
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled degradation of low-fouling poly(oligo(ethylene glycol)methyl ether methacrylate) hydrogels†
Muhammad M. Shoaib a,
Vincent Huynha,
Yousuf Shada,
Rashik Ahmedb,
Alexander H. Jesmera,
Giuseppe Melaciniab and
Ryan G. Wylie*ac
a,
Vincent Huynha,
Yousuf Shada,
Rashik Ahmedb,
Alexander H. Jesmera,
Giuseppe Melaciniab and
Ryan G. Wylie*ac
aDepartment of Chemistry and Chemical Biology, McMaster University, Hamilton, Ontario L8S 4M1, Canada. E-mail: wylier@mcmaster.ca
bDepartment of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario L8S 4M1, Canada
cSchool of Biomedical Engineering, McMaster University, Hamilton, Ontario L8S 4M1, Canada
First published on 17th June 2019
Abstract
Degradable low-fouling hydrogels are ideal vehicles for drug and cell delivery. For each application, hydrogel degradation rate must be re-optimized for maximum therapeutic benefit. We developed a method to rapidly and predictably tune degradation rates of low-fouling poly(oligo(ethylene glycol)methyl ether methacrylate) (P(EG)xMA) hydrogels by modifying two interdependent variables: (1) base-catalysed crosslink degradation kinetics, dependent on crosslinker electronics (electron withdrawing groups (EWGs)); and, (2) polymer hydration, dependent on the molecular weight (MW) of poly(ethylene glycol) (PEG) pendant groups. By controlling PEG MW and EWG strength, P(EG)xMA hydrogels were tuned to degrade over 6 to 52 d. A 6-member P(EG)xMA copolymer library yielded slow and fast degrading low-fouling hydrogels suitable for short- and long-term delivery applications. The degradation mechanism was also applied to RGD-functionalized poly(carboxybetaine methacrylamide) (PCBMAA) hydrogels to achieve slow (∼50 d) and fast (∼13 d) degrading low-fouling, bioactive hydrogels.
1. Introduction
Degradable low-fouling hydrogels are being developed as implantable vehicles for drug and cell delivery to decrease the incidence rate of adverse events (e.g. foreign body response (FBR)) by minimizing non-specific protein adsorption and cell binding.1,2 The FBR cascade impedes drug release or cell egress from hydrogels by surrounding implants in dense fibrous capsules.3 Due to their strong hydration shells, non-specific protein adhesion to low-fouling polymers such as poly(oligo(ethylene glycol)xmethyl ether methacrylate) (P(EG)xMA) and poly(carboxybetaine methacrylamide) (PCBMAA) is energetically unfavorable.4 P(EG)4–5MA coated surfaces resist protein fouling5 and platelet binding.6 Moreover, carboxybetaine coated surfaces have been shown to prevent non-specific protein adsorption in serum7 and resist the FBR for up to 3 months in vivo.8The degradation of low-fouling hydrogels must be tuned to match requirements for short- and long-term delivery applications. Hydrogel drug delivery applications, such as cancer therapies,9–11 wound healing,12 pain management,13,14 and retinal degenerative disease treatments,15,16 often require drug release profiles that span a wide-distribution of timeframes, ranging from as little as a days to several weeks. We therefore require low-fouling hydrogels with highly tunable degradation timeframes. Predictable degradation rates are particularly important for long-term drug delivery (∼4 weeks) wherein uncontrolled hydrogel degradation can limit efficacy by increasing the initial burst release.17 Thus far, degradation mechanisms for low-fouling gels have focused on endogenous triggers (e.g. reduction of disulfide bonds,18–20 enzyme cleavage sites21), or hydrolytic bonds (e.g. esters, hydrazones).22 In situ crosslinking P(EG)xMA copolymers with aldehyde and hydrazide repeats yield hydrogels that crosslink through reversible hydrazone bonds with degradation rates proportional to pH and copolymer molecular weight (MW).23,24 Carboxybetaine copolymers with zwitterionic thiol repeats have been developed to achieve biodegradable hydrogels in the presence of reducing agents such as glutathione.19,20,25 A detailed description of hydrogel degradation mechanisms is provided in the referenced review.26
Endogenous triggered degradation is dependent on dynamic biological environments, which may result in unpredictable degradation rates, and current methods to tune hydrolysis rates require extensive synthetic modifications. To improve therapies requiring low-fouling hydrogels, there is a great need to develop a versatile method to easily tune degradation rates that are independent of endogenous triggers and don't require additional synthetic steps. To achieve an accessible library of low-fouling hydrogels with varied degradation profiles, hydrogels should be formed by simply mixing a limited number of pre-defined polymers for in situ crosslinking. The combination of low-fouling P(EG)xMA hydrogels of varying hydration levels with different crosslinkers for irreversible base-catalyzed degradation27,28 is expected to provide a method to rapidly tune degradation over clinically relevant timeframes. Irreversible base-catalyzed crosslink degradation27,28 is solely dependent on the pH of the implantation site, a known value, and not reliant on dynamic endogenous triggers. Furthermore, hydrogels composed of P(EG)xMA with different PEG pendant group molecular weights (MWs) will exhibit different hydration levels to further tune base-catalyzed crosslinker degradation rates.29 By simply mixing pre-defined polymers, a library of P(EG)xMA copolymers is expected to yield low-fouling hydrogels with highly tunable degradation kinetics.
2. Materials and methods
2.1 Materials
Triethylene glycol methyl ether methacrylate 93% ((EG)3MA), poly(ethylene glycol)methyl ether methacrylate number average molecular weight (Mn) 300 ((EG)4–5MA), poly(ethylene glycol)methyl ether methacrylate average Mn 500 ((EG)8–9MA), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CTP), 4,4′-azobis(4-cyanovaleric acid) (V-501), N-[3-(dimethylamino)propyl]-methacrylamide (DMAPMA), t-butyl bromoacetate, trifluoroacetic acid, 6-chloro-1-hexanol, sodium azide, trichloroisocyanuric acid, TEMPO, sodium bicarbonate, 1.6 M n-butyllithium in hexane, 4-(methylsulfonyl)toluene, pyridine, triphosgene, N-hydroxysuccinimide (NHS), fluorescamine, triethylamine, picrylsulfonic acid 5% (w/v), sodium dodecyl sulfate (SDS), bovine serum albumin (BSA), and fluorescein sodium salt were purchased from Sigma-Aldrich (Oakville, ON). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) was purchased from Chem-Impex International (Wood Dale, IL). 4-Chlorophenyl methyl sulfone was purchased from Alfa Aesar (Ward Hill, MA). N-(3-Aminopropyl)methacrylamide hydrochloride, >98% (APMA) was purchased from Polysciences, Inc. (Warrington, PA). 1,2-Bis(2-(4,5-dihydro-1H-imidazol-2-yl)propan-2-yl)diazene dihydrochloride (VA-044) was purchased from Toronto Research Chemicals (Toronto, ON). CGRGDS >95% was purchased from GenScript (Piscataway, NJ). Calcein AM fluorescent dye was purchased from Corning, New York, USA. PrestoBlue™ cell viability reagent, Hoechst 33342, HyClone™ calf bovine serum (CBS) and DMEM/F12 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were purchased from Thermo Fisher Scientific (Whitby, ON). Methylcellulose (MC; METOLOSE® SM-4000) was purchased from Shin-Etsu Corp (Tokyo, Japan). Solvents were reagent grade and obtained from Caledon Laboratories (Georgetown, ON) and Thermo Fisher Scientific (Whitby, ON). Dibenzylcyclooctyne-NHS ester (DBCO-NHS) was gifted by Dr Alex Adronov at McMaster University (Hamilton, ON).
:1 were purchased from Thermo Fisher Scientific (Whitby, ON). Methylcellulose (MC; METOLOSE® SM-4000) was purchased from Shin-Etsu Corp (Tokyo, Japan). Solvents were reagent grade and obtained from Caledon Laboratories (Georgetown, ON) and Thermo Fisher Scientific (Whitby, ON). Dibenzylcyclooctyne-NHS ester (DBCO-NHS) was gifted by Dr Alex Adronov at McMaster University (Hamilton, ON).
2.2 Synthesis and characterization of P(EG)xMA copolymers
:1 water:dioxane (7 mL). For P(EG)4–5MA–APMA, (EG)4–5MA (1 g, 3.3 mmol), APMA (48 mg, 0.27 μmol), CTP (5.8 mg, 21 μmol) and VA-044 (1.35 mg, 4.2 μmol) were dissolved in 2:1 water:dioxane (2.5 mL). For P(EG)8–9MA–APMA, (EG)8–9MA (1 g, 2 mmol) APMA (29 mg, 0.16 mmol), CTP (5.8 mg, 21 μmol) and VA-044 (1.35 mg, 4.2 μmol) were dissolved in 2:1 water:dioxane (1.1 mL). All reaction solutions were freeze–pump–thawed with a nitrogen backfill (3 times), and acidified to pH ∼ 3.5 using 0.1 M HCl prior to polymerization at 40 °C for ∼16 h. Polymers were purified by dialysis (MWCO 12–14k) against pH 3 water and lyophilized to yield a pink paste (P(EG)3MA–APMA: 1.325; P(EG)4–5MA–APMA: 0.904; P(EG)8–9MA–APMA: 0.948 g).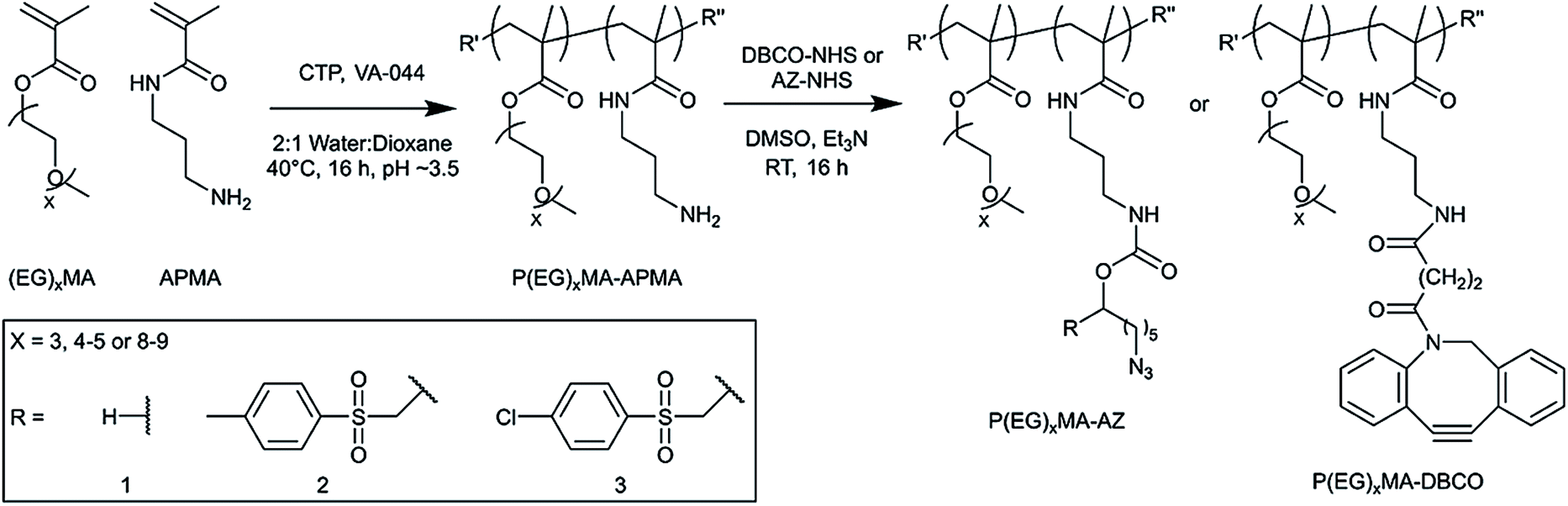 | ||
| Scheme 1 Synthesis of P(EG)xMA–AZ and P(EG)xMA–DBCO copolymers for in situ crosslinking. P(EG)xMA–APMA copolymers were synthesized by RAFT polymerization for subsequent derivatization with NHS-AZ derivatives or NHS-DBCO. For each PEG MW, three different P(EG)xMA–AZ copolymers were synthesized with a non-degradable crosslinker (R1 = H), and two degradable crosslinkers with EWGs (R2 = 4-methylpheyl sulfone, and R3 = 4-chlorophenyl sulfone). | ||
2.3 Gelation and characterization of P(EG)xMA hydrogels
Gelation time was measured via gravitational flow analysis. Hydrogels (100 μL) were made with equal volumes and concentrations (50 mg mL−1) of azide and DBCO copolymers. Vials were tilted periodically until flow was no longer observed. The number of crosslinks was determined by tracking the absorbance of 100 μL hydrogels (5 wt%) at 309 nm over time to measure consumption of DBCO over the first 24 h; gels were formed in a 96-well polypropylene plate. After 24 h, a 10-fold excess of sodium azide was added to react all remaining DBCO groups to determine background hydrogel absorbance. Hydrogel (5 wt%) cloud points were measured using the same procedure as for copolymer LCSTs.Hydrogel swelling was determined by incubating 100 μL hydrogels (5 wt%) in pH 7.4 PBS at 37 °C for 22 h. The initial hydrogel wet weight was recorded. After selected time intervals, buffer was removed from hydrogel surfaces and their wet weight was recorded.
2.4 1H NMR kinetic analysis of P(EG)xMA–AZ–R3 copolymer degradation
1H NMR spectra were recorded at 37 °C with 128 scans, 32 K complex points and spectral widths of 20 ppm on a Bruker 700 MHz spectrometer equipped with a TCI cryoprobe. All spectra were processed and analyzed in TopSpin 3.2.1. The base-catalyzed degradation of the polymers was assessed through the time-dependent increase in the 1H-NMR signal intensity of the nascent hydrolyzed product centred around ∼7.8 ppm. For the purpose of comparison, the enhancement in the hydrolyzed product intensity was measured relative to the final intensity of the P(EG)8–9MA–AZ–R3 hydrolyzed product at the end of the 3 h period. The resulting experimental points were used to create the kinetic profiles shown in Fig. 3D. The initial hydrolysis rate for P(EG)8–9MA–AZ–R3 was determined by linear regression to compute the slope over the first 1 h period. To determine the initial hydrolysis rates of P(EG)3MA–AZ–R3 and P(EG)4–5MA–AZ–R3 copolymers, a linear regression was used to compute the slopes over the first 1 h period post lag phase (0.6 h); lag phase is defined as the initial period with no detectable hydrolysis.2.5 Protein adsorption and cell adhesion on P(EG)xMA hydrogels
3. Results and discussion
We developed low-fouling P(EG)xMA hydrogels with tunable degradation rates by controlling two interdependent variables: (1) base-catalysed crosslink degradation kinetics, dependent on the strength of the incorporated electron withdrawing group (EWG; Fig. 1A); and, (2) polymer hydration, dependent on the MW of PEG pendant groups (Fig. 1B). By combining P(EG)xMAs with different PEG pendant groups (x = 3, 4–5, and 8–9) with two different crosslinkers (EWG = 4-methylphenyl sulfone or 4-chlorophenyl sulfone), we developed a 6-member copolymer library to rapidly tune P(EG)xMA hydrogel degradation over 6, 13, 31, or 52 d, yielding gels suitable for short- and long-term applications (Fig. 1C). We also demonstrated the degradation of RGD functionalized low-fouling PCBMAA hydrogels over 13 and 52 d to produce bioactive, low-fouling gels for cell delivery applications.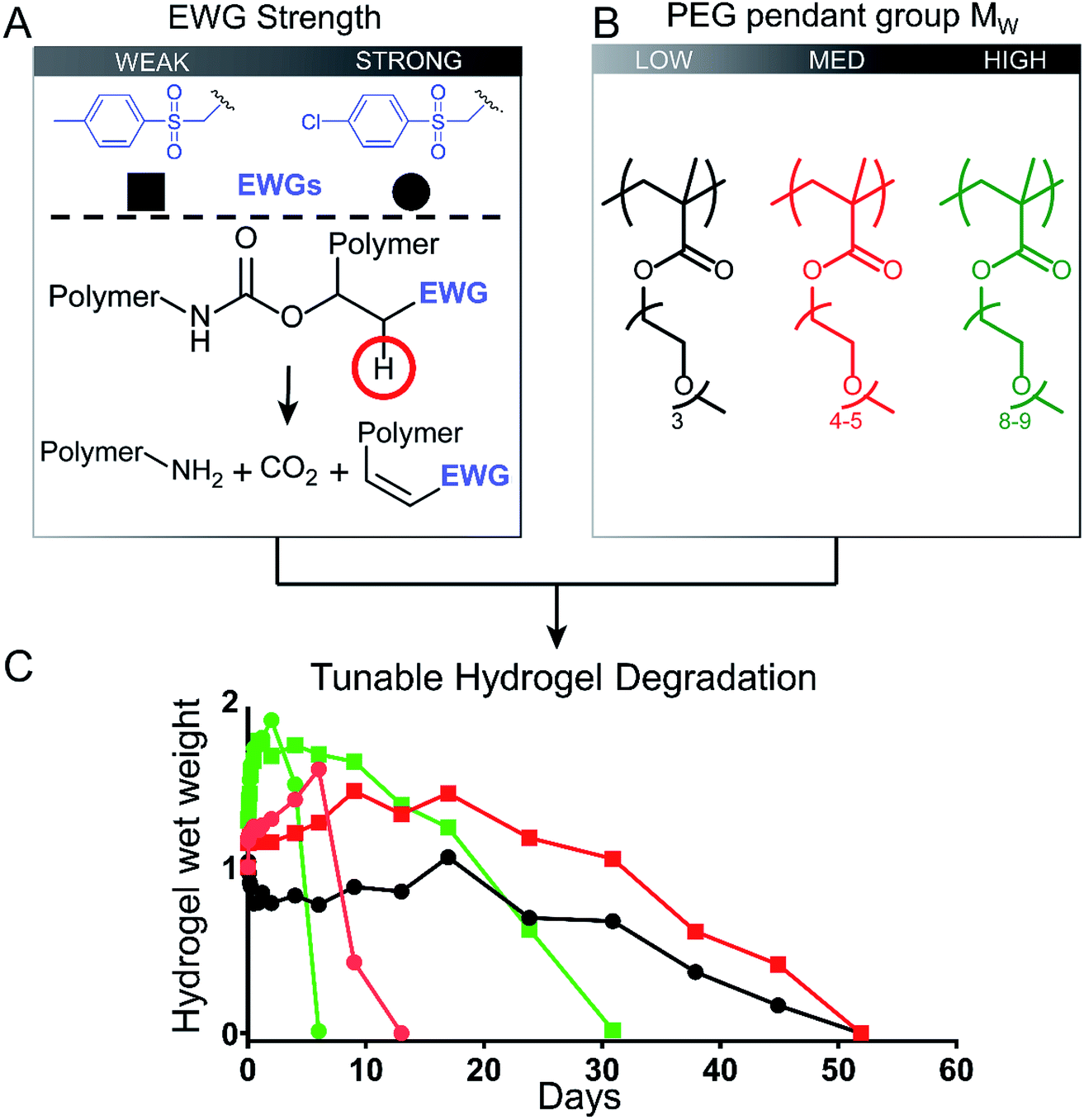 | ||
| Fig. 1 Schematic highlighting the short- and long-term degradation of low-fouling hydrogels from a library of P(EG)xMA copolymers with two different crosslinkers. (A) Irreversible base-catalyzed hydrolysis of carbamate crosslinks tuned with two different EWGs (4-methylphenyl sulfone and 4-chlorophenyl sulfone). Deprotonation site is highlighted by a red circle. (B) The MW of PEG pendant groups in P(EG)xMA influenced hydrogel degradation rates. (C) Tunable hydrogel degradation from 6 to 52 days was achieved by creating a library consisting of 3 P(EG)xMA copolymers and two different EWGs. Lower PEG MW (less hydrated) P(EG)xMAs and weaker EWGs produced slower degradation rates. | ||
3.1 Synthesis and chemical characterization of P(EG)xMA copolymers
Because P(EG)xMA is synthesized by controlled radical polymerization, monomers with reactive functional groups can be polymerized into P(EG)xMA copolymers for in situ crosslinking30 and controlled degradation.28 We first synthesized random P(EG)xMA copolymers with N-(3-aminopropyl)methacrylamide (APMA) using reversible addition–fragmentation chain-transfer (RAFT) polymerization to yield P(EG)xMA–APMA with MWs and dispersities (Đ) between 30–40 kDa and 1.06 to 1.35, respectively (Scheme 1; Table 1). P(EG)xMA–APMA copolymers with different PEG pendant groups (x = 3, 4–5, and 8–9), were prepared to determine the influence of polymer hydration on hydrogel degradation.The (EG)xMA to APMA composition in copolymers was optimized to ensure similar crosslink densities (crosslinks per g of polymer) in all P(EG)xMA hydrogels, which allowed for the comparison of polymer hydration on degradation rates. To this end, P(EG)3MA–APMA, P(EG)4–5MA–APMA, and P(EG)8–9MA–APMA were synthesized with 2, 5 and 10 APMA mol%, respectively, as confirmed by 1H NMR upon comparing integrations of methylene peaks in (EG)xMA and APMA (Table 1; Fig. S1–S3†). Due to sterics associated with PEG pendant groups, greater crosslinker mole fractions, and thus APMA, were required for P(EG)xMAs with higher PEG MWs to standardize crosslink density.
P(EG)xMA–APMAs were further functionalized with NHS-AZ molecules resulting in copolymers that contained a non-degradable (R1; H, no EWG), slow-degrading (R2; 4-methylphenyl sulfone), or fast-degrading (R3; 4-chlorophenyl sulfone) carbamate bond for base-catalysed crosslink degradation. Carbamate bond half-lives have been previously reported to vary between 14 h to 437 d by substituting an adjacent EWGs for base-catalyzed degradation.27 P(EG)xMA–APMAs were also modified with NHS-DBCO to yield P(EG)xMA–DBCO for in situ crosslinking with P(EG)xMA–AZ copolymers. All APMA amines were fully reacted with NHS-AZ or NHS-DBCO, as confirmed by 1H NMR (Fig. S4–S15†) and an amine quantification assay (fluorescamine).
To compare polymer hydration through solvation, we studied the solubility of P(EG)3MA and P(EG)4–5MA copolymers as a function of temperature and determined LCSTs (Fig. 2, S21A and B†). A homopolymer (HP) of P(EG)3MA had an LCST of 45 °C (25 g L−1), defined as the onset of cloudiness. In comparison to the HP, all P(EG)3MA–AZ and P(EG)3MA–DBCO copolymers demonstrated lower LCSTs near physiological temperature (36–37 °C) due to increased hydrophobic content. The HP of P(EG)4–5MA and P(EG)4–5MA–AZ copolymers had LCSTs of 62 and 52–54 °C, respectively. Interestingly, P(EG)4–5MA–DBCO's LCST was 2 °C lower than the P(EG)4–5MA HP and 6–8 °C higher than P(EG)4–5MA–AZs. Therefore, the increased hydrophobic content from DBCO did not substantially influence P(EG)4–5MA's temperature-dependent solubility, indicating a DBCO composition greater than 5 mol% is required to influence P(EG)4–5MA's LCST. Due to larger PEG side chains, P(EG)8–9MA copolymers did not exhibit an aqueous LCST (Fig. S21C†) when heated to 65 °C.31 Copolymer LCSTs will also predict hydrogel fouling properties as copolymers with LCSTs near or above body temperature will maintain their low-fouling properties; hydrogels are fouling at temperatures above their LCSTs due to increased hydrophobic interactions with proteins.32
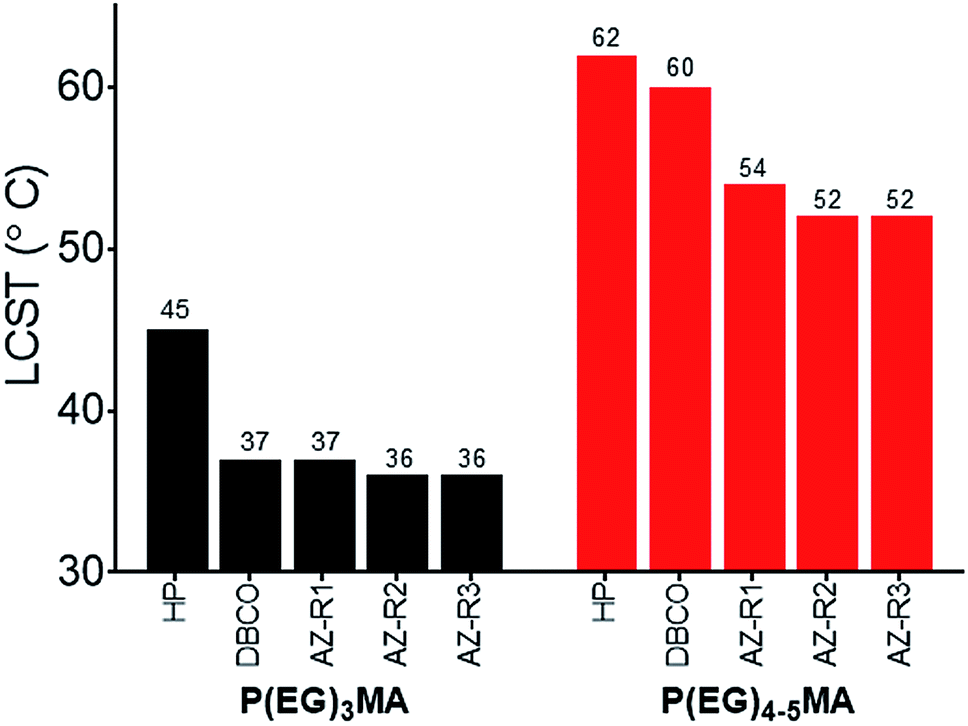 | ||
| Fig. 2 P(EG)3MA and P(EG)4–5MA copolymer LCSTs. LCSTs (cloud points) of P(EG)3MA and P(EG)4–5MA homopolymers (HP) and AZ/DBCO copolymers (25 g L−1 in PBS) were determined by measuring solution turbidity at 600 nm as a function of temperature. LCSTs were defined as the lowest temperature that increased turbidity. No LCST was observed for P(EG)8–9MA copolymers. | ||
3.2 1H NMR kinetic analysis of P(EG)xMA–AZ carbamate degradation
To probe the influence of PEG MW on carbamate degradation kinetics, 1H NMR spectra of P(EG)xMA–AZ copolymers with the 4-chlorophenyl sulfone EWG (P(EG)xMA–AZ–R3) were acquired in real time for 3 h in borate buffer at pH 9.3, which increased reactions rates for efficient NMR analysis. Degradation was monitored by peak intensity changes of aromatic protons in 4-chlorophenyl sulfone, which is cleaved from the polymer upon hydrolysis. Over the course of the 3 h experiment, time-dependent losses in signal intensity were observed for polymer bound 4-chlorophenyl sulfone (aromatic protons, 7.8–8.1 ppm) and intensity gains for cleaved 4-chlorophenyl sulfone (aromatic protons, dashed box, sharp doublet, ≤7.8 ppm; Fig. 3A–C).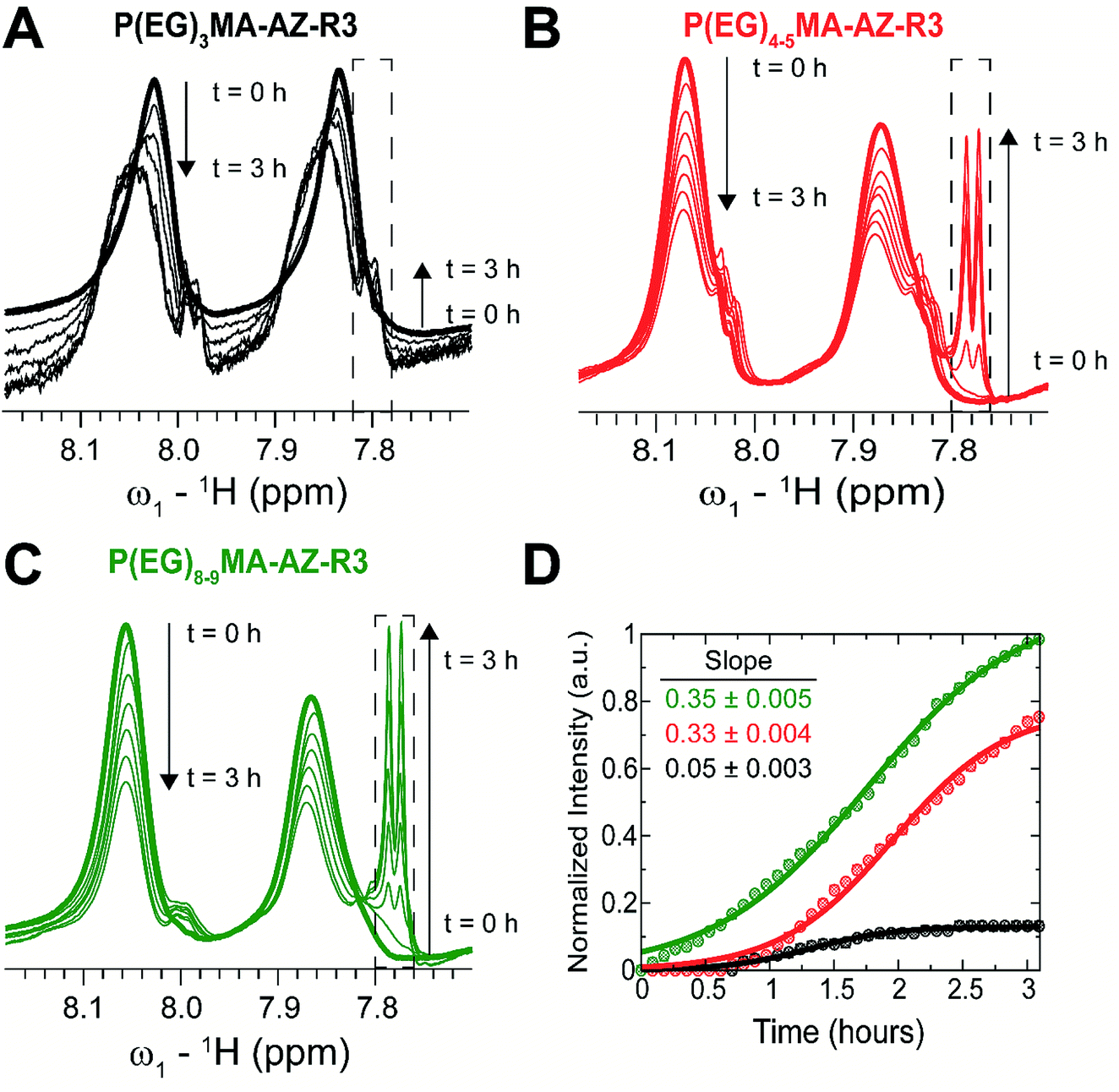 | ||
| Fig. 3 1H NMR kinetic analysis of P(EG)xMA–AZ–R3 copolymer crosslinker degradation at pH 9.3 and 37 °C. Upon base-catalysed degradation, 4-chlorophenyl sulfone is cleaved from the polymer resulting in the loss in signal intensity between 7.8–8.1 (two broad peaks) and appearance of a sharp doublet (≤7.8 ppm, dashed box). The rate of (A) P(EG)3MA–AZ–R3, (B) P(EG)4–5MA–AZ–R3, and (C) P(EG)8–9MA–AZ–R3 degradation was followed over 3 h. (D) Degradation profiles as probed by changes in intensity of 4-chlorophenyl sulfone's aromatic protons. All intensities were normalized to the intensity of the P(EG)8–9MA decomposition signature peak (dashed box) at the end of the 3 h period. Slopes were determined by linear regression of the normalized intensities; slopes were computed from 0 to 1 h for P(EG)8–9–AZ–R3 and from ∼0.6 to 1.6 h for P(EG)3MA–AZ–R3 and P(EG)4–5MA–AZ–R3. | ||
The degradation rate of P(EG)xMA–AZ–R3 copolymers were dependent on the PEG MW. P(EG)8–9MA–AZ–R3 degraded immediately due to its higher solubility (LCST > 65 °C; Fig. 3D). In contrast, P(EG)4–5MA–AZ–R3 exhibited a lag phase (∼0.6 h) with no detectable degradation followed by a degradation rate similar to P(EG)8–9MA–AZ–R3 (slopes of 0.33 and 0.35, respectively; Fig. 3D). The biphasic degradation profile of P(EG)4–5MA–AZ–R3 is a function of polymer solubility over time; the cleavage of hydrophobic 4-chlorophenyl sulfone increases P(EG)4–5MA–AZ–R3 solubility. Only minor P(EG)3MA–AZ–R3 degradation was observed over the 3 h time period (Fig. 3A and D) because of its lower solubility at 37 °C, which is supported by LCST data (Fig. 2 and S22A†). From P(EG)4–5MA–AZ–R3 data, we can conclude that PEG MW mainly influences initial degradation rates.
3.3 Cytotoxicity of P(EG)xMA copolymers
All P(EG)xMA copolymers were non-cytotoxic according to NIH 3T3 fibroblast cell viability assays (Fig. 4). Cells were cultured in DMEM/F12 media with 10% calf bovine serum (CBS) in the presence of a single copolymer (1 mg mL−1). After 24 h, cell viability was determined using the PrestoBlue assay and compared to a positive control, cells cultured without polymer. All copolymer conditions were indistinguishable from the positive control, indicating copolymers are non-cytotoxic and suitable for hydrogel fabrication.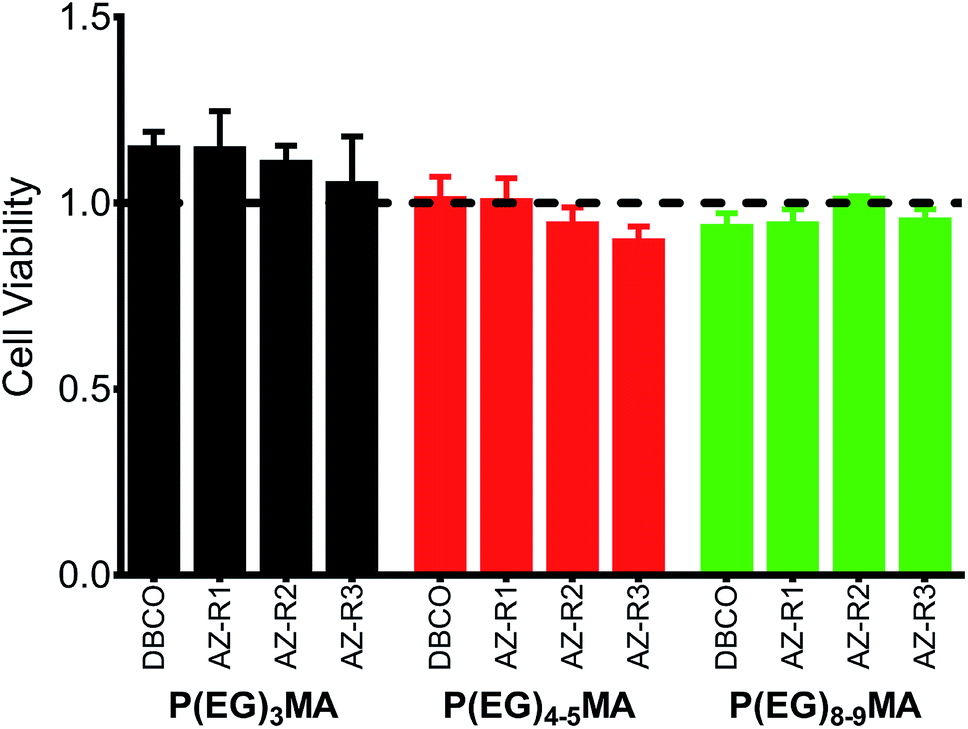 | ||
| Fig. 4 P(EG)xMA copolymers are non-cytotoxic. NIH 3T3 fibroblasts were cultured in the presence of 1 mg mL−1 of each copolymer for 24 h. Cell viability was assessed by the PrestoBlue assay and normalized to cells cultured in the absence of polymer (positive control, dashed line). No significant difference (p < 0.05) was observed between all polymer conditions and the positive control (mean + standard deviation, n = 3). | ||
3.4 Hydrogel gelation and characterization
Non-degradable P(EG)xMA–AZ copolymers were mixed with corresponding P(EG)xMA–DBCO copolymers for in situ gelation and characterization of hydrogel crosslink density, swelling and protein fouling. Hydrogels were prepared by mixing equal volumes of corresponding azide and DBCO copolymer solutions (5 wt% in PBS) for strain-promoted alkyne–azide cycloaddition (SPAAC) crosslinking, and gelation times were determined by gravitational flow analysis (Fig. 5A); gelation time increased with larger PEGs, which limits crosslinking rates due to sterics.33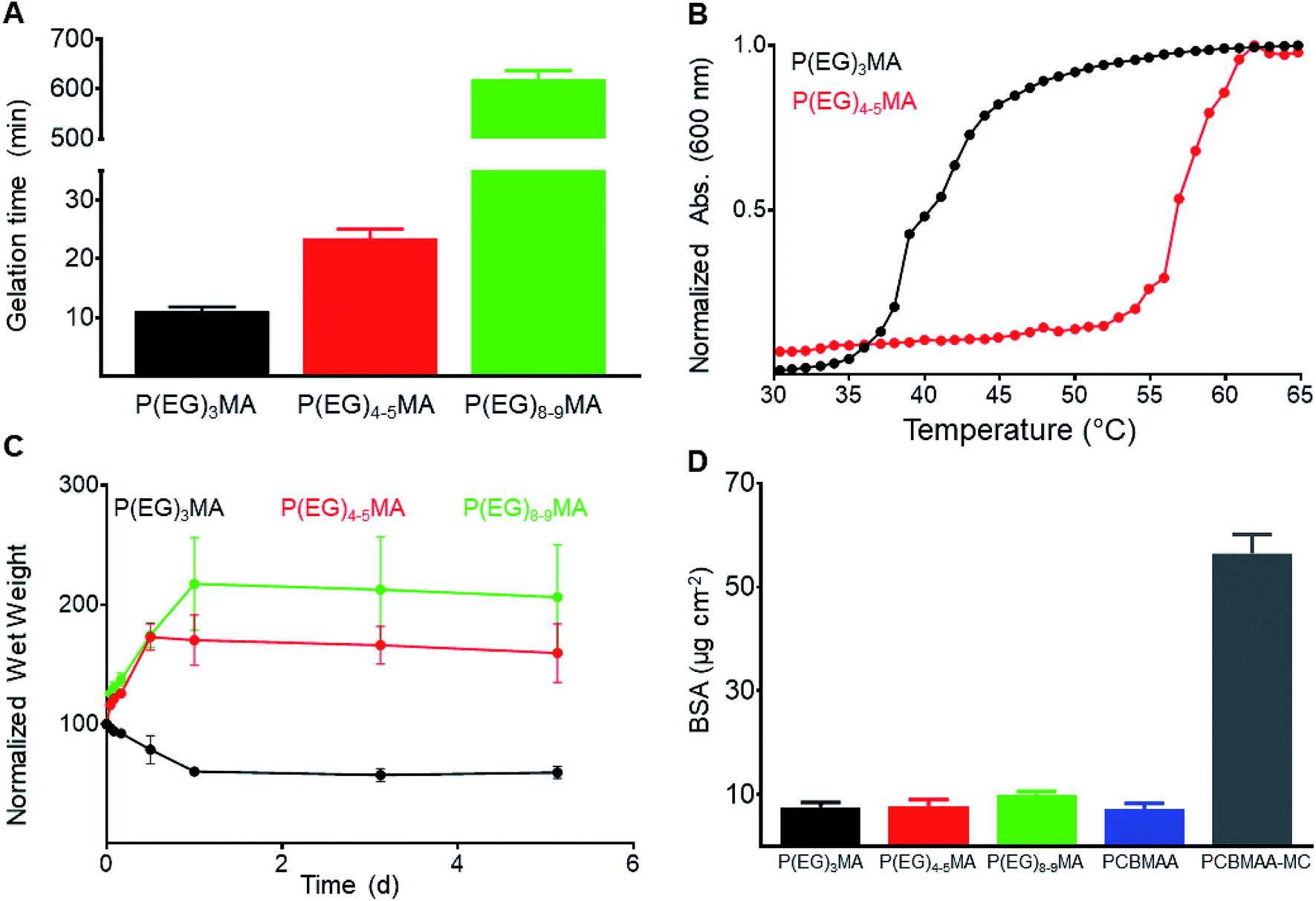 | ||
| Fig. 5 Characterization of non-degradable P(EG)xMA hydrogels: gelation time, hydration and protein fouling. (A) Hydrogel (5 wt%) gelation time, determined by gravitational flow analysis (mean + standard deviation, n = 6). (B) Cloud point of P(EG)3MA and P(EG)4–5MA hydrogels (5 wt% in PBS) defined as the onset of turbidity (600 nm). No cloud point was observed for P(EG)8–9MA gels. (C) Equilibrium swelling of non-degradable gels. After overnight gelation, hydrogels (5 wt%) were submerged in PBS and their wet weight was determined at specified time points (mean ± standard deviation, n = 3). (D) Adsorbed fluorescent BSA (μg cm−2) on hydrogel surfaces compared to low-fouling PCBMAA gels (6 mol% AZ/DBCO) and fouling PCBMAA–MC gels (mean + standard deviation, n = 3). | ||
To study the influence of polymer hydration (PEG MW) on hydrogel degradation, all P(EG)xMA hydrogels (P(EG)3MA, P(EG)4–5MA, and P(EG)8–9MA) required similar crosslink densities. The density of hydrogel crosslinks (μmol per g of polymer) was determined by quantifying DBCO consumption after SPAAC crosslinking (Table 2); unreacted DBCO absorbs light at 309 nm with an extinction coefficient of 12000 M−1 cm−1.34 To achieve P(EG)xMA hydrogels with similar crosslink densities, copolymer precursors with different AZ and DBCO mole fractions were required due to unique crosslinking kinetics. P(EG)3MA, P(EG)4–5MA, and P(EG)8–9MA required crosslinker mole fractions of 2, 5, and 10 mol%, respectively, to yield hydrogels with similar crosslink densities (47–52 μmol of crosslinks per g of polymer). Crosslink densities were determined by measuring unreacted DBCO concentrations (absorbance at 309 nm) after an overnight incubation to ensure maximum crosslinking. To calculate hydrogel background absorbance, all gels were subsequently exposed to excess sodium azide to consume remaining DBCOs.35
To compare hydration of P(EG)xMA hydrogels, hydrogel cloud points and swelling ratios were determined. The cloud points of non-degradable (R1) and degradable (R2, Fig. 5B, S23A and B†) P(EG)3MA and P(EG)4–5MA hydrogels were similar to their corresponding P(EG)xMA–AZ and P(EG)xMA–DBCO copolymers; P(EG)3MA and P(EG)4–5MA hydrogels had cloud points of 36 and 54 °C, respectively. As expected from P(EG)8–9MA copolymer LCSTs, P(EG)8–9MA gels did not exhibit a cloud point (Fig. S23C†). In agreement with hydrogel cloud points, the equilibrium swelling ratio of P(EG)xMA hydrogels increased with greater PEG MW (Fig. 5C). P(EG)3MA gels incubated at 37 °C shrunk by expelling ∼40% of their initial water content, due to the promotion of polymer–polymer interactions at temperatures near its cloud point (36 °C). P(EG)4–5MA (cloud point 54 °C) and P(EG)8–9MA hydrogels swelled to ∼160 and 200%, respectively, of their initial wet weight.
To ensure P(EG)xMA retained their low-fouling properties, we quantified the non-specific adsorption of fluorescent BSA to hydrogel surfaces. Non-degradable hydrogels were incubated in 0.5 mg mL−1 fluorescent BSA solutions for 2 h and rinsed with PBS. Adsorbed fluorescent BSA was extracted from the hydrogels with a sodium dodecyl sulfate (SDS) solution for quantification.22 All gels bound between 5 and 10 μg cm−2 of fluorescent BSA, similar to other low-fouling hydrogels22 (Fig. 5D). PCBMAA hydrogels with 6 mol% AZ/DBCO content and PCBMAA gels with 0.5 wt% methylcellulose (PCBMAA–MC) were included as controls; PCBMAA gels remain non-fouling with AZ/DBCO content below 10 mol%36 and PCBMAA–MC gels non-specifically absorbed BSA due to MC hydrophobic interactions. PCBMAA and PCBMAA–MC non-specifically bound ∼5 and 60 μg cm−2 of BSA, respectively. Therefore, all P(EG)xMA gels remained non-fouling towards BSA.
3.5 Tunable degradation of P(EG)xMA hydrogels
By developing a library of P(EG)xMA–AZ and P(EG)xMA–DBCO copolymers with R2 and R3 crosslinkers, we were able to easily achieve hydrogel degradation over 6 to 52 d (Fig. 6). Degradation rates were controlled by polymer hydration (P(EG)3MA, P(EG)4–5MA, and P(EG)8–9MA) and the acidity of the crosslinker's β-hydrogen by exchanging EWG groups (4-methylphenyl sulfone or 4-chlorophenyl sulfone). Because PEG MW influenced degradation rates (Fig. 3), each crosslinker (R2 or R3) yielded three different hydrogel degradation profiles. Hydrogels with higher PEG MWs degraded faster due to greater polymer solvation and initial degradation rates, as demonstrated by hydrogel swelling (Fig. 5C) and 1H NMR kinetic studies (Fig. 3). For example, P(EG)3MA–R3, P(EG)4–5MA–R3 and P(EG)8–9MA–R3 gels degraded over 52, 13 and 6 d, respectively (Fig. 6). As expected, P(EG)xMA hydrogels degraded faster with crosslinks containing the stronger EWG (4-chlorophenyl sulfone, R3).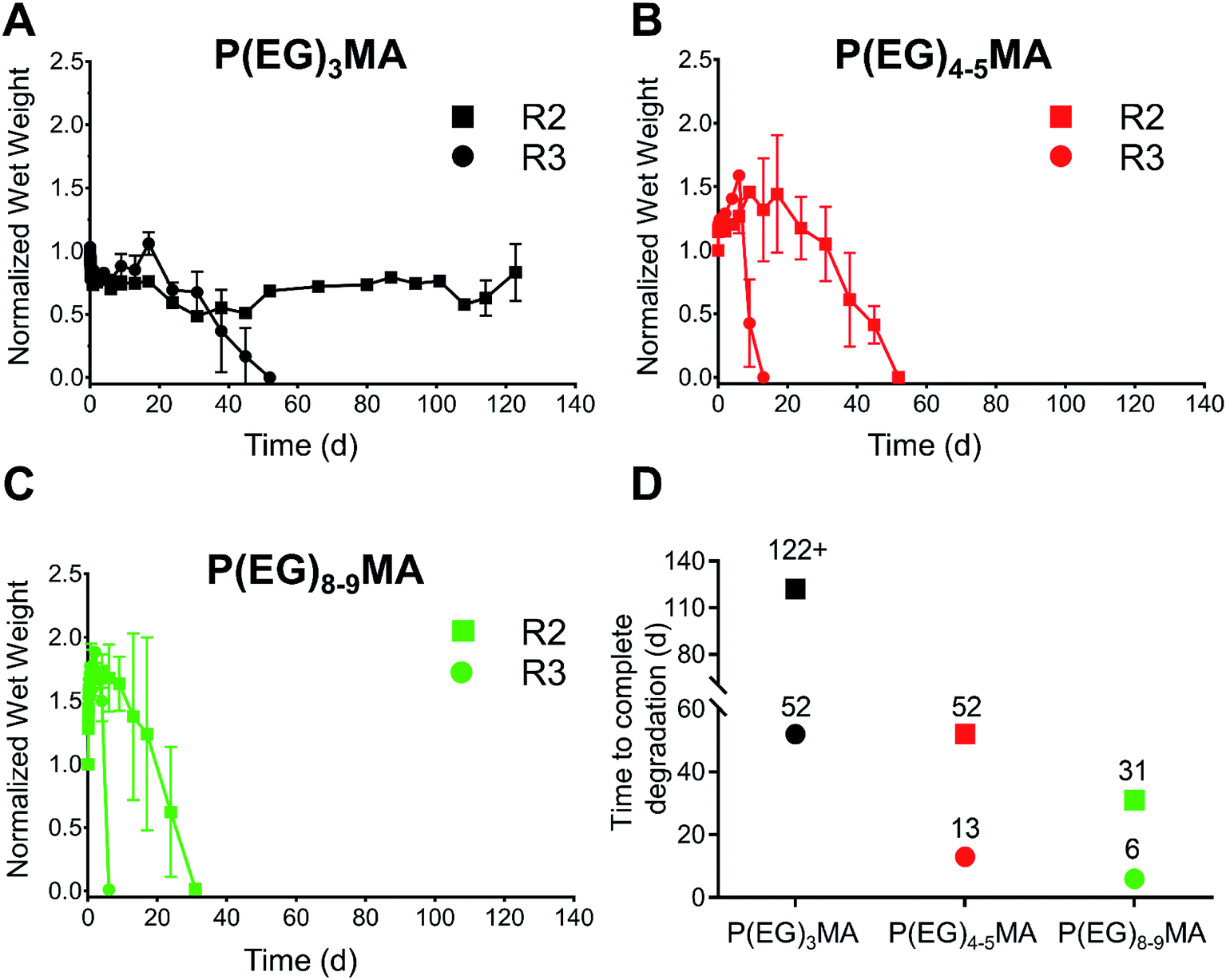 | ||
| Fig. 6 Tunable degradation of P(EG)xMA hydrogels at pH 7.4. The combination of different P(EG)xMA copolymers with R2 or R3 crosslinkers yields low-fouling hydrogels that degrade over 6 to 52 d. The degradation of (A) P(EG)3MA, (B) P(EG)4–5MA, and (C) P(EG)8–9MA hydrogels (100 μL, 5 wt%) with R2 (4-methylphenyl sulfone) or R3 (4-chlorophenyl sulfone) crosslinkers was followed over time in pH 7.4 PBS at 37 °C (mean ± standard deviation, n = 3). (D) Illustration summarizing time required for complete degradation of each hydrogel. | ||
P(EG)3MA hydrogels, which shrunk over time (Fig. 5C), produced the slowest degradation profiles (Fig. 6A and D). P(EG)3MA–R3 gels degraded over 52 d, 4-fold slower than P(EG)4–5MA–R3 gels. Therefore, PEG MW has a significant impact on hydrogel degradation timeframes. Interestingly, P(EG)3MA–R2 gels remained intact for >120 d. We expect P(EG)3MA–R2 gels to eventually degrade because P(EG)3MA–R3 gels degraded, which indicates that crosslink deprotonation and cleavage occur in P(EG)3MA gels. Therefore, the design of P(EG)3MA gels that degrade over 6 to >120 d may be possible using the developed copolymer library. Because P(EG)3MA–R3 and P(EG)4–5MA–R2 gels both degraded over 52 d, low-fouling P(EG)xMA hydrogels can be tuned to degrade over 6, 13, 31 or 52 d from a library of 6 P(EG)xMA copolymers: (1) P(EG)4–5MA–AZ–R2; (2) P(EG)4–5MA–AZ–R3; (3) P(EG)4–5MA–DBCO; (4) P(EG)8–9MA–AZ–R2; (5) P(EG)8–9MA–AZ–R3; and, (6) P(EG)8–9MA–DBCO.
3.6 P(EG)xMA gels are non-cell adhesive
Non-specific binding of cells to hydrogels can impede drug and cell delivery. To demonstrate P(EG)xMA gels are resistant to non-specific cell adhesion, non-degradable P(EG)xMA gels were exposed to fibroblasts in 10% CBS in DMEM/F12 media. After 24 h, gel surfaces were gently washed with PBS to remove non-adhered cells. After staining with Calcein AM and Hoechst, micrographs of cells on gel surfaces were collected. No adhered cells were detected on any P(EG)xMA gel (Fig. 7A), indicating the gels are low-fouling towards cells. PCBMAA gels formed through in situ PCBMAA–AZ and PCBMAA–DBCO (6 mol% AZ/DBCO) crosslinking were included as controls; PCBMAA gels are known to resist non-specific cell binding36 (Fig. 7A).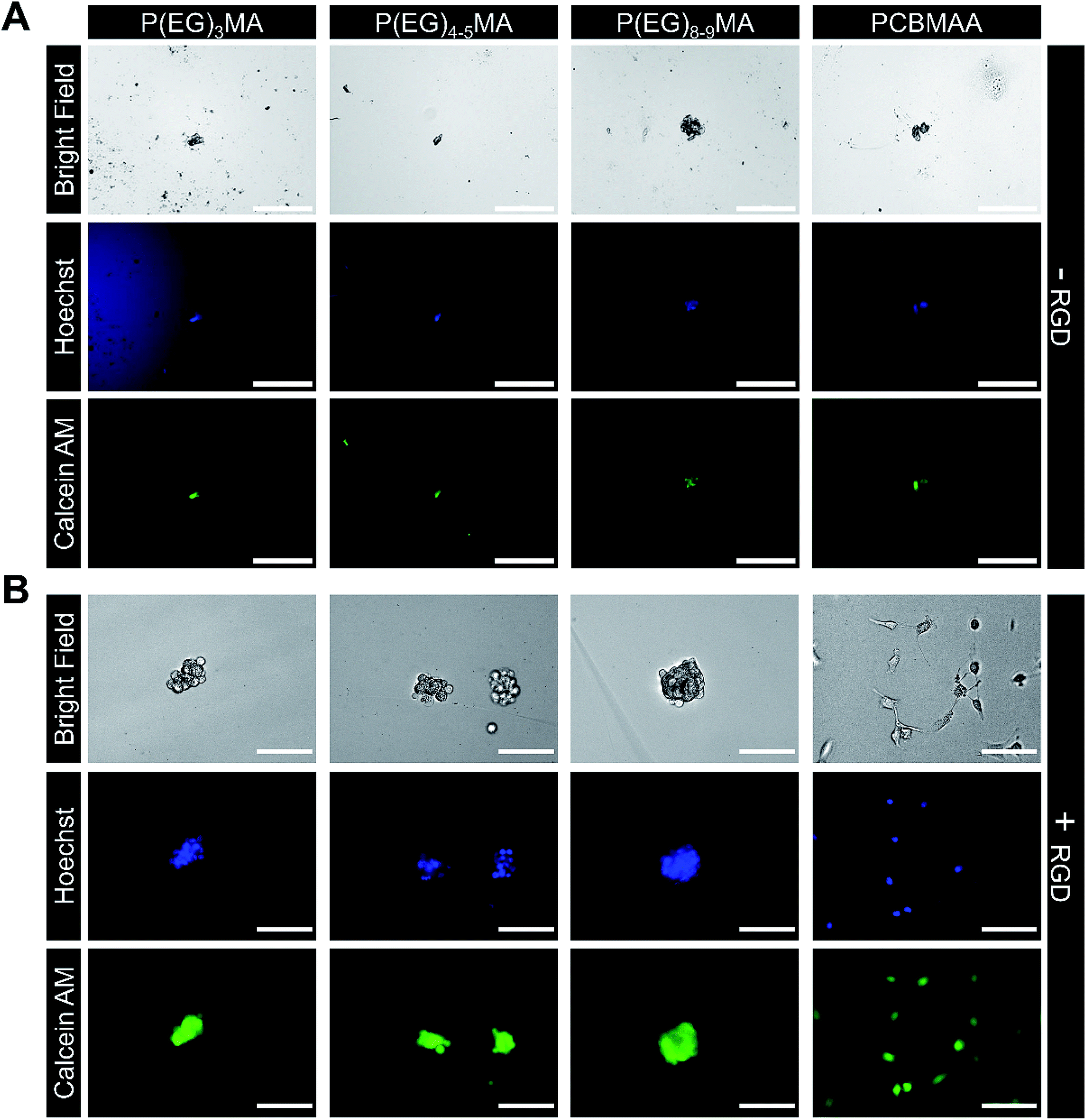 | ||
| Fig. 7 Cell adhesion to P(EG)xMA and PCBMAA hydrogels with and without immobilized RGD. Fibroblasts were seeded on gel surfaces and incubated for 24 h in DMEM/F12 with 10% CBS, gels were gently washed to remove non-adhered cells and stained with Calcein AM and Hoechst. (A) No cell adhesion was observed on P(EG)xMA and PCBMAA gel surfaces without RGD. (B) Only a small number of cell clusters were observed on the surface of all P(EG)xMA gels modified with RGD, indicating limited cell adhesion. In contrast, cell attachment and spreading on PCBMAA gels modified with RGD was similar to TCP controls (scale bars: 200 μm for −RGD and 100 μm for +RGD micrographs; n = 3). Micrographs of cells adhered to TCP are presented in Fig. S25.† | ||
P(EG)xMA gels modified with RGD cell adhesion peptides demonstrated limited cell adhesion, indicating P(EG)xMA gels also hindered integrin mediated adhesion. Excess DBCO groups in P(EG)xMA gels were modified with an RGD-AZ peptide; the reaction was monitored by decreasing DBCO absorbance (309 nm; data not shown). Fibroblasts were seeded on hydrogel surfaces and incubated for 24 h. Cells were then stained with Calcein AM and Hoechst gels, gently washed with PBS to remove non-adhered cells, and gel surfaces were imaged. Only small cell clusters were observed on RGD modified P(EG)xMA gels indicating weak RGD mediated cell–hydrogel interactions (Fig. 7B). In contrast, cells adhered to RGD modified PCBMAA gels (Fig. 7B) with morphologies similar to cells on tissue culture plastic (TCP; Fig. S25†). PCBMAA polymers are known to promote protein–ligand interactions,37 whereas the PEG pendant groups in P(EG)xMA may hinder protein–ligand complexation; PEG polymers are known to decrease enzymatic activity.37 Because all hydrogels contained a large excess of DBCO groups (>100 nmol cm−2 on the surface) for RGD immobilization, all hydrogel surfaces had sufficient RGD for cell adhesion (>1 fmol cm−2).38 Therefore, P(EG)xMA gels are ideal for applications that require minimal protein and cell binding.
For some stem cell delivery applications, degradable hydrogels functionalized with adhesive peptide have been shown to improve cell survival.39 Therefore, we incorporated the two crosslinkers (R2 and R3) into PCBMAA hydrogels (Scheme S1†), which degraded over 13 and 52 d (Fig. S24C†). Due to the high solubility of PCBMAA copolymers, the rate of PCBMAA hydrogel degradation was expected to be similar to P(EG)8–9MA gels, which degraded over 6 and 31 d. The slower degradation rate was attributed to PCBMAA's greater crosslink density than P(EG)8–9MA gels (128 vs. 48 μmol g of polymer). Therefore, the PCBMAA hydrogel can be used for short- and long-term applications that require bioactive hydrogels.
3.7 Further discussion
The developed degradation mechanism for low-fouling P(EG)xMA gels tunes the deprotonation rate of the carbamate crosslinker independently from endogenous triggers by changing EWGs and PEG MW, which provides a method to reliably achieve different hydrogel degradation timeframes. All components, except for released CO2, remain covalently bound to degraded non-cytotoxic copolymers, we therefore expect minimal in vivo toxicity for future applications. Moreover, all degradation timeframes (6, 13, 31, and 52 d) can be achieved from highly soluble P(EG)4–5MA and P(EG)8–9MA copolymers (LCSTs > physiological temperature) and will therefore remain soluble after hydrogel degradation to further improve biocompatibility.Controlled degradation of hydrogels is important for drug and cell delivery. For example, the sustained release of antibody checkpoint inhibitors for ∼1 week from degradable poly(vinyl alcohol) hydrogels improved survival in a melanoma mouse model by 50% over intravenous injections.40 Adoptive cell therapies are improved by creating a local cell depot with injectable hydrogels that degrade to allow for cell egress. Hydrogels have been shown to improve transplantation efficiency of neural stem cells41 and T cell infiltration42 into tumors for cancer immunotherapies. The ability to control hydrogel degradation will improve adoptive cell therapies by controlling the rate of cell egress from the hydrogel into surrounding tissue. Therefore, we demonstrated that encapsulated fibroblasts in P(EG)xMA gels retained high viability to ensure the crosslinking mechanism is not cytotoxic (Fig. S26†); SPAAC crosslinking has previously been demonstrated to be non-cytotoxic.43 To avoid unwanted adverse events (e.g. FBR), low-fouling hydrogels that degrade over several days to weeks are required, such as those developed here.
The degradation rate of P(EG)xMA gels will influence both drug and cell delivery. For the developed P(EG)xMA gels, cells seeded on the surface of RGD modified hydrogels did not migrate into the gel, indicating hydrogel pore sizes are sub-micron and prevent cell migration. Therefore, the degradation rate of the P(EG)xMA gels will be the main determinant for cell delivery rates. However, hydrogel degradation will not be required for drug efflux because P(EG)xMA gels with similar and higher crosslink densities have previously been demonstrated to release proteins for drug delivery applications.44 Although, faster degradation rates will increase rates of drug release.
4. Conclusions
The developed 6-member P(EG)xMA copolymer library allows for the rapid fabrication of low-fouling hydrogels that degrade over 6 to 52 d, which will be useful for short- and long-term drug and cell delivery applications. Furthermore, P(EG)3MA–R2 may result in hydrogels that degrade over >120 d. The combination of tunable base-catalyzed crosslink degradation and P(EG)xMA hydration provides a simple method to rapidly tune degradation rates. Given that the non-cytotoxic P(EG)xMA gels remained low-fouling towards proteins and cells, it is expected that they will help mitigate adverse immune responses (e.g. FBR) upon implantation. Interestingly, P(EG)xMA gels modified with RGD remained non-cell adhesive by preventing integrin mediated adhesion. The established low-fouling P(EG)xMA hydrogel library that easily achieves different degradation timeframes will expedite the establishment of drug and cell delivery therapies.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC: 2015-05429, 506209-16), Canada Foundation for Innovation: John R. Evans Leaders Fund (CFI-JELF: 34107), Ontario Research Fund – Research Infrastructure (ORF-RI: 34107), and McMaster University. We would also like to thank Dr Alex Adronov and Stuart McNelles for donation of NHS-DBCO.References
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- R. Klopfleisch and F. Jung, J. Biomed. Mater. Res., Part A, 2017, 105, 927–940 CrossRef CAS PubMed.
- W. Chen, B. C. Yung, Z. Qian and X. Chen, Adv. Drug Delivery Rev., 2018, 127, 20–34 CrossRef CAS PubMed.
- C. Leng, S. Sun, K. Zhang, S. Jiang and Z. Chen, Acta Biomater., 2016, 40, 6–15 CrossRef CAS PubMed.
- W. Feng, S. Zhu, K. Ishihara and J. L. Brash, Biointerphases, 2006, 1, 50–60 CrossRef CAS PubMed.
- W. Feng, X. Gao, G. McClung, S. Zhu, K. Ishihara and J. L. Brash, Acta Biomater., 2011, 7, 3692–3699 CrossRef CAS PubMed.
- W. Yang, H. Xue, W. Li, J. Zhang and S. Jiang, Langmuir, 2009, 25, 11911–11916 CrossRef CAS PubMed.
- L. Zhang, Z. Cao, T. Bai, L. Carr, J. R. Ella-Menye, C. Irvin, B. D. Ratner and S. Jiang, Nat. Biotechnol., 2013, 31, 553–556 CrossRef CAS PubMed.
- A. Singh and N. A. Peppas, Adv. Mater., 2014, 26, 6530–6541 CrossRef CAS PubMed.
- C. Bastiancich, P. Danhier, V. Préat and F. Danhier, J. Controlled Release, 2016, 243, 29–42 CrossRef CAS PubMed.
- M. Norouzi, B. Nazari and D. W. Miller, Drug Discovery Today, 2016, 21, 1835–1849 CrossRef CAS PubMed.
- S. V. Bharambe, A. B. Darekar and R. B. Saudagar, Int. J. Pharm. Technol., 2013, 5, 2764–2786 Search PubMed.
- N. Gerwin, C. Hops and A. Lucke, Adv. Drug Delivery Rev., 2006, 58, 226–242 CrossRef CAS.
- M. F. Rai and C. T. Pham, Curr. Opin. Pharmacol., 2018, 40, 67–73 CrossRef CAS PubMed.
- S. G. Schwartz, I. U. Scott, H. W. Flynn and M. W. Stewart, Expert Opin. Drug Delivery, 2013, 11, 61–68 CrossRef PubMed.
- V. Huynh and R. G. Wylie, Angew. Chem., Int. Ed., 2018, 57, 3406–3410 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- L. Zhang, H. Xue, Z. Cao, A. Keefe, J. Wang and S. Jiang, Biomaterials, 2011, 32, 4604–4608 CrossRef CAS PubMed.
- H. W. Chien, X. Xu, J. R. Ella-Menye, W. B. Tsai and S. Jiang, Langmuir, 2012, 28, 17778–17784 CrossRef CAS PubMed.
- P. Jain, H.-C. C. Hung, B. Li, J. Ma, D. Dong, X. Lin, A. Sinclair, P. Zhang, M. B. O'Kelly, L. Niu and S. Jiang, Langmuir, 2019, 35, 1864–1871 CrossRef CAS PubMed.
- G. Cheng, F. Cheng, H. Wang, H. Wu and F. Xu, RSC Adv., 2016, 6, 30862–30866 RSC.
- D. Dong, J. Li, M. Cui, J. Wang, Y. Zhou, L. Luo, Y. Wei, L. Ye, H. Sun and F. Yao, ACS Appl. Mater. Interfaces, 2016, 8, 4442–4455 CrossRef CAS PubMed.
- E. Bakaic, N. M. B. Smeets, M. Badv, M. Dodd, O. Barrigar, E. Siebers, M. Lawlor, H. Sheardown and T. Hoare, ACS Biomater. Sci. Eng., 2018, 4, 3713–3725 CrossRef CAS.
- I. Urosev, E. Bakaic, R. J. Alsop, M. C. Rheinstädter and T. Hoare, J. Mater. Chem. B, 2016, 4, 6541–6551 RSC.
- H.-W. W. Chien, W.-B. B. Tsai and S. Jiang, Biomaterials, 2012, 33, 5706–5712 CrossRef CAS PubMed.
- J. Leijten, J. Seo, K. Yue, G. Trujillo-de Santiago, A. Tamayol, G. U. Ruiz-Esparza, S. R. Shin, R. Sharifi, I. Noshadi, M. M. Álvarez, Y. S. Zhang and A. Khademhosseini, Mater. Sci. Eng., R, 2017, 119, 1–35 CrossRef PubMed.
- D. V. Santi, R. Reid, E. L. Schneider, G. W. Ashley and L. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6211–6216 CrossRef CAS PubMed.
- G. W. Ashley, J. Henise, R. Reid and D. V Santi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2318–2323 CrossRef CAS PubMed.
- Z. Cui, B. H. Lee, C. Pauken and B. L. Vernon, J. Biomed. Mater. Res., Part A, 2011, 98, 159–166 CrossRef PubMed.
- S. M. Hodgson, E. Bakaic, S. A. Stewart, T. Hoare and A. Adronov, Biomacromolecules, 2016, 17, 1093–1100 CrossRef CAS PubMed.
- J. F. Lutz, J. Polym. Sci. Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- B. Yang, C. Wang, Y. Zhang, L. Ye, Y. Qian, Y. Shu, J. Wang, J. Li and F. Yao, Polym. Chem., 2015, 6, 3431 RSC.
- N. M. B. Smeets, E. Bakaic, M. Patenaude and T. Hoare, Acta Biomater., 2014, 10, 4143–4155 CrossRef CAS PubMed.
- N. Kotagiri, A. Nehorai, Z. Li, S. Achilefu, X. Xu and S. Mondal, Bioconjugate Chem., 2014, 25, 1272–1281 CrossRef CAS PubMed.
- M. Bjerknes, H. Cheng, C. D. McNitt and V. V. Popik, Bioconjugate Chem., 2017, 28, 1560–1565 CrossRef CAS PubMed.
- V. Huynh, A. Jesmer, M. M. Shoaib and R. G. Wylie, Langmuir, 2019, 35, 1631–1641 CrossRef CAS PubMed.
- A. J. Keefe and S. Jiang, Nat. Chem., 2012, 4, 59–63 CrossRef CAS PubMed.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089–1100 CrossRef CAS PubMed.
- S. S. Ho, K. C. Murphy, B. Y. K. Binder, C. B. Vissers and J. K. Leach, Stem Cells Transl. Med., 2016, 5, 773–781 CrossRef CAS PubMed.
- C. Wang, J. Wang, X. Zhang, S. Yu, D. Wen, Q. Hu, Y. Ye, H. Bomba, X. Hu, Z. Liu, G. Dotti and Z. Gu, Sci. Transl. Med., 2018, 10, eaan3682 CrossRef PubMed.
- L. R. Nih, S. T. Carmichael and T. Segura, Curr. Opin. Biotechnol., 2016, 40, 155–163 CrossRef CAS PubMed.
- S. B. Stephan, A. M. Taber, I. Jileaeva, E. P. Pegues, C. L. Sentman and M. T. Stephan, Nat. Biotechnol., 2015, 33, 97–101 CrossRef CAS PubMed.
- S.-S. Han, H. Y. Yoon, J. Y. Yhee, M. O. Cho, H.-E. Shim, J.-E. Jeong, D.-E. Lee, K. Kim, H. Guim, J. H. Lee, K. M. Huh and S.-W. Kang, Polym. Chem., 2018, 9, 20–27 RSC.
- E. Bakaic, N. M. B. Smeets, H. Dorrington and T. Hoare, RSC Adv., 2015, 5, 33364–33376 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03441b |
| This journal is © The Royal Society of Chemistry 2019 |