the basis of EPA-6 |
| EPA-9-PU | Polyurethanes synthesized on the basis of EPA-9 |
| PPG-PU | Polyurethanes synthesized on the basis of PPG and PIC |
Acknowledgements
This work was supported by the Russian Science Foundation (grant no. 19-19-00136). NMR measurements were performed at the Center of Shared Facilities of Kazan Federal University.References
- I. Capek, Adv. Colloid Interface Sci., 2004, 112, 1 CrossRef CAS PubMed.
- A. Eisenberg and M. King, Ion-Containing Polymers: Physical Properties and Structure, Academic Press, New York, 1977, vol. 7, p. 3 Search PubMed.
- J. Ro, S. J. Huang and R. A. Weiss, J. Biomed. Mater. Res., Part A, 2008, 49, 422 Search PubMed.
- H. Tachino, H. Hara, E. Hirasawa, S. Kutsumizu and S. Yano, Polymer, 1994, 26, 1170 CAS.
- S. Wu, Q. Guo, M. Kraska, B. Stuhn and Y. W. Mai, Macromolecules, 2013, 46, 8190 CrossRef CAS.
- R. Dolog and R. A. Weiss, Macromolecules, 2013, 46, 7845 CrossRef CAS.
- M. Grande, L. Castelnovo, L. D. Landro, C. Giacomuzzo, A. Francesconi and M. A. Rahman, J. Appl. Polym. Sci., 2013, 130, 1949 CrossRef.
- F. Pierre, B. Commarieu, A. C. Tavares and J. Claverie, Polymer, 2016, 86, 91 CrossRef CAS.
- G. Polizos, A. Kyritsis, P. Pissis, V. V. Shilov and V. V. Shevchenko, Solid State Ionics, 2000, 136, 1139 CrossRef.
- C.-L. Xu, J.-B. Zeng and Y.-Z. Wang, Compos. Sci. Technol., 2014, 96, 109 CrossRef CAS.
- G. C. Bazuin and A. Eisenberg, Ind. Eng. Chem. Prod. Res. Dev., 1981, 20, 271 CrossRef.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901 CrossRef CAS.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780 CrossRef CAS PubMed.
- T. J. Peckham and S. Holdcroft, Adv. Mater., 2010, 22, 4667 CrossRef CAS PubMed.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29 CrossRef CAS.
- T. J. Peckham, J. Schmeisser, M. Rodgers and S. Holdcroft, J. Mater. Chem., 2007, 17, 3255 RSC.
- T. Weissbach, E. M. W. Tsang, A. C. C. Yang, R. Narimani, B. J. Frisken and S. Holdcroft, J. Mater. Chem., 2012, 22, 24348 RSC.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175 CrossRef CAS.
- G. He, Z. Li, J. Zhao, S. Wang, H. Wu, M. D. Guiver and Z. Jiang, Adv. Mater., 2015, 27, 5280 CrossRef CAS PubMed.
- C. C. Yang, R. Narimani, B. J. Frisken and S. Holdcroft, J. Membr. Sci., 2014, 469, 251 CrossRef.
- Y. A. Elabd and M. Hickner, Macromolecules, 2011, 44, 1 CrossRef CAS.
- J. Ding, C. Chuy and S. Holdcroft, Chem. Mater., 2001, 13, 2231 CrossRef CAS.
- J. R. Rowlett, V. Lilavivat, A. T. Shaver, Y. Chen, A. Daryaei, H. Xu, C. Mittelsteadt, S. Shimpalee, J. S. Riffle and J. E. McGrath, Polymer, 2017, 122, 296 CrossRef CAS.
- Y. A. Elabd, E. Napadensky, C. W. Walker and K. I. Winey, Macromolecules, 2006, 39, 399 CrossRef CAS.
- J. Peron, A. Mani, X. Zhao, D. Edwards, M. Adachi, T. Soboleva, Z. Shi, Z. Xie, T. Navessin and S. Holdcroft, J. Membr. Sci., 2010, 356, 44 CrossRef CAS.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759 CrossRef CAS PubMed.
- U. Plawky and W. Wenig, J. Mater. Sci., 1998, 33, 1611 CrossRef CAS.
- D. Basu, A. Das, K. W. Stockelhuber and S. Wiebner, Designing of Elastomer Nanocomposites, 2016, vol. 275, p. 235 Search PubMed.
- D. G. Peiffer, B. L. Hager, R. A. Weiss, P. K. Agarwal and R. D. Lundberg, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 1869 CrossRef CAS.
- J. T. Koberstein, A. F. Galambos and L. M. Leung, Macromolecules, 1992, 25, 6195 CrossRef CAS.
- S. Chen and W. Chan, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 1499 CrossRef CAS.
- G. Nelson, ACS Symposium Fire and polymers, ACS Symposium Series, Washington, 1990 Search PubMed.
- R. Lyon, Polym. Degrad. Stab., 1998, 61, 201 CrossRef CAS.
- V. D. Krevelen, Properties of polymers, Elsevier, New York, ch. 26B, 1976 Search PubMed.
- W. Bassett, Proc. fire retardant chemicals association meeting, San Antonio, USA, 1989 Search PubMed.
- C. Tsonos, L. Apekis, K. Viras, L. Stepanenko, L. Karabanova and L. Sergeeva, J. Macromol. Sci., Part B: Phys., 2000, 39(2), 155 CrossRef.
- C. Tsonos, L. Apekis, K. Viras, L. Stepanenko, L. Karabanova and L. Sergeeva, Solid State Ionics, 2001, 143, 229 CrossRef CAS.
- C. Tsonos, L. Apekis, C. Zois and G. Tsonos, Acta Mater., 2004, 52, 1319 CrossRef CAS.
- H. S. Egboh, A. Ghaffar, M. H. George, J. A. Barrie and D. J. Walsh, Polymer, 1982, 23, 1167 CrossRef CAS.
- T. Y. T Chui, A. S Coote, C. Butler, M. H. George and J. A. Barrie, Polym. Commun., 1988, 29, 40 Search PubMed.
- T. Y. T. Chui, P. K. H. Lam, M. H. George and J. A. Barrie, Polym. Commun., 1998, 29, 317 Search PubMed.
- P. K. H. Lam, M. H. George and J. A. Barrie, Polym. Commun., 1989, 30, 2321 Search PubMed.
- S. Shahgaldi, I. Alaefour and X. Li, Appl. Energy, 2018, 217, 295 CrossRef CAS.
- M. Kuramoto, M. Sakamoto, T. Teshirogi, J. Komiyama and Y. Iijima, J. Appl. Polym. Sci., 1984, 29, 977 CrossRef.
- B. K. Kim, J. S. Yang, S. M. Yoo and J. S. Lee, Colloid Polym. Sci., 2003, 281, 461 CrossRef CAS.
- X. Wei and X. Yu, J. Polym. Sci., 1997, 35, 225 CrossRef CAS.
- M. Davletbaeva, O. Yu. Emelina, I. V. Vorotyntsev, R. S. Davletbaev, E. S. Grebennikova, A. N. Petukhov, A. I. Ahkmetshina, T. S. Sazanova and V. V. Loskutov, RSC Adv., 2015, 5, 65674 RSC.
- M. Davletbaeva, G. R. Nurgaliyeva, A. I. Ahkmetshina, R. S. Davletbaev, A. A. Atlaskin, T. S. Sazanova, S. V. Efimov, V. V. Klochkov and I. V. Vorotyntsev, RSC Adv., 2016, 6, 111109 RSC.
- P. Johnston and R. Adhikari, Eur. Polym. J., 2017, 95, 138 CrossRef CAS.
- D. K. Kakati and M. H. George, Polymer, 1993, 34, 4319 CrossRef CAS.
- J.-S. Kim, Y. H. Nah, S. S. Jarng, W. Kim, Y. Lee and Y. W. Kim, Polymer, 2000, 41, 3099 CrossRef CAS.
- K. Mequanint, R. Sanderson and H. Pasch, Polym. Degrad. Stab., 2002, 77, 121 CrossRef CAS.
- Z. Petrovic, Z. Zavargo, J. Flynn and W. MacKnight, J. Appl. Polym. Sci., 1994, 51, 1087 CrossRef CAS.
- K. Troev, R. Tsevl, T. Bourova, S. Kobayashi, H. Uayama and D. Roundhill, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 621 CrossRef CAS.
- L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley-Interscience, New York, 2000 Search PubMed.
- R. Engel and J. I. Cohen, Synthesis of Carbon–Phosphorus Bonds, Florida, CRC Press LLC, 2004 Search PubMed.
- K.-J. Fest and C. Schmidt, The Chemistry of Organophosphorus, Springer-Verlag, Berlin, Heidelberg, New York, 1982 Search PubMed.
- A. Sakakura, M. Katsukawa, T. Hayashib and K. Ishihara, Green Chem., 2007, 9, 1166 RSC.
- A. Sakakura, M. Katsukawa and K. Ishihara, Org. Lett., 2005, 10, 1999 CrossRef PubMed.
- A. Sakakura, M. Katsukawa and K. Ishihara, Angew. Chem., Int. Ed., 2007, 46, 1423 CrossRef CAS PubMed.
- K. J. Powell, P. L. Brown, R. H. Byrne, T. Gajda, G. Hefter, S. Sjöberg and H. Wanner, Pure Appl. Chem., 2005, 77(4), 739 CAS.
- E. Malmstrom and A. Hult, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1997, 37, 555 CrossRef.
- B. Romagnoli and W. Hayes, J. Mater. Chem., 2002, 12, 767 RSC.
| This journal is © The Royal Society of Chemistry 2019 |