
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt(II)-catalyzed benzylic oxidations with potassium persulfate in TFA/TFAA†
Tianlei Li‡
 a,
Jishun Li‡abc,
Zihao Zhua,
Weidong Pan*b and
Song Wu*a
a,
Jishun Li‡abc,
Zihao Zhua,
Weidong Pan*b and
Song Wu*a
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, 100005, China. E-mail: ws@imm.ac.cn
bState Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Medcial University, Guiyang 550014, China. E-mail: wdpan@163.com
cGuizhou University of Traditional Chinese Medicine, Huaxi University Town, Guiyang, Guizhou 550025, China
First published on 4th July 2019
Abstract
A cobalt-catalyzed C(sp3)–H oxygenation reaction to furnish aldehyde was herein reported. This transformation demonstrated high chemo-selectivity, and tolerated various methylarenes bearing electron-withdrawing substituents. This reaction provided rapid access to diverse aldehydes form methylarenes. Notably, TFA/TFAA was used for the first time as a mixed solvent in cobalt-catalyzed oxygenation of benzylic methylenes.
Introduction
Benzylic oxidative reaction is one of the most useful and important C–H functionalization in synthetic organic chemistry due to its wide application in the manufacture of pharmaceuticals and chemicals,1 and these transformations are able to construct various functional groups such as ketone, aldehyde, hydroxyl, carboxylic, ester etc. Numerous approaches for the C(sp3)–H benzylic oxygenation to the corresponding carbonyl derivatives have been developed.1,2 Traditional methods involving the use of stoichiometric amounts of metallic reagents such as KMnO4, Cr(VI) or potassium dichromate2a are common. In the past few decades, many efforts have been devoted to develop oxidation processes catalyzed by transition metals, such as Mn,3 Cr,4 Fe,5 Cu,6 Bi,7 Rh,8 Ru,9 Pd,10 Re,11 Au12 and V13 species among others.14, and fruitful progresses have been achieved.With the development of C–H functionalization that catalyzed by low-cost and environmentally benign first-row transition metals,15 various cobalt-catalyzed direct benzylic or allylic oxidation reactions have been reported (Scheme 1).16–21 Pioneered by Ishii et. al., cobalt-catalyzed C–H(sp3) oxidation with N-hydroxyphthalimide (NHPI) has proven to be a valuable method for the preparation of ketones from alkylarenes and benzoic acids form methylarenes.16 Following this strategy, Stahl's group reported that benzylic methylene groups in pharmaceutically relevant heterocyclic substrates could be effectively converted into the corresponding ketones by the cobalt(II)/N-hydroxyphthalimide (NHPI) catalyst system.17 Recently, Pappo and co-workers also revealed the unique chemoselectivity for aerobic autoxidation of methylarenes to benzaldehydes based on N-hydroxyphthalimide (NHPI) and cobalt(II) acetate in 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP). The fluorinated alcohol and benzaldehyde may form a H-bond adduct that markedly slow down H-abstraction of the aldehydic C–H bond.18 It is worth mentioning that this strategy address the long-standing selectivity problem of generating benzaldehydes directly from methylarenes under sustainable conditions.
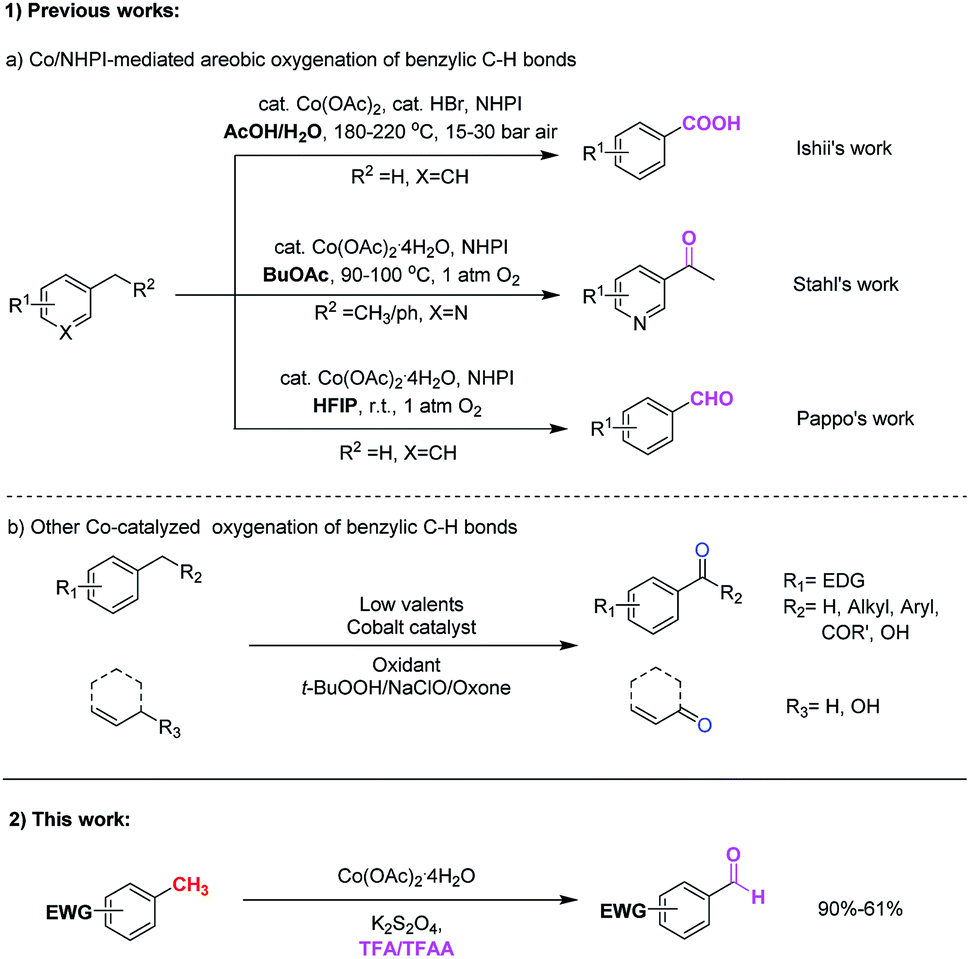 | ||
| Scheme 1 Cobalt-catalyzed C–H functionalization (benzylic and allylic oxidation reactions). | ||
Other cobalt-catalyzed systems such as TEMPO/Co(OAc)2,19 tert-butyl hydroperoxide/Co(acac)2,20 and oxone/Co(ClO4)2 (ref. 21) were developed for the allylic and benzylic oxidation of alkylarenes and methylarenes. The desired ketones derivatives are effectively obtained under these conditions. However, the large excess of oxidants were often required. In addition, the typically more reactive aldehydes are generally difficult to prepare directly from the corresponding methylarenes under most of the above methods.
Despite these significant advancements made in the area of low-valent cobalt-catalyzed benzylic oxygenation, but the selective oxidation of methylarenes to form benzaldehydes is still very challenging. Moreover, the efficiency of C(sp3)–H benzylic oxygenation were severely restricted by the electronic property of additional substituents on the aryl ring. With the aryl rings bearing electron-donating groups, the corresponding C(sp3)–H benzylic oxygenation smoothly occurred. While electron-withdrawing groups on the aryl rings generally hampered this transformation.16–21 Up to date, the direct C(sp3)–H benzylic oxygenation reactions for the preparation of benzaldehyde that could tolerate with electron-withdrawing groups on the aryl ring are still rare. In consequence, the discovery of a new method for direct benzylic oxidation tolerated with electron-withdrawing groups on the aryl ring would be of considerable importance. Herein, we report a new method for the direct benzylic C(sp3)–H bond oxidation through cobalt catalysis to afford a series of corresponding aldehydes. This method manifests highly chemo-selectivity, and tolerates with various methylarenes bearing electron-withdrawing substituents. The reaction employs the low cost Co(OAc)2·4H2O as the catalyst, K2S2O8 as the oxidant and TFA/TFAA as the co-solvent.
Results and discussion
We commenced our study with the benzylic oxidation reaction of 4-methylbenzophenone (1a) in the presence of 20 mol% of Co(OAc)2·4H2O as the catalyst, 2.0 equiv. of K2S2O8 as the oxidant in TFA/TFAA (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) co-solvent system at 100 °C. Under this condition, 2a could be obtained in 12% isolated yields. Initially, we examined this reaction in the presence of a mixted solvents of TFA/TFAA, the aldehyde product 2a was observed in less than 5% yield with the ratio 5:5 of TFA/TFAA (entry 2) and 36% yield with the ratio 9:1 of TFA/TFAA at 40 °C (entry 5). But the reaction did not occur in TFA as single solvent (entry 6). The results suggest that the ratio of TFA/TFAA play a key role in this reaction. The high proportion of TFA could dramatically improve the conversion of benzylic oxidation, and TFAA might be involved in the initiation step of cobalt-catalyzed C–H bond cleavage process.8c,22 When the reaction was performed at 80 °C, the yield dramatically increased to 78% (entry 9). Further experiments revealed that other cobalt catalysts such as CoBr2 and CoF3 were inferior to Co(OAc)2·4H2O (entry 10–12), and the benzylic bromination product was observed when CoBr2 was used as a catalyst. As a control reaction, the benzylic oxidation could not occur in the absence of Co(OAc)2·4H2O (entry 13). Among the oxidants investigated, Na2S2O8 was less efficient than K2S2O8 (entry 10). (NH4)2S2O8 was much more powerful than K2S2O8. However, the decomposition was observed when (NH4)2S2O8 was used instead of K2S2O8 (entry 11). The commonly applied oxidants NFSI and PhI(OAc)2 were proved to be inactive (entry 7–8). In general, 20 mol% of Co(OAc)2·4H2O was enough to catalyze this transformation, the process typically went to completion within 10 h with 2.0 equivalents of K2S2O8 at 80 °C Table 1.
:4) co-solvent system at 100 °C. Under this condition, 2a could be obtained in 12% isolated yields. Initially, we examined this reaction in the presence of a mixted solvents of TFA/TFAA, the aldehyde product 2a was observed in less than 5% yield with the ratio 5:5 of TFA/TFAA (entry 2) and 36% yield with the ratio 9:1 of TFA/TFAA at 40 °C (entry 5). But the reaction did not occur in TFA as single solvent (entry 6). The results suggest that the ratio of TFA/TFAA play a key role in this reaction. The high proportion of TFA could dramatically improve the conversion of benzylic oxidation, and TFAA might be involved in the initiation step of cobalt-catalyzed C–H bond cleavage process.8c,22 When the reaction was performed at 80 °C, the yield dramatically increased to 78% (entry 9). Further experiments revealed that other cobalt catalysts such as CoBr2 and CoF3 were inferior to Co(OAc)2·4H2O (entry 10–12), and the benzylic bromination product was observed when CoBr2 was used as a catalyst. As a control reaction, the benzylic oxidation could not occur in the absence of Co(OAc)2·4H2O (entry 13). Among the oxidants investigated, Na2S2O8 was less efficient than K2S2O8 (entry 10). (NH4)2S2O8 was much more powerful than K2S2O8. However, the decomposition was observed when (NH4)2S2O8 was used instead of K2S2O8 (entry 11). The commonly applied oxidants NFSI and PhI(OAc)2 were proved to be inactive (entry 7–8). In general, 20 mol% of Co(OAc)2·4H2O was enough to catalyze this transformation, the process typically went to completion within 10 h with 2.0 equivalents of K2S2O8 at 80 °C Table 1.
|
|||||
|---|---|---|---|---|---|
| Entry | Cobalt source (mol%) | Oxidants | TFA/TFAA ratio | Temperature | Yield |
| a Note: Reaction condition: 1 (0.1 mmol), cobalt catalyst (0.02 mmol), oxidant (0.15 mmol, 1.5 eq.), TFA and TFAA (1 mL).b Isolated yield.c When the conversion are less than 50%, the conversion ratio was detected by 1H-NMR.d N.O. means no observation. | |||||
| 1 | Co(OAc)2·4H2O | K2K2O8 | 6:4 |
100 °C | 12%b |
| 2 | Co(OAc)2·4H2O | K2S2O8 | 5:5 |
40 °C | <5%c |
| 3 | Co(OAc)2·4H2O | K2S2O8 | 7:3 |
40 °C | 21%c |
| 4 | Co(OAc)2·4H2O | K2S2O8 | 8:2 |
40 °C | 29%c |
| 5 | Co(OAc)2·4H2O | K2S2O8 | 9:1 |
40 °C | 36%b |
| 6 | Co(OAc)2·4H2O | K2S2o8 | 10:0 |
50 °C | <5%c |
| 7 | Co(OAc)2·4H2O | NFSI | 9:1 |
80 °C | N.O.d |
| 8 | Co(OAc)2·4H2O | Phl(OAc)2 | 9:1 |
80 °C | N.O.d |
| 9 | Co(OAc)2·4H2O | K2S2o8 | 9:1 |
80 °C | 78%b |
| 10 | Co(OAc)2·4H2O | Na2S2O8 | 9:1 |
80 °C | 41%b |
| 11 | Co(OAc)2·4H2O | (NH4)2S2O8 | 9:1 |
80 °C | 52%c |
| 12 | CoCl2 | K2S2O8 | 9:1 |
80 °C | 19%c |
| 13 | CoF3 | K2S2O8 | 9:1 |
80 °C | <5%c |
| 14 | CoBr2 | K2S2O8 | 9:1 |
80 °C | 15%c |
| 15 | Without catalysis | K2S2O8 | 9:1 |
80 °C | N.O.d |

Having identified the optimal conditions for the direct C(sp3)–H benzylic oxygenation, we set out to explore the substrates scope for this new reaction. As illustrated in Scheme 2, a range of substituted benzylic substrates were investigated. The scope of methylarenes was broad, and the transformation was smoothly occurred to selectively generate the corresponding aldehydes in moderate to good yields. Aryl groups with different substituent groups, such as Cl, Br, NO2, acetyl, were tolerated under the optimal conditions. The various substituted methylbenzophenones (2a–2d) were successfully oxidized into the desired aldehydes with good yields (78–86%). Interestingly, we found that 4,4′-dimethyl-benzophenone (2d) and methyl 3,5-dimethylbenzoate (2k) were selectively oxidized to give mono-oxidation products in 74% and 65% yields, respectively. To our delight, a range of methylbenzoates and methylpropiophenones bearing with electron-withdrawing groups (–NO2, –Br, –Cl) were smoothly transformed into the corresponding aldehydes in moderate to good yields (61–70%). Unfortunately, the electron-rich substituted methylarene substrates, such as p-toluidine, 1-methoxy-4-methylbenzene and p-cresol, could not be converted the aromatic aldehydes (2u, 2v and 2w) due to some side-reactions (Friedel–Crafts reaction, acylation etc. see ESI†) under the optimal conditions. It was revealed that the electronic property of substituents on methylarene derivatives displayed important effects on the reaction efficiency, and this strategy prefers to the electron-deficient methylarenes. As mentioned before, the electron-deficient methylarenes are difficult to be oxidized under the previous reported conditions,2,18–21 because the electron-withdrawing groups lead to the benzylic C(sp3)–H more inert. It is worth pointing out that our method provides a new access for the selective oxidation of the electron-deficient methylarenes to prepare benzaldehydes. To explore the practical utility of this C(sp3)–H benzylic oxygenation reaction, a gram-scale reaction of methyl 5-bromo-2-methylbenzoate oxidation was performed (Scheme 2) under the standard conditions. The oxidation product 2i could be obtained in 69% yield with 20 mol% of cobalt catalyst, which demonstrated scalable and practical of this protocol.
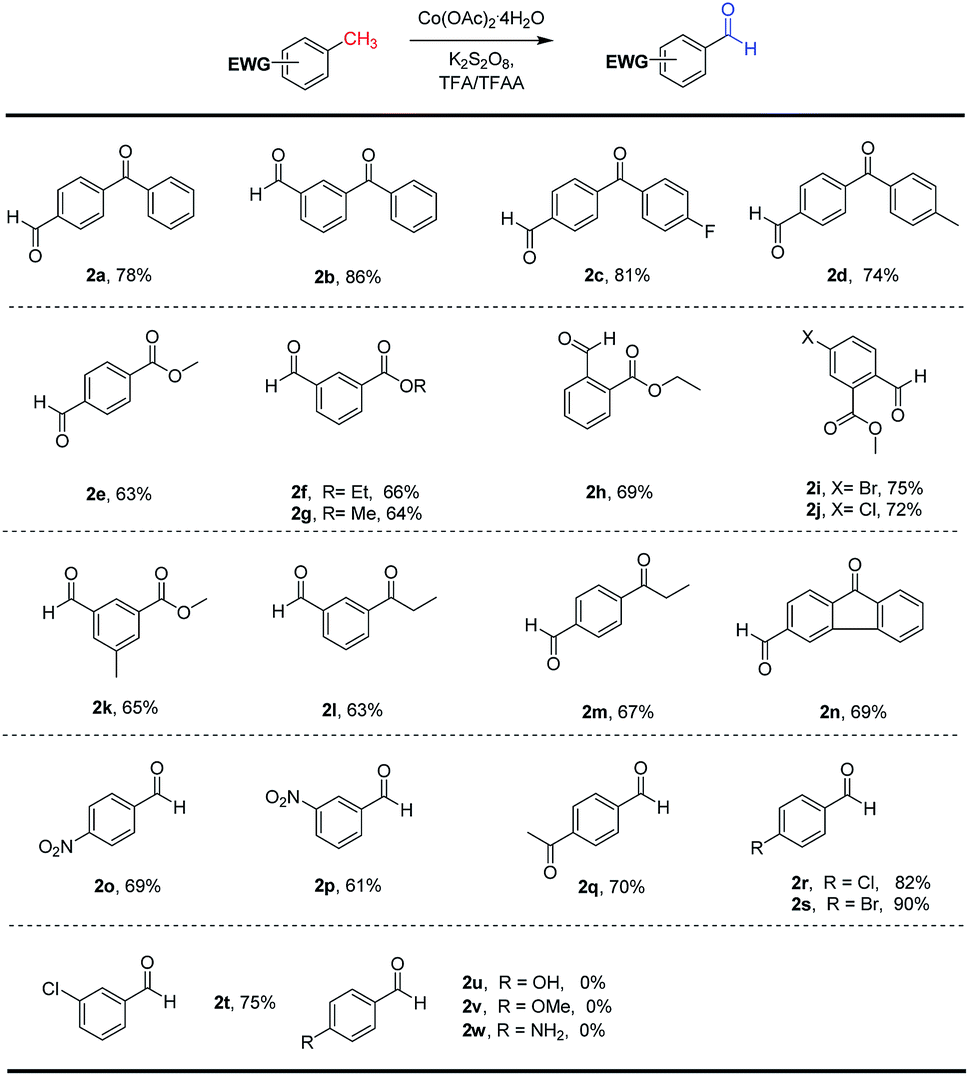 | ||
| Scheme 2 Substrate scope of cobalt(II)-catalyzed C(sp3)–H benzylic oxygenation. | ||
Previous reports proposed that a SO4˙− radical was generated in situ in the presence of cobalt.21,23 Based on literatures,23 a plausible mechanism for this oxidation process was proposed as shown in Scheme 3. Firstly, the reactions involved the oxidation of Co2+ with peroxydisulfate (S2O82−) to generate a SO4˙˙ radical in situ.23a-b Then the radical reacted with methylarenes to give the radical intermediate A radical assisted by the TFA/TFAA solvents, followed by the reduction of Co3+ to provide the intermediate B that could be detected when the reaction was proceeded at ambient temperature (see ESI†). Following with the second C–H oxidation, the intermediate C was generated, which could be further converted to the desired product after a hydrolytic process. Very recently, Fyokin and co-workers demonstrated highly polar trifluoroacetic acid could be an efficient solvent for the metal-free aerobic NHPI-catalyzed oxidations of toluene.24 In our study, the reaction cannot occur when TFA/TFAA solvent system was replaced with AcOH/Ac2O under the standard conditions.
 | ||
| Scheme 3 Proposed mechanism cobalt(II)-catalyzed C(sp3)–H oxygenation. | ||
Conclusions
In summary, we developed a new protocol for the preparation of benzylaldehydes from the corresponding methylarenes by the direct oxygenation of benzylic C(sp3)–H. The method shows high chemo-selectivity, and tolerates with various electron-withdrawing substituted arenes, esters and ketones. The TFA/TFAA solvent is first used as a mixed solvent in cobalt-catalyzed oxidation of methylarenes to benzaldehydes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No: 81703364), Chinese Academy of Medical Sciences-CAMS Innovation Fund for Medical Sciences, Grant No: 2017-I2M-1-011, China Postdoctoral Science Foundation (Grant No: 2016M590107). The authors thank Dr Tianlong LAN and Dr Chao Zhang for helpful suggestions during the preparation of this manuscript.Notes and references
- (a) R. A. Sheldon and J. K. Kochi, Metal-catalyzed Oxidation of Organic Compounds, Academic Press, New York, 1981 Search PubMed; (b) J. E. Backvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, 2010, 2nd edn CrossRef; (c) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed; (d) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- (a) M. Hudlicky, in Oxidations in Organic Chemistry; ACS Monograph No. 186, American Chemical Society, Washington, DC, 1990, For metal-free conditions Search PubMed; (b) K. K. Park, L. K. Tsou and A. D. Hamilton, Synthesis, 2006, 3617 CrossRef CAS; (c) C. Qi, H. Jiang, L. Huang, Z. Chen and H. Chen, Synthesis, 2011, 387 CAS; (d) C. Zhang, Z. Xu, L. Zhang and N. Jiao, Tetrahedron, 2012, 68, 5258 CrossRef CAS; (e) A. Dos Santos, L. El Kaim and L. Grimaud, Org. Biomol. Chem., 2013, 11, 3282 RSC; (f) L. Ren, L. Wang, Y. Lv, G. Li and S. Gao, Org. Lett., 2015, 17, 2078 CrossRef CAS PubMed; (g) K. Bao, F. Li, H. Liu, Z. Wang, Q. Shen, J. Wang and W. Zhang, Sci. Rep., 2015, 5, 10360 CrossRef PubMed.
- (a) J. F. Pan and K. Chen, J. Mol. Catal. A: Chem., 2001, 176, 19–22 CrossRef CAS; (b) Y. Li, T. B. Lee, T. Wang, A. V. Gamble and A. E. V. Gorden, J. Org. Chem., 2012, 77, 4628–4633 CrossRef CAS PubMed; (c) M. Milan, G. Carboni, M. Salamone, M. Costas and M. Bietti, ACS Catal., 2017, 7, 5903–5911 CrossRef CAS.
- (a) T. K. Das, K. Chaudhari, E. Nandanan, A. J. Chandwadkar, A. Sudalai, T. Ravindranathan and S. Sivasanker, Tetrahedron Lett., 1997, 38, 3631–3634 CrossRef CAS; (b) S. Yamazaki, Org. Lett., 1999, 1, 2129–2132 CrossRef CAS; (c) S. Boitsov, J. Songstad and J. Muzart, J. Chem. Soc., Perkin Trans. 2, 2001, 2318–2323 RSC.
- (a) C. T. Mbofana, E. Chong, J. Lawniczak and M. S. Sanford, Org. Lett., 2016, 18, 4258–4261 CrossRef CAS PubMed; (b) J. C. Cooper, C. Luo, R. Kameyama and J. F. Van Humbeck, J. Am. Chem. Soc., 2018, 140, 1243–1246 CrossRef CAS PubMed; (c) I. Bauer and H.-J. Knoelker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- (a) Y. Li, T. B. Lee, T. Wang, A. V. Gamble and A. E. V. Gorden, J. Org. Chem., 2012, 77, 4628–4633 CrossRef CAS PubMed; (b) J. Liu, X. Zhang, H. Yi, C. Liu, R. Liu, H. Zhang, K. Zhuo and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 1261–1265 CrossRef CAS PubMed; (c) W. J. Ang and Y. Lam, Org. Biomol. Chem., 2015, 13, 1048–1052 RSC; (d) H. Sterckx, J. De Houwer, C. Mensch, I. Caretti, K. A. Tehrani, W. A. Herrebout, S. Van Doorslaer and B. U. W. Maes, Chem. Sci., 2016, 7, 346–357 RSC; (e) L. C. Finney, L. J. Mitchell and C. J. Moody, Green Chem., 2018, 20, 2242–2249 RSC.
- Y. Bonvin, E. Callens, I. Larrosa, D. A. Henderson, J. Oldham, A. J. Burton and A. G. M. Barrett, Org. Lett., 2005, 7, 4549–4552 CrossRef CAS PubMed.
- (a) A. J. Catino, J. M. Nichols, H. Choi, S. Gottipamula and M. P. Doyle, Org. Lett., 2005, 7, 5167–5170 CrossRef CAS PubMed; (b) Y. Wang, Y. Kuang and Y. Wang, Chem. Commun., 2015, 51, 5852–5855 RSC; (c) Y. Lin, L. Zhu, Y. Lan and Y. Rao, Chem.–Eur. J., 2015, 21, 14937–14942 CrossRef CAS PubMed; (d) J. A. S. Coelho, A. F. Trindade, R. Wanke, B. G. M. Rocha, L. F. Veiros, P. M. P. Gois, A. J. L. Pombeiro and C. A. M. Afonso, Eur. J. Org. Chem., 2013, 2013, 1471–1478 CrossRef CAS.
- (a) S.-I. Murahashi and D. Zhang, Chem. Soc. Rev., 2008, 37, 1490–1501 RSC; (b) C. S. Yi, K.-H. Kwon and D. W. Lee, Org. Lett., 2009, 11, 1567–1569 CrossRef CAS PubMed; (c) B. B. Shingate, B. G. Hazra, D. B. Salunke and V. S. Pore, Tetrahedron Lett., 2011, 52, 6007–6010 CrossRef CAS; (d) S.-F. Hsu and B. Plietker, ChemCatChem, 2013, 5, 126–129 CrossRef CAS.
- (a) J.-Q. Yu and E. J. Corey, Org. Lett., 2002, 4, 2727–2730 CrossRef CAS PubMed; (b) J.-Q. Yu and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 3232–3233 CrossRef CAS PubMed; (c) J.-Q. Yu, H.-C. Wu and E. J. Corey, Org. Lett., 2005, 7, 1415–1417 CrossRef CAS PubMed.
- (a) H. Peng, A. Lin, Y. Zhang, H. Jiang, J. Zhou, Y. Cheng, C. Zhu and H. Hu, ACS Catal., 2012, 2, 163–167 CrossRef CAS; (b) X. Geng and C. Wang, Org. Lett., 2015, 17, 2434–2437 CrossRef CAS PubMed; (c) H. Jin, J. Xie, C. Pan, Z. Zhu, Y. Cheng and C. Zhu, ACS Catal., 2013, 3, 2195–2198 CrossRef CAS.
- (a) H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed; (b) H. Li, Z. Li and Z. Shi, Tetrahedron, 2009, 65, 1856–1858 CrossRef CAS.
- J.-B. Xia, K. W. Cormier and C. Chen, Chem. Sci., 2012, 3, 2240–2245 RSC.
- (a) I. Bauer and H.-J. Knoelker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed; (b) J. Liu, K.-F. Hu, J.-P. Qu and Y.-B. Kang, Org. Lett., 2017, 19, 5593–5596 CrossRef CAS PubMed; (c) G. Pandey, R. Laha and D. Singh, J. Org. Chem., 2016, 81, 7161–7171 CrossRef CAS PubMed; (d) B. Muehldorf and R. Wolf, Angew. Chem., Int. Ed., 2016, 55, 427–430 CrossRef CAS PubMed; (e) H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed.
- (a) P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed; (b) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed; (c) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (d) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (e) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed.
- (a) Y. Ishii, K. Nakayama, M. Takeno, S. Sakaguchi, T. Iwahama and Y. Nishiyama, J. Org. Chem., 1995, 60, 3934–3935 CrossRef CAS; (b) Y. Ishii, T. Iwahama, S. Sakaguchi, K. Nakayama and Y. Nishiyama, J. Org. Chem., 1996, 61, 4520–4526 CrossRef CAS PubMed; (c) Y. Yoshino, Y. Hayashi, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1997, 62, 6810–6813 CrossRef CAS; (d) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393–427 CrossRef CAS; (e) N. Sawatari, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2003, 44, 2053–2056 CrossRef CAS.
- D. P. Hruszkewycz, K. C. Miles, O. R. Thiel and S. S. Stahl, Chem. Sci., 2017, 8, 1282–1287 RSC.
- E. Gaster, S. Kozuch and D. Pappo, Angew. Chem., Int. Ed., 2017, 56, 5912–5915 CrossRef CAS PubMed.
- C. Jin, L. Zhang and W. Su, Synlett, 2011, 10, 1435–1438 Search PubMed.
- X. Han, Z. Zhou, C. Wan, Y. Xiao and Z. Qin, Synthesis, 2013, 45, 615–620 CrossRef CAS.
- Y. Yang and H. Ma, Tetrahedron Lett., 2016, 57, 5278–5280 CrossRef CAS.
- (a) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070–13074 CrossRef CAS PubMed; (b) Y. Yang, Y. Lin and Y. Rao, Org. Lett., 2012, 14, 2874–2877 CrossRef CAS PubMed; (c) Y. Rao, Synlett, 2013, 24, 2472–2476 CrossRef CAS.
- (a) J. G. Muller, P. Zheng, S. E. Rokita and C. J. Burrows, J. Am. Chem. Soc., 1996, 118, 2320–2325 CrossRef CAS; (b) Y. Yang and H. Ma, Tetrahedron Lett., 2016, 57, 5278–5280 CrossRef CAS; (c) G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- P. A. Gunchenko, J. Li, B. Liu, H. Chen, A. E. Pashenko, V. V. Bakhonsky, T. S. Zhuk and A. A. Fokin, Mol. Catal., 2018, 447, 72–79 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03346g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |