
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface amino-functionalization of Sn-Beta zeolite catalyst for lactic acid production from glucose†
Zheng Shen ab,
Ling Konga,
Wei Zhangb,
Minyan Gua,
Meng Xiab,
Xuefei Zhoub and
Yalei Zhang*ab
ab,
Ling Konga,
Wei Zhangb,
Minyan Gua,
Meng Xiab,
Xuefei Zhoub and
Yalei Zhang*ab
aNational Engineering Research Center of Protected Agriculture, Tongji University, Shanghai, 200092, China. E-mail: zhangyalei@tongji.edu.cn
bState Key Laboratory of Pollution Control and Resource Reuse, Tongji University, Shanghai, 200092, China
First published on 17th June 2019
Abstract
The catalytic conversion of glucose into lactic acid (LA) provides an alternative approach for LA production to bio-fermentation. Nevertheless, during the process, the dehydration of glucose and fructose into 5-hydroxymethylfurfural catalyzed by Brønsted acid exists as the main side reaction that leads to the decrease in LA yield. In order to promote the yield and selectivity of LA, a series of acid–base bifunctional Sn-Beta-NH2 catalysts were prepared by post-grafting aminopropyl groups with the surface silanol groups of Sn-Beta zeolite. The catalysts were systematically characterized by X-ray diffraction, N2 adsorption–desorption, elemental analysis, X-ray photoelectron spectroscopy, Fourier transform infrared spectra analysis following pyridine adsorption, and CO2 temperature-programmed desorption. The as-prepared Sn-Beta-NH2 catalysts exhibiting both Lewis acid and moderate base sites facilitated the conversion of glucose to LA in competition with undesirable side reactions. In addition, effects of reaction parameters including reaction temperature and time, catalyst dosage, and glucose concentration were investigated. A high LA yield up to 56% was achieved under optimized hydrothermal conditions (190 °C, 2 h), along with a complete conversion of glucose and a 5-hydroxymethylfurfural yield of 7%. The result indicated an alternative modification method of Sn-Beta zeolite for a more favorable LA yield.
Introduction
Biomass, as the most important and perhaps the only sustainable and clean carbon source, is the most abundant and widespread renewable resource. It is being intensively studied worldwide as raw materials for the production of high value-added chemicals in order to solve the problems of energy crisis and environmental pollution.1–9 Lactic acid (LA) is a versatile chemical in industry and is regarded as one of the most promising platform molecules because of its wide applications in the production of alkyl lactates, propylene glycol, biodegradable plastics, amino acids, etc.10–12 LA can be manufactured either by carbohydrate fermentation or by chemical synthesis approaches. However, the commercial biotechnological routes for LA production are mainly based on the use of grains including rice, wheat and corn through anaerobic fermentation in the presence of Lactobacillus.10,13 The growth of Lactobacillus relies on the nutrition including carbohydrates, amino acids, nucleotides and minerals, as well as a pH value of 5.5–5.8. The fermentation process of lactic acid production and its subsequent separation process are all complex and of low efficiency. Additionally, the inevitable use of lime as the neutralization agent may lead to the production of calcium sulfate waste. Compared to bio-fermentation, chemical catalytic reactions provide a more acceptable approach for LA production from biomass instead of grain because of its potential for large-scale production and higher efficiency.14,15Up to date, great efforts have been devoted to synthesizing multiple catalysts for the conversion of carbohydrates into LA, including homogenous and heterogeneous catalysts. Homogenous catalysts, such as metal salts (Pb2+),16 alkaline-earth metal hydroxides (Ca(OH)2 and lanthanide catalysts (ErCl3 and Er(OTf)3)),17–19 always exhibit outstanding catalytic performance in the process of biomass conversion to produce LA. However, the separation and recovery of these catalysts may hinder their practical application. Another widely studied route is the synthesis of heterogeneous catalysts, such as metal oxides (Nb2O5, CuO and Al–Zr mixed oxides),20–22 solid acid catalysts (AlW and Er/K10)23,24 and zeolite catalysts (Sn-Beta).14,25
Among numerous heterogeneous catalysts, Sn-Beta zeolite has shown benign catalytic performance because of its stability and various Lewis acid sites generated from Sn.25–27 Therefore, much attention has been paid to the modification of Sn-based zeolites. In order to accelerate the diffusion of glucose and fructose molecules, Zhang et al. synthesized a hierarchical Sn-Beta zeolite catalyst and a 72% alkyl lactate yield was obtained from sucrose.28 Wan et al. found that Sn-Beta supported gold catalyst could readily convert acetol to LA with a yield up to 73% under hydrothermal conditions.29
During the reaction process of glucose conversion, there exist some side reactions that may lead to the decrease of LA yield, including the dehydration of glucose and fructose to 5-hydroxymethylfurfural (HMF) and the rehydration of HMF to formic acid and levulinic acid. Moreover, the generated LA may inevitably act as a Brønsted acid to catalyze these side reactions. In order to suppress the side reactions, researchers attempted to add base into the reaction solution, followed by the recovery of lactic acid with the aid of acid neutralization.30 But this approach brings additional cost and may lead to corrosion to the reactor. To avoid the use of liquid base, Dong et al. firstly developed a Sn–Zn-Beta catalyst that contained both Lewis acid and base sites and a 48% LA yield was obtained.14 It was a novel thought to modify the Sn-Beta zeolite with another metal to form heterogeneous catalysts with both acidic and basic catalytic sites. Apart from the introduction of metal atoms, the plenty silanol groups on the surface of Beta zeolite are also favorable for modification by organic functional groups. Post-grafting method is a common method for organic functionalization. By the reaction of silanol groups and functionalization reagents to form corresponding covalent bond, the surface hydrophilicity/hydrophobicity or acidity/basicity of Beta zeolite could be adjusted. In addition, compared to metal modification, surface modification with covalently bound organic functional groups could also promote the interaction and collision opportunity with organic substrates, leading to a further increase in reaction efficiency.
Here, we developed a new method to modify Sn-Beta zeolite with amino groups, in order to strengthen its basicity while not decrease its acidity drastically. By post-grafting organoamines with the surface silanol groups of the zeolite materials, highly stable catalysts containing solid acid and base catalytic sites could be synthesized. The surface amino-functionalization of supporters has already been conducted to the surface modification of silica,31 MCM-48,32,33 SBA-15,34,35 MCM-41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 and ion exchange resins.37 The structural and physicochemical properties of the catalysts were systematically characterized upon functionalization and the functionalized catalysts exhibited improved activity for the self-condensation of cyclohexanone, Knoevenagel reaction, and Henry reaction.32–37 Considering that the Sn-Beta zeolite contains surface silanol groups, it could also be modified by surface amino-functionalization. Herein, we report that glucose can be efficiently converted to LA under hydrothermal conditions over amino groups functionalized Sn-Beta catalyst. A systematical optimization of reaction parameters is conducted and the possible reaction pathway is also proposed based on structural and acid–base characterization.
36 and ion exchange resins.37 The structural and physicochemical properties of the catalysts were systematically characterized upon functionalization and the functionalized catalysts exhibited improved activity for the self-condensation of cyclohexanone, Knoevenagel reaction, and Henry reaction.32–37 Considering that the Sn-Beta zeolite contains surface silanol groups, it could also be modified by surface amino-functionalization. Herein, we report that glucose can be efficiently converted to LA under hydrothermal conditions over amino groups functionalized Sn-Beta catalyst. A systematical optimization of reaction parameters is conducted and the possible reaction pathway is also proposed based on structural and acid–base characterization.
Experimental
Materials
All reagents were used without further purification. Beta zeolite was obtained from the Catalyst Plant of Nankai University. Tin(II) acetate (95%), lactic acid (LA) (1.0 N), 5-hydroxymethylfurfural (HMF) (97%), levulinic acid (LeA) (98%) were obtained from Alfa Aesar. 3-Aminopropyltrimethoxysilane (APTMS) (98%) was obtained from Wako. Formic acid (FA) (99%) was obtained from J&K China Chemical Co., Ltd. Phosphoric acid (HPLC) was obtained from Sigma-Aldrich. Nitric acid (GR), ethanol (AR), D-(+)-glucose (AR) were obtained from Sinopharm Chemical Reagent Co., Ltd.Catalysts synthesis
Commercial Beta zeolite was dealuminated by stirring in HNO3 solution (13 M) at 80 °C for 20 h with a HNO3 (mL)/Beta zeolite (g) ratio of 20. After filtrating, washing with a large amount of deionized water until the supernatant was neutral, drying at 150 °C overnight, the dealuminated Beta zeolite was obtained and denoted as deAl-Beta. Solid-state ion exchange (SSIE) was then performed by grinding 0.2 g of tin(II) acetate with 1.0 g of dealuminated zeolite for 30 min. Finally, the sample was calcined in an air flow at 550 °C for 6 h with a ramp of 5 °C min−1 and was denoted as Sn-Beta.38As to the synthesis of surface amino-functionalizated Sn-Beta zeolite (Sn-Beta-NH2(v), v represents the volume of APTMS used (μL)), in a typical run, 0.5 g of Sn-Beta zeolite and a certain volume of APTMS were dissolved into 250 mL of ethanol in a 500 mL round-bottomed flask equipped with a reflux condenser. After stirring at 80 °C for 6 h, the white precipitates were collected by centrifugation and followed by washing with a large amount of ethanol. Then, the sample was dried at 80 °C overnight to obtain Sn-Beta-NH2 catalyst.
Catalysts characterizations
The X-ray diffraction (XRD) patterns of the catalysts were recorded on a Bruker D8 Advance X-ray powder diffractometer with Cu Kα radiation of λ = 1.54 Å (40 kV and 40 mA) from 2θ 5= 10° to 90° with a scan speed of 2° min−1. N2 adsorption–desorption isotherms measurements were performed using a Micromeritics ASAP 2460 analyzer at −196 °C. Prior to the analysis, samples were degassed at 150 °C for 6 h under vacuum. The total surface area was calculated by employing the multipoint BET method and the microporous pore volume was determined using the t-plot method. The C and N contents of the Sn-Beta-NH2 catalysts were determined on Elementar Vario EL III elemental analyzer at 1000 °C. The contents of Si and Al of the catalysts were measured by inductively coupled plasma optical emission spectroscopy (ICP-OES, PerkinElmer Optima 2100DV). Prior to the measurements, the samples were digested in an acidic mixture (HCl–HNO3–HF) at 150 °C for 12 h. X-ray photoelectron spectroscopy (XPS) studies were performed on a RBD upgraded PHI-5000C ESCA system (PerkinElmer) with Al Kα radiation (hν = 1486.6 eV). Charge referencing was done against adventitious carbon (C1s at 284.6 eV). The acidic properties of the catalysts were tested by adsorption, followed by temperature programmed desorption (TPD) of pyridine using Fourier transform infrared spectroscopy (Py-IR). The Py-IR spectra were recorded on a PerkinElmer Frontier FT-IR in the range of 1400–1700 cm−1 with a resolution of 2 cm−1. A 10 mg of sample was pressed into a self-supported wafer with a diameter of 13 mm. Then, the wafer was set in a quartz IR cell which was sealed with CaF2 windows and connected to a vacuum system. The samples were evacuated at 450 °C for 2 h under vacuum. After cooling, pyridine vapor was employed into the cell and the adsorption was performed for 0.5 h. Subsequently, desorption steps were conducted at different temperatures (25 °C, 150 °C and 250 °C) for 1 h. The alkaline properties of the samples were performed on Micromeritics AutochemII 2920 chemisorption analyzer using the CO2-TPD method. The sample was first purged with Ar and then heated to 550 °C at a rate of 10°C min−1 for 1 h and finally cooled to 45 °C. The surface of the catalyst was saturated with CO2 for 30 min at this temperature, and then, the excess CO2 was purged with Ar blowing. The TPD curve of the sample was obtained at the temperature range from 45 to 800 °C with a heating rate of 10 °C min−1 under an Ar flow and a flowing rate of 30 cm3 min−1.Catalytic reactions and products analysis
The catalytic conversion of glucose to LA was carried out in a Teflon-lined stainless steel autoclave reactor with an inner volume of 50 mL. In a typical procedure, 225 mg of glucose (7.5 mmol of carbon) and 160 mg of catalyst were mixed in 10 mL of H2O in the autoclave, followed by heating to the desired reaction temperature (190 °C) and keeping for the appropriate time (2 h). After reaction, the autoclave was cooled, and the liquid mixture was separated by centrifugation and filtration. The concentrations of organic acids, HMF and sugars were analyzed on an Agilent 1200 series HPLC. For organic acids and HMF, a Gemini-NX 5u C18 column was used with a UV detector (210 nm) using a 0.1% aqueous phosphoric acid as the mobile phase with a flow rate of 1 mL min−1. The column temperature was 30 °C. The glucose concentration was analyzed using a RI detector (Bio-Rad HPX-87H column) and the column and RI detector temperature were 55 °C and 45 °C, respectively. The mobile phase was a 5 mM aqueous sulphuric acid solution with a flow rate of 0.5 mL min−1. All the concentrations were determined based on the external standard. The conversion of sugars was defined as the moles of sugars reacted divided by initial sugars moles used in the system and the product yields were determined based on carbon. All experiments were performed for more than three times and the quantitative data were the average values of the analytical results. The relative errors were less than 10% for all experiments.Results and discussion
Catalysts preparation and characterizations
The Sn-Beta-NH2 catalyst was synthesized from a combined strategy of both Sn loading and organoamines functionalization.32,33,36,37 As proposed in Fig. 1, commercial Beta zeolite was pretreated using HNO3 to remove most of Al species in the framework. Then, the Sn sites were incorporated into the dealuminated zeolite by SSIE. The synthesis of the bifunctional catalyst, or the post-grafting of organoamine groups, was carried out by stirring a certain amount of APTMS with Sn-Beta zeolite in a polar solvent ethanol. Thus, organoamines were grafted on the Sn-Beta zeolites surface in formation of Si–O–Si bonds.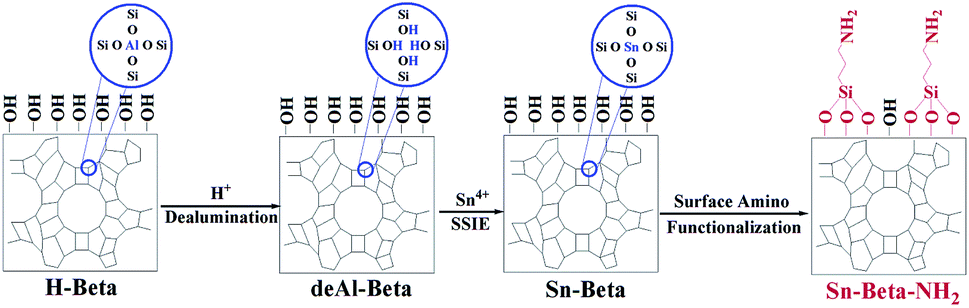 | ||
| Fig. 1 Proposed procedures for preparation of the Sn-Beta-NH2 catalyst. | ||
The XRD patterns in Fig. 2A confirmed that the topology of Beta zeolite was well preserved for the prepared deAl-Beta in HNO3 solution. Then the structure of deAl-Beta changed during the process of incorporation of Sn species. The intensity of the peak located at 2θ = 22.5–22.6° decreased after Sn incorporation, indicating a lower crystallinity. The Sn-Beta-NH2 possessed a similar crystallinity to Sn-Beta, showing that the further post-grafting of organoamines process did not destroy the Sn-Beta zeolite structure. Meanwhile, a slight amount of tin oxide was observed, as evidenced by the occurrence of the peaks at 26°, 34° and 51°, possibly due to the high dosage of tin(II) acetate used during the SSIE procedure.39 Fig. 2B presents the N2 adsorption–desorption isotherms of all catalysts. Typical type-I and type-IV isotherms with two steep steps at the relative pressure of P/P0 < 0.01 and 0.6 < P/P0 < 0.9, corresponding respectively to micropore filling and capillary condensation in the mesopores, were observed. It can also be confirmed that the most probable pore sizes of all catalysts were about 3.9 nm (Fig. S1†), indicating that the post-grafting of organic functional groups did not influence the mesopore size of Sn-Beta zeolite.
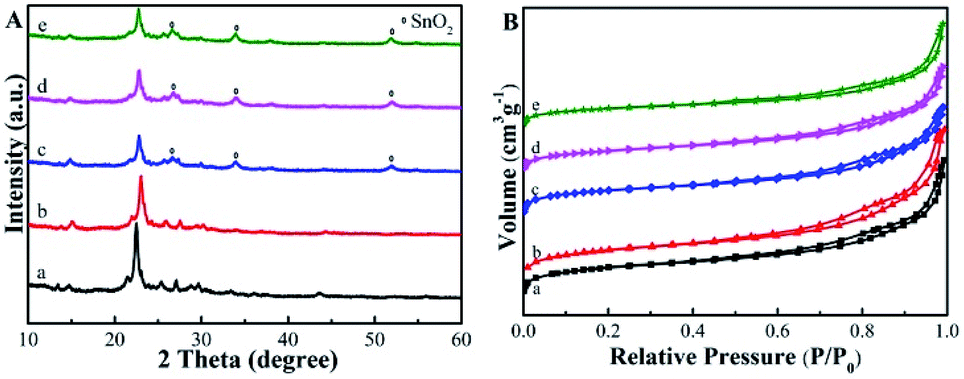 | ||
| Fig. 2 (A) Powder XRD patterns of the samples. (B) N2 sorption isotherms of the samples. The samples are: (a) Beta, (b) deAl-Beta, (c) Sn-Beta, (d) Sn-Beta-NH2(30) and (e) Sn-Beta-NH2(200). | ||
Table 1 summarizes the physicochemical properties of the Beta-type zeolites. The N contents of Sn-Beta-NH2(30) and Sn-Beta-NH2(200) catalysts measured by CHN elemental analysis were approximately 0.2 wt% and 0.8 wt%, respectively. The former value was similar to the theoretical value on the grounds of the dosage of APTMS, while the latter one was lower than the theoretical value. The results of ICP-OES show that the elemental ratio of Si/Al increased from 25 for commercial Beta zeolite to over 1700 for Sn-Beta and Sn-Beta-NH2, indicating that Al was almost completely removed during the nitric treatment process. Fig. 3A gives the XPS spectra of Sn-Beta-NH2(30) catalyst with five main elements including Si, C, N, Sn and O. The peaks that were observed in the Fig. 3B at 487.1 and 495.5 eV could be assigned to Sn 3d3/2 and Sn 3d5/2, respectively. These two peaks were obviously different from the peaks of extra-framework octahedral Sn species, as often observed at 494.5 and 486.1 eV.40 Combined with the XRD results, it can be confirmed that Sn-Beta zeolite synthesized by SSIE method contained some tetrahedrally coordinated Sn species different from SnO2. The C1s spectra of Sn-Beta-NH2(30) catalyst was shown in Fig. 3C. The most intensive peak observed at 284.6 eV was attributed to C–H in APTMS. The presence of amine groups (–NH2) was evident from the N1s spectra shown in Fig. 3D. The standard binding energy for the free amine group fell in the region of 399–401 eV. Thus, the peak at 399.9 eV was assigned to the –NH2, while the peak at 401.9 eV may be attributed to protonated amine group.41
| Samples | SBETa | Vmicrob | Vtotalc | DV(d)d | Be | Le | Nf |
|---|---|---|---|---|---|---|---|
| (m2 g−1) | (cm3 g−1) | (nm) | (mmol g−1) | (wt%) | |||
| a BET surface area.b Micropore volume, calculated by the t-plot method.c Single point total pore volume at P/P0 = 0.95.d The most probable aperture.e The number of Brønsted acid and Lewis acid sites based on pyridine IR adsorption at 423 K.f Determined by CHN elemental analysis. | |||||||
| Beta | 509 | 0.17 | 0.39 | 3.89 | 0.26 | 0.15 | 0 |
| deAl-Beta | 576 | 0.21 | 0.50 | 3.89 | 0 | 0 | 0 |
| Sn-Beta | 451 | 0.15 | 0.35 | 3.91 | 0 | 0.47 | 0 |
| Sn-Beta-NH2(30) | 348 | 0.11 | 0.29 | 3.90 | 0 | 0.40 | 0.2 |
| Sn-Beta-NH2(200) | 320 | 0.11 | 0.26 | 3.89 | 0 | 0.20 | 0.8 |
 | ||
| Fig. 3 XPS spectra of (A) Sn-Beta-NH2(30), (B) Sn3d, (C) C1s and (D) N1s. | ||
The pyridine-probed FTIR analysis can distinguish the Brønsted and Lewis acid sites readily. Shown in Fig. 4A are the IR spectra of pyridine adsorbed on various catalysts at 150 °C. For deAl-Beta zeolite, all bands corresponding to the Brønsted acid (bands at 1544 and 1633 cm−1) and Lewis acid sites (bands at 1448 and 1610 cm−1) disappeared after dealumination by HNO3. The subsequent incorporation of Sn species enhanced the Lewis acid sites, confirmed by the strong absorption band at 1448 cm−1. After post-grafting organoamine groups, the Lewis acidity was weakened due to the alkalinity of the amine groups and the screening of Sn by the amine groups. When the dosage of APTMS was 30 μL, the Lewis acid sites (0.40 mmol g−1) decreased slightly compared to Sn-Beta zeolite (0.47 mmol g−1). Meanwhile, when the dosage was further increased to 200 μL, the quantity of Lewis acid sites decreased to 0.20 mmol g−1 (Table 1). The decrease in active sites was ascribed to the bulky aminopropyl groups, which were spatially spaced on the surface of catalysts.34 Actually, as the distances between the post-grafted aminopropyl groups were small on average,36 it may lead to a negative effect on the functionality of Sn species both in the characterization and reaction, especially under the APTMS dosage of 200 μL. But the Lewis acid sites quantity of Sn-Beta-NH2(30) catalyst was only a little less than the one of Sn-Beta catalyst, which would not influence the process to yield LA obviously. The CO2-TPD profiles of the Beta zeolites and APTMS-functionalized catalysts were presented in Fig. 4B. It can be seen that the APTMS-functionalized catalysts exhibited the new strong desorption peak located at about 400 °C associated with moderate base sites at 300–600 °C,22 while the zeolites unmodified with amino groups possessed no basic sites obviously. These phenomena confirmed the incorporation of basic groups (–NH2).
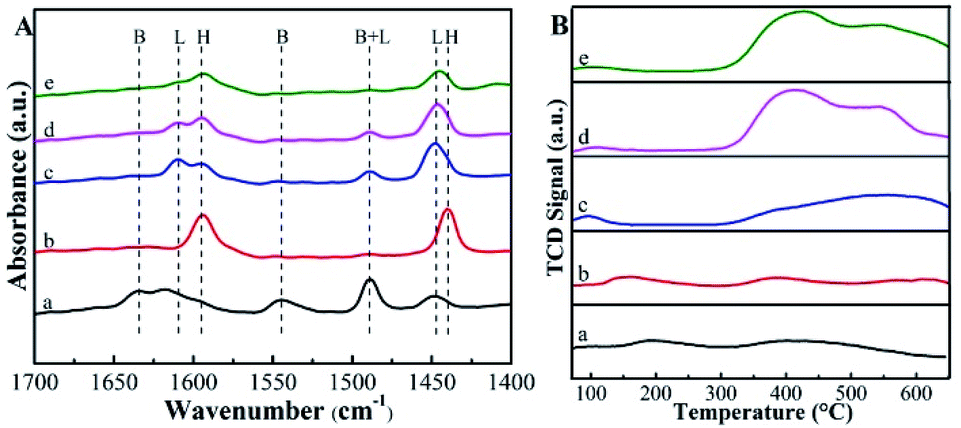 | ||
| Fig. 4 (A) Py-IR spectra of the samples at 150 °C. (B) CO2-TPD profiles of the samples. The samples are (a) Beta, (b) deAl-Beta, (c) Sn-Beta, (d) Sn-Beta-NH2(30) and (e) Sn-Beta-NH2(200). | ||
Catalytic conversion of glucose to lactic acid
According to above discussion, we discovered that Sn-Beta zeolite, equipped with Lewis acid sites, could be an acid–base bifunctional catalyst by simply modifying with APTMS. Fig. 5 showed the glucose conversion and product yields using different Beta zeolite catalysts. When unmodified commercial Beta zeolite was used as the catalyst, the yields of main products consisting of LA, FA, LeA and HMF were all negligible while the glucose conversion was about 80%. The poor catalytic activity of the commercial Beta zeolite should be attributed to its plentiful Brønsted acid sites and trifling Lewis acid sites internally. While in the presence of deAl-Beta zeolite, the yields of the main products were still low (LA yield 4.78%) and the conversion of glucose decreased to 40% simultaneously. By modifying the deAl-Beta zeolite with APTMS to obtain deAl-Beta-NH2(30) zeolite, the LA yield slightly changed to 6.54%. The extremely low LA yields obtained by deAl-Beta-NH2(30) indicated that the amino group exhibited no catalytic efficiency towards the main reaction to produce LA. Using Sn-Beta zeolite, however, the conversion of glucose reached up to 90% and the yield of LA increased to 23%, along with larger amounts of HMF (16%), which is similar to the results reported in literatures.14,25,29 The catalytic activity of the Sn-Beta zeolite should be ascribed to the incorporation of Sn species into the deAl-Beta zeolite, and the resulted generation of various Lewis acid sites. For comparison, when the Sn-Beta zeolite was modified with 30 μL of APTMS to form Sn-Beta-NH2(30), the reaction proceeded with a surprising LA yield (56%) and a decreasing HMF (7%) yield. The LA yield using Sn-Beta-NH2(30) was also slightly higher than that using the bimetallic Zn–Sn-Beta catalyst in our previous work (48%).14 The HMF yield decreased obviously compared to which was obtained by Sn-Beta zeolite. These results suggested that the LA yield was improved significantly upon small addition of APTMS, accompanied by a complete conversion of glucose. Furthermore, the introduction of organoaminos drastically restricted the formation of the main side product HMF.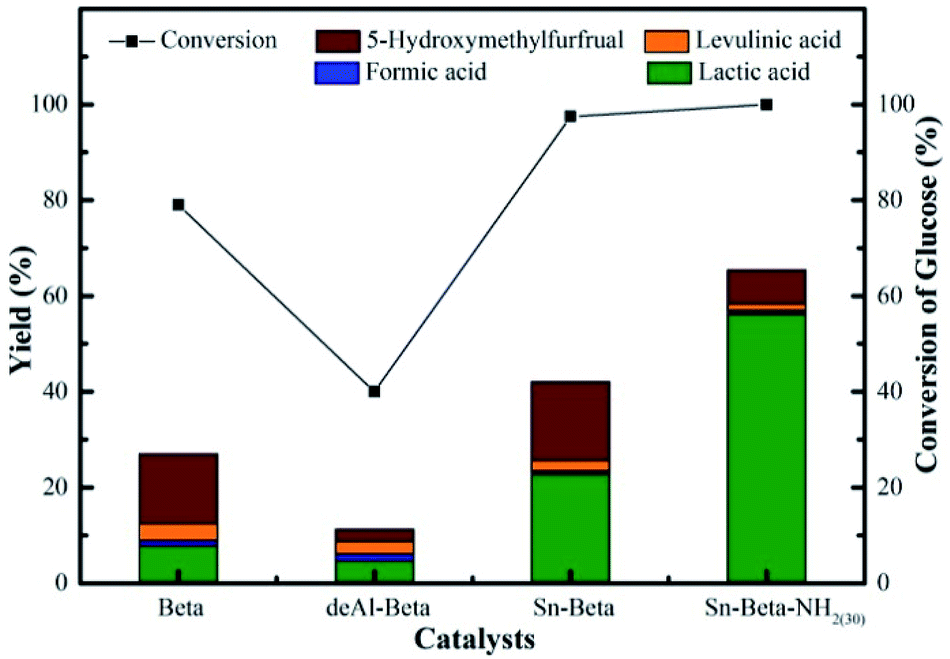 | ||
| Fig. 5 Conversion of glucose and yield of LA, FA, LeA and HMF over Beta, deAl-Beta, Sn-Beta and APTMS-functionalized catalysts. Reaction conditions: 225 mg of glucose, 160 mg of catalyst, 10 mL of H2O, 190 °C, 2 h. | ||
It is generally known that the glucose-to-LA proceeds through multiple steps of isomerization of glucose to fructose and then conversion of fructose to LA, together with side reaction of the formation of HMF and other subsequent side products.14,28 The main reaction to produce LA, is significantly enhanced by the generation of Lewis acid sites. In the meantime, the side reaction to form HMF, could be restrained by the basic –NH2 group (Fig. 6).
 | ||
| Fig. 6 Proposed reaction pathways in the conversion of glucose to lactic acid. | ||
Reaction parameter studies
The performances of Sn-Beta-NH2 catalysts with various dosages of APTMS were studied, as shown in Fig. 7. Without APTMS, only 23% yield of LA was obtained and the yield of HMF was about 16%. The presence of APTMS with a volume of 30 μL remarkably promoted the LA yield to 56% with glucose conversion up to 100%. Meanwhile, the HMF yield decreased to 7% and this result indicated that the post-grafted organoamines indeed inhibit the formation of side products. With a further increase in the APTMS dosage, the glucose conversion remained 100% but LA yield slightly decreased. When the volume of APTMS used was 200 μL, the yield of LA decreased to 30%, and the HMF yield was still in decline, down to 4% approximately. As the APTMS dosage increased, the amount of Lewis acid sites decreased. The present Lewis acid sites could still lead to a glucose conversion of 100%. Meanwhile, the production of LA is closely related to the amount of Lewis acid sites. The sharp decrease of LA yield could be attributed to the excessive alkalinity and the screening of Lewis acid sites caused by amino group on the surface, which also led to the decline in HMF yield. Based on these results, the Sn-Beta-NH2(30) catalyst was selected for subsequent studies since this system resulted in the highest LA yield from glucose.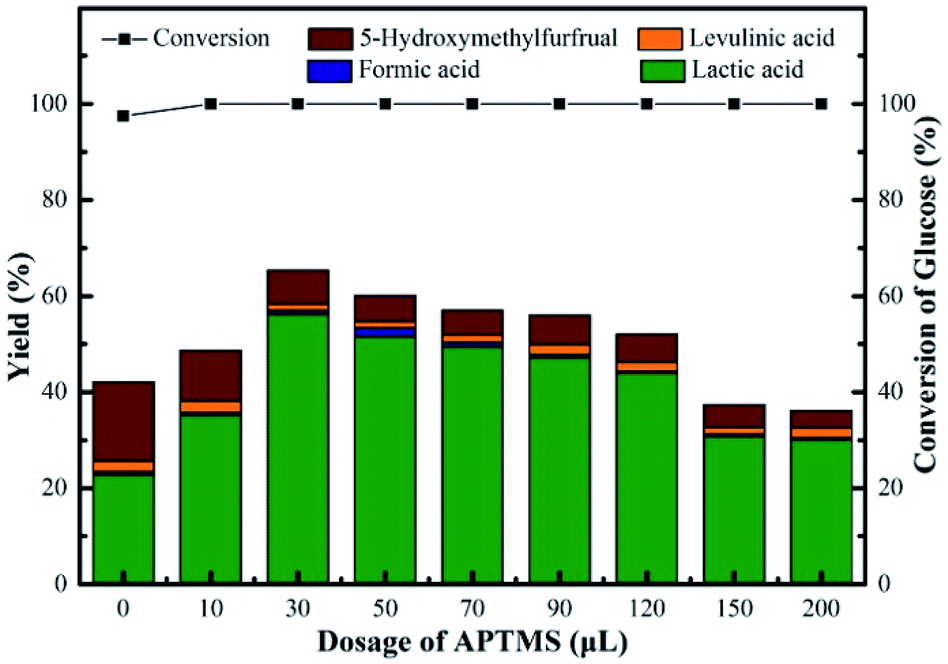 | ||
| Fig. 7 Conversion of glucose and yield of LA, FA, LeA and HMF over various NH2-supporting catalysts. Reaction conditions: 225 mg of glucose, 160 mg of catalyst, 10 mL of H2O, 190 °C, 2 h. | ||
Fig. 8 and S2† revealed that the reaction time and reaction temperature had a great effect on both the conversion of glucose and LA yield. At a relatively low reaction temperature (below 190 °C), the yield of LA first increased and then became almost constant with the increase of reaction time. When the reaction temperature was 150 °C, the LA yield increased from 10 to 32%, in parallel with the increase in the conversion of glucose (91–98%) as the reaction time prolonged (Fig. S2†). The poor yields should be attributed to the unfavorable temperature condition. When the reaction temperature increased to 190 °C, a remarkable LA yield of 56% was achieved for a 2 h reaction. As the reaction proceeded, no significant differences in the yield of LA were observed for a longer reaction time. However, a sharp decrease in LA yield was observed when the reaction temperature was over 190 °C, which might be caused by the inactivation of the supporting amino groups. It was suggested that 190 °C and 2 h were sufficient for the catalytic reaction, and LA was stable in the system. Moreover, it could be seen that an increase in the reaction temperature not only increased glucose conversion and maximum LA yield, but also shortened the reaction time needed to reach the maximum yield of LA.
 | ||
| Fig. 8 Effect of reaction temperature and reaction time on yield of LA over Sn-Beta-NH2(30) catalyst. Reaction conditions: 225 mg of glucose, 160 mg of catalyst, 10 mL of H2O. | ||
Fig. 9 showed the changes in the glucose conversion and LA yield with catalyst dosage over the Sn-Beta-NH2(30) system. The conversion of glucose increased from 98 to 100% when the dosage of catalyst increased from 40 to 160 mg for a 2 h reaction, indicating that the influence of increasing catalyst amount to glucose conversion was negligible. Nevertheless, a sharp increase in LA yield (29–56%) was observed. When the catalyst dosage was 40 mg, the conversion of glucose was 98%, while the LA yield was 29%. In this case, the amount of catalytic sites was relatively insufficient, so it might took a very long period of reaction time to reach a high LA yield. Obviously, compared to other catalyst dosages, a reaction time of 2 h was not enough for the catalyst dosage of 40 mg. In the reaction process, the glucose was nearly completely converted, and some intermediates were formed, mainly including pyruvaldehyde and dihydroxyacetone according to our previous work.14 Further increase in catalyst amount (>160 mg), an inconspicuous change in LA yield was observed. The maximum LA yield of 56% was obtained in the presence of 160 mg of catalyst. The promoted LA yield was attributed to the increase in the accessibility of active sites. Overall, the conversion of glucose increased from 98 to 100%, the yield of LA increased from 29 to 56% and the change in yields of other products was insignificant with increasing catalyst dosage. Additionally, as part of catalyst efficiency, the effect of initial glucose concentration on LA yield was also investigated. As the glucose dosage increased from 45 to 675 mg, glucose conversion decreased slightly from 100 to 96%, and LA yield decreased from 67 to 23% in the presence of 160 mg of Sn-Beta-NH2(30) catalyst (Fig. S3†). The possible reason accounting for the decrease in LA yield was the reduced availability of active sites to convert glucose to LA. For practical application, apart from LA yield, the products distribution, the reaction parameters optimization and the cost of catalyst recycling are also of vital importance, thus more systematical investigation is still needed.
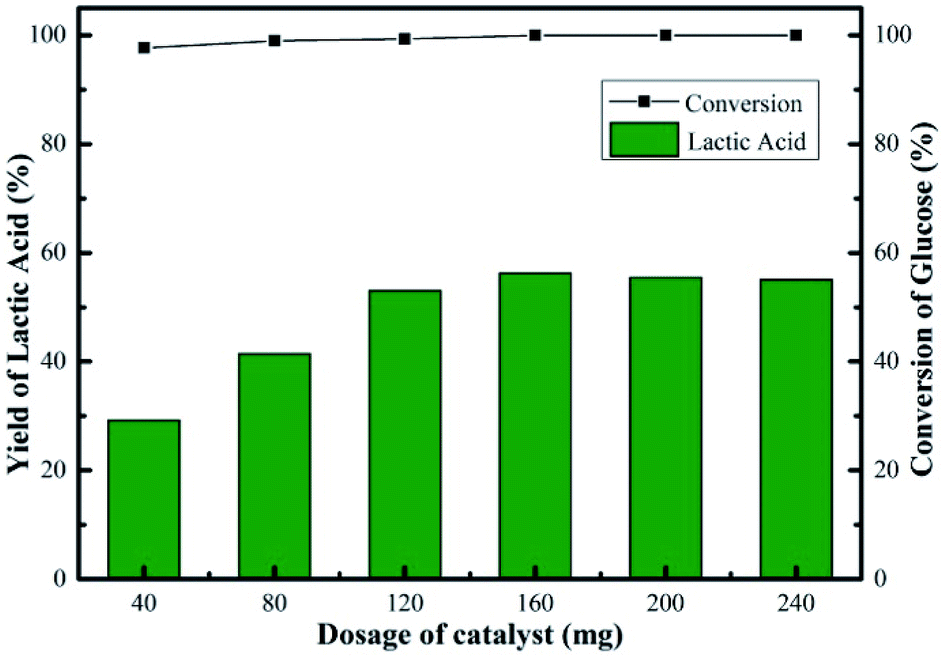 | ||
| Fig. 9 Effect of catalyst dosage on yield of LA over Sn-Beta-NH2(30) catalyst. Reaction conditions: 225 mg of glucose, 10 mL of H2O, 190 °C, 2 h. | ||
Following the study of the effect of reaction parameters, the regeneration and reuse of Sn-Beta-NH2(30) catalyst were also examined (Fig. S4†). After each run, the catalysts were separated by centrifugation and regenerated by washing with ethanol several times. Although the recycled catalysts exhibited similar crystalline structure according to their XRD patterns (Fig. S5†), the yield of LA decreased notably to 22% after recycling for 3 runs. In order to gain further insight into the reason for the decreasing LA yield, Sn content in the reaction solution after reaction and centrifugation. The result of ICP test showed that the Sn content was about 2 mg L−1, indicating that the leaking percent of Sn was below 0.3%, so the decrease of LA yield was not caused by Sn leaking. The decrease might be attributed to the screening of active sites and the prevention of glucose diffusion caused by the coke and humins attached on the surface of catalysts and the leaching of amine groups, which was in accordance with the C and N contents shown in Table S1.† Notably, the surface coke and humins of the catalysts cannot be efficiently removed even by the hydrothermal method as mentioned in the catalytic reaction above. Moreover, after the catalyst was calcined at the range of 300–550 °C for 3–6 h, the yield of LA was still 20–25%. Therefore, more systematic studies on this issue are required.
Conclusions
In conclusion, we synthesized a series of Sn-Beta-NH2 catalysts by means of SSIE and post-grafting organoamines. The catalysts contained both Lewis acid sites and moderate base sites according to the Py-IR and CO2-TPD characterization. The acid–base bifunctional catalyst could efficiently convert glucose to LA and the side reaction was well suppressed. Using Sn-Beta-NH2(30) catalyst, 56% LA yield was obtained under optimized hydrothermal conditions (190 °C, 2 h) along with only 7% HMF yield. It was evidenced that by the introduction of base sites into the Lewis acid catalyst, such a high yield of LA could be achieved. We hope that this work would be helpful for synthesizing highly efficient heterogeneous catalysts for biomass conversion.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21676205), the National Science Fund for Distinguished Young Scholars (51625804), the National Key R&D Program of China (2018YFD1100503) and the Fundamental Research Funds for the Central Universities (22120190012, 22120190054).Notes and references
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. J. Straathof, Chem. Rev., 2014, 114, 1871–1908 CrossRef CAS PubMed.
- S. Dutta, J. Kim, Y. Ide, J. H. Kim, M. S. A. Hossain, Y. Bando, Y. Yamauchi and K. C. W. Wu, Mater. Horiz., 2017, 4, 522–545 RSC.
- S. Dutta, A. Bhaumik and K. C. W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- J. Kuo, N. Suzuki, Y. Yamauchi and K. C. W. Wu, RSC Adv., 2013, 3, 2028–2034 RSC.
- Y. Y. Li and K. C. W. Wu, Phys. Chem. Chem. Phys., 2012, 14, 13914–13917 RSC.
- W. H. Hsu, Y. Y. Lee, W. H. Peng and K. C. W. Wu, Catal. Today, 2011, 174, 65–69 CrossRef CAS.
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- P. Maki-Arvela, I. L. Simakova, T. Salmi and D. Y. Murzin, Chem. Rev., 2014, 114, 1909–1971 CrossRef CAS PubMed.
- W. P. Deng, Y. Z. Wang, S. Zhang, K. M. Gupta, M. J. Hülsey, H. Asakura, L. M. Liu, Y. Han, E. M. Karp, G. T. Beckham, P. J. Dyson, J. W. Jiang, T. Tanakac, Y. Wang and N. Yan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5093–5098 CrossRef CAS PubMed.
- X. Y. Lv, B. Yu, X. W. Tian, Y. Chen, Z. J. Wang, Y. P. Zhuang and Y. H. Wang, J. Taiwan Inst. Chem. Eng., 2016, 61, 124–131 CrossRef CAS.
- W. J. Dong, Z. Shen, B. Y. Peng, M. Y. Gu, X. F. Zhou, B. Xiang and Y. L. Zhang, Sci. Rep., 2016, 6, 26713 CrossRef PubMed.
- W. P. Deng, Y. Wang and N. Yan, Curr. Opin. Green Sustain. Chem., 2016, 2, 54–58 CrossRef.
- T. L. Wang, W. P. Deng, B. J. Wang, Q. H. Zhang, X. Y. Wan, Z. C. Tang, Y. Wang, C. Zhu, Z. X. Cao, G. C. Wang and H. L. Wan, Nat. Commun., 2013, 4, 2141 CrossRef.
- X. Lei, F. F. Wang, C. L. Liu, R. Z. Yang and W. S. Dong, Appl. Catal., A, 2014, 482, 78–83 CrossRef CAS.
- S. Lux and M. Siebenhofer, Catal. Sci. Technol., 2013, 3, 1380–1385 RSC.
- F. F. Wang, C. L. Liu and W. S. Dong, Green Chem., 2013, 15, 2091–2095 RSC.
- D. Cao, W. J. Cai, W. T. Tao, S. Y. Zhang, D. Z. Wang and D. Z. Huang, Catal. Lett., 2017, 147, 926–933 CrossRef CAS.
- Y. Q. Wang, F. M. Jin, M. W. Sasaki, F. W. Wang, Z. Z. Jing and M. Goto, AIChE J., 2013, 59, 2096–2104 CrossRef CAS.
- W. Zeng, D. G. Cheng, F. Q. Chen and X. L. Zhan, Catal. Lett., 2009, 133, 221–226 CrossRef CAS.
- F. Chambon, F. Rataboul, C. Pinel, A. Cabiac, E. Guillon and N. Essayem, Appl. Catal., B, 2011, 105, 171–181 CrossRef CAS.
- F. F. Wang, J. Liu, H. Li, C. L. Liu, R. Z. Yang and W. S. Dong, Green Chem., 2015, 17, 2455–2463 RSC.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- J. Dijkmans, D. Gabriëls, M. Dusselier, F. Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 15, 2777–2785 RSC.
- M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- J. Zhang, L. Wang, G. X. Wang, F. Chen, J. Zhu, C. T. Wang, C. Q. Bian, S. X. Pan and F. S. Xiao, ACS Sustainable Chem. Eng., 2017, 5, 3123–3131 CrossRef CAS.
- Y. Wan, M. Q. Zhuang, S. P. Chen, W. D. Hu, J. Sun, J. D. Lin, S. L. Wan and Y. Wang, ACS Catal., 2017, 7, 6038–6047 CrossRef CAS.
- X. C. Wang, Y. L. Song, C. P. Huang, F. B. Liang and B. H. Chen, Green Chem., 2014, 16, 4234–4240 RSC.
- L. D. White and C. P. Tripp, J. Colloid Interface Sci., 2000, 232, 400–407 CrossRef CAS PubMed.
- J. Yang, B. Tang, W. L. Dai, L. D. Li, G. J. Wu and N. J. Guan, Sci. Sin.: Chim., 2015, 45, 396–404 CAS.
- S. G. Wang, Catal. Commun., 2003, 4, 469–470 CrossRef CAS.
- J. C. Hicks, R. Dabestani, A. C. Buchanan and C. W. Jones, Chem. Mater., 2006, 18, 5022–5032 CrossRef CAS.
- M. W. McKittrick and C. W. Jones, J. Am. Chem. Soc., 2004, 126, 3052–3053 CrossRef CAS PubMed.
- K. K. Sharma and T. Asefa, Angew. Chem., Int. Ed., 2007, 119, 2937–2940 CrossRef.
- B. Xu, Y. Q. Liu, J. G. Wang, X. Q. Ren, W. M. Ma and W. L. Wu, J. Chem. Eng. Chin. Univ., 2015, 29, 335–340 CAS.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- P. Wolf, M. Valla, A. J. Rossini, A. Comas-Vives, F. Nunez-Zarur, B. Malaman, A. Lesage, L. Emsley, C. Coperet and I. Hermans, Angew. Chem., Int. Ed., 2014, 53, 10179–10183 CrossRef CAS.
- H. Y. Luo, L. Bui, W. R. Gunther, E. Min and Y. Román-Leshkov, ACS Catal., 2012, 2, 2695–2699 CrossRef CAS.
- M. Chong and X. S. Zhao, J. Phys. Chem. B, 2003, 103, 12650–12657 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01264h |
| This journal is © The Royal Society of Chemistry 2019 |