
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiospecific synthesis of polysubstituted furans with mono- to tricarboxylates from various sulfonium acylmethylides and acetylenic esters†
Jun Dong,
Hongguang Du and
Jiaxi Xu*
and
Jiaxi Xu*
State Key Laboratory of Chemical Resource Engineering, Department of Organic Chemistry, College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, People's Republic of China. E-mail: jxxu@mail.buct.edu.cn
First published on 12th August 2019
Abstract
Polysubstituted furans were prepared in moderate to good yields from various sulfur ylides and alkyl acetylenic carboxylates. The direct reactions of dimethylsulfonium acylmethylides with dialkyl acetylenedicarboxylates afforded dialkyl furan-3,4-dicarboxylates through a tandem sequence of Michael addition, intramolecular nucleophilic addition, 4π ring opening, intramolecular Michael addition, and elimination. The method was extended to synthesize furan-3-carboxylate, -2,4-dicarboxylates, and -2,3,4-tricarboxylates as well. The current method provides a direct and simple strategy in the synthesis of structurally diverse polysubstituted furans with mono to tricarboxylate groups from safe and readily available dimethylsulfonium acylmethylides and different alkyl acetylenic carboxylates.
Introduction
Furan derivatives are not only important building blocks in organic chemistry, but also natural products found in various natural sources, most in plants, algae, and microorganisms, and structural motifs in biologically drug molecules (Fig. 1).1 Polysubstituted furans represent important cores and moieties of some biological compounds as well as acting as useful intermediates in organic synthesis.2 The development of efficient methods for their preparation has been an important research area in organic chemistry.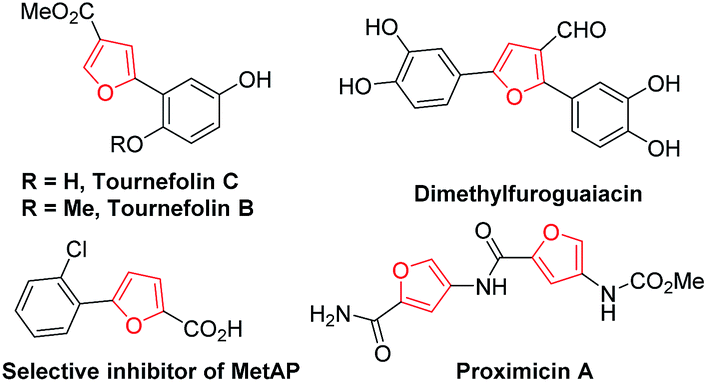 | ||
| Fig. 1 Furan-derived natural products and drugs. | ||
Historically, the classical approaches such as Paal–Knorr and Feist–Benary syntheses of furan derivatives have been widely applied. The Paal–Knorr method relies on an acid-catalyzed intramolecular cyclization of 1,4-dicarbonyl compounds,3 while the Feist–Benary method provides a strategy for the efficient preparation of polysubstituted furan derivatives via intermolecular annulation of β-dicarbonyl compounds and α-haloketones.4 In the past few decades, many efforts for the synthesis of polysubstituted furans have been made to explore the annulation of alkynes and other unsaturated compounds.
Such as, oxazoles, 1,3,4-oxadiazoles, and furobenzopyran undergo inter- or intramolecular Diels–Alder/retro-Diels–Alder reactions with electron-poor alkynes to generate polysubstituted furans.5 The addition/oxidative cyclization of alkyl alkynoates with carbonyl compounds in the presence of metal leads to polysubstituted furans as well.6
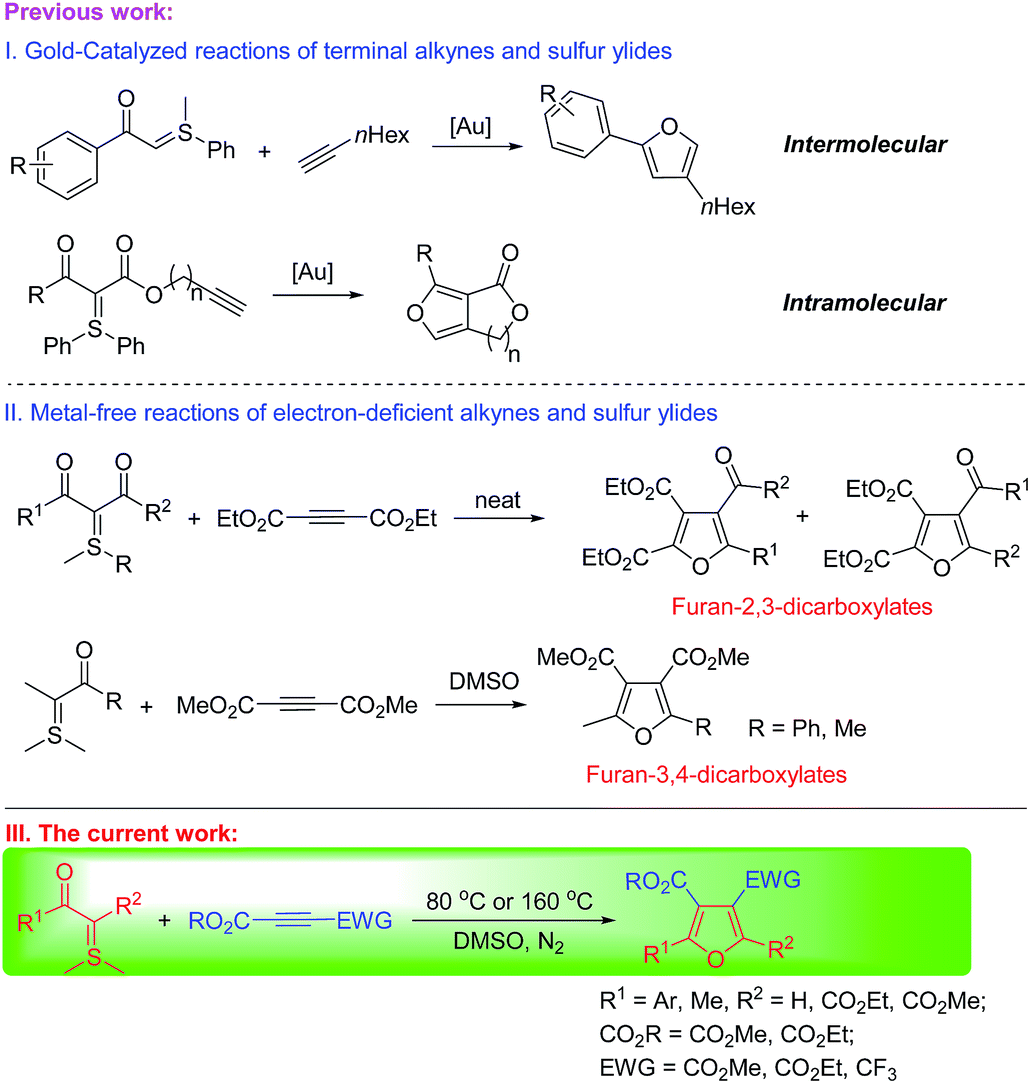
Sulfur ylides (sulfonium and sulfoxonium methylides) have been used as universal synthetic precursors for various chemical transformations.7 The reactions of sulfur ylides and alkynes were also applied in the synthesis of furan derivatives. One interesting and powerful method to construct the furan motif was designed through the precious metal gold-catalyzed addition of sulfur ylides to terminal alkynes in an inter- or intramolecular reaction. The reactions were the electrophilic addition of metal gold-carbenes to electron-rich alkynes (Scheme 1(I)).8 Another method for the synthesis of furan derivatives was the reaction of sulfonium ylides with dialkyl acetylenedicarboxylates. The reactive intermediate sulfonium acylmethylides were trapped by acetylenic esters to yield substituted furans in an inter- or intramolecular reaction.9 Mostly, furan-2,3-dicarboxylic acid derivatives were obtained with this method10 and only two examples were reported on the synthesis of furan-3,4-dicarboxylic acid derivatives (Scheme 1(II)).11 We, herein, report the regioselective synthesis of structurally diverse polysubstituted furans with mono to tricarboxylates from various acetylenic esters and dimethylsulfonium acylmethylides and the mechanistic rationale on the selective generation of dialkyl furan-3,4-dicarboxylates.
 | ||
| Scheme 1 Synthesis of polysubstituted furans from sulfur ylides and alkynes. | ||
Results and discussion
To understand the mechanism and to explore the substrate scope of the reaction of dialkyl acetylenedicarboxylates and dimethylsulfonium acylmethylides, dimethyl acetylenedicarboxylate (DMAD) (1a) and 2-(dimethyl-λ4-sulfanylidene)-1-phenylethan-1-one (dimethylsulfonium benzoylmethylide) (2a) were employed as the model substrates to optimize the reaction conditions in 1,2-dichloroethane (DCE) under nitrogen atmosphere. We first optimized the reactant ratio (Table 1, entries 1–3). When the ratio of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a was increased from 1:2 to 1:2.4, the yield of product dimethyl 2-phenylfuran-3,4-dicarboxylate (3aa) decreased from 44% to 34% (Table 1, entries 1 and 2). The yield dropped sharply to 15% when the ratio of 1a:2a was 2:1 (Table 1, entry 3). Solvents were also screened. Product 3aa was obtained in 57% in polar DMF as solvent (Table 1, entry 4). The reactions with MeCN and DMSO as solvents gave product 3aa in 50% and 81% yields, respectively (Table 1, entries 5 and 6). The results indicated that DMSO was the best choice. The yield decreased to 70% when the reaction was conducted without nitrogen protection (Table 1, entry 7). It was found that the extension of the reaction time had no significant effect on the yield of product 3aa (Table 1, entry 8). When the reaction time was shortened from 4 h to 2 h, the yield was slightly dropped to 73% (Table 1, entry 6 vs. 9). At last, the yield of product 3aa was 80% when the reaction was conducted at 110 °C (Table 1, entry 10).
:2a was increased from 1:2 to 1:2.4, the yield of product dimethyl 2-phenylfuran-3,4-dicarboxylate (3aa) decreased from 44% to 34% (Table 1, entries 1 and 2). The yield dropped sharply to 15% when the ratio of 1a:2a was 2:1 (Table 1, entry 3). Solvents were also screened. Product 3aa was obtained in 57% in polar DMF as solvent (Table 1, entry 4). The reactions with MeCN and DMSO as solvents gave product 3aa in 50% and 81% yields, respectively (Table 1, entries 5 and 6). The results indicated that DMSO was the best choice. The yield decreased to 70% when the reaction was conducted without nitrogen protection (Table 1, entry 7). It was found that the extension of the reaction time had no significant effect on the yield of product 3aa (Table 1, entry 8). When the reaction time was shortened from 4 h to 2 h, the yield was slightly dropped to 73% (Table 1, entry 6 vs. 9). At last, the yield of product 3aa was 80% when the reaction was conducted at 110 °C (Table 1, entry 10).
|
||||
|---|---|---|---|---|
| Entry | 1a:2a |
Solvent | Time (h) | Yield (%)b |
| a All reactions were conducted on a 0.125 mmol scale of 1a in 1 mL of solvent at 80 °C.b NMR yield of the crude product using 1,3,5-trimethoxybenzene as an internal standard.c Yield of the isolated product.d Without nitrogen protection.e The reaction conducted at 110 °C. | ||||
| 1 | 1:2 |
DCE | 4 | 44 |
| 2 | 1:2.4 |
DCE | 4 | 34 |
| 3 | 2:1 |
DCE | 4 | 15 |
| 4 | 1:2 |
DMF | 4 | 57 |
| 5 | 1:2 |
MeCN | 4 | 50 |
| 6 | 1:2 |
DMSO | 4 | 81(79)c |
| 7 | 1:2 |
DMSO | 4 | 70d |
| 8 | 1:2 |
DMSO | 6 | 80 |
| 9 | 1:2 |
DMSO | 2 | 73 |
| 10e | 1:2 |
DMSO | 4 | 80 |
With the optimized reaction conditions, the reaction scope was then evaluated (Table 2). Various sulfonium ylides 2 were examined in the reaction with dimethyl acetylenedicarboxylate (DMAD) (1a). We were pleased to find that the application of various sulfonium ylides 2 led to the corresponding furan-3,4-dicarboxylates 3 in moderate to good yields. Various functional groups such as methyl, fluoro, chloro, bromo, trifluoromethyl, and cyano on the aryl group of the sulfur ylides 2 were well tolerated under optimized reaction conditions (Table 2, 3ab–3ah). For electron-deficient 2-(dimethyl-λ4-sulfanylidene)-1-(4-nitrophenyl)ethan-1-one (2i), the desired furan-3,4-dicarboxylate 3ai was isolated in 45% yield under the optimized reaction conditions. In comparison with dimethylsulfonium monosubstituted benzoylmethylides 2a–2i, the sulfur ylide with more substituted on the aryl group was tested as well. 1-(3,4-Dichlorophenyl)-2-(dimethyl-λ4-sulfanylidene)ethan-1-one (1j) gave rise to the corresponding furan-3,4-dicarboxylate 3aj in 74% yield. After replacing the phenyl group with naphthyl, the corresponding products 3ak and 3al were obtained in 85% and 75% yields, respectively. After investigating the reaction of different dimethylsulfonium 2-aryl-2-oxoethylides 2 with dimethyl acetylenedicarboxylate, diethyl acetylenedicarboxylate (1b) was evaluated with three representative sulfur ylides 2a, 2b, 2f, affording the desired products 3ba, 3bb, and 3bf in 56%, 53%, and 68% yields, respectively, under the optimized reaction conditions.
|
|---|
| a Reaction conditions A: acetylenic ester 1 (0.125 mmol) and sulfur ylide 2 (0.250 mmol) in DMSO (1 mL) were stirred under nitrogen at 80 °C for 4 h.b Reaction conditions B: acetylenic ester 1 (0.125 mmol) and sulfur ylide 2 (0.250 mmol) in DMSO (1 mL) were stirred under nitrogen at 160 °C for 15 min under microwave irradiation. |
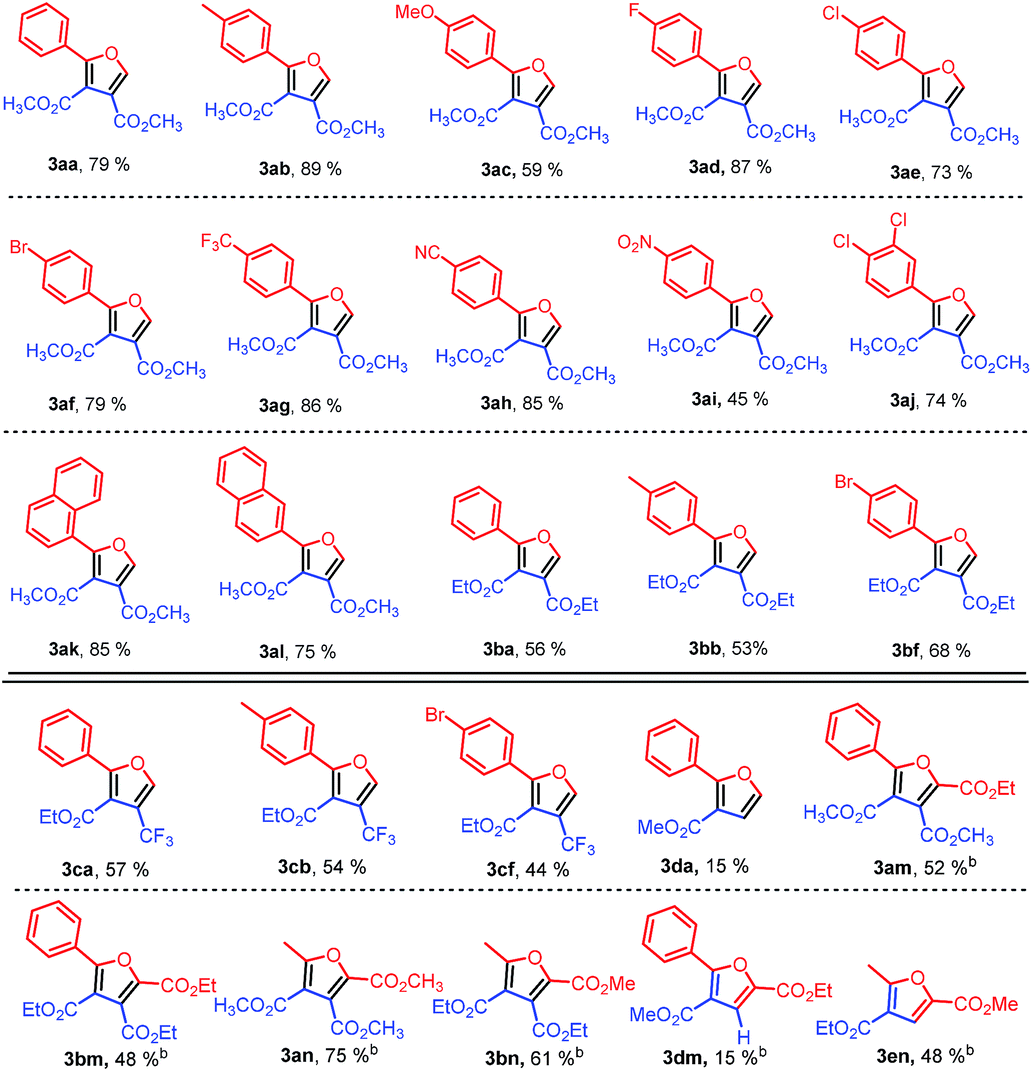 |
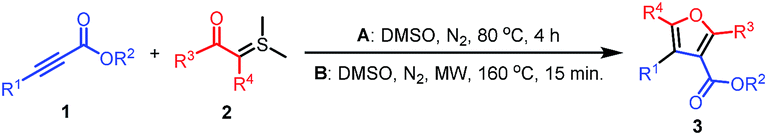
To extend the application of the synthetic method, several alkyl propynoates were attempted to prepare alkyl 2-substituted furan-3-carboxlyates (Table 2). Gratifyingly, ethyl 4,4,4-trifluorobut-2-ynoate (1c) was also successful for the synthesis of the corresponding trifluoromethylated furan-3-carboxylate derivatives. The desired 4-trifluoromethylfuran-3-carboxylates 3ca–3cf were obtained in moderate yields of 44–57%. However, methyl propynoate (1d) only produced the corresponding furan-3-carboxylate 3da in a low yield of 15% when it was reacted with sulphur ylide 2a.
Further extension of the synthetic strategy in the preparation of trialkyl furan-2,3,4-tricarboxylates was performed (Table 2). It was worth noting that no desired products were observed for diacyl stabilized sulfonium ylides 2m and 2n at 80 °C when they were applied in the reaction. To our delight, the desired trialkyl furan-2,3,4-tricarboxylates 3am, 3bm, 3an, and 3bn were obtained in moderate to good yields of 48% to 75% when the reaction temperature was increased from 80 °C to 160 °C under microwave irradiation due to the microwave assistance.11,12
Finally, the synthetic strategy was tested in the preparation of dialkyl furan-2,4-dicarboxylates as well (Table 2). The reaction of diacyl stabilized sulfonium ylide 2m and methyl propynoate (1d) afforded the desired furan-2,4-dicarboxylate 3dm in a low yield of 15%, while diacyl stabilized sulfonium ylide 2n reacted with ethyl propynoate (1e), giving the desired furan-2,4-dicarboxylate 3en in a moderate yield of 48%. However, in a previous report,10a the reaction of diacyl stabilized sulfonium ylide 2n and ethyl propynoate (1e) produced 2-ethyl 4-methyl 5-methylfuran-2,4-dicarboxylate (4en) only in 73% yield in 1,4-dioxane at 160 °C, showing a different reaction regioselectivity from our results. To verify the different selectivity, we conducted the same reaction in 1,4-dioxane at 160 °C under microwave irradiation, affording a mixture of 4-ethyl 2-methyl 5-methylfuran-2,4-dicarboxylate (3en) and 2-ethyl 4-methyl 5-methylfuran-2,4-dicarboxylate (4en) in a ratio of 1:1.81 in 53% total yield. However, under our optimal conditions, only 4-ethyl 2-methyl 5-methylfuran-2,4-dicarboxylate (3en) was obtained regiospecifically in DMSO as solvent. The results revealed that solvent and temperature played important effects on the reaction pathways, resulting in regioselective formation of different furan derivatives. The exact reason for the selective control is not clear now possible it is attributed the stability of intermediates in different solvents during reaction.
The reactions of both sulfur ylides 2a and 2m with methyl propynoate (1d) gave rise to the corresponding products 3da and 3dm in low yields because methyl propynoate (1d) was not electron-deficient enough compared with other acetylenic esters and then it was difficult for sulfonium ylides 2a or 2m to undergo Michael addition to methyl propynoate (1d).
After obtaining the above information, we proposed the following reaction mechanism (Scheme 2). Dimethyl acetylenedicarboxylate (1a) and 2-(dimethyl-λ4-sulfanylidene)-1-phenylethan-1-one (2a) are used to illustrate the proposed mechanism. First, 2-(dimethyl-λ4-sulfanylidene)-1-phenylethan-1-one (2a) undergoes Michael addition to dimethyl acetylenedicarboxylate (1a) to generate intermediate A, which further takes place an intramolecular nucleophilic addition to the carbonyl group of the benzoyl group to produce zwitterionic intermediate B. The intermediate B undergoes a 4π ring opening to form intermediate enolate C. The enolate in intermediate C takes place an intramolecular Michael addition followed by the elimination of dimethyl sulfide to give furan 3aa. In the previous report,11 the conversion of intermediates A to C was assumed as the benzoyl group shift followed by tautomerization. In our viewpoint, it is an intramolecular nucleophilic addition followed by a 4π ring opening process. This is a more reasonable conversion pathway.
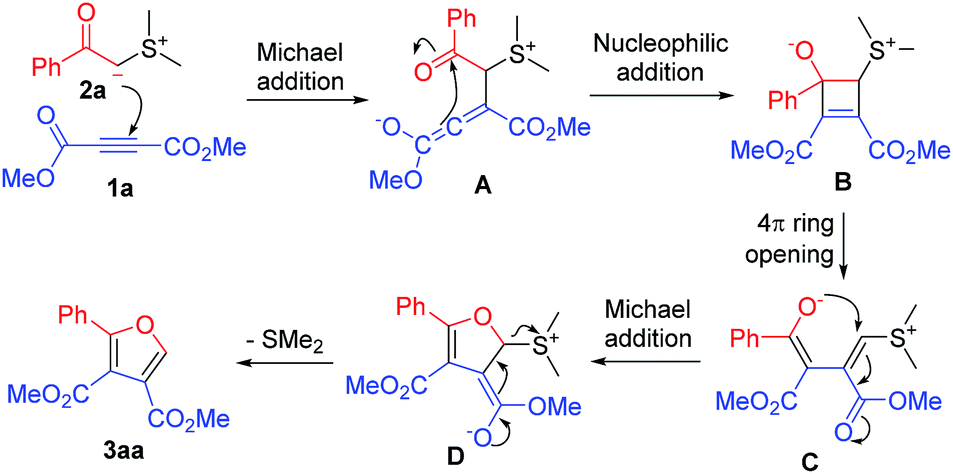 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Selected applications of polysubstituted furans, dimethyl 2-phenylfuran-3,4-dicarboxylates, from the literature are listed in Scheme 3,13 One for synthesis of diaryldihydrofuranones,13a the other for furanmethanol.13b
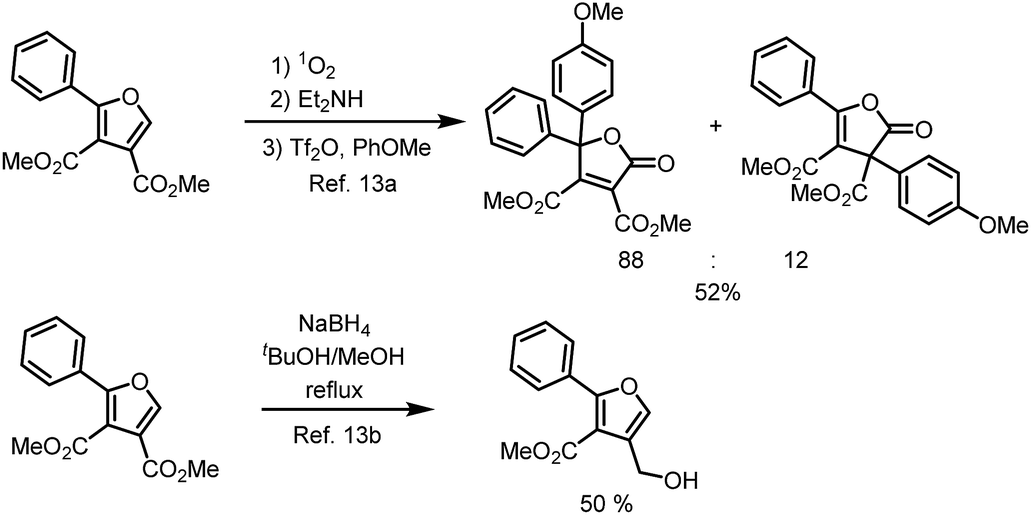 | ||
| Scheme 3 Selected reported application. | ||
Conclusions
Polysubstituted furans with carboxylate(s) were prepared in moderate to good yields from dimethylsulfonium acylmethylides with alkyl acetylenecarboxylates through a tandem sequence of Michael addition, intramolecular nucleophilic addition, 4π ring opening, intramolecular Michael addition, and elimination. The current method provides a direct and simple strategy in the efficient preparation of dialkyl 2-substituted furan-3,4-dicarboxylates from safe and readily available dimethylsulfonium acylmethylides and dialkyl acetylenedicarboxylates. In addition, the current method can also be applied for the synthesis of alkyl 2-arylfuran-3-carboxylates, dialkyl 5-substituted furan-2,4-dicarboxylates, and trialkyl 5-substituted furan-2,3,4-tricarboxylates, even alkyl 4-trifluoromethylfuran-3-carboxylates, showing versatile application. A new reasonable reaction mechanism is proposed as well.Experimental
Unless otherwise noted, all materials were purchased from commercial suppliers. Flash column chromatography was performed using silica gel (normal phase, 200–300 mesh) from Branch of Qingdao Haiyang Chemical Industry. The petroleum ether (PE) used for column chromatography is the 60–90 °C fraction, and the removal of residue solvent was accomplished under rotovap. The reactions were monitored by thin-layer chromatography (TLC) on silica gel GF254 coated 0.2 mm plates from Institute of Yantai Chemical Industry. The plates were visualized under UV light, as well as the other TLC stains (10% phosphomolybdic acid in ethanol; 1% potassium permanganate in water; 10 g of iodine absorbed on 30 g of silica gel). 1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer in CDCl3 with TMS as an internal standard, and the chemical shifts (δ) are reported in parts per million (ppm). All coupling constants (J) in 1H NMR spectra are absolute values given in hertz (Hz) with peaks labelled as singlet (s), broad singlet (brs), doublet (d), triplet (t), quartet (q), and multiplet (m). The IR spectra (KBr pellets, v [cm−1]) were recorded on a Nicolet 5700 FTIR spectrometer. HRMS measurements were carried out on an Agilent LC/MSD TOF mass spectrometer. LRMS measurements were carried out on a Thermo Trace 1300/ISQ QD system. Melting points were obtained on a Yanaco MP-500 melting point apparatus and are uncorrected. All the microwave reactions were conducted in a CEM Discover SP microwave system equipped with an infrared temperature detector.General procedure for the synthesis of sulfur ylides 2a–2l
To a solution of halomethyl carbonyl compound 4 (10 mmol) in acetone (15 mL) was added dimethyl sulfide (620 mg, 10 mmol). After the mixture was stirred for 12 h, the residue was filtered and washed with acetone. The solid product was used as sulfonium halide without further purification. The corresponding sulfonium halide was added to a solution of NaOH (400 mg, 10 mmol) in water (10 mL) at 0 °C. The solution was stirred for 30 min and then extracted several times with dichloromethane. The combined organic layers were washed with water and brine sequentially, dried over Na2SO4, filtered, and concentrated. The sulfur ylide 2 was obtained and can be used directly without further purification.General procedure for the synthesis of sulfur ylides 2m and 2n14
To a suspension of NCS (3.472 g, 26 mmol) in anhydrous dichloromethane (110 mL) was added dimethyl sulfide (2.7 mL, 36.9 mmol) at −78 °C under nitrogen. The mixture was stirred for 1 h at the same temperature. A solution of an active methylene compound (20 mmol) was added at the same temperature. After addition of triethylamine (4.2 mL, 30.3 mmol), the resulting mixture was stirred for another 1 h. The reaction mixture was treated with cold brine (60 mL) and extracted with ether (180 mL). The organic layer was washed with brine (60 mL) three times, dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography with chloroform and methanol (20:1, v/v) as eluent to afford the desired yilde 2m or 2n.
General procedure for the synthesis of furan derivatives 3
:1, v/v) as eluent to afford furan 3.:1, v/v) as eluent to afford furan 3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported partially from the National Natural Science Foundation of China (no. 21702014 and 21572017) and the Fundamental Research Funds for the Central Universities (XK1802-6).References
- (a) Q.-Z. Ye, S.-X. Xie, M. Huang, W.-J. Huang, J.-P. Lu and Z.-Q. Ma, J. Am. Chem. Soc., 2004, 126, 13940–13941 CrossRef CAS PubMed; (b) Y.-L. Lin, Y.-L. Tsai, Y.-H. Kuo, Y.-H. Liu and M.-S. Shiao, J. Nat. Prod., 1999, 62, 1500–1503 CrossRef CAS PubMed; (c) J. F. Kratochvil, R. H. Burris, M. K. Seikel and J. M. Harkin, Phytochemistry, 1971, 10, 2529–2531 CrossRef CAS.
- A. Blanc, V. Beneteau, J. M. Weibel and P. Pale, Org. Biomol. Chem., 2016, 14, 9184–9205 RSC.
- (a) L. Knorr, Chem. Ber., 1884, 17, 2863–2870 CrossRef; (b) C. Paal, Chem. Ber., 1884, 17, 2756–2767 CrossRef.
- (a) F. Feist, Chem. Ber., 1902, 35, 1545 CrossRef CAS; (b) E. Benary, Chem. Ber., 1911, 44, 489–493 CrossRef CAS.
- (a) J. Wang, S. Luo, J. Huang, T. Mao and Q. Zhu, Chem, 2014, 20, 11220–11224 CrossRef CAS PubMed; (b) E. O. Onyango and P. A. Jacobi, J. Org. Chem., 2012, 77, 7411–7427 CrossRef CAS PubMed; (c) T. Aoyama, Y. Hari and T. Iguchi, Synthesis, 2004, 1359–1362 CrossRef; (d) P. C. Ducepta and S. P. Marsden, ARKIVOC, 2002, 22–34 Search PubMed; (e) G. E. Daia, C. D. Gabbutt, J. D. Hepworth, B. M. Heron, D. E. Hibbs and M. B. Hursthouse, Tetrahedron Lett., 2002, 43, 4507–4510 CrossRef CAS; (f) W. Pei, J. Pei, S. Li and X. Ye, Synthesis, 2000, 2069–2077 CrossRef CAS; (g) T. Ibata and K. Fukushima, Chem. Lett., 1992, 21, 2197–2200 CrossRef; (h) H. Gotthardt, R. Huisgen and H. O. Bayer, J. Am. Chem. Soc., 1970, 92, 4340–4344 CrossRef CAS.
- (a) Z. Luo, Y. Fang, Y. Zhao, P. Liu, X. Xu, C. Feng, Z. Li and J. He, RSC Adv., 2016, 6, 5436–5441 RSC; (b) S. Manna and A. P. Antonchick, Org. Lett., 2015, 17, 4300–4303 CrossRef CAS PubMed; (c) W.-B. Liu, C. Chen and Q. Zhang, Synth. Commun., 2013, 43, 951–960 CrossRef CAS; (d) W. Liu, H. Jiang, M. Zhang and C. Qi, J. Org. Chem., 2010, 75, 966–968 CrossRef CAS PubMed; (e) Y. Liang, G. Huang, R. Yan, J. Huang, J. Luo and P. Wen, Synlett, 2010, 1071–1074 Search PubMed.
- (a) L. Q. Lu, T. R. Li, Q. Wang and W. J. Xiao, Chem. Soc. Rev., 2017, 46, 4135–4149 RSC; (b) A. C. B. Burtoloso, R. M. P. Dias and I. A. Leonarczyk, Eur. J. Org. Chem., 2013, 5005–5016 CrossRef CAS; (c) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611–620 CrossRef CAS PubMed; (d) A. H. Li, L. X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341–2372 CrossRef CAS; (e) J. Dong and J. X. Xu, Org. Biomol. Chem., 2017, 15, 836–844 RSC.
- (a) S. Kramer and T. Skrydstrup, Angew. Chem., Int. Ed., 2012, 51, 4681–4684 CrossRef CAS PubMed; (b) X. Huang, B. Peng, M. Luparia, L. F. Gomes, L. F. Veiros and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 8886–8890 CrossRef CAS PubMed.
- (a) M. Hamaguchi, N. Tomida and Y. Iyama, J. Org. Chem., 2007, 72, 1326–1334 CrossRef CAS PubMed; (b) M. Anary-Abbasinejad and N. Rostami, J. Chem. Res., 2006, 589–590 CrossRef CAS; (c) J. Azizian, M. R. Mohammadizadeh, A. A. Mohammadi and A. R. Karimi, Heteroat. Chem., 2005, 16, 259–262 CrossRef CAS; (d) V. Nair, S. Bindu and L. Balagopal, Tetrahedron Lett., 2001, 42, 2043–2044 CrossRef CAS; (e) T. Johnson, D. R. Cheshire, M. J. Stocks and V. T. Thurston, Synlett, 2001, 0646–0648 CrossRef CAS; (f) P. Clawson and D. A. Whiting, Tetrahedron Lett., 1987, 28, 3155–3158 CrossRef CAS; (g) M. Hamaguchi and T. Nagai, J. Chem. Soc., Chem. Commun., 1985, 190–191 RSC; (h) J. F. Gisch and J. A. Landgrebe, J. Org. Chem., 1985, 50, 2050–2054 CrossRef CAS; (i) H. S. Gill and J. A. Landgrebe, J. Org. Chem., 1983, 48, 1051–1055 CrossRef CAS; (j) H. S. Gill and J. A. Landgrebe, Tetrahedron Lett., 1982, 23, 5099–5102 CrossRef CAS; (k) H. C. Berk, K. E. Zwikelmaier and J. E. Franz, Synth. Commun., 1980, 10, 707–710 CrossRef CAS; (l) H. Gotthardt, C. M. Weisshuhn and K. Dörhöfer, Chem. Ber., 1978, 111, 3336–3345 CrossRef CAS.
- (a) Y. Hayasi, M. Kobayasi and H. Nozaki, Tetrahedron, 1970, 26, 4353–4360 CrossRef CAS; (b) M. Takaku, Y. Hayasi and H. Nozaki, Tetrahedron Lett., 1969, 10, 2053–2056 CrossRef.
- M. Higo and T. Mukaiyama, Tetrahedron Lett., 1970, 11, 2565–2568 CrossRef.
- (a) X. H. Li and J. X. Xu, Curr. Microwave Chem., 2017, 4, 339–346 CAS; (b) X. H. Li and J. X. Xu, Tetrahedron, 2016, 72, 5515–5520 CrossRef CAS; (c) X. H. Li and J. X. Xu, Curr. Microwave Chem., 2017, 4, 158–162 CrossRef CAS; (d) L. B. Hu, Y. K. Wang, B. N. Li, D. M. Du and J. X. Xu, Tetrahedron, 2007, 63, 9387–9392 CrossRef CAS; (e) D. Q. Yang and J. X. Xu, Curr. Microwave Chem., 2018, 5, 120–127 CrossRef CAS.
- (a) M. DellaGreca, S. Zuppolini, A. Zarrelli, M. R. Iesce and L. Previtera, Tetrahedron, 2013, 69, 4725–4730 CrossRef CAS; (b) M. R. Iesce, R. Sferruzza, F. Cermola and M. DellaGreca, Helv. Chim. Acta, 2016, 99, 296–301 CrossRef CAS.
- S. Katayama, T. Watanabe and M. Yamauchi, Chem. Lett., 1989, 18, 973–976 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Analytic data and copies of 1H and 13C NMR spectra of furan products 3, and copies of HR-MS spectra of unknown furan products 3. See DOI: 10.1039/c9ra03563j |
| This journal is © The Royal Society of Chemistry 2019 |