
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructured spinel cobalt ferrites: Fe and Co chemical state, cation distribution and size effects by X-ray photoelectron spectroscopy†
Marzia
Fantauzzi
 *a,
Fausto
Secci
ac,
Marco
Sanna Angotzi
ab,
Cristiana
Passiu
c,
Carla
Cannas
ab and
Antonella
Rossi
ab
*a,
Fausto
Secci
ac,
Marco
Sanna Angotzi
ab,
Cristiana
Passiu
c,
Carla
Cannas
ab and
Antonella
Rossi
ab
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Campus di Monserrato S.S. 554, Italy. E-mail: fantauzzi@unica.it
bINSTM, UdR Cagliari, Italy
cDepartment of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, Zurich, Switzerland
First published on 18th June 2019
Abstract
Nanostructured spinel cobalt ferrite samples having crystallite size ranging between 5.6 and 14.1 nm were characterized by X-ray photoelectron spectroscopy and X-ray induced Auger electron spectroscopy in order to determine the chemical state of the elements, the iron/cobalt ratio and the cation distribution within tetrahedral and octahedral sites. The presence of size-dependent trends in the binding energy of the main photoelectron peaks and in the kinetic energy of the X-ray induced O KLL signal was also investigated. The results showed that iron is present as FeIII and cobalt is present as CoII. The iron/cobalt ratio determined by XPS ranges between 1.8 and 1.9 and it is in very good agreement, within experimental uncertainty, with the expected 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The percentage of Fe in octahedral sites ranges between 62% and 64% for all samples. The kinetic energy of the O KLL signals increases with crystallite size. These results are explained in terms of changes in the ionicity of the metal–oxygen bonds. The results of this investigation highlight how the XPS technique represents a powerful tool to investigate the composition, the chemical state and inversion degree of cobalt spinel ferrites, contributing to the comprehension of their properties.
:1 ratio. The percentage of Fe in octahedral sites ranges between 62% and 64% for all samples. The kinetic energy of the O KLL signals increases with crystallite size. These results are explained in terms of changes in the ionicity of the metal–oxygen bonds. The results of this investigation highlight how the XPS technique represents a powerful tool to investigate the composition, the chemical state and inversion degree of cobalt spinel ferrites, contributing to the comprehension of their properties.
Introduction
Nanostructured spinel cobalt ferrite (CoFe2O4), because of its unique properties, has received great attention in a wide range of applications,1,2 which include magnetic recording,3,4 catalysis,5,6 biomedicine7–11 and sensors.12,13 Among the cubic ferrites, CoFe2O4 is the material with the highest magnetocrystalline anisotropy and reasonably high magnetization14 and it shows excellent chemical and thermal stabilities, good mechanical properties and is also easily synthesized by different approaches.15–18 In the spinel structure, oxygen ions are close-packed in a cubic arrangement in which metallic cations fill one out of the eight tetrahedral interstices (commonly identified as Td or with round brackets) and half of the octahedral ones (Oh or square brackets). Tetrahedral sites are occupied by divalent cations in a direct or normal spinel (MII)[MIII]2O4, whereas trivalent cations replace them in an inverse spinel (MIII)[MII;MIII]2O4.19The chemical and physical properties are directly correlated to the composition and to the structure. Bulk cobalt ferrite shows an inverse spinel structure: all CoII cations occupy the octahedral sites, and the FeIII cations are equally distributed in Td and Oh sites (γ = 1, γ is the inversion degree or the fraction of divalent cations in octahedral sites). The coupling of the magnetic moments associated with the ions in Td and Oh sites justifies the ferrimagnetic behavior of the cobalt ferrite below 860 K.19 Conversely, when prepared in form of nanostructured material CoII and FeIII are randomly distributed (γ = 0.66). In the literature, different techniques (57Fe-Mössbauer spectroscopy, EXAFS, neutron diffraction) have been applied in order to determine the inversion degree of nanostructured CoFe2O4 synthesized with various approaches and the values fall in the 0.68–0.76 range.10,20–22
It is clear that among all the factors that mainly influence the properties of the ferrites, besides the size and the shape of the particles, the stoichiometry and the cation distribution play a crucial role. Therefore, the knowledge of the chemical composition, of the structure and of the properties is the key to design a material for a specific application.
In this context, X-ray photoelectron spectroscopy is acknowledged to be a state-of-art tool for providing elemental, chemical state and quantitative information also when investigating nanostructured materials.23 Due to the above mentioned features it is often used as a complementary technique in papers dealing with nanostructured ferrites.24–26
The chemical state analysis of XPS data usually starts with the curve fitting of the most intense photoelectron signal. For the transition metals this procedure is not straightforward being the line shapes of the 2p peaks complicated by peak asymmetry, multiplet splitting, by the presence of shake-up satellites and plasmon loss structures. Furthermore, if iron and cobalt are simultaneously present in a sample, as in CoFe2O4, the photoelectron Fe 2p and Co 2p peaks overlap the Auger Co L3M45M45 and Fe L3M45M45 respectively, when the Al Kα X-ray source is used.
Among the scientific literature on ferrites, only few papers provide details on the curve fitting procedure.27,28 Some of the papers report XPS spectra, tables listing the binding energy values and the quantitative composition without details on the spectra processing.29–31 The present work focuses on the use of XPS for the characterization of nanostructured cobalt ferrites samples with crystallite size ranging between 5.6 and 14.1 nm.
In order to determine the iron and cobalt chemical states, their most intense photoelectron signals were resolved in components following two different approaches. The first one is based on the results of the calculation of the multiplet structure expected for 2p core levels of iron and cobalt free ions, proposed by Gupta and Sen32 and the strategy developed in ref. 25 for first row transition metals oxides and hydroxides. This model was successfully applied for curve fitting the iron signals in various applications ranging from the environmental33 to the biological ones.34 According to the curve fitting strategy proposed in literature27 the iron of a nickel ferrite is composed of four components that were chosen as starting point for curve fitting the spectra in this paper.
The magnetic properties of the ferrites are influenced by disorder in the cation distribution between octahedral and tetrahedral sites, which can influence saturation magnetization, exchange couplings, and ferrimagnetic ordering temperatures;35 in order to determine the spinel inversion degree of the cobalt ferrite, a second curve fitting strategy, based on that proposed by Aghavniana and co-workers,36 has been adopted in this paper.
Since it is known that the binding energy of photoelectron lines is a size dependent parameter,37–40 in this paper it is also evaluated if a systematic chemical shift of the binding energy of iron Fe 2p3/2, cobalt Co 2p3/2, oxygen O 1s and of the kinetic energy of the X-ray induced O KLL signal is observable, changing the cobalt ferrite crystallite size. Since initial and final state effects influence the chemical shift of both photoelectron and Auger lines, an explanation of the observed trends is provided.
Experimental
Cobalt ferrite samples (labelled as Co1-5 in Table S1†) with different crystallite size have been synthesized following a solvothermal procedure similar to the one described elsewhere.41,42 In order to tune the crystallite size, the amount of the mixed CoII–FeIII oleate precursor, the solvents as well as the temperature were properly modified. In details: the amount of precursor for the 5.6 nm nanoparticles (NPs) was 3 mmol in 20 cm3 of 1-pentanol and 10 cm3 of distilled water. The synthesis was conducted at 180 °C for 10 h. The same temperature and reaction time were used for the synthesis of the 6.7 nm NPs: in this case 6 mmol of precursor were added to 10 cm3 of 1-pentanol, 10 cm3 of octanol, and 5 cm3 of distilled water. With the same amount of precursor and solvents, but at 220 °C for 10 h, 8.8 nm NPs were obtained. 11.2 nm NPs were synthetized starting with 2.5 mmol of precursor, 10 cm3 of 1-pentanol, 10 cm3 of toluene and 5 cm3 of distilled water at 230 °C for 10 h. The largest nanoparticles (14.1 nm) were obtained by a seed-mediated approach using a solvothermal procedure set up for core–shell nanoparticles having a CoFe2O4 core and MnFe2O4 or Fe3O4/γ-Fe2O3 shell.42Table S1† summarises the synthesis conditions adopted in this work. The first column reports the sample notation.
X-ray diffraction, using a PANalytical X'Pert PRO with Cu Kα radiation (1.5418 Å), secondary monochromator, and a PIXcel position-sensitive detector was used for characterizing the crystalline phases. Calibration of the peak position and instrumental width was carried out using powder LaB6 from NIST. The hexane dispersions were dried on a glass plate and measured in the angular range of 10–90° with a step of 0.039°. The XRD patterns aiming to the identification of the crystalline phase, after background subtraction, were processed using PANalytical X'Pert HighScore software. The most intense X-ray peaks [(220), (311), (400), (422), (511), (440)] were fitted with Origin Software by PseudoVoigt function, using a 1:1 Gaussian/Lorentzian ratio. In reciprocal space for the cubic system, the reciprocal of the interplanar distance q is defined as
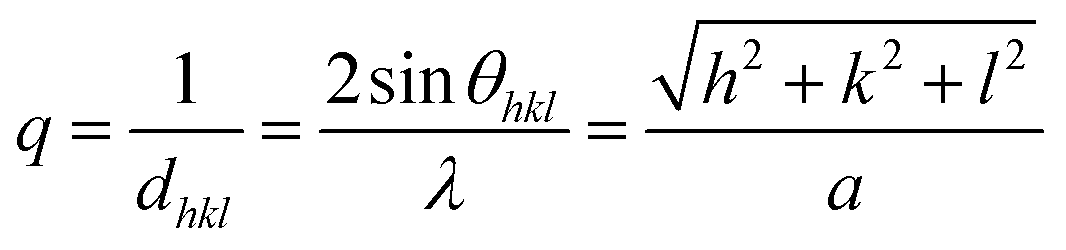
where
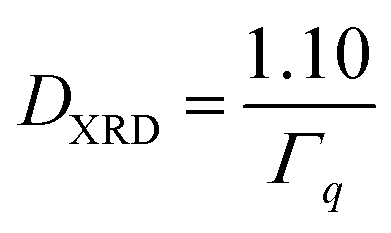
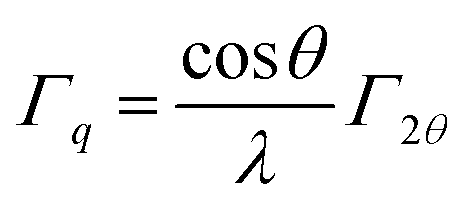
In this equation, the value 1.10 takes into account the information on size distribution and shape of the particles and the model derived from Repko et al.41 based on the fitting of the peak of Fourier-transformed solid spheres with volume-weighted log-normal size distribution with σ = 0.18, which are almost Gaussian. For each sample, the crystallite size is given as average of the values calculated on the most intense peak and the associated error is the standard deviation.
AFM analysis was performed on spin-coated silicon wafer-samples to observe the changes occurring on their surfaces when varying the spin-coating conditions.
To obtain a uniform layer, the ideal suspension concentration was 4 mg cm−3. 100 μL of each solution were deposited on each silicon wafer and the spin-coating conditions were 4000 rpm for one minute.
AFM images were acquired using an Icon Dimension (Bruker, Camarillo, California, USA) with a silicon cantilever (BudgetSensors, Bulgaria) with a spring constant of about 26 N m−1 (manufacturer's value).
XPS measurements were carried out using a Sigma2 XPS and a Thetaprobe manufactured by ThermoFisher Scientific Inc., East Grinstead, UK. Sigma 2 is equipped with an Al/Mg Kα twin source. A non-monochromatic Mg Kα source (1253.6 eV) operated at 200 W was used in order to eliminate the overlap between Fe 2p and Co LMM peaks and between Co 2p and Fe LMM peaks, which is observed if an Al Kα source is used. The residual pressure during the analysis was always lower than 10−6 to 10−7 Pa. The emitted electrons are collected by an Alpha 110 hemispherical analyzer, and a multichannel detector consisting of seven channeltrons is used. The analyzer was operated in fixed-analyzer transmission mode setting the pass energy at 25 eV for collecting the high-resolution spectra and at 50 eV for the survey spectra; a step size of 0.05 eV and 1 eV was respectively used. The instrument was used in large-area mode (diameter of the analyzed area: 8 mm). The energy resolution in large-area XPS mode, expressed as the full width at half maximum (FWHM) of the Ag 3d5/2 signal was 1.1 eV; the intensity was 3.7 Mcps using an Al window less than 1 μm thick to limit the contribution of backscattered electrons and to reduce the contribution of Bremsstrahlung radiation.
The Thetaprobe spectrometer is equipped with a monochromatic Al Kα X-ray source; the residual pressure during the analysis was always lower than 10−7 Pa. Spectra were collected using a monochromatic beam (Al Kα1,2E=1486.6 eV) operated at 4.7 mA and 15 kV (70 W), with a nominal spot size of 300 μm. The analyzer was operated in fixed analyzer transmission mode at 100 eV pass energy for collecting narrow scan spectra and at 200 eV for the survey spectra with a step size of 0.05 eV and 1 eV, respectively. The linearity of the binding energy scale of both spectrometers was monthly checked according to the literature43 and to ISO 15472:2010, using Cu, Au and Ag reference samples. Data were acquired under computer control. Binding energy values were corrected by referencing to the aliphatic carbon at 285.0 eV. Thetaprobe was used to acquire the O 1s and O KLL spectra since the lower FWHM allowed obtaining a better-resolved O KLL signal.
Magnetic nanoparticles dispersed and dried on silicon wafer can be characterized by XPS taking care of checking the shape of the survey spectrum at the low kinetic energy side i.e. below 200 eV. This part of the spectrum is reported to be affected in the presence of stray magnetic fields.44 No changes at the low kinetic energy values were observed during this investigation. Fe/Co ratios were calculated correcting the experimental areas for the relative sensitivity factors. The details are reported in the ESI.†
Results
For all the samples XRD results indicated the presence of a spinel cobalt ferrite nanophase, as confirmed by the comparison with the CoFe2O4 PDF-Card 0221086 (Fig. S1†).42 The absence of other XRD peaks is a clear indication of the presence of a unique spinel phase, within the detection limit of the technique. The crystallites size determined by XRD is shown in Table S1.† Cobalt ferrite samples were dispersed in hexane and deposited on silicon wafer by spin coating, dried, and analyzed by XPS. To check the homogeneity of the covering of the silicon wafer substrate, atomic force microscopy was used. The optimum concentration for achieving the best coverage of the silicon wafer was found to be 4 mg of ferrites in 1 cm3 of hexane. At lower concentration of the dispersion (1–2 mg cm−3) disordered and not homogeneous depositions were obtained (Fig. S2†). Together with the signals due to the presence of iron and cobalt, the survey spectra (Fig. S3†) showed the signals due to oxygen from both the ferrite and the capping agent, and the peaks due to carbon deriving from both the capping agent and a possible organic contamination due to the exposure of the samples to the laboratory environment. The absence of signals at about 100 eV and 150 eV allowed excluding the presence of the silicon contribution from the wafer, confirming a proper coverage.Fe 2p peaks (Fig. 1a) exhibit a doublet separated by 13.5 eV due to spin–orbit coupling. For the curve-fitting purpose only the Fe 2p3/2 peak was taken into account.
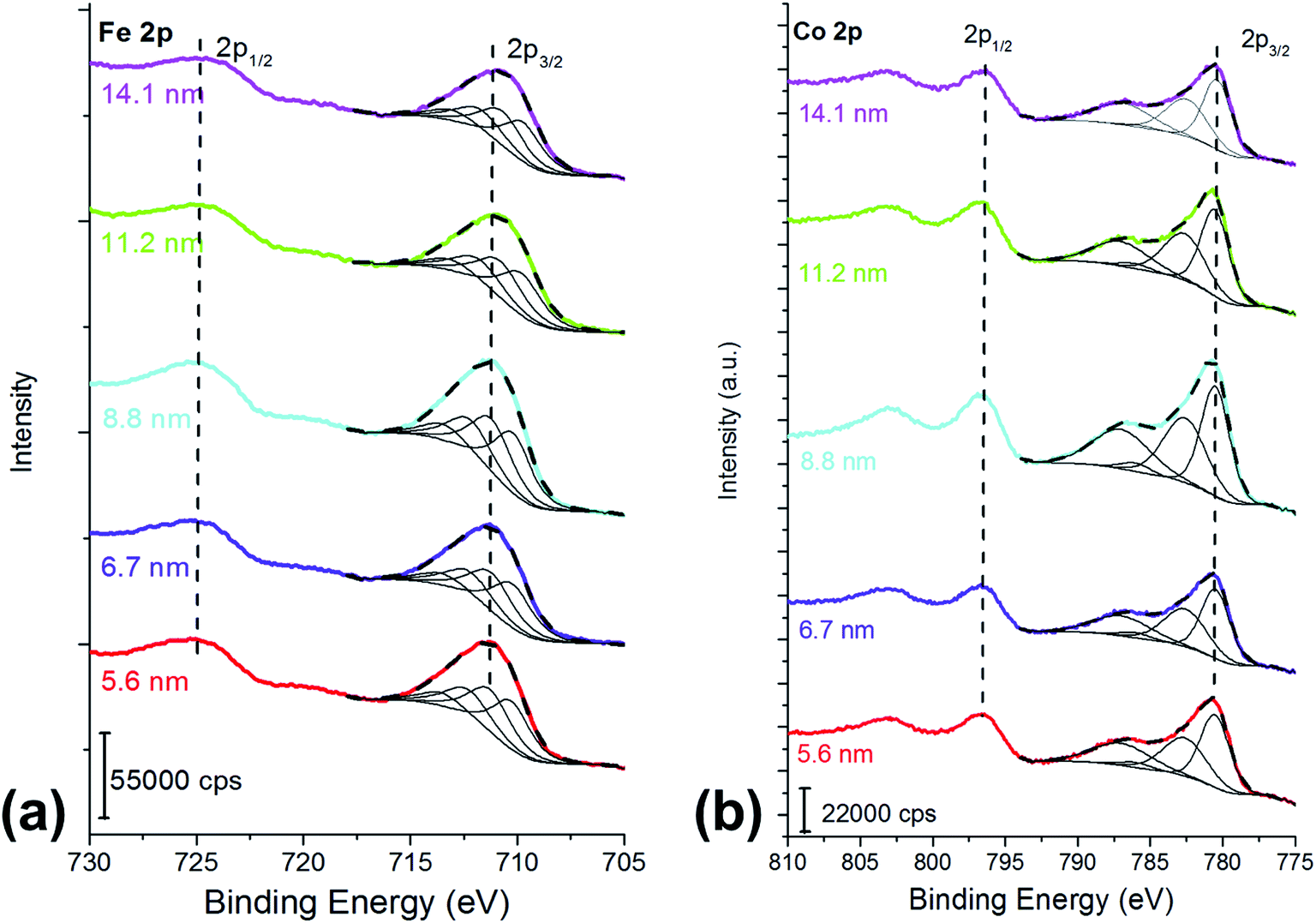 | ||
| Fig. 1 (a) Fe 2p and (b) Co 2p peaks of the cobalt ferrite samples following the curve-fitting procedure based on the multiplet splitting approach.27 The crystallite size is reported in the graph. X-ray source: Mg Kα. | ||
Fe 2p3/2 peaks were resolved in four components due to multiplet splitting phenomena, following what is reported in literature27 for NiFe2O4. The binding energy of the four components are listed in Table 1.
| Crystallite size (nm) | Fe/Co ratio | Fe 2p I | Fe 2p II | Fe 2p III | Fe 2p IV | Co 2p I | Co 2p II | Co 2p III | Co 2p IV |
|---|---|---|---|---|---|---|---|---|---|
| 5.6 (2) | 1.8 | 710.3 (1) | 711.3 (1) | 712.5 (1) | 713.9 (1) | 780.5 (1) | 782.6 (1) | 786.0 (1) | 787.0 (1) |
| 6.7 (1) | 1.9 | 710.2 (1) | 711.2 (1) | 712.4 (1) | 713.8 (1) | 780.5 (1) | 782.6 (1) | 786.0 (1) | 787.0 (1) |
| 8.8 (2) | 1.9 | 710.1 (1) | 711.1 (1) | 712.4 (1) | 713.8 (1) | 780.5 (1) | 782.6 (1) | 786.0 (1) | 787.0 (1) |
| 11.2 (6) | 1.9 | 710.1 (1) | 711.1 (1) | 712.4 (1) | 713.8 (1) | 780.5 (1) | 782.6 (1) | 786.0 (1) | 787.0 (1) |
| 14.1 (8) | 1.9 | 710.0 (1) | 711.0 (1) | 712.2 (1) | 713.6 (1) | 780.4 (1) | 782.5 (1) | 785.9 (1) | 786.9 (1) |
The curve-fitting parameters are shown in the ESI (Table S2†): the full width at half-maximum of the peak height and the area ratios were kept constant during curve synthesis, while the position of the components was let free to change. Despite this degree of freedom, the binding energy difference between the first peak (Fe 2p I) and the other peaks was reproducible, and it is reported in Table S2.† The binding energy of the signals (Table 1) is typical for FeIII27 and no evidence of FeII is observed. Fig. 1a and Table 1 show a small binding energy shift of the Fe 2p3/2 components with the crystallite size: the smaller the cobalt ferrites particles size, the higher the binding energy of the peak.
Co 2p peak is a doublet due to spin–orbit coupling (Fig. 1b); the binding energy separation between Co 2p3/2 and Co 2p1/2 was 16 eV and it allowed us to exclude the presence of CoIII, thus the experimental lines were curve-fitted starting from the fitting parameters of CoO reported in Biesinger et al.27 Also in this case, for curve-fitting purposes, only the Co 2p3/2 was taken into account (Fig. 1b). The shape of the Co 2p3/2 signal, which shows an intense satellite separated by 6.5 eV from the main most intense peak, is typical of CoII.27 No significant shifts were observed for the different samples (Table 1). The curve-fitting parameters are listed in Table S3† of the ESI.†
O 1s is a multicomponent peak (Fig. 2a): the most intense component is found at about 530 eV (Table 2) and it is ascribed to oxygen in the oxide lattice.27 The component at about 531.5 eV can be ascribed to both hydroxides and defective oxygen.27 The peak at the highest binding energy value might be due to water and/or to the oleic acid used as capping agent.27
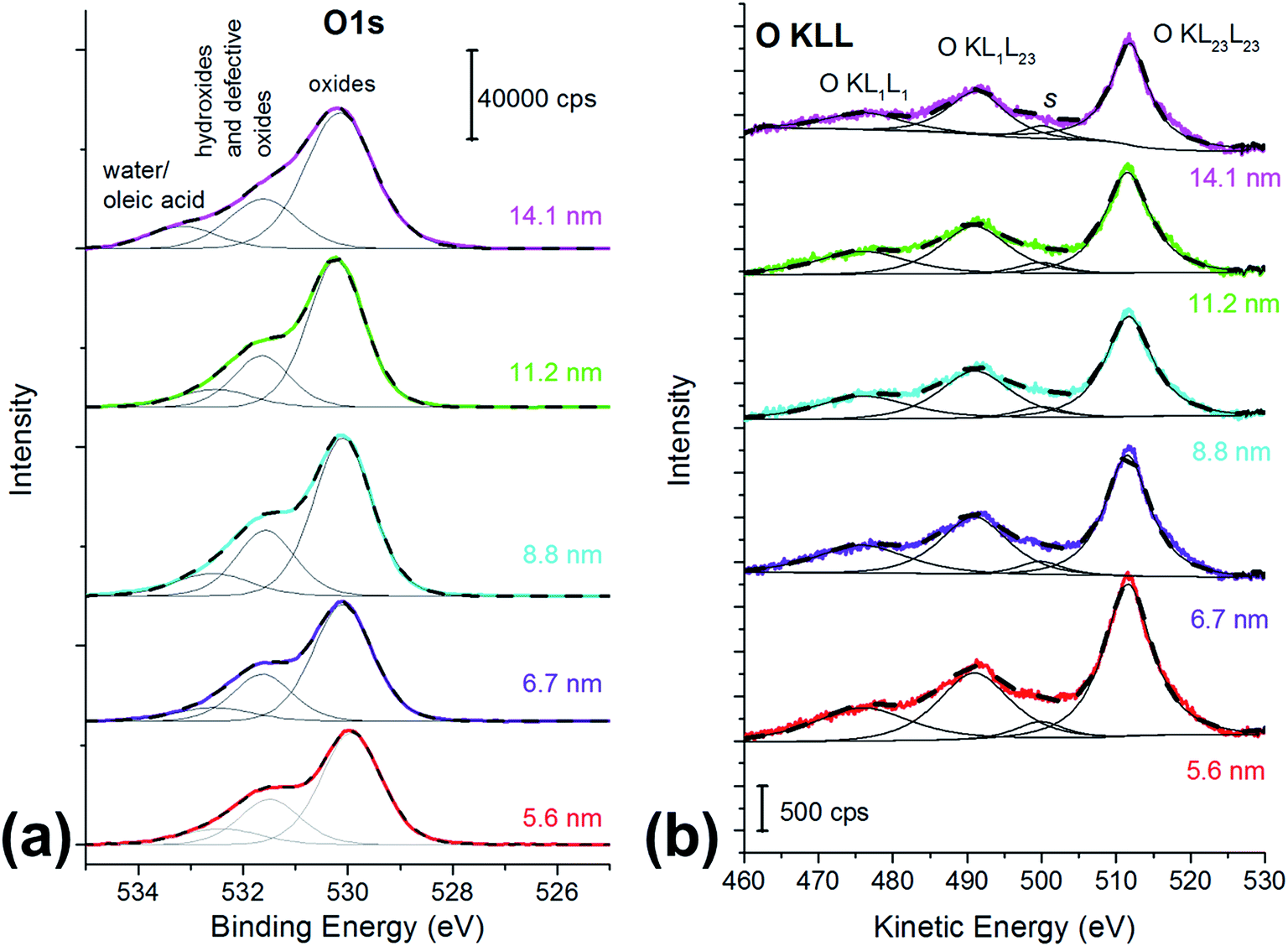 | ||
| Fig. 2 (a) O 1s spectra and (b) O KLL spectra of the cobalt ferrite nanoparticles samples. The particle size is reported in the graph. X-ray source: monochromatic Al Kα. | ||
| Crystallite size (nm) | O1s oxide | O1s hydroxide or defective oxygen | O1s water | O KL23L23 | O KL1L23 | ΔKEOKLL | I KL23L23/IKL1L23 | α′a | Δα′b | ΔReac |
|---|---|---|---|---|---|---|---|---|---|---|
| a α′ = (BEO 1s + KEO KL23L23). b Δα′= (α′CoFe2O4 NPs − α′Fe2O3 taken at 1042 eV). c ΔRea = Δα′. | ||||||||||
| 5.6 (2) | 530.0 (1) | 531.5 (1) | 532.4 (1) | 511.2 (1) | 490.5 (1) | 20.7 (1) | 1.75 | 1041.2 (2) | −0.8 | −0.4 |
| 6.7 (1) | 530.0 (1) | 531.6 (1) | 532.6 (1) | 511.4 (1) | 490.8 (1) | 20.6 (1) | 1.78 | 1041.4 (2) | −0.6 | −0.3 |
| 8.8 (2) | 530.1 (1) | 531.5 (1) | 532.4 (1) | 511.5 (1) | 491.0 (1) | 20.5 (1) | 1.80 | 1041.6 (2) | −0.4 | −0.2 |
| 11.2 (6) | 530.1 (1) | 531.6 (1) | 532.3 (1) | 511.7 (1) | 491.3 (1) | 20.4 (1) | 1.89 | 1041.8 (2) | −0.2 | −0.1 |
| 14.1 (8) | 530.1 (1) | 531.5 (1) | 532.5 (1) | 511.9 (1) | 491.3 (1) | 20.4 (1) | 2.04 | 1042.0 (2) | 0 | 0 |
O KLL spectrum showed the presence of two prominent peaks, assigned to O KL23L23 and O KL1L23 (Fig. 2b),45 together with a component at about 477 eV assigned to the KL1L1 Auger transition45 and a small peak at 501 eV (peak s in Fig. 2b) that may be due to shake-up and shake-off contribution to the Auger peak.45
The kinetic energy values of the O KL23L23 components are reported in Table 2, together with KEO KL23L23 − KEO KL1L23 difference, which is related to the ionicity of the oxides, in agreement with literature.46 The identification of the three different oxygen species (oxide, hydroxides or defective oxygen, and water or oleic acid) by curve-fitting of the O KLL signals was not attempted since it was beyond the scope of this work. The O KLL signals were resolved in their main components in order to calculate the modified Auger parameter (α′) and to evaluate any possible size effect in the kinetic energy values of the peak maxima. The values of the modified Auger parameter47α′ = BEO1s + KEOKL23L23 are also reported in Table 2.
Iron/cobalt ratios of the CoFe2O4 samples are found to be close to 2:1 (Table 1) for all the samples, in agreement with the ICP-OES (inductively coupled plasma–optical emission spectrometry) results obtained on CoFe2O4 samples synthesized by an identical synthesis route.42 To evaluate the ratio by XPS data, the Tougaard's background-subtracted Fe 2p and Co 2p peaks (both 2p3/2 and 2p1/2 components) were considered. Tougaard's background was chosen since it is less sensitive to the choice of the minimum and maximum of the selected energy range than the Shirley background, especially if large energy ranges are taken into account.48 In the present case the energy range was 35 eV for Co 2p and 25 eV for Fe 2p.
Discussion
The curve-fitting approach adopted for Fe 2p3/2 and for Co 2p3/2 (Fig. 1), following the calculated components and their intensity ratio presented by Gupta and Sen32 and in agreement with the curve-fitting reported in literature27 provided the evidence of the presence of FeIII and of CoII. Gupta and Sen32 calculated the multiplet structure of core p vacancy levels for the 3d-transition metal ions. McIntyre and coworkers,49 since 1977, adopted Gupta and Sen32 prediction for resolving Fe 2p and, later, Co 2p spectra of bulk powder.27 The curve-fitting showed in this work is in agreement not only with Gupta and Sen32 but also with successive publications by McIntyre and co-workers.49 Thus in this paper it is demonstrated that the fit predicted for the isolated ions,32 tested on bulk powders,27,49,50 can be successfully used also for nanostructured cobalt ferrites. The multiplet splitting of Fe 2p3/2 signals from different iron oxides was investigated by Grosvenor and coworkers50 and it was shown that the FeIII octahedrally-coordinated (α-Fe2O3) can be distinguished from the same signal in γ-Fe2O3, in which 75% of FeIII cations are octahedrally-coordinated and 25% of the cations are tetrahedrally coordinated. The main differences between the two Fe 2p3/2 signals are the different area ratios between the multiplet splitting components for α-Fe2O3 and γ-Fe2O3 and their binding energy (BE) separation. However, the proposed curve-fitting did not allow distinguishing, in the case of γ-Fe2O3, between octahedral and tetrahedral iron.50 The results obtained in this work for Fe 2p are comparable with those obtained for NiFe2O4:27 the presence of FeII can be ruled out since no peaks at about 708 eV and 709 eV27,50 are observed but nothing can be said about cation distribution.
The curve-fitting strategy of Co 2p peaks proposed by other authors36 was thus followed in order to estimate the spinel inversion parameter from XPS data. The estimation strategy starts with distinguishing between CoII in octahedral sites (Oh) and CoII in tetrahedral (Td) sites on Co 2p peaks (Fig. 3). Both Co 2p3/2 and Co 2p1/2 are taken into account after Shirley background subtraction, in agreement with literature.36 Co 2p3/2 was curve-fitted with a peak located at 780.5 eV, which is ascribed to cobalt in Oh sites, with a peak at 782.9 eV due to CoII in Td sites, and with a satellite at 786.3 eV for all the samples (Table 3 and Fig. 3).
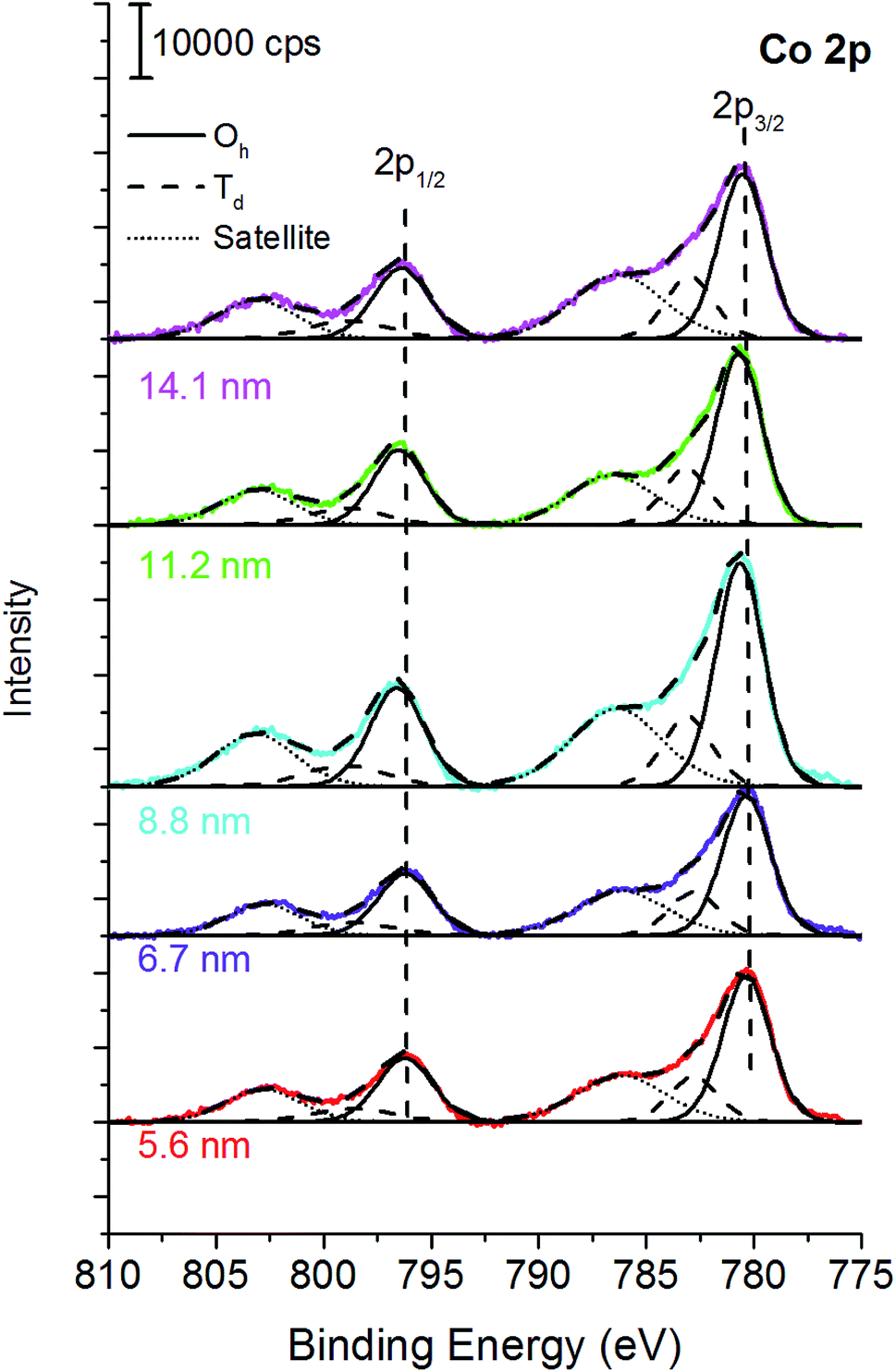 | ||
| Fig. 3 Octahedral and tetrahedral CoII from XPS. | ||
| Crystallite size (nm) | Co 2p3/2 (Oh) | Co 2p3/2 (Td) | Co 2p3/2 SAT | ΔBE 2p3/2 − 2p1/2 (eV) | γ | % Fe (Oh) | |||
|---|---|---|---|---|---|---|---|---|---|
| BE (eV) | FWHM (eV) | BE (eV) | FWHM (eV) | BE (eV) | FWHM (eV) | ||||
| 5.6 (2) | 780.4 (1) | 2.6 (1) | 782.8 (1) | 2.6 (1) | 786.2 (1) | 4.9 (1) | 16.0 (1) | 0.76 | 62 |
| 6.7 (1) | 780.4 (1) | 2.6 (1) | 782.8 (1) | 2.6 (1) | 786.2 (1) | 4.9 (1) | 15.9 (1) | 0.75 | 62 |
| 8.8 (2) | 780.5 (1) | 2.6 (1) | 782.9 (1) | 2.6 (1) | 786.4 (1) | 5.0 (1) | 15.9 (1) | 0.76 | 62 |
| 11.2 (6) | 780.5 (1) | 2.5(1) | 783.0 (1) | 2.5(1) | 786.6 (1) | 4.7 (1) | 15.8 (1) | 0.75 | 63 |
| 14.1 (8) | 780.5(1) | 2.6 (1) | 782.9(1) | 2.6 (1) | 786.3 (1) | 5.0 (1) | 16.0 (1) | 0.72 | 64 |
Despite the larger number of oxygen atoms near the CoII in octahedral sites, its BE is lower than the BE of CoII in tetrahedral sites. This is explained in terms of effective charge on the cobalt atoms and of oxygen polarizability.51 The BE separation between CoII in Oh and Td sites is fixed.36 The area ratio between CoII in Oh and Td in Co 2p1/2 was constrained to be equal to their ratio in Co 2p3/2. Assuming a general formula: (FeyCo1−y)Td[Fe2−yCoy]OhO4 the percentages of area of the CoII components in Oh and Td sites allow the estimation of y, thus the fraction of cobalt in Oh sites. The percentage of iron in octahedral sites is thus calculated with the formula %Fe (Oh) = (y × 0.5 + (1 − y) × 1) × 100.36 The percentage of Fe in octahedral sites calculated following this approach for all the samples ranges between 62 and 64% (Table 3) and it is in very good agreement with the cation distribution formula obtained by 57Fe Mössbauer spectroscopy and for nanostructured CoFe2O4:10,20,21(FeIII0.7CoII0.3)[FeIII1.3CoII0.7]O4.
On the basis of these results it seems that this curve synthesis based on two symmetric peaks for distinguishing between differently coordinated iron and cobalt is effective to estimate the cation distribution.
Due to the presence of the oleic acid shell, which is estimated to be about 1 nm thick in agreement with Shard,52 the signals from cobalt ferrite are attenuated. The sampling depth of an electron travelling in a ferrite nanoparticle can be estimated by the formula 3λferritecosθ where λferrite is the inelastic mean free path determined for inorganic compounds, according to Seah and Dench,53 and θ is the emission angle. For iron Fe 2p, for example, the sampling depth was found to be 4.6 nm for electrons travelling in pure cobalt ferrite. Due to the presence of the 1 nm-thick oleic acid layer, the sampling depth is reduced and it can be estimated by the formula 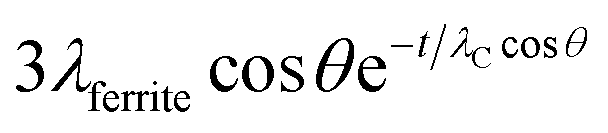 , where t = 1 nm is the thickness of the organic layer and λC is the inelastic mean free path of an electron from Fe 2p travelling in an organic layer, determined according to Seah and Dench.53
, where t = 1 nm is the thickness of the organic layer and λC is the inelastic mean free path of an electron from Fe 2p travelling in an organic layer, determined according to Seah and Dench.53
The sampling depth for an Fe 2p electron starting from the cobalt ferrite nanoparticle and attenuated by the oleic acid layer is estimated to be 4.0 nm.
The agreement between the results obtained by XPS (cation distribution and the Fe/Co ratios) on the surface and those extracted by bulk techniques (57Fe Mössbauer spectroscopy and ICP-OES)42 supports a compositional and structural homogeneity throughout the nanoparticle, also for the larger NPs, for which only 1/3 of their volume is sampled by XPS.
It is known that the binding energy of photoelectron peaks can be strongly influenced by the particle size. In many papers dealing with metal nanoparticles a positive shift of the binding energy values is observed with decreasing particle size.38,54 In other papers a negative shift is observed for metal supported nanoparticles and it might be attributed to the interaction between the metal nanoparticles and the support rather than to an intrinsic particle size effect.37,38 The observed chemical shift, due to both size effects and to metal/support interactions, has been explained taking into account both initial- and final-state effects. The reduced coordination number of surface atoms, which is more significant for small NPs, is an initial-state effect; the electron exchange between the support and the NPs and the screening of the core hole on the photo-emitting atom (relaxation energy) are final-state effects and are influenced by particle size as well.38
Concerning the oxides, a positive shift of Cu 2p3/2 from CuO with decreasing nanoparticles size was observed and it was explained on the basis of the quantum-size effect.39 The shift toward higher BE for smaller NPs was interpreted with the increase of Cu–O bond ionicity. A study on the effect of particle size of ZnO showed an opposite trend in the binding energy of the Zn 2p signal: the smaller the particle the lower the binding energy.40 The authors explained this shift with a decrease of the number of oxygen atoms in nanocrystalline ZnO that reduces the charge transfer from Zn to oxygen and increases the shielding effect of the valence electrons in Zn ions, resulting in the decrease of the Zn 2p binding energy. In this paper only a small positive shift (0.3 eV) of the binding energy of the Fe 2p3/2 peak was observed, when decreasing the particle size from 14.1 to 5.6 nm (Table 1): these results seem to be in agreement with the results obtained on CuO.
If there is a size-dependent effect on the ionicity of the iron-involving bonds (Fe–O) the same effect should be observed on oxygen. For this reason, the effect of the particle size on O 1s and the O KLL signals was also evaluated (Table 2). The binding energy of the photoelectron O 1s signal does not show significant differences with the ferrite particle size, being the chemical shift between the smallest and the largest particles within the experimental uncertainty (0.1 eV). On the contrary, a trend in the kinetic energy of the most intense Auger peak, O KL23L23, is observed. Auger O KLL lines provide information not only on the chemical state of oxygen, but also on the ionicity of the oxygen-involving bonds and on their polarizabilities.46,55 The ΔKEOKLL difference between the kinetic energies of O KL23L23 and O KL1L23 and the ratio between their intensity were calculated (Table 2). Both these parameters are influenced by the ionicity of the M–O bond.46,56 A correlation was found between the ΔKEOKLL value and the charge on the oxygen atom (q) calculated after Pauling's electronegativity scale, following a previous study.55 The lower was q (lower ionicity) the higher was ΔKEOKLL. Similarly, a correlation between the IOKL23L23/IOKL1L23 intensity ratio and the ΔKEOKLL value was found, and it was observed that highly ionic oxides (MgO) showed higher values of the intensity ratios and lower values of ΔKEOKLL.46 They proposed a phenomenological model for oxygen Auger spectra that allows justifying this finding. The authors considered an oxygen ion having a charge q immersed in a dielectric oxide and proposed an equation that relates the ΔKEOKLL shift to the charge on the oxygen atom.46 In this paper ΔKEOKLL decreases with increasing crystallite size (Table 2), thus we can deduce that the charge on oxygen atom is higher and thus the ionicity of the M–O bond. Furthermore, the smallest particles (5.6 ± 0.2 nm) showed the lowest IOKL23L23/IOKL1L23 intensity ratio and the highest ΔKEOKLL value. This result suggests that the ionicity of the metal–oxygen bond decreases with the particle size. Further information can be obtained from the oxygen Auger parameter shifts. It is known that differences in the Auger parameter values (Δα′) reflect differences in the extra-atomic relaxation energies:57
| Δα′ = ΔRea |
In this work Δα′ was calculated referring it to bulk Fe2O3. The values are reported in Table 2. Δα′ increases with the decrease of the particle size (Fig. 4) and it reaches the maximum value for the smallest particles (5.6 nm). This results in lower relaxation energies than those of bulk Fe2O3 (similar results are obtained if Co3O4 is taken into account). Lower values of relaxation energy indicate lower polarizabilities of the chemical environment: for the smallest particles, the polarizability of the electronic cloud towards the hole is less effective than in bulk oxides.
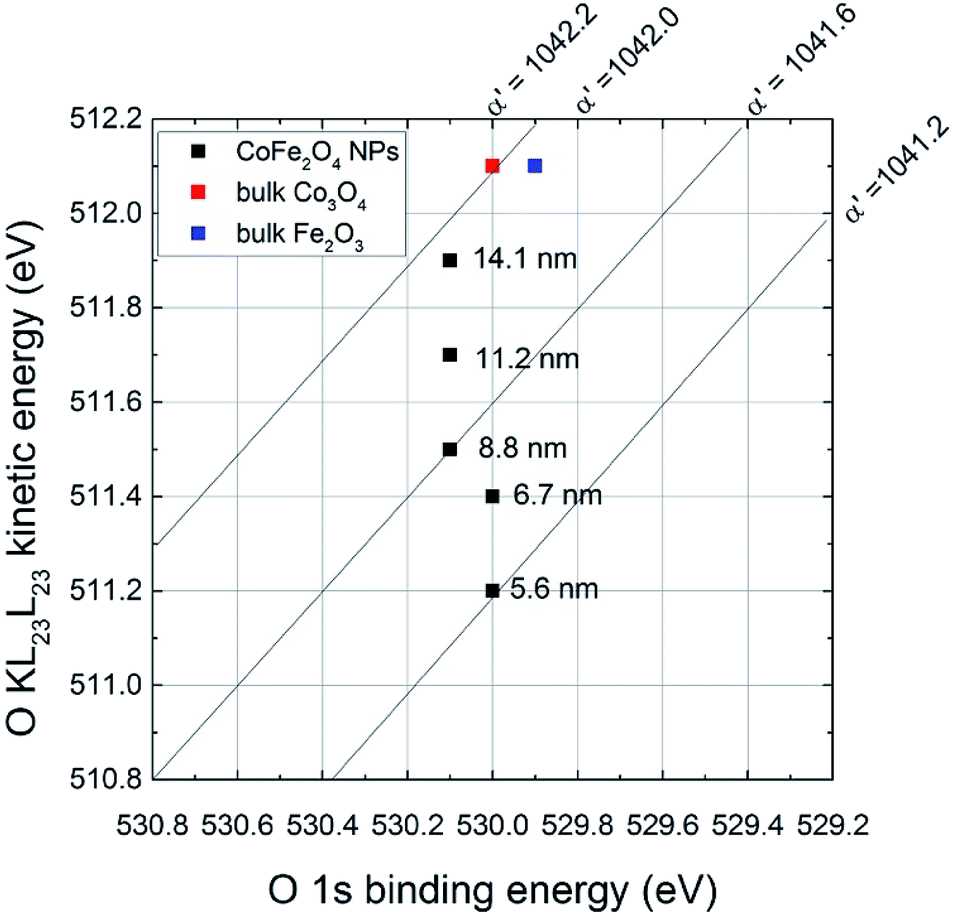 | ||
| Fig. 4 Oxygen chemical state plot. Data from this work are reported (black dots) together with data from literature55 (colored squares). | ||
Conclusions
In this work a surface chemical characterization by X-ray photoelectron spectroscopy and X-ray induced Auger electron spectroscopy of nanostructured cobalt ferrite CoFe2O4 synthesized by solvothermal route is presented. The results show that XPS can provide very useful information about the composition of nanostructured ferrite samples, highlighting the possibility not only to univocally determine the cation chemical states and their ratio but also correlate the peak position with the crystallite size. Furthermore, the cation distribution between the tetrahedral and octahedral sites was determined by fitting the Co 2p3/2 signals: the results are in very good agreement with those obtained by 57Fe Mössbauer spectroscopy. It is proposed that the crystallite size, which likely affects the ionicity of the metal–oxygen bond, influences the kinetic energy of the X-ray induced Auger O KLL signal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. N. D. Spencer for providing access to the X-ray photoelectron spectrometers and the AFM facilities during the ERASMUS stage of Mr Fausto Secci. The authors wish to express their gratitude to Mr Giovanni Cossu for the technical support in the XPS measurements. The financial support of Fondazione di Sardegna and Regione Autonoma della Sardegna Progetti Biennali di Ateneo Annualità 2016, Fondazione Sardegna CUP F72F16003070002 is gratefully acknowledged.References
- L. Wu, A. Mendoza-Garzia, Q. Li and S. Sun, Chem. Rev., 2016, 116, 10473–10512 CrossRef CAS.
- S. Jauhar, J. Kaur, A. Goyal and S. Singhal, RSC Adv., 2016, 6, 97694–97719 RSC.
- E. Manova, B. Kunev, D. Paneva, I. Mitov and L. Petrov, Chem. Mater., 2004, 16, 5689–5696 CrossRef CAS.
- A. Hutlova, D. Niznansky and M. Kurmoo, Adv. Mater., 2003, 15, 1622–1625 CrossRef CAS.
- M. Nasrollahzadeh, M. Bagherzadeh and H. Karimi, J. Colloid Interface Sci., 2016, 465, 271–278 CrossRef CAS PubMed.
- J. Deng, Y. Shao, C. Tan, S. Zhou and X. Hu, J. Hazard. Mater., 2013, 15, 836–844 CrossRef.
- N. V. Long, Y. Yang, T. Teranishi, C. M. Thi, Y. Cao and M. Nogami, J. Nanosci. Nanotechnol., 2015, 15, 10091–10107 CrossRef PubMed.
- C. Tudisco, M. T. Cambria, and G. G. Condorelli, in Biomedical Applications of functionalized nanomaterials. Concepts, development and clinical translation, ed. B. Sarmento and J. das Neves, Elsevier, Amsterdam, Netherlands, 2018, ch. 12, pp. 335–361 Search PubMed.
- S. Zhang, and S. Sun, in Nanotechnology for biomedical imaging and Diagnostics. From nanoparticles design to clinical applications, ed. M. Y. Berezin, John Wiley and Sons, Inc., Hoboken (NJ), 2015, ch. 2, pp. 27–48 Search PubMed.
- V. Mameli, A. Musinu, A. Ardu, G. Ennas, D. Peddis, D. Nižňanský, C. Sangregorio, C. Innocenti, N. T. K Thanh and C. Cannas, Nanoscale, 2016, 8, 10124–10137 RSC.
- L. T. T. M. Karoline, T. N. E. Taveira, H. A. A. Winkler, F. Hatem and P. E. A. Gòmez, J. Colloid Sci. Biotechnol., 2016, 5, 45–54 CrossRef.
- M. A. Mohamed, F. M. El-Badawy, H. S. El-Desoky and M. M. Ghoneim, New J. Chem., 2017, 41, 11138–11147 RSC.
- Y. Yuan, B. Wang, C. Wang, X. Li, J. Huang, H. Zhang, F. Xia and J. Xiao, Sens. Actuators, B, 2017, 239, 462–466 CrossRef CAS.
- B. D. Cullity and C. D. Graham, Introduction to magnetic materials, John Wiley and Sons, Inc., Hoboken (NJ), 2009 Search PubMed.
- C. Cannas, A. Ardu, A. Musinu, L. Suber, G. Ciasca, H. Amenitsch and G. Campi, ACS Nano, 2015, 28, 7277–7286 CrossRef PubMed.
- C. Cannas, A. Musinu, D. Peddis and G. Piccaluga, Chem. Mater., 2006, 18, 3835–3842 CrossRef CAS.
- C. Cannas, A. Musinu, A. Ardu, F. Orrù, D. Peddis, M. Casu, R. Sanna, F. Angius, G. Diaz and G. Piccaluga, Chem. Mater., 2010, 22, 3353–3361 CrossRef CAS.
- V. Mameli, M. S. Angotzi, C. Cara and C. Cannas, Liquid Phase Synthesis of Nanostructured Spinel Ferrites—A Review, J. Nanosci. Nanotechnol., 2019, 19, 4857–4887 CrossRef PubMed.
- A. R. West, Solid State Chemistry and its applications, John Wiley and Sons, Inc., Hoboken (NJ), 2nd edn, 2014 Search PubMed.
- D. Peddis, N. Yaacoub, M. Ferretti, A. Martinelli, G. Piccaluga, A. Musinu, C. Cannas, G. Navarra, J. M. Greneche and D. Fiorani, J. Phys.: Condens. Matter, 2011, 23, 426004–426011 CrossRef CAS PubMed.
- G. Concas, G. Spano, C. Cannas, A. Musinu, D. Peddis and G. Piccaluga, J. Magn. Magn. Mater., 2009, 321, 1893–1897 CrossRef CAS.
- D. Carta, M. F. Casula, A. Falqui, D. Loche, G. Mountjoy, C. Sanregorio and A. Corrias, J. Phys. Chem. C., 2009, 113, 8606–8611 CrossRef CAS.
- D. R. Baer and M. H. Engelhard, J. Electron Spectrosc. Relat. Phenom., 2010, 178–179, 415–432 CrossRef CAS.
- S. Diodati, L. Pandolfo, A. Caneschi, S. Gialanella and S. Gross, Nano Res., 2014, 7, 1027–1042 CrossRef CAS.
- J. Bennet, R. Tholkappiyan, K. Vishista, N. V. Jaya and F. Hamed, Appl. Surf. Sci., 2016, 383, 113–125 CrossRef CAS.
- A. S. Albuquerque, M. V. C. Tolentino, J. D. Ardisson, F. C. C. Moura, R. de Mendonca and W. A. A. Macedo, Ceram. Int., 2012, 38, 2225–2231 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson, R. St and C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- E. Kester and B. Gillot, J. Phys. Chem. Solids, 1998, 59, 1259–1269 CrossRef CAS.
- D. Zhao, X. Wu, H. Guan and E. Han, J. Supercrit. Fluids, 2007, 42, 226–233 CrossRef CAS.
- N. V. Long, Y. Yang, T. Teranishi, C. M. Thi, Y. Cao and M. Nogami, RSC Adv., 2015, 5, 56560–56569 RSC.
- Y. Sun, G. Ji, M. Zheng, X. Chang, S. Li and Y. Zhang, J. Mater. Chem., 2010, 20, 945–952 RSC.
- R. P. Gupta and S. K. Sen, Phys. Rev., 1975, 12, 15–19 CAS.
- M. Fantauzzi, A. Pacella, D. Atzei, A. Gianfagna, G. B. Andreozzi and A. Rossi, Anal. Bioanal. Chem., 2010, 396, 2889–2898 CrossRef CAS PubMed.
- Y. Shen, L. Posavec, S. Bolisetty, F. M. Hilty, G. Nyström, J. Kohlbrecher, M. Hilbe, A. Rossi, J. Baumgartner, M. B. Zimmermann and R. Mezzenga, Nat. Nanotechnol., 2017, 12, 642–647 CrossRef CAS.
- M. A. Willard, Y. Nakamura, D. E. Laughlin and M. E. McHenry, J. Am. Ceram. Soc., 1999, 82, 3342–3346 CrossRef CAS.
- T. Aghavniana, J.-B. Moussy, D. Stanescu, R. Belkhou, N. Jedrecy, H. Magnan, P. Ohresser, M. A. Arrio, P. Sainctavit and A. Barbier, J. Electron Spectrosc. Relat. Phenom., 2015, 202, 16–21 CrossRef.
- A. M. Venezia, A. Rossi, D. Duca, A. Martorana and G. Deganello, Appl. Catal., A, 1995, 125, 113–128 CrossRef CAS.
- J. Radnik, C. Mohr and P. Claus, Phys. Chem. Chem. Phys., 2003, 5, 172–177 RSC.
- K. Borgohain, J. B. Singh, M. V. Rama Rao, T. Shripathi and S. Mahamuni, Phys. Rev. B, 2000, 61, 11093–11096 CrossRef CAS.
- Y. Y. Tay, S. Li, C. Q. Sun and P. Chen, Appl. Phys. Lett., 2006, 88, 173118–173120 CrossRef.
- A. Repko, J. Vejpravová, T. Vacková, D. Zákutná and D. Nižňanský, J. Magn. Magn. Mater., 2015, 390, 142–151 CrossRef CAS.
- M. Sanna Angotzi, A. Musinu, V. Mameli, A. Ardu, C. Cara, D. Nižňanský, H. L. Xin and C. Cannas, ACS Nano, 2017, 11, 7889–7900 CrossRef CAS PubMed.
- M. P. Seah, Surf. Interface Anal., 2001, 31, 721–723 CrossRef CAS.
- M. P. Seah, Surf. Interface Anal., 1993, 20, 243–266 CrossRef CAS.
- D. E. Ramaker, J. S. Murday, G. Moore, M. G. Lagally and J. Houston, Phys. Rev. B, 1979, 19, 5375–5387 CrossRef CAS.
- P. Ascarelli and G. Moretti, Surf. Interface Anal., 1985, 7, 8–12 CrossRef CAS.
- C. D. Wagner, L. H. Gale and R. H. Raymond, Anal. Chem., 1979, 51, 466–482 CrossRef CAS.
- G. C. Smith, in Handbook of Surface and Interface Analysis: Methods for Problem-Solving, ed. J. C. Riviere and S. Myhra, CRC Press, Taylor and Francis Group, Boca Raton, FL, 2nd edn, 2009, ch. 5, pp. 159–208 Search PubMed.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- J. P. Bonnelle, J. Grimbolt and A. D. Huysser, J. Electron Spectrosc. Relat. Phenom., 1975, 7, 151–162 CrossRef CAS.
- A. G. Shard, J. Phys. Chem. C, 2012, 116, 16806–16813 CrossRef CAS.
- M. P. Seah and W. A. Dench, Surf. Interface Anal., 1979, 1, 2–11 CrossRef CAS.
- M. Carrus, M. Fantauzzi, F. Riboni, M. Makosch, A. Rossi, E. Selli and J. van Bokhoven, Appl. Catal., A, 2616, 519, 130–138 CrossRef.
- C. D. Wagner, D. A. Zatko and R. H. Raymond, Anal. Chem., 1980, 52, 1445–1451 CrossRef CAS.
- R. Weiβmann, Solid State Commun., 1979, 31, 347–349 CrossRef.
- C. D. Wagner, Faraday Discuss. Chem. Soc., 1975, 60, 291–300 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03488a |
| This journal is © The Royal Society of Chemistry 2019 |