
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZnO nanorod arrays grown on g-C3N4 micro-sheets for enhanced visible light photocatalytic H2 evolution
Hengfei Qina,
Yuanhui Zuoa,
Jutao Jina,
Wenlong Wanga,
Youlong Xub,
Lifeng Cui *a and
Haifeng Dang*a
*a and
Haifeng Dang*a
aSchool of Materials Science and Engineering, Dongguan University of Technology, Dongguan 523808, PR China. E-mail: lifeng.cui@gmail.com; cehfdang@163.com
bElectronic Materials Research Laboratory, Key Laboratory of the Ministry of Education & International Center for Dielectric Research, Xi'an Jiaotong University, Xi'an, 710049, PR China
First published on 7th August 2019
Abstract
The designed synthesis of noble-metal-free photocatalysts with hierarchical heteroassemblies in a facile, mild and eco-friendly way becomes more and more important, because we can explore the novel properties and applications of novel heterostructures via this method. Herein we report a two-step aqueous strategy for novel hierarchical heterostructures of ZnO nanorod (NR) arrays grown on graphitic carbon nitride (g-C3N4). The novel g-C3N4/ZnO NR heterostructures that integrate g-C3N4 and ZnO NR via high-quality g-C3N4–ZnO heterojunctions have beneficial properties such as high specific surface area (SSA), open spatial architecture, good electronic conductivity, and effective charge transfer interfaces, and are promising in many related areas such as water splitting, solar cells, etc. As a noble-metal-free and visible-light-responsive photocatalytic material, a typical g-C3N4/ZnO NR photocatalytic system exhibits enhanced photocatalytic activity toward H2 evolution, almost 3.5 times higher than that of pure g-C3N4. The superior photocatalytic property can be ascribed to the synergistic effect of the unique g-C3N4/ZnO NR heterostructures.
Introduction
With the aggravation of increasing global energy demands and environment issues, the development of clean and renewable energy becomes a promising way to improve the situation.1 Photocatalysis has attracted increasing attention in the area of photocatalytic water splitting for H2 evolution.2 Until now, for water splitting photocatalysts, a huge number of semiconductor materials and nanoheterostructures of materials have been searched out.3–9 Newly, g-C3N4, a metal-free photocatalyst only consisting of carbon and nitrogen, has been followed closely with worldwide focus due to its various excellent merits, for example, visible light response, high photochemical stability and easily-modified textural/electronic structure.10 Moreover, ZnO is an important wide-band gap (3.37 eV) and non-toxic semiconductor which has been extensively studied owing to its abundant easy-to-prepare morphologies and high optical, electrical and catalytic properties.11–20 However, the photocatalytic performance of pristine g-C3N4 and ZnO are mediocre, mainly because the high recombination ratio of photo-induced electron–hole pairs, the insufficient optical absorption, the small SSA and photocorrosion have limited the practical photocatalytic activity.21 In order to ameliorate the target materials' photocatalytic properties, many approaches have been presented. Just like the widely reported methods such as doping, morphological-controlled synthesis, coupling and sensitization, have been frequently used for modifying inorganic photocatalysts.10 Recently, ZnO hybridized with a conjugative π structure material has received special attention. For example, ZnO hybridized with g-C3N4 has been proved to be an effective method for enhancing photocatalytic activity.12 Owing to their superior physical/chemical properties, g-C3N4/ZnO heterostructures have shown higher photocatalytic properties than the pristine. It is generally recognized that the π-conjugated structure promotes separation of charge and transfer efficiency of photoinduced electron–hole pairs at interfaces of semiconductors.22 As we know, a variety of g-C3N4/ZnO NR heterostructures with different structures and dimensions have been synthesized including g-C3N4/ZnO nanoparticles (NPs),23–25 g-C3N4/ZnO core–shell structures26 or g-C3N4/ZnO core–shell nanoplates (NPLs).27 However, these heterojunctions mainly consist of isolated phases of the two components. It will certainly create plenty of crystal interfaces with many defects and electron traps, which are detrimental to the transport and output of carriers and the performance of these heterostructure-based devices.11,28–32 To solve this problem, here we refine the synthetic strategy to a novel advanced hierarchical g-C3N4/ZnO heterostructure where ZnO NR arrays are growing on the surface of g-C3N4 which has been realized through a two-step process involving ZnO seed coating and subsequent heteroepitaxial growth of ZnO NRs (see Scheme 1).11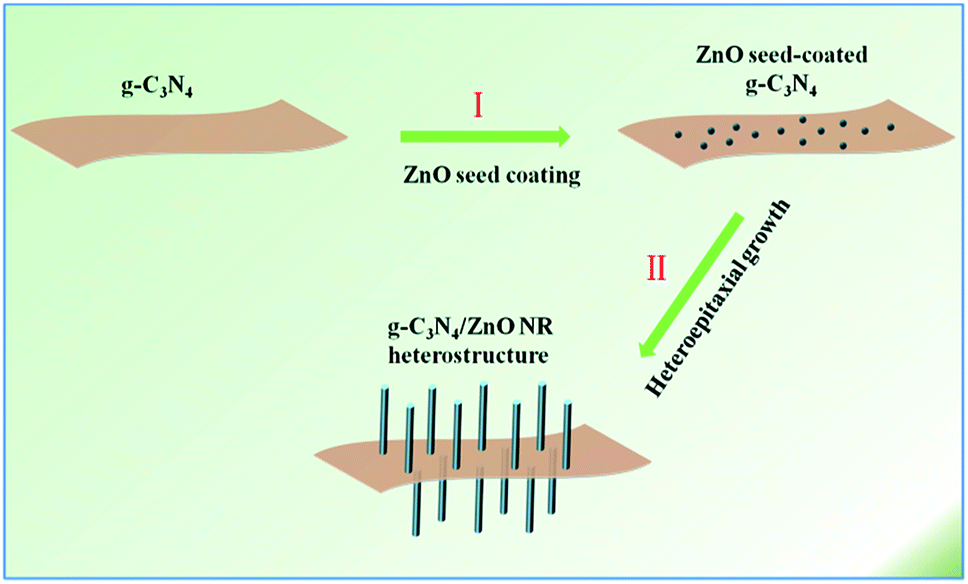 | ||
| Scheme 1 Schematic illustration of the procedure of hierarchical g-C3N4/ZnO heterostructure. | ||
Results and discussion
Fig. 1 shows the typical heterostrucures of g-C3N4/ZnO NR heterostructures. From an overview SEM image in Fig. 1a, it can be observed that a delicate herbosa-like heteroassemblies have been fabricated. The g-C3N4 denoted with the red dotted arrow is the symmetry plane covered by ZnO NR arrays on its both sides. In Fig. 1b, g-C3N4/ZnO NR heterostructure outlined in yellow is selected for close observation. However, it is difficult to find g-C3N4 in the heterostructure due to dense covering of ZnO NR arrays on their surfaces. Fig. 1c presents the typical TEM image of g-C3N4 nanosheet with soft edges. Fig. 1d shows the TEM image of g-C3N4/ZnO NR heterostructure. It can be seen that ZnO NRs are closely coupled with g-C3N4. | ||
| Fig. 1 SEM (a and b), TEM (d) images of g-C3N4/ZnO NR heterostructure; SEM (c) of pure g-C3N4. | ||
The XRD pattern of g-C3N4, ZnO, and g-C3N4/ZnO heterostructures samples were shown in Fig. 2. It can be seen clearly a strong peak at 2θ = 27.5° corresponding to the characteristic (002) crystal planes of g-C3N4 and distinct diffraction peak at 31.8°, 34.0°, 36.3°, 47.5°, 56.9°, 62.9° and 68.0° corresponding to the hexagonal-structured wurtzite ZnO (JCPDS, no. 36-1451), which confirms that the heterostructures are composed of g-C3N4 and ZnO.
 | ||
| Fig. 2 XRD patterns of the prepared g-C3N4, ZnO, g-C3N4/ZnO NR heterostructure. | ||
The surface chemical composition and chemical state of g-C3N4/ZnO NRs heterostructure was measured using XPS analysis. As shown in Fig. 3, the survey spectrum shows the distinctive peaks corresponding to carbon (C), nitrogen (N), zinc (Zn), oxygen (O), indicate the formation of g-C3N4/ZnO heterostructure. The high resolution spectra of the prepared g-C3N4/ZnO NRs heterostructure was composed of ZnO and g-C3N4. The spectra of Zn 2p exhibited two major peaks at 1023.63 eV and 1046.47 eV aroused from Zn 2p3 and Zn 2p1, respectively (Fig. 3a). The position of these peaks illustrates the presence of Zn in the form of ZnO. The broad peak centered at 531.68 eV was related to core level of O in ZnO (Fig. 3b). In case of C 1s, four different peaks were found in deconvoluted spectra (Fig. 3c). The major peak at 284.6 eV stemmed from graphitic carbon nitride phase with low graphitic value. Relatively low intense peak at 287.96 eV originated from sp2-hybridized carbon in N-containing aromatic rings C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C g-C3N4 in network. Additional, two peaks at 286.16 eV and 289.26 eV correspond to CN coordination and OH–CO bonds, respectively. Finally, high resolution spectra of N 1s displayed three peaks at 398.19 eV, 399.66 eV and 401 eV which are attributed to sp2 nitrogen in triazine ring. The bridging N atoms in N(–C)3 and terminal amino groups (C–N–N) from incomplete condensation during the thermal polymerization process. From these we could say that the heterostructure exhibited ZnO as well as g-C3N4 phases.
C g-C3N4 in network. Additional, two peaks at 286.16 eV and 289.26 eV correspond to CN coordination and OH–CO bonds, respectively. Finally, high resolution spectra of N 1s displayed three peaks at 398.19 eV, 399.66 eV and 401 eV which are attributed to sp2 nitrogen in triazine ring. The bridging N atoms in N(–C)3 and terminal amino groups (C–N–N) from incomplete condensation during the thermal polymerization process. From these we could say that the heterostructure exhibited ZnO as well as g-C3N4 phases.
 | ||
| Fig. 3 XPS spectrum of g-C3N4/ZnO NRs heterostructure (a) survey spectrum, (b) Zn 2p, (c) O 1s, (d) C 1s, (e) N 1s. | ||
The UV-vis diffuse reflection spectra of dispersed ZnO NRs, bare g-C3N4 and g-C3N4/ZnO NRs heterostructures are shown in Fig. 4. Pure g-C3N4 has photoabsorption from UV light to visible light, with an absorption edge of 471 nm, originating from a band gap of 2.63 eV. In the case of ZnO, only UV light can be absorbed, the band gap is determined to be 3.04 eV. The addition of ZnO shows little effect on the optical property of g-C3N4. As shown in Fig. 4, the UV-vis spectrum of g-C3N4/ZnO NRs heterostructure is nearly the same as pure g-C3N4. The result suggests that g-C3N4/ZnO NRs heterostructure can absorb visible light.
 | ||
| Fig. 4 UV-vis diffuse reflection spectra of the prepared g-C3N4, ZnO, g-C3N4/ZnO NR heterostructure. | ||
Photocatalytic H2 production catalyzed by different catalysts was evaluated via visible light (λ > 400 nm) irradiation in aqueous solution containing 0.2 M Na2S and 0.5 M Na2SO3 as sacrificial agents. It can be seen that (Fig. 5), when g-C3N4 alone was used as photocatalyst, the H2-production rate is not high, which could be ascribed to the rapid recombination of photoinduced carriers generated under visible light. After the combination g-C3N4 with ZnO, the H2 production performance is significantly enhanced. The H2-production activity of g-C3N4/ZnO NRs heterostructure under visible light was 3.5 times higher than that of pure g-C3N4. We also conducted a comparative experiment using a mechanical mixture of g-C3N4 and ZnO with the optimum composition ratio as the photocatalyst. The result showed that the H2 production rate was much lower than that of g-C3N4/ZnO sample. We think this remarkable improvement is likely resulted from the unique 2D structure of g-C3N4 which promotes a faster and more efficient electron transport and export, and significantly enhance the chance for electron acceptors to grasp electron. In brief, firstly, g-C3N4 connects more ZnO NRs, offering more integrated g-C3N4–ZnO heterojunctions for better photoinduced hole–electron separation. Secondly, g-C3N4 can be equivalent to 2D planes consisted of large number of interwoven and serve as spatially-extended electron active centers for electron acceptors (SSA: 31.6 m2 g−1). It can be imaged that photogenerated electrons can be transferred directly from the conduction band (CB) of g-C3N4 to the CB of double-side ZnO NR arrays and the reductive half reactions can be performed, which actually improve the reaction chances and thus prompt the catalysis proceeding.
 | ||
| Fig. 5 Photocatalytic H2 evolution on g-C3N4, ZnO, mixture of g-C3N4 and ZnO, g-C3N4/ZnO NR heterostructure. | ||
Recycling performance was carried out to investigate the stability of the composition-optimized product. The H2 generation in different runs is displayed in Fig. 6. No remarkable degradation of H2 generation is observed in the repeated runs for the photocatalytic reaction of 9 h under visible light illumination.
 | ||
| Fig. 6 Reusability experiment for photocatalytic H2 generation by the g-C3N4/ZnO NR heterostructure under visible light. | ||
To uncover the separation, transfer of photoinduced eletron–hole pairs at the interfaces of heterostrucures, photoluminescence (PL) spectra was collected to investigate the photogenerated electron–hole pairs separation and electron transfer performance between g-C3N4 and ZnO.34,35 The PL spectrum of pure g-C3N4, mixture of g-C3N4 and ZnO, and g-C3N4/ZnO NRs heterostructure were characterized respectively (Fig. 7). It can be observed that the PL spectrum of pure g-C3N4 excited at 380 nm showed a strong emission peak at around 445 nm. In the PL spectrum of g-C3N4/ZnO NRs heterostructure, a weaker emission peak in the same position was detected, suggesting that the addition of ZnO significantly inhibits the recombination of photogenerated electron–hole pairs.36 For further purposes of comparison, the PL spectrum of the physical mixture of g-C3N4 and ZnO was also presented. The result suggests that the peak of mixture of g-C3N4 and ZnO is weaker than that of pure g-C3N4, but stronger than g-C3N4/ZnO NRs heterostructure. It confirms that the charge transfer between g-C3N4 and ZnO would further suppress the recombination of photogenerated electron–hole pairs, which is consistent with the previously described mechanism by Cao et al.33
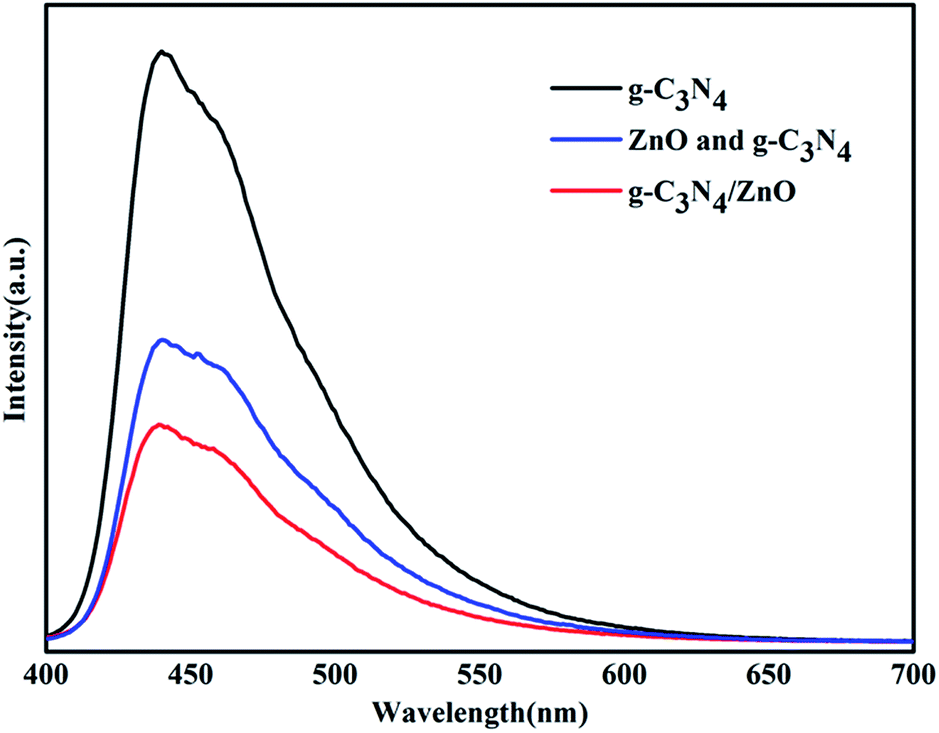 | ||
| Fig. 7 PL spectra of the prepared g-C3N4, mixture of g-C3N4 and ZnO, g-C3N4/ZnO NR heterostructure. | ||
Electrochemical impedance spectroscopy tests of pure g-C3N4, ZnO, and g-C3N4/ZnO NRs heterostructure were performed to further validate the superior effect of embedded ZnO for improving charge transfer in the samples, and the results are displayed in Fig. 8. The Nyquist plots showed in Fig. 8 were analyzed by a non-linear least squares (NLLS) analysis program. It is well known that the radius of the semicircular arcs on the EIS Nyquist plots reflects the interfacial charge transfer processes occurring on the electrode surface. The larger the arc radius on EIS Nyquist plots, the higher the charge transfer resistance of the sample is. As shown in Fig. 8, the size of the semicircle arc radius of g-C3N4/ZnO NRs electrode was obviously smaller than that of g-C3N4 and ZnO electrode. The decreased arc radius on the EIS Nyquist plot of g-C3N4/ZnO NRs sample implied more efficient separation of photoinduced electron–hole pairs and higher interfacial charge transfer efficiency in the g-C3N4/ZnO NRs product.
 | ||
| Fig. 8 EIS Nyquist plots of pure ZnO, g-C3N4 and g-C3N4/ZnO. | ||
According to the aforementioned results, the proposed mechanism for H2 evolution by the typical g-C3N4/ZnO NRs sample is demonstrated in Fig. 9. Under visible light irradiation, photo-induced electron–hole pairs are generated on the surface of g-C3N4, while ZnO itself cannot be excited by visible light. Because the CB position of ZnO (−0.2 eV vs. SHE) is lower than that of the conduction bond (CB) of g-C3N4 (−1.1 eV vs. SHE), the photoelectrons generated from g-C3N4 will inject quickly into the CB of double-side ZnO NR arrays by the well developed interface while photoholes leave in g-C3N4. During the process, ZnO NR arrays and g-C3N4 work as both 1D/2D spatial pathways for fast electron/hole transport and spatial active sites for efficient catalytic reductive and oxidative reactions. The rapidly proceeding of the oxidative and reductive half reactions leads to the high solar-energy conversion efficiency of photocatalysis. We emphasize here that double-side ZnO NR arrays with opened structure can offer more accessible active sites for H2 formation. As a result, the high photocatalytic H2 production activity was achieved over g-C3N4/ZnO NRs heterostructures.
 | ||
| Fig. 9 (a) Schematic illustrations for the photo-induced charge separation over g-C3N4/ZnO NRs heterostructures. (b) The speculated photocatalytic mechanism over g-C3N4/ZnO NRs heterostructures. | ||
Conclusions
In summary, the novel hierarchical g-C3N4/ZnO NRs with double-side ZnO NR arrays grown on g-C3N4 have been fabricated via a ZnO-seeded heteroepitaxial growth of ZnO NRs on g-C3N4. The typical noble-metal-free heterostructure of g-C3N4/ZnO NRs showed enhanced photocatalytic activity in photocatalytic measurements of H2 evolution compared with the other catalysts under visible light irradiation. The H2 evolution rate of g-C3N4/ZnO NRs is about 3.5 times higher than that of bare g-C3N4 catalysts and about 1.8 times relative to bare ZnO catalysts. The remarkable improvement in photocatalytic efficiency is ascribed to the synergistic contribution of the unique structure of the g-C3N4/ZnO NRs, such as high SSA supplying abundant active sites for effective catalyzing, 1D/2D ZnO NR/g-C3N4 structures with integrated contact for effective charge separation and high rate 1D/2D electron transport, and special spatial configuration consisting of double side NR arrays and accessible architecture for effective diffusion. With these structural advantages, the novel g-C3N4/ZnO heterostructures when made some suitable modifications also are promising in many related areas such as energy conversion and storage, electro-chemistry and so on.Experimental
Materials
The urea (99%) used in the experiment was supplied by Sigma-Aldrich. All of the other chemical reagents were of analytical grade without further purification, and all the water used in the experiment was deionized water.Preparation of g-C3N4
About 15 g of urea was put in a quartz crucible with a cover and was heated at 550 °C for 2 h in air. In addition, the heating rate was 2 °C min.Preparation of g-C3N4/ZnO heterostructure
For a typical synthesis, firstly, the g-C3N4 powder were dispersed in 1 g L−1 poly-(vinylpyrrolidone) (PVP) aqueous solution for 2 h, followed by washing with ethanol several times to remove excessive adsorbed PVP molecules. Next, the PVP-treated g-C3N4 were soaked in the pre-prepared ZnO seed solution.9 Four hours later, the g-C3N4 with coated ZnO seeds were centrifugalized and washed with ethanol twice. Then, the obtained ZnO seed-coated g-C3N4 sample were scattered around in a 40 mL aqueous solution composed of equimolar zinc acetate (Zn(CH3COO)2, 0.02 M) and hexamethylenetetramine (HMT, 0.02 M). After incubation at 85 °C for 6 h, the product was separated with the centrifuge method, washed by deionized water several times, dried in stove and reserved for further use.Characterization
The morphology of g-C3N4/ZnO NR heterostructures was characterized using a field emission scanning electron microscope (FESEM, JSM-7000F, 5 kV) and high-resolution transmission electron microscopy (HR-TEM, JEM 2011, acceleration voltage 200 kV). The crystal structure was determined by X-ray diffraction (XRD) using a D8 Advance X-ray diffractometer with Cu Kα radiation with a 1.5418 Å wavelength (Bruker AXS, German). The accelerating voltage of 40 kV and a 40 mA applied current beam were used. X-ray photoelectron spectroscopy (XPS) was characterized on a Thermo Fisher Scientific Escalab 250 spectrometer with monochromatized Al Kα radiation. UV-vis diffuse reflectance spectra of the samples were performed on a Cary-50 UV-vis spectrophotometer. Photoluminescence (PL) spectra were recorded at room temperature with a fluorescence spectrophotometer (F-7000, Hitachi, Japan). The excitation wavelength was 380 nm. The Brunauer–Emmett–Teller (BET) surface area of the sample was evaluated by N2 adsorption–desorption measurements at 77 K in an automated surface area and porosity analyzer (ASAP2020, Micromeritics, USA).Evaluation of photocatalytic H2 evolution
The photocatalytic reactions were carried out in a quartz reaction vessel connected to a closed gas circulation and evacuation system. The diameter of the quartz container was 7.5 cm, and the height was 12 cm. A 300 W Xe lamp (HSX-F/UV 300, Bjnbet Co., Ltd., China) equipped with a UV cut-off filter (λ > 400 nm) was used as the visible light source and the integrated light intensity on the reaction reactor was about 60 mW cm−2. The temperature of the reaction system was maintained at ca. 293 K through programmed temperature control device. Before each experiment, the closed system was degassed with N2 for 0.5 h to drive away the O2. For each experiment, 0.1 g catalyst was suspended into 250 mL aqueous solution with 0.2 M Na2S and 0.5 M Na2SO3 as sacrificial reagent. The gas product was examined with an on-line gas chromatography with a thermal conductivity detector (GC2600, N2 carrier, 5A molecular sieve column).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Startup Research Fund of Dongguan University of Technology (KCYKYQD2017015) and Training Program of Innovation and Entrepreneurship for Undergraduates of Dongguan University of Technology (201911819133).Notes and references
- Z. A. Huang, Q. Sun, K. L. Lv, Z. H. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420 CrossRef CAS.
- X. Q. Fan, L. X. Zhang, R. L. Cheng, M. Wang, M. L. Li, Y. J. Zhou and J. L. Shi, ACS Catal., 2015, 5, 5008 CrossRef CAS.
- S. E. Habas, P. D. Yang and T. J. Mokari, J. Am. Chem. Soc., 2008, 130, 3294 CrossRef CAS PubMed.
- C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2004, 43, 4774 CrossRef CAS PubMed.
- F. R. Fan, Y. Ding, D. Y. Liu, Z. Q. Tian and Z. L. Wang, J. Am. Chem. Soc., 2009, 131, 12036 CrossRef CAS PubMed.
- K. Sun, Y. Jing, N. Park, C. H. Li, Y. S. Bando and D. L. Wang, J. Am. Chem. Soc., 2010, 132, 15465 CrossRef CAS PubMed.
- J. S. Lee, E. V. Shevchenko and D. V. Talapin, J. Am. Chem. Soc., 2008, 130, 9673 CrossRef CAS PubMed.
- L. Wu, B. G. Quan, Y. L. Liu, R. Song and Z. Y. Tang, ACS Nano, 2011, 5, 2224 CrossRef CAS PubMed.
- J. Yang and J. Y. Ying, Angew. Chem., Int. Ed., 2011, 50, 4637 CrossRef CAS PubMed.
- Y. M. He, Y. Wang, L. H. Zhang, B. T. Teng and M. H. Fan, Appl. Catal., B, 2015, 168, 1 Search PubMed.
- Y. H. Zuo, Y. Qin, C. Jin, Y. Li, S. D. Shi, Q. S. Wu and J. H. Yang, Nanoscale, 2013, 5, 4388 RSC.
- A. I. Hochbaum and P. D. Yang, Chem. Rev., 2010, 110, 527 CrossRef CAS PubMed.
- Z. W. Pan, Z. R. Dai and Z. L. Wang, Science, 2001, 291, 1947 CrossRef CAS PubMed.
- Z. L. Wang and J. H. Song, Science, 2006, 312, 242 CrossRef CAS PubMed.
- J. Yin, Y. S. Zang, C. Yue, Z. M. Wu, S. T. Wu, J. Li and Z. H. Wu, J. Mater. Chem., 2012, 22, 7902 RSC.
- M. H. Huang, S. Mao, H. Feick, H. Q. Yan, Y. Y. Wu, H. Kind, E. Weber, R. Russo and P. D. Yang, Science, 2001, 292, 1897 CrossRef CAS PubMed.
- S. H. Ko, D. Lee, H. W. Kang, K. H. Nam, J. Y. Yeo, S. J. Hong, C. P. Grigoropoulos and H. J. Sung, Nano Lett., 2011, 11, 666 CrossRef CAS PubMed.
- M. H. Hsu, C. J. Chang and H. T. Weng, ACS Sustainable Chem. Eng., 2016, 4, 1381 CrossRef CAS.
- M. H. Hsu and C. J. Chang, Int. J. Hydrogen Energy, 2014, 39, 16524 CrossRef CAS.
- M. H. Hsu and C. J. Chang, J. Hazard. Mater., 2014, 278, 444 CrossRef CAS PubMed.
- D. C. Chen, K. W. Wang, D. G. Xiang, R. L. Zong, W. Q. Yao and Y. F. Zhu, Appl. Catal., B, 2014, 147, 554 CrossRef CAS.
- Y. J. Wang, R. Shi, J. Lin and Y. F. Zhu, Energy Environ. Sci., 2011, 4, 2922 RSC.
- C. Liu, C. M. Li, X. D. Fu, F. Z. Raziq, Y. Qu and L. Q. Jing, RSC Adv., 2015, 5, 37275 RSC.
- W. Liu, M. L. Wang, C. X. Xu and S. F. Chen, Chem. Eng. J., 2012, 5, 386 CrossRef.
- W. Liu, M. L. Wang, C. X. Xu, S. F. Chen and X. L. Fu, J. Mol. Catal. A: Chem., 2013, 368, 9 CrossRef.
- J. Wang, Y. Xia, H. Y. Zhao, G. F. Wang, L. Xiang and J. L. Xu, Appl. Catal., B, 2006, 63, 305 CrossRef.
- S. Kumar, A. Baruah, S. Tonda, B. Kumar, V. Shanker and B. Sreedhar, Nanoscale, 2014, 6, 4830 RSC.
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Dalton Trans., 2012, 41, 6756 RSC.
- R. Hao, G. H. Wang, H. Tang, L. L. Sun, C. Xu and D. Y. Han, Appl. Catal., B, 2016, 187, 47 CrossRef CAS.
- J. W. Zhou, M. Zhang and Y. F. Zhu, Phys. Chem. Chem. Phys., 2015, 17, 3647 RSC.
- D. M. Chen, K. W. Wang, T. Z. Ren, H. Ding and Y. F. Zhu, Dalton Trans., 2014, 43, 13105 RSC.
- L. Zu, Y. Qin and J. Yang, J. Mater. Chem. A, 2015, 3, 10209 RSC.
- S. W. Cao, X. F. Liu, Y. P. Yuan, Z. Y. Zhang, Y. S. Liao, J. Fang, S. C. Joachim Loo, T. C. Sun and C. Xue, Appl. Catal., B, 2014, 147, 940 CrossRef CAS.
- W. K. Jo, J. Y. Lee and N. C. S. Selvam, Chem. Eng. J., 2016, 289, 306 CrossRef CAS.
- H. Dang, G. Tan, W. Yang, F. Su, H. Fan, X. Dong and L. Ye, J. Taiwan Inst. Chem. Eng., 2017, 78, 185 CrossRef CAS.
- J. Wang, Z. Yang, X. Gao, W. Yao, W. Wei, X. Chen, R. Zong and Y. Zhu, Appl. Catal., B, 2017, 217, 169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |