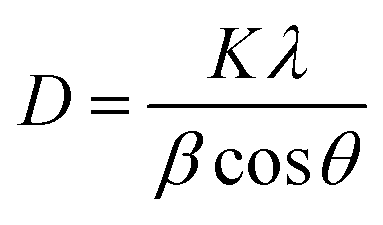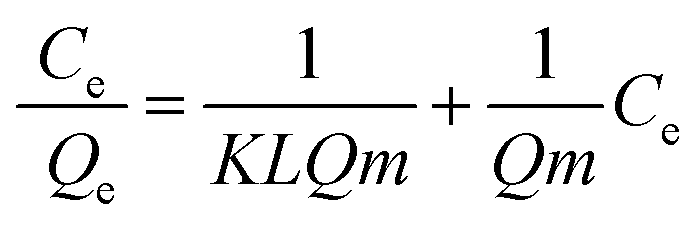DOI:
10.1039/C9RA03252E
(Paper)
RSC Adv., 2019,
9, 23129-23141
Magnetically retrievable Ce-doped Fe3O4 nanoparticles as scaffolds for the removal of azo dyes†
Received
1st May 2019
, Accepted 5th July 2019
First published on 26th July 2019
Abstract
Considering the significant impact of magnetically retrievable nanostructures, herein, Fe3O4 and Ce-doped Fe3O4 nanoparticles were employed as scaffolds for the removal of the Reactive Black 5 (RB5) azo dye. We synthesized the Ce-doped Fe3O4 nanoparticles via hydrothermal treatment at 120 °C for 10 h with varying cerium concentrations (1.5–3.5%) and characterized them using basic techniques such as FTIR and UV-visible spectroscopy, and XRD analysis. The retention of their magnetic behaviors even after cerium amalgamation was demonstrated and confirmed by the VSM results. FESEM and EDX were used for the morphological and purity analysis of the synthesized nanoabsorbents. XPS was carried out to determine the electronic configuration of the synthesized samples. The porosity of the magnetic nanoparticles was investigated by BET analysis, and subsequently, the most porous sample was further used in the adsorption studies for the cleanup of RB5 from wastewater. The dye adsorption studies were probed via UV-visible spectroscopy, which indicated the removal efficiency of 87%. The prepared Ce-doped Fe3O4 nanoabsorbent showed the high adsorption capacity of 84.58 mg g−1 towards RB5 in 40 min. This is attributed to the electrostatic interactions between the nanoabsorbent and the dye molecules and high porosity of the prepared sample. The adsorption mechanism was also analyzed. The kinetic data well-fitted the pseudo-first-order model, and the adsorption capability at different equilibrium concentrations of the dye solution indicated monolayer formation and chemisorption phenomena. Furthermore, the magnetic absorbent could be rapidly separated from the wastewater using an external magnetic field after adsorption.
1. Introduction
Synthetic dyes have provided us with the advantage of achieving fast and brighter colors. Dyestuffs require a large amount of water for dyeing. About 80% of dyestuffs stay on the substrate, whereas the rest just go down the drain.1 With the development of the printing and dyeing industry, wastewater pollution is posing a threat with dyes as typical organic pollutants. The toxic nature of dyes has become a cause of serious concern to environmentalists. The use of synthetic dyes has an adverse effect on all life forms such as humans, animals or plants, and the grave outcomes necessitate their effective disposal; azo dyes bear an azo (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) functional group together with complex chemical moieties comprising phenyl (C6H5CH2), naphthyl (C10H7CH2) or phenylamine (C6H5–NH2) groups;2 these moieties are highly complex for treatment via traditional biological methods. Thus, various chemical and physical treatment methods, such as coagulation–flocculation,3 advanced oxidation,4 membrane filtration,5 adsorption on activated carbon,6 and electrochemical7 and catalysis approaches,8 have long been established for the decolorization of textile wastewater. However, most of these methods suffer from high cost of experimentation, use of sophisticated instruments and manpower and inability to meet the permissible disposal levels. Most importantly, they have unsatisfactory recyclability and inefficient regeneration, which restrict their application in an industrial setup.
N) functional group together with complex chemical moieties comprising phenyl (C6H5CH2), naphthyl (C10H7CH2) or phenylamine (C6H5–NH2) groups;2 these moieties are highly complex for treatment via traditional biological methods. Thus, various chemical and physical treatment methods, such as coagulation–flocculation,3 advanced oxidation,4 membrane filtration,5 adsorption on activated carbon,6 and electrochemical7 and catalysis approaches,8 have long been established for the decolorization of textile wastewater. However, most of these methods suffer from high cost of experimentation, use of sophisticated instruments and manpower and inability to meet the permissible disposal levels. Most importantly, they have unsatisfactory recyclability and inefficient regeneration, which restrict their application in an industrial setup.
Adsorption has been commonly used as an effective technique for the removal of dyes. Activated carbon,9 rice husk,10 metal hydroxide sludge,11 zeolites12 and other compounds derived from natural sources have been used as adsorbents. Moreover, the adsorption process is preferred over the degradation of azo linkages since various reactive species, i.e. hydroxyl and hydrogen radicals, are involved in the latter process that lead to various degradation products (e.g. arylamines), which are potentially more toxic than the dyestuff itself.13
Magnetic nanoparticle (Np) adsorption has unfolded as an auxiliary method to usual adsorption for the treatment of effluent wastewater;14 hence, magnetic nanoadsorbents can serve as rapid dye adsorbing agents for the adsorption of dyes from wastewater effluent due to the presence of hydroxyl groups on their surfaces and consequently can be easily retrieved from the medium using a magnetic field post implementation. Some studies have been reported on the adsorption of pollutants on magnetite-based composites.15–17 Magnetic lignin-based adsorbents have been synthesized, and their adsorption capacity has been found to be more than 90% for azo dyes.18 Elwakeel et al.19 used magnetic chitosan resins to remove Reactive Black 5 (RB5) from aqueous solutions and were able to efficiently elute the adsorbate back. Chatterjee et al.20 used modified zero-valent iron with various surfactants, such as cetyltrimethylammonium bromide and sodium dodecyl sulfate, for the removal of RB5 from wastewater. Pure magnetite (Fe3O4) exhibits excellent magnetic properties; however, its adsorption efficiency towards pollutants is low.21 Thus, doping of other metals/metal ions, such as zinc and cobalt, into the Fe3O4 matrix increases the availability of its surface sites; this accordingly improves the adsorption capacity of Fe3O4.22,23 Furthermore, Su et al.24 reported the relationship between manganese doping and adsorption properties. The maximum adsorption capacity was found to be 84.54 mg g−1 in the case of doped hematite as compared to the case of undoped hematite (33.02 mg g−1). Vanadium-doped Fe3O4 promoted the adsorption of methylene blue with a high degree of decolouration. The improved adsorption efficiency was due to the increase in superficial hydroxyl groups.25
In the present study, we fabricated Fe3O4 and Fe3O4 doped with cerium (Ce), Ce–Fe3O4, via a hydrothermal treatment. Ce exhibits a facile transition between the +3 and +4 oxidation states, and the differences in the oxidation states of the host and dopant metal leads to the generation of octahedral (Ov) defects, which act as charge compensating vacancies and tend to influence various properties. The synthesized samples were well characterized by XRD, UV-vis spectroscopy, N2 adsorption/desorption, FTIR spectroscopy, FE-SEM and XPS. The concentration of cerium as a dopant was varied, and XRD was conducted to confirm the doping. The magnetic behavior was analyzed using VSM studies. Furthermore, the effect of cerium concentration on various properties, such as magnetic and surface area properties, was investigated. Reactive Black 5 (RB5), an anionic dye that belongs to the category of azo-reactive dyes and is extensively used in textile industries for dyeing, has been selected as a model dye. This study highlights the potential application of Ce-doped Fe3O4 Nps as efficient RB5 adsorbents and their magnetic retrieval with the retention of their properties, which widens their scope for application in environmental remediation processes.
2. Experimental
2.1. Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O, 97%), iron(II) sulphate hexahydrate (FeSO4·6H2O, ≥99%) and Reactive Black 5 were purchased from Sigma-Aldrich. Aqueous ammonia (25%) was purchased from Merck. Cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, ≥99%) was obtained from HPLC Pvt. Ltd. All chemicals were of analytical grade and used without further purification. Deionised water (DW) was used for the synthesis and sample preparation.
2.2. Synthesis of Fe3O4 and Ce–Fe3O4 Nps through the hydrothermal treatment
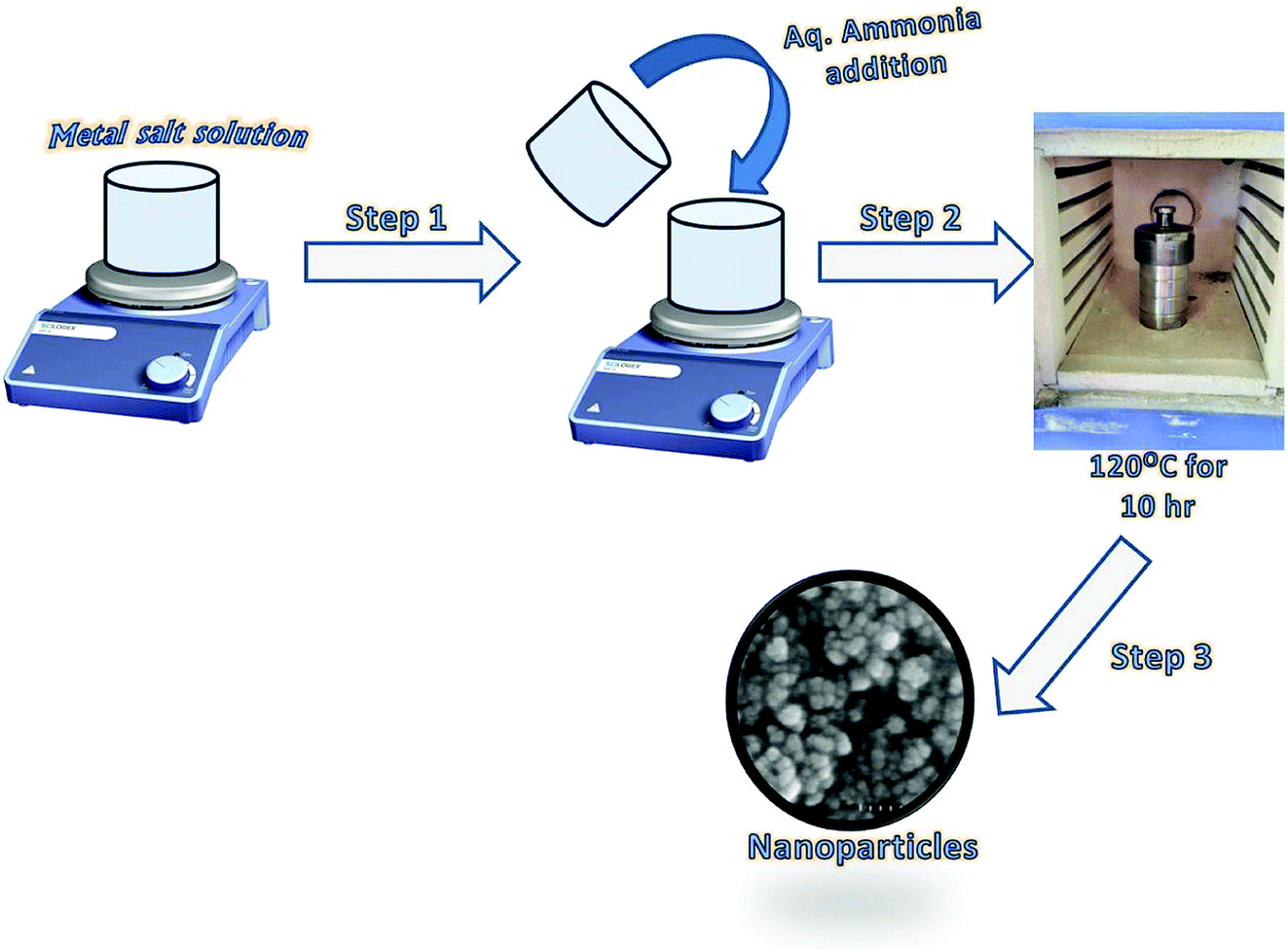
The synthesis of Fe3O4 and Ce–Fe3O4 Nps was performed by a hydrothermal method (Scheme 1). In a typical reaction process, FeCl3·6H2O (0.270 g) and 0.139 g of FeSO4·6H2O solutions were dissolved in 10 mL distilled water. The mixture was then stirred for 5 min, followed by the dropwise addition of 5 mL of NH3 solution (25%). To this mixture, Ce(NO3)3·6H2O at different concentrations, obtained by dissolving different amounts of it ranging from 1.5 to 3.5 wt% in 10 mL distilled water, was added followed by stirring for 30 min at room temperature. The obtained solution was then transferred to a Teflon-lined stainless-steel autoclave and placed in a furnace at 120 °C for 10 h. The autoclave was allowed to naturally cool down to room temperature after the hydrothermal treatment. The precipitate was recovered, washed several times with distilled water and finally with ethanol to remove the impurities and dried in an oven at 60 °C.
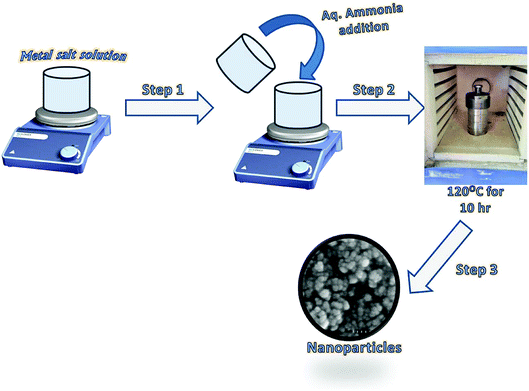 |
| | Scheme 1 Schematic for the fabrication of Fe3O4 and Ce–Fe3O4 Nps. | |
2.3. Characterization techniques
X-ray diffraction studies were performed using the PANalytical X'pert PRO XRD at the scanning speed of 8° min−1 with Cu-Kα radiation (1.5418 Å) in the 2θ range of 10–80°. The FTIR spectra were obtained using a thermally controlled diode laser in the transmittance mode in the spectral region of 4000–500 cm−1 using Thermo Scientific, Nicolet iS50 FTIR. The UV-visible absorption spectra were obtained using the JASCO V 530 spectrophotometer (4-21, Sennin-cho 2-chome, Hachioji, Tokyo 193-0835, Japan model). The spectral range of 190–450 nm was covered with the precision of ±0.2 nm using quartz cuvettes having 1 cm path length. The Lake Shore Vibrating Sample Magnetometer (VSM) was used to measure the magnetization as a function of the applied field and temperature. Malvern Zeta Nano S90 (Malvern Instruments, Malvern, UK), which calculates intensity weighted mean particle size (Z-average, nm ± S.D.), has been used to obtain the particle size distribution of the sample. The surface morphology of the sample was analyzed using the Hitachi-SU8010 field-emission electron microscope (FE-SEM), operating at the voltage of 15 kV. Electron micrographs were obtained by transmission electron microscopy (TEM-TECNAI 200 kV). Energy dispersive X-ray spectroscopy analysis was carried out using Bruker-Xflash. The BET surface areas of the samples were analyzed by nitrogen adsorption–desorption using a N2 adsorption analyzer, BELSORP. After degassing all the samples at 423 K for 15 hours, the BET surface area was determined using the multipoint BET method. The Barrett–Joyner–Halenda (BJH) method was used to analyse the pore-size distribution using the adsorption branch of the isotherm. X-ray photoelectron spectroscopy (XPS) was performed using the Omicron energy analyzer (EA-125) with an Al Kα (1486.6 eV) X-ray source. The sample was sputtered with Ar+ for 5 min before the spectra were obtained. The background vacuum in the analyzer chamber was of the order of 10−10 Torr during the XPS measurement.
2.4. Dye adsorption experiments
All experiments were performed at room temperature under light. For adsorption studies, at first, 10 mg of the synthesized sample was added to 50 ppm of dye solution, i.e. RB5, to confirm the adsorption capability of the synthesized nanomaterials. The dye concentration was 30 ppm for all the dyes employed herein. The pH value was maintained between 3 and 4. The color of the RB5 solution faded with time, confirming adsorption. Initially, various optimization parameters were investigated by analysing the effect of contact time, pH and dosage on adsorption. For adsorption capacity calculations, the UV-visible absorbance was measured every 5 min by pipetting out 1 mL solution and then filtering it. The equilibrium adsorption amount (Qe) of the 3.5% Ce–Fe3O4 Nps for the RB5 dye was calculated using the following eqn (1):where Co and Ce are the initial and equilibrium concentrations (mg L−1) of the dye solution, respectively, v corresponds to the volume of the dye solution, and m refers to the weight of the Nps.
3. Results and discussion
3.1. Physical characterization of the synthesized samples
3.1.1. X-ray diffraction. The XRD patterns of the Fe3O4 and Ce–Fe3O4 samples are shown in Fig. 1. The X-ray diffractograms of the polycrystalline samples reveal the generation of an unambiguous single-phase inverse spinel structure. The XRD pattern of the undoped Fe3O4 sample exhibits diffraction peaks at about 30.39°, 35.66°, 43.15°, 53.96°, 57.30° and 62.94°, corresponding to the (220), (311), (400), (422), (511) and (440) planes; this is agreement with literature (JCPDF card, file no. 74-0748); all the observed peaks have been indexed to pure Fe3O4, and no diffraction peaks corresponding to oxidized cerium species are found; the XRD peaks provide the requisite information about the location of the dopant in the crystal lattice. The ion distribution between the tetrahedral and octahedral sites is determined by the relative size and charge on/of cations and the size of the interstices.26,27 In the Ce–Fe3O4 system, the Fe2+ ions show high crystal field stabilization energy (CFSE), whereas the Fe3+ and Ce4+ ions have zero CFSE at both the octahedral and the tetrahedral sites. Therefore, considering the CFSE and ionic radii, it is reasonable that Ce4+ enters the octahedral sites. The substitution can change the 2θ values, as shown in the inset of Fig. 1; this is indicative of cation substitution since upon substitution, the crystallite size changes, resulting in a change in scattering.35
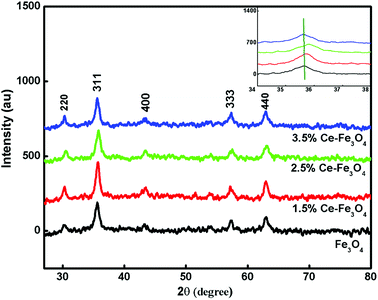 |
| | Fig. 1 XRD pattern of the pure Fe3O4 and doped synthesized samples. | |
With an increase in the dopant concentration, an increase in crystallinity was observed; this was indirect evidence of the decrease in the crystallite size of the synthesized samples. The crystallite size was calculated using the Debye–Scherrer equation as follows:
| |
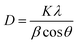 | (2) |
where the parameter
D is the crystallite size,
λ is the wavelength of X-rays used (0.154 nm),
θ is the Bragg's diffraction angle and
β is the full-width-at-half maxima (FWHM). The average crystallite size was calculated and is presented in
Table 1. The crystallite size was found to increase; this could be ascribed to the slight difference in the ionic radii of both cations, which made the substitution possible. If the ionic radii of the two ions differ by less than 14%, crystal chemistry permits the miscibility and substitution of ions.
28 Therefore, we can conclude that substitution of ions in the crystal lattice occurs without the formation of any other phase.
Table 1 Crystallite size calculated from the XRD pattern
| System |
Crystallite size (nm) |
| Fe3O4 |
25.33 |
| 1.5% Ce–Fe3O4 |
24.88 |
| 2.5% Ce–Fe3O4 |
23.22 |
| 3.5% Ce–Fe3O4 |
23.09 |
3.2. Fourier transform infrared spectroscopy (FTIR)
FTIR spectra provide details about the molecular structure and its environment since it is sensitive to various chemical bonds and functional groups present in a molecule. The FTIR spectra of Fe3O4 and Ce–Fe3O4 Nps are depicted in Fig. 2. Fe3O4 exhibits29 only two bands between 400 and 800 cm−1. The peak at 1614 cm−1 and the broad band between 3200 and 3500 cm−1 are attributed to the stretching and bending vibrations of the hydroxyl groups,30,31 which indicate the existence of water molecules adsorbed on the surface of Fe3O4 and Ce–Fe3O4, respectively. This is mainly due to chemical co-precipitation, where particles are naturally covered with hydroxyl groups. The characteristic peak at 540 cm−1 with a shoulder at 625 cm−1 corresponds to the intrinsic stretching vibrations of metal–oxygen at the tetrahedral site.32
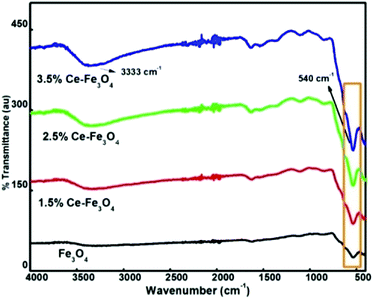 |
| | Fig. 2 FTIR spectra of pure Fe3O4 and doped Fe3O4. | |
In addition, the low frequency peak observed at around 445 cm−1 is assigned to the metal–oxygen stretching vibration at the octahedral site. The band disturbances below 1500 cm−1 are due to the lowering of the local symmetry as a result of distortion due to the presence of different cations at different sites33 (Fig. 2). Moreover, the bands near 540 cm−1 become less intense and broader because of the disturbance of the local symmetry via the introduction of Ce4+ into the lattice. The absorption band at 540 cm−1 gradually shifted; this was attributed to the revamped bonding force between the cations and oxygen anions because of the presence of cerium ions.
3.3. UV-visible spectra
The UV-vis analysis helps to understand the optical behaviour of materials. Thus, in this study, the absorbance of Fe3O4 and Ce–Fe3O4 Nps was monitored at ambient temperature (Fig. S1†). Magnetite exhibits thermally induced electron delocalization between adjacent Fe2+ and Fe3+ ions, and its electronic transitions are assigned to intervalence charge transfer (IVCT) transitions in the visible and near-IR region.34 The optical properties were enhanced by the introduction of Ce into the lattice of Fe3O4, as can be observed by the intensity enhancement. This can be accredited to the fact that new defects are introduced after the substitution of Ce cations into the lattice unit.
3.4. Vibrating sample magnetometer (VSM)
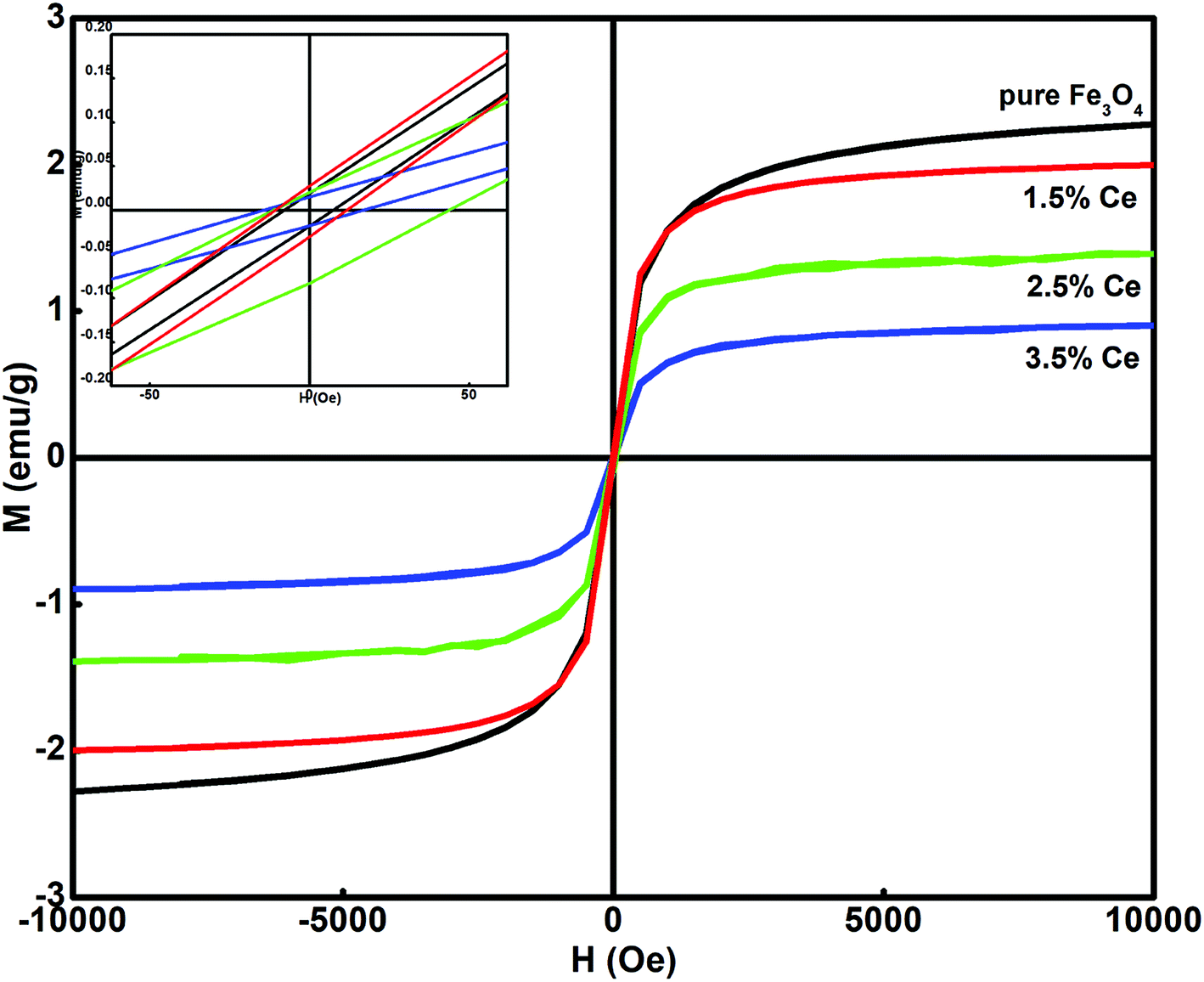
Fig. 3 shows the dependence of magnetization on the applied field (M–H) for all the synthesized samples at room temperature. All the samples displayed ferromagnetic behaviour. The hysteresis loop was used to calculate the magnetism saturation (Ms) and coercivity (Hc), as listed in Table 2. The undoped Fe3O4 sample displayed better Ms than the doped samples. The Ce-doped Fe3O4 sample exhibited decreased magnetism due to the presence of surface defects, which caused a decrease in the exchange coupling between neighbouring iron ions on the surface.35 This occurs due to the lack of an oxygen super exchange mechanism. Surface spin disorder is caused by the broken exchange bonds on the surface of the particles, which influences magnetism.36 It has been reported that spin canting reduces after rare earth doping and thus increases the magnetization.37 The decrease in Ms with an increase in the doping level is attributed to the pronounced effect of the f-electrons in cerium.38 As f-electrons are deep- seated, they produce an opposite magnetic field, which cancels the effect of the overall magnetic field produced by a domain, and hence, the saturation magnetization becomes smaller. Another reason may be that with a decrease in particle size, an increase in surface to volume ratio occurs, i.e. the dead surface layers contribute more to the magnetism than the internal layers. Therefore, due to the dominant role of the dead surface layers in the particle gross magnetic property, smaller particle size results in a decrease in saturation magnetization; all the synthesized samples exhibit magnetic behavior; this makes their separation easy after treatment.
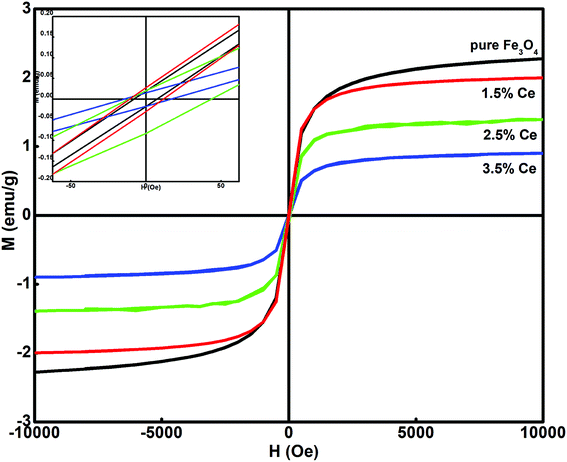 |
| | Fig. 3 Hysteresis loop at room temperature for the synthesized samples. | |
Table 2 Effect of cerium doping on the synthesized samples
| System |
Saturation magnetization, Ms (emu g−1) |
Magnetic moment |
Coercivity, Hc (Oe) |
| Fe3O4 |
11.970 |
0.507 |
7.004 |
| 1.5% Ce–Fe3O4 |
7.298 |
0.309 |
8.083 |
| 2.5% Ce–Fe3O4 |
4.326 |
0.183 |
10.776 |
| 3.5% Ce–Fe3O4 |
2.959 |
0.125 |
15.567 |
3.5. Morphological analysis: field-emission scanning electron microscopy (FE-SEM), transmission electron microscopy and energy dispersive X-ray (EDX) analysis
The morphological characterization of the synthesized samples was carried out by obtaining their FE-SEM (Fig. 4) and TEM images (Fig. S2†). The results of both analysis reveal that the synthesized samples have a spherical, loose, aggregated structure with the average size of 24 ± 2 nm in the case of the undoped sample and 29 ± 0.9 nm for the 3.5% doped sample (Fig. S2†). As the dopant concentration increased, although the morphology remained unchanged, the particle size increased. This is also indicative of a slight change in aggregation. The results obtained via FESEM and TEM support those obtained via the XRD analysis; this reveals that an increase in size occurs with an increase in dopant concentration.
 |
| | Fig. 4 FE-SEM analysis of the (a) undoped and (b) 1.5%, (c) 2.5%, and (d) 3.5% cerium-doped samples. | |
The EDX spectrum of Fe3O4 shows the presence of Fe and O and that of Ce–Fe3O4 shows the presence of Ce along with Fe and O (Fig. S3a†).
3.6. Brunauer–Emmett–Teller (BET) isotherm analysis
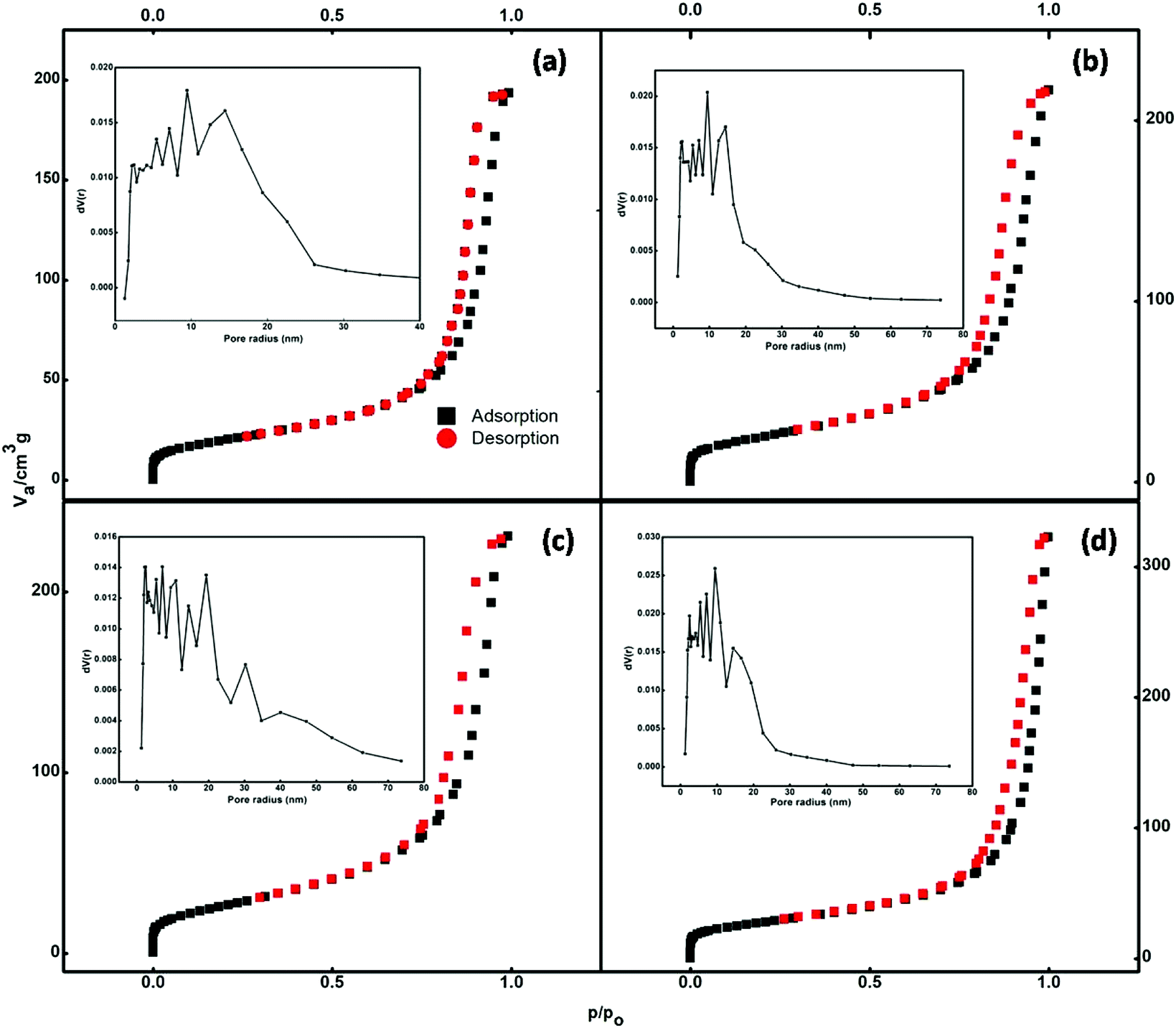
The nitrogen adsorption–desorption isotherms of the prepared samples are illustrated in Fig. 5. According to the IUPAC classification, the isotherms shown in Fig. 5 can be classified as type IV. Type IV isotherms are particularly observed in the case of mesoporous adsorbents, which have a pore size between 2 and 50 nm. The adsorbent adsorptive interactions along with the interactions between molecules determine the adsorption behaviour in the mesopores in the condensed state. In the pore condensation phenomenon, gas condenses into a liquid-like phase in a pore at the pressure p, less than the saturation pressure p0 of the bulk liquid.39 Thus, final saturation plateaus of variable length are a typical feature of type IV isotherms.
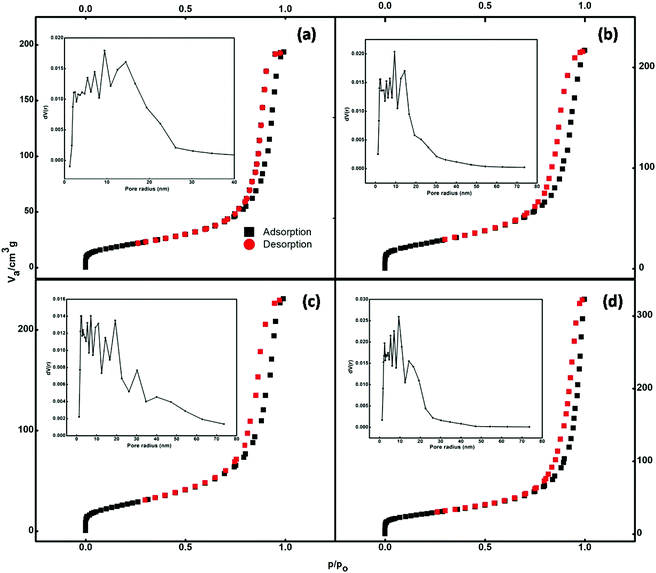 |
| | Fig. 5 BET curves of (a) undoped and (b) 1.5%, (c) 2.5%, and (d) 3.5% cerium-doped samples. | |
Hysteresis is accompanied by the phenomenon of capillary condensation in the case of type IV isotherms.40 The mean pore diameter of the synthesized samples ranged between 14 and 19 nm (Table S1†). The void spaces between nanoparticles that constitute the nanospheres create these pores.
The pore size distributions of the catalysts were calculated by the Barrett–Joyner–Halenda (BJH) curve (inset of Fig. 5). It is evident that the adsorption capacities of the pure Fe3O4 and Ce-doped Fe3O4 samples increase with equilibrium pressure; this may be attributed to the condensation of N2 molecules in mesopores at high pressures. Doping with Ce increased the specific surface area (Table S1†) available for adsorption from 71.6 to 97.6 cm3 g−1, implying that 3.5% Ce–Fe3O4 could facilitate the diffusion and adsorption of the target pollutant; this would be helpful for the elevation of adsorption capacity. The BET specific surface area, which is an important property for adsorption studies, is high; this may be due to porosity resulting from the aggregation of magnetic Nps. Therefore, the system with maximum surface area (3.5% Ce–Fe3O4) is favorable for adsorption applications and has been chosen for further studies, i.e. adsorption of RB5 from wastewater.
3.7. X-ray photoelectron spectroscopy (XPS) analysis
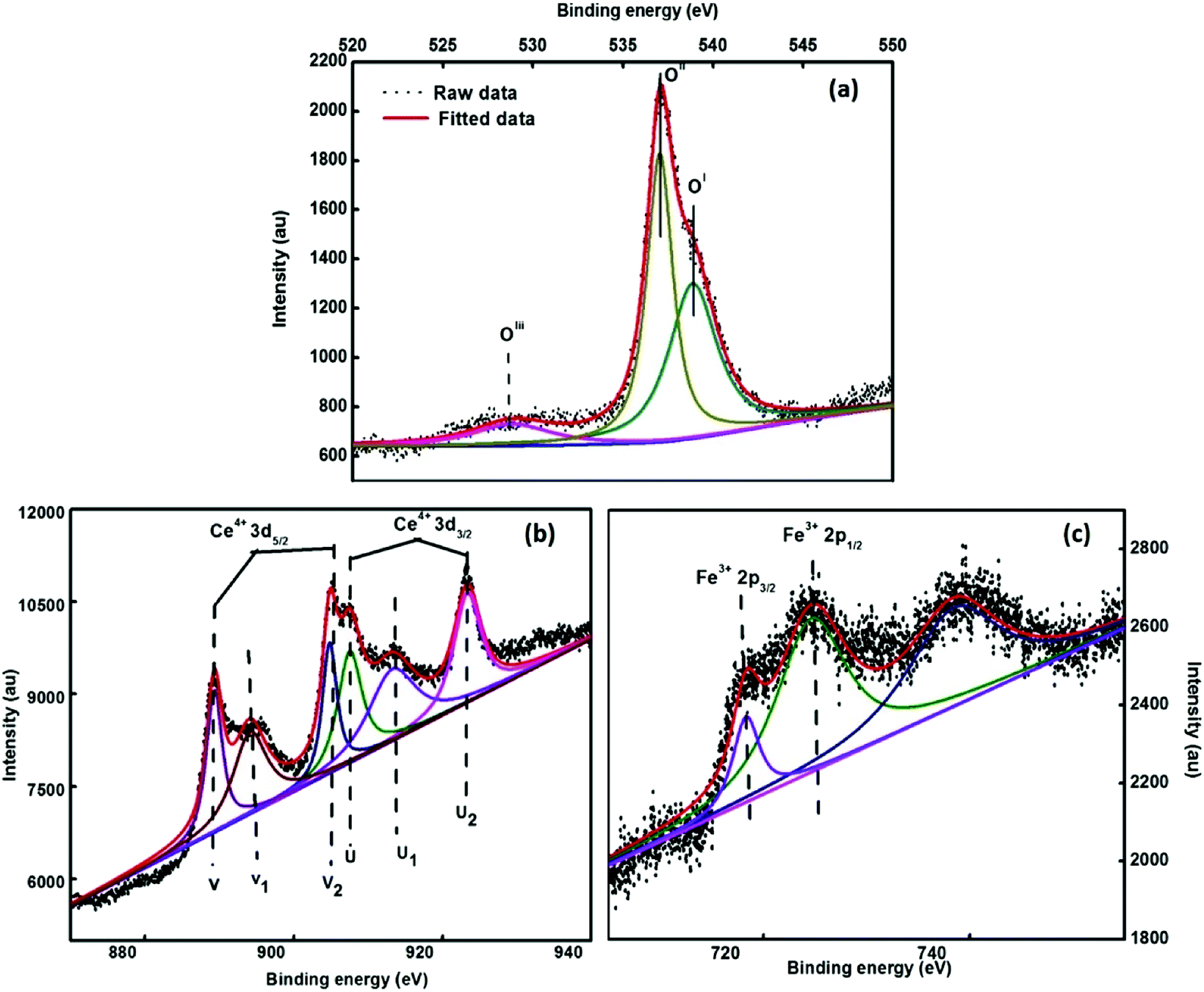
To evaluate the elemental composition, chemical environment and oxidation states of the various elements present in the synthesized system, the XPS spectra were obtained. Fig. 6 depicts the deconvoluted XPS spectra of 3.5% Ce–Fe3O4, i.e. Ce 3d, O 1s and Fe 2p states of the elements present in the examined sample. The peak centered around 537.3 eV (OII) is attributed to the O2− ions attached to the Fe atoms (Fig. 6a). The intense peak at 538.1 eV (OI) results from the configuration and photo ionization of the 1s core level in the case of molecular oxygen.41
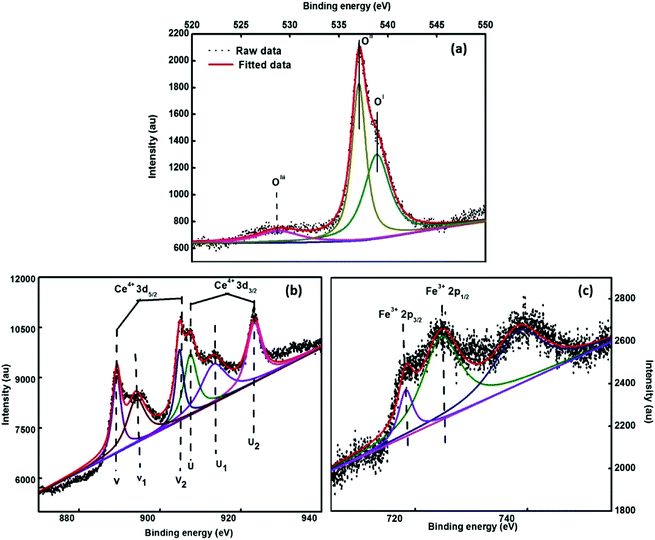 |
| | Fig. 6 XPS spectra of the 3.5% cerium-doped sample (a) O 1s, (b) Fe 2p and (c) Ce 3d. | |
The peak around 528.06 eV (OIII) is due to oxygen present in the cerium oxide lattice,42 which indicates the presence of cerium oxide as an impurity. The ratio of the oxygen peaks (OII/OIII) in the iron and cerium lattice depicts that the amount of cerium oxide is very less. The peaks observed at 883.4 eV and 901.6 eV (Fig. 6b) are due to the spin–orbit coupling of the 3d5/2 and 3d3/2 levels, respectively. The peaks located at 883.4 eV, 889.9 eV, 899 eV, 901.6 eV, 908.4 eV and 917.5 eV are ascribed to the 3d104f1 electronic state of Ce4+, and the peaks positioned at 885.06 eV and 902.8 eV are attributed to the 3d104f1 states of the Ce3+ ion.43 The Fe 2p core level XPS spectrum reveals the peaks of Fe 2p3/2 and Fe 2p1/2 at 718.3 eV and 724.5 eV, which are attributed to the Fe3+ state of Fe44 (Fig. 6c). Fe 2p3/2 does not have a satellite peak;44 this confirms the formation of Fe3O4 in the present study.
3.8. Adsorption studies of 3.5% Ce–Fe3O4
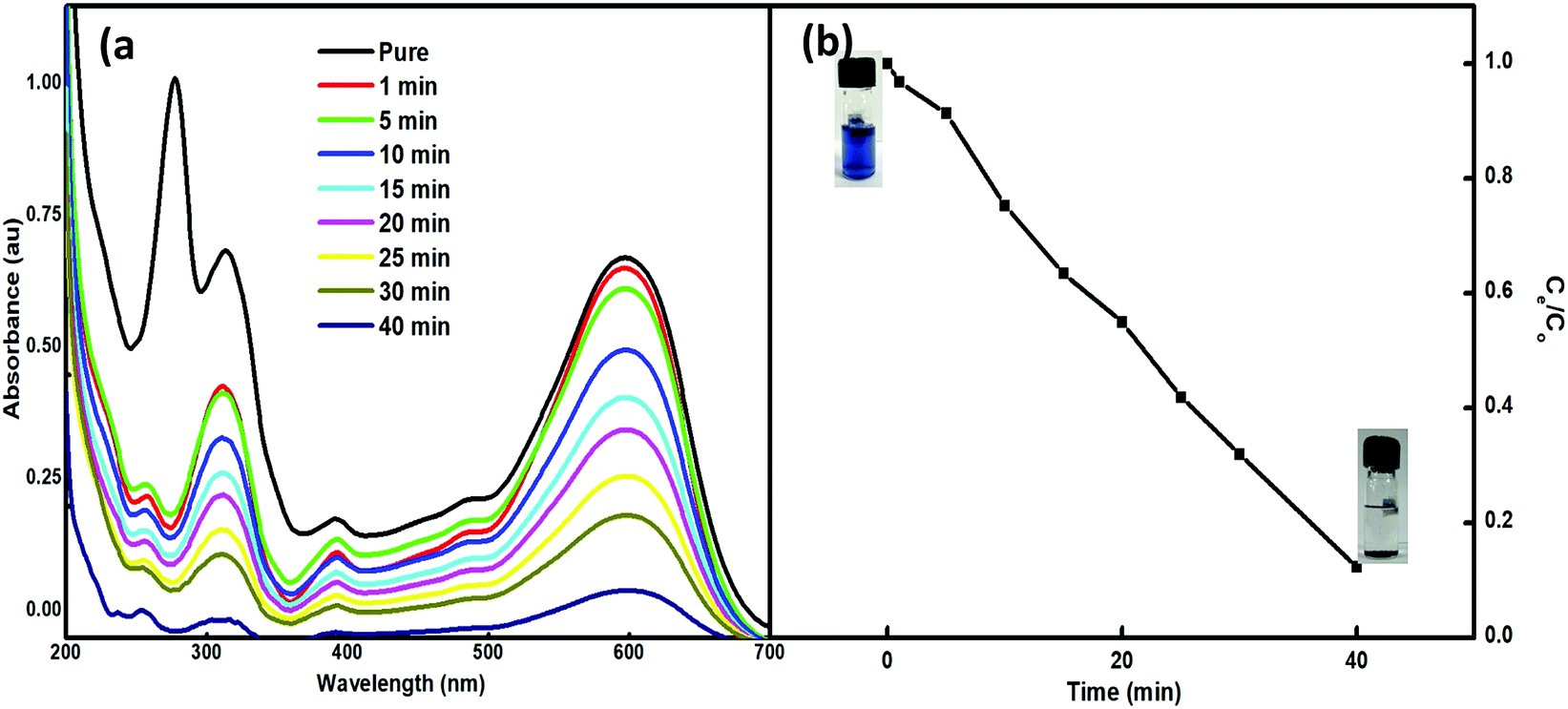
The feasibility of the synthesized nanoparticles as proficient nanoabsorbents for the removal of Reactive Black 5 (RB5), a textile azo dye, was explored. The strong adsorption capacity of Nps is ascribed to the ionic interactions between the dye and the nano-absorbent. The higher charge and porosity of the nanoabsorbent led to high removal efficiency. The adsorption peak at 600 nm in the visible spectrum of an aqueous solution of RB5 was obtained using 5 mg of Nps and found to decrease with time (1–90 min) (Fig. 7). For this, the absorbance of RB5 was monitored in the presence of the nanoadsorbent. The higher charge and porosity of the presented nanoadsorbent facilitated the removal of the dye through strong ionic interactions between the dye and Ce–Fe3O4. Fig. 7 shows the absorbance spectra in the presence and absence of the nanoadsorbent.
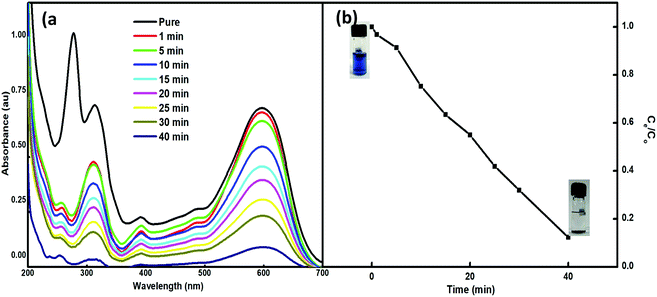 |
| | Fig. 7 (a) UV-visible spectra of RB5 in the presence of 5 mg of Nps within time interval of 1–40 min and (b) adsorption profile of RB5 with time. | |
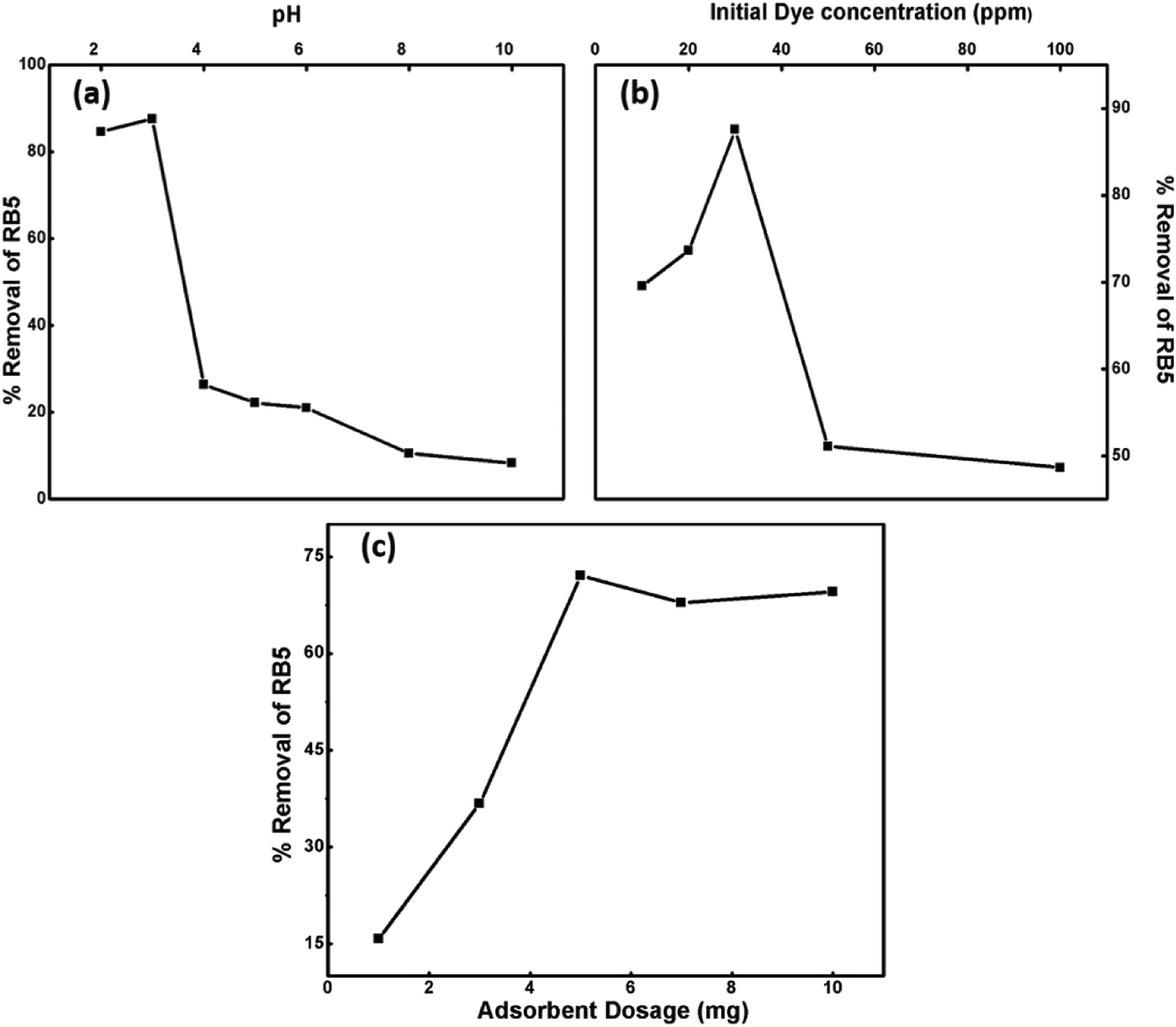
3.8.1. Effect of operational parameters on the removal of dye. To optimize the adsorption ability of the nanoadsorbent for RB5, the effect of various operational parameters, such as pH, dosage, dye concentration and contact time, on the removal of the dye was examined at room temperature.
3.8.1.1. Effect of pH. pH is a vital parameter for contaminant removal as the degree of RB5 adsorption is initially controlled by the surface charge on Ce–Fe3O4, which is affected by the solution pH. Thus, the effects of the solution pH on RB5 adsorption were investigated in the pH range from 3 to 10 (Fig. 8a). The optimum pH for the removal of RB5 was determined via batch mode experiments performed on the RB5 dye with the concentration of 30 ppm and Nps dosage of 5 mg. Significant adsorption took place at acidic pH than that at alkaline pH. The maximum removal was found at pH 3. The results obtained were interpreted on the basis of zero point charge (zpc). The zpc generally lies between 6 and 6.8 mV.45 The effect of pH on the surface of the synthesized Nps is presented in Fig. S4,† which depicts that the zpc lies between 6 and 6.9 mV. A pH lower than the zpc generates a positive surface charge due to the accumulation of H+ ions or association (FeOH + H+ → FeOH2+) of protons. Contrarily, on moving to a higher pH, the presence of excessive OH− ions or dissociation (FeOH → FeO− + H+) makes the surface negatively charged. Being an anionic dye, RB5 is adsorbed more cogently at lower pH values. Therefore, the higher removal efficacy of the nanoadsorbent at low pH can be attributed to the electrostatic attraction between RB5 and Nps, and the reduction in removal at higher pH is linked to the electrostatic force of repulsion between these two (Fig. S5†). The maximum removal was observed for pH = 3, and therefore, this value was maintained for the remaining batch adsorption experiments.
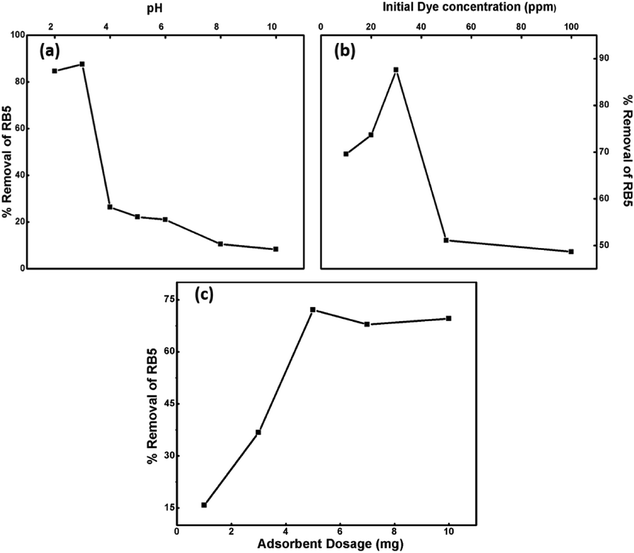 |
| | Fig. 8 Influence of different operational parameters: (a) pH, (b) Initial dye concentration and (c) nanoparticle dosage on percentage removal of RB5. | |
3.9. Kinetic model studies
Adsorption is a physicochemical process that involves the mass transfer of solution from the liquid phase to the adsorbent surface. To study the kinetics of Nps, RB5 solution with the initial concentration 30 ppm was used under fixed adsorbent conditions. To understand the adsorption process, the pseudo-first-order model and pseudo-second-order model were analysed using eqn (3) and (4), respectively:where Co and Ce are the initial and equilibrium concentrations of dye in solution, respectively, and K is the rate constant (min−1) (eqn (3))| | |
t/qt = 1/K2qe2 + t/qe
| (4) |
where qt and qe denote the absorbed amount at any time and equilibrium. K2 (g mg−1 min−1) is the rate constant for the pseudo-second-order model. The best fitted results of models are shown in Fig. S6,† and the data calculated using equation (eqn (4)) is depicted in Table 3. The system follows the pseudo-second-order kinetics model with high value of correlation coefficient (R2), which may be due to chemical interaction between the nanoabsorbent and dye, thus resulting in the removal of the dye from wastewater.
Table 3 The fitting results of the kinetic models
| First order kinetics |
Second order kinetics |
| qe (mg g−1) |
K1 (min−1) |
R2 |
qe (mg g−1) |
K2 × 10−4 (g mg−1 min−1) |
R2 |
| 69.98 |
0.023 |
0.957 |
370.37 |
1.19 |
0.978 |
3.10. Adsorption isotherms
The dependency of the adsorbed amount of RB5 on its equilibrium concentration in solution was investigated using the batch technique described earlier in the Experimental section, and the experimental data was analysed according to the Langmuir, Freundlich and DR isotherms to examine the type of dye molecule adsorption on the Nps surface. For wastewater treatment applications, the adsorption isotherms were analysed for evaluating the adsorption mechanism. The Langmuir model is valid for monolayer sorption on a surface with a finite number of identical sites. The distribution of dye at the solid solution interface has been described by the Langmuir equation. The well-known Langmuir model is presented by eqn (5) as follows:| |
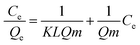 | (5) |
| |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe = logKf + 1/nlogCe Qe = logKf + 1/nlogCe
| (6) |
where Qe is the adsorbed amount of dye at equilibrium concentration, Ce is the equilibrium solute concentration, Qm is the maximum adsorption capacity and KL is the equilibrium constant for the Langmuir fit. Kf and 1/n are the Freundlich constants related to the adsorption capacity and adsorption intensity, respectively. Fig. S7a and b† show the fitting results of the Langmuir and Freundlich adsorption models, respectively. The calculated values are presented in Table 4. The Qm value obtained from the Langmuir isotherm adsorption model is supported by the experimental Qm (81.11 mg g−1). The R2 values of both the Langmuir and the Freundlich model indicate that the adsorption site can be occupied by only one or more RB5 molecules; this results in the formation of a monolayer/multilayer.
Table 4 The fitting results of different models
| Isotherm |
Parameters |
| Langmuir |
R2 = 0.988, Qm = 84.585 mg g−1, b= 0.337 L g−1 |
| Freundlich |
R2 = 0.983, n = 2.46, Kf = 3.227 |
| Dubinin–Radushkevich (D–R) |
R2 = 0.726, β = 2.28 × 10−7 mol2 J−2, Qs = 28.19 mg g−1 |
The adsorption process is mainly controlled by two methods: (a) film diffusion and (b) intraparticle (surface or pore) diffusion. The Weber–Morris equation (eqn (7)) was evaluated to determine the rate-controlling adsorption step via the intra-particle diffusion kinetic model.
where
Ki is the intraparticle diffusion rate constant for adsorption at each step, and
C is the intercept that symbolizes the boundary thickness.
46 In Fig. S8b,
† the graph of adsorption amount
versus the square of time is presented, which depicts the intra-particle diffusion rate constants for each adsorption step, influencing the rate-limiting steps. The calculated values are listed in
Table 4. The adsorption process can be interpreted based on the analysis of the abovementioned data: (i) the slope
K1 indicates that there is an electrostatic interaction between Nps and RB5 molecules, (ii) the second step indicates that the dye molecule diffuses into the inner structure of the nanoabsorbent, which is fast diffusion, and (iii) last step represents the adsorption equilibrium process. Another well-known method to determine the type of adsorption,
i.e. physisorption or chemisorption, is the Dubinin–Radushkevich (D–R) isotherm (Fig. S8a
†) method, which is presented by the following equation:
| | |
logQe = logQs − βε2
| (8) |
where
ε is the Polanyi potential presented by
| | |
ε = RTln(1 + 1/Ce)
| (9) |
where
Qs is the D–R constant and
β is related to the free energy (
E) of adsorption per molecule of adsorbate by the following equation:
where
R is the gas constant and
T is the temperature. If the energy value is more than 8 kJ mol
−1, it must be a chemical reaction, and if it is less than this value, it must be physical adsorption.
47 In our studies, the energy value was determined to be 1.48 × 10
3 J mg
−1, revealing multilayer adsorption formation, which is based on the assumption that the adsorption sites are distributed exponentially. It also indicates that physical adsorption is the dominant phenomenon. The surface charge and hydroxyl groups are responsible for the removal of dye using the nanoabsorbent. The values of n lie between 1 and 10, which indicate the favorability of the adsorption of dye over the nanoabsorbent. Therefore, the nanoabsorbent can be considered an efficient absorbent for RB5.
3.11. Adsorption of a mixture of dyes onto the absorbent
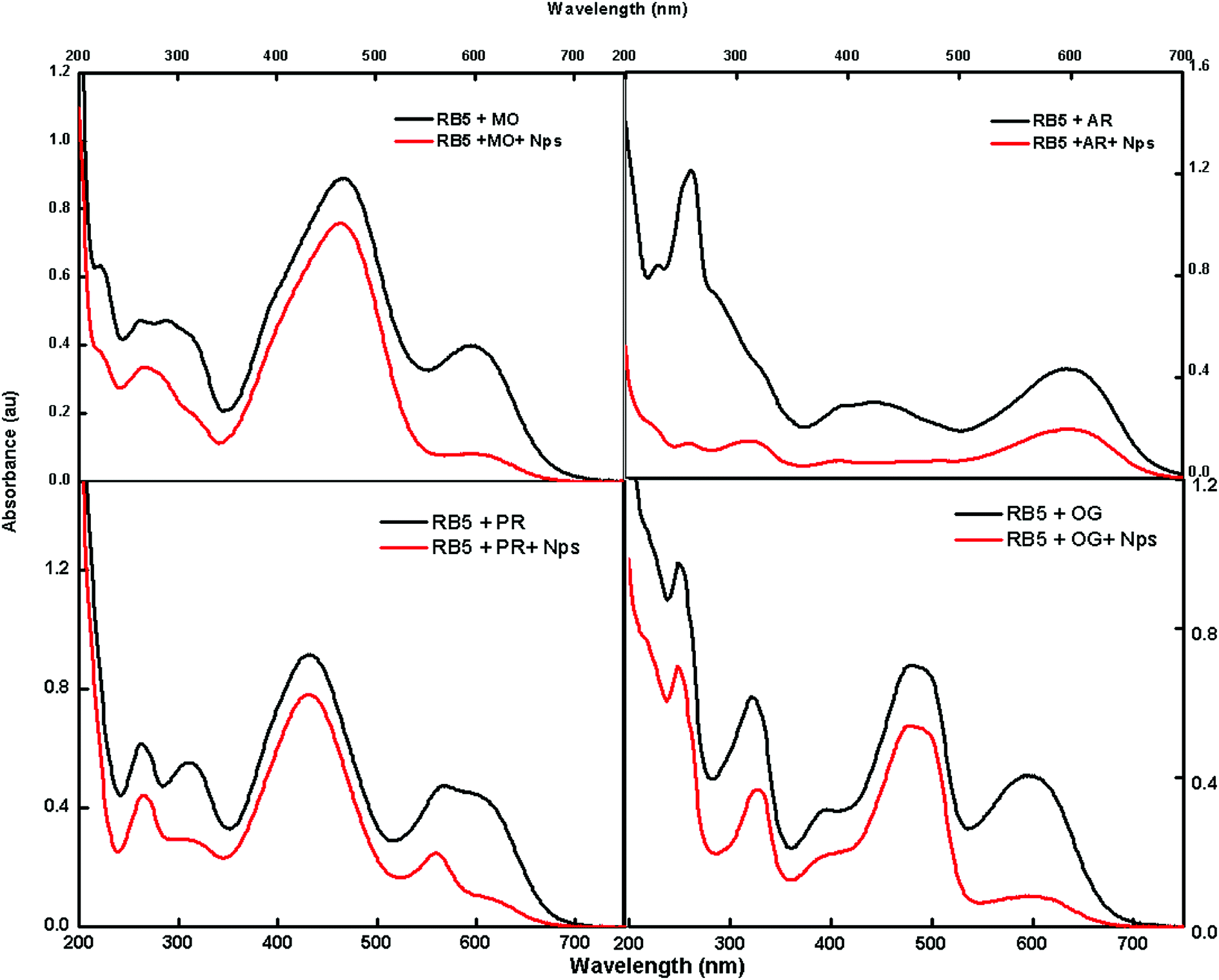
The feasibility of the absorbent towards a mixture of dyes was also tested using methylene orange (MO), Alizarin red 5 (AR), phenol red (PR) and Orange G (OG); these dyes were selected to cover the diverse range of commercially available dyes of the azo nature for the adsorption studies. Fig. 9 shows that there is a decrease in intensity after exposure to Nps.
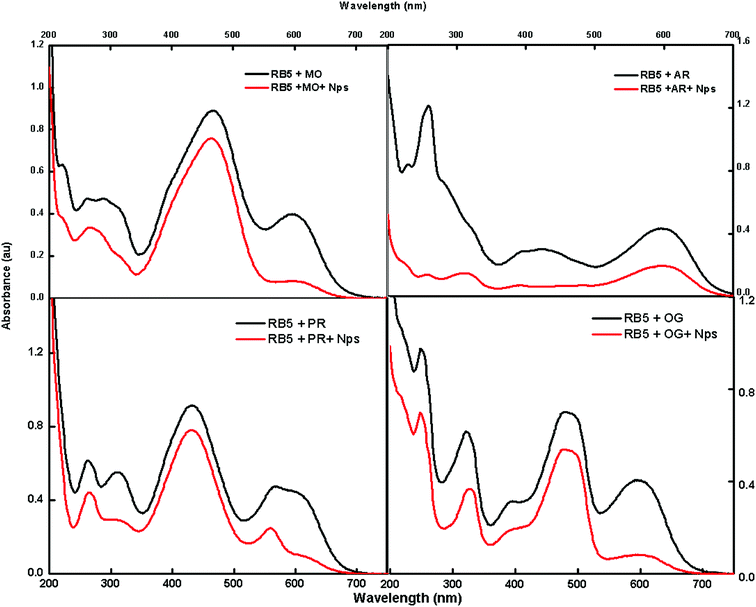 |
| | Fig. 9 UV spectra of a mixture of dyes before and after the adsorption of dyes. | |
The fast elimination capability of the nanoabsorbent further opens a new path for utilizing this type of nanomaterials for the removal of multianalytes, which can act as a good environmental remediating agent.
3.12. Desorption studies
The recyclability of Nps was investigated using sodium hydroxide solution as the recovery solvent. The adsorbed dye was removed by shaking in sodium hydroxide solution for 1 min. The recovered Nps were washed with water followed by ethanol. The recovered Nps were reused and the nanoabsorbent was found to be efficient after 5 cycles, i.e. the removal efficiency was reduced by only 5% of its initial removal (Fig. S9†). The identity of the recovered Nps was confirmed by FTIR (Fig. S10†). This study indicates the robustness of the nanoabsorbent after the treatment procedure.
3.13. Comparison with other adsorbents
The experiments were performed at room temperature under light. The dye concentration was 30 ppm for all the dyes employed. The pH was maintained between 3–4. A clear judgement can be made by comparing the prepared absorbent with the persisting absorbents in the literature. A comparison study was done and summarized in Table 5. It can be clearly seen that adsorption capacity (Qm; mg g−1) of Ce–Fe3O4 was overall greater than the previously reported magnetic systems to remove dye from wastewater.
Table 5 Comparison with other absorbents reported in the literature
| Adsorbent |
Dye |
Qm (mg g−1) |
Reference |
| Fe3O4 nanosphere |
Bismarck brown |
33.80 |
48 |
| CuO |
Coomassie Brilliant blue R-250 |
31.75 |
49 |
| Direct Red 81 |
48.49 |
|
| α-Fe2O3 nanoparticles |
Methyl orange |
28.90 |
50 |
| Sphere-like Mn2O3 |
Reactive black 5 |
14.6 |
51 |
| TiO2 nanoparticles |
Acid Red-27 |
38.88 |
52 |
| ZnO nanoparticles |
Reactive black 5 |
80.90 |
53 |
| Ce–Fe3O4 |
Reactive black 5 |
84.58 |
Present work |
The advantage of the present system is its adsorption capacity with magnetic retention accompanied by heterogeneous active sites, which lead to its potential application in wastewater remediation. The recyclability and easy synthesis procedure are additional advantages of this nanoabsorbent.
4. Conclusions
In summary, magnetite (Fe3O4) Nps with different cerium concentrations as dopants were synthesized using the hydrothermal method. The XRD analysis confirmed the presence of a single phase with an inverse final structure at all doping levels. The crystallite size was found to increase with an increase in doping concentration. The XRD results revealed the doping of Ce ions into the crystal lattice of magnetite. The saturation magnetization decreased with an increase in Ce content. The structural modification depends on the cation occupancy among the octahedral and tetrahedral sites, which influences different properties of the prepared nanoparticles. BET analysis was performed to determine the surface area of the nanoparticles, and the system with highest surface area was considered for wastewater treatment. The adsorption behavior of the 3.5% Ce–Fe3O4 was examined in detail by varying the operational parameters including pH, concentration of Nps, dye concentration and contact time. The kinetic studies and adsorption isotherms were employed to determine the mechanism of docking of the dye over the Nps surface. The removal efficiency was found to be 87%, and the efficiency was maintained even after 5 cycles. Thus, the results confirm the validity of the synthesized Nps in environmental applications i.e. efficacy in the removal of toxic pollutants from the environment.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors gratefully acknowledge the DST-PURSE-II and SAIF/CIL, Panjab University, Chandigarh, for providing characterization facility.
References
- M. T. Yagub, T. K. Sen, S. Afroze and H. M. Ang, Adv. Colloid Interface Sci., 2014, 209, 172–184 CrossRef CAS PubMed.
- N. N. Nassar, N. N. Marei, G. Vitale and L. A. Arar, Can. J. Chem. Eng., 2015, 93, 1965–1974 CrossRef CAS.
- Y. Lau, Y. Wong, T. Teng and N. Morad, RSC Adv., 2015, 5, 34206–34215 RSC.
- N. Azbar, T. Yonar and K. Kestioglu, Chemosphere, 2004, 55, 35–43 CrossRef CAS PubMed.
- T. Kim, C. Park and S. Kim, J. Cleaner Prod., 2005, 13, 776–786 CrossRef.
- K. Narcisse, Y. Augustin, K. Benjamin, D. Gone and A. Trokourey, J. Environ. Manage., 2015, 156, 10–14 CrossRef PubMed.
- C. Araújo, G. R. Oliveira, N. S. Fernandes and C. A. Martínez-huitle, Environ. Sci. Pollut. Res., 2014, 21, 9777–9784 CrossRef PubMed.
- M. Blanco, A. Martinez, A. Marcaide, E. Aranzabe and A. Aranzabe, Am. J. Anal. Chem., 2014, 5, 490–499 CrossRef CAS.
- D. Pathania, S. Sharma and P. Singh, Arabian J. Chem., 2017, 10, S1445–S1451 CrossRef CAS.
- T. G. Chuah, A. Jumasiah, I. Azni, S. Katayon and T. Choong, Desalination, 2005, 175, 305–316 CrossRef CAS.
- S. Netpradit, P. Thiravetyan and S. Towprayoon, Water Res., 2004, 38, 71–78 CrossRef CAS PubMed.
- C. Li, H. Zhong, S. Wang, J. Xue, Z. Zhang, J. Xue and Z. Zhang, J. Ind. Eng. Chem., 2015, 23, 344–352 CrossRef CAS.
- M. Cai, M. Jin and L. K. Weavers, Ultrason. Sonochem., 2011, 18, 1068–1076 CrossRef CAS PubMed.
- J. Gong, B. Wang, G. Zeng, C. Yang, C. Niu and Q. Niu, J. Hazard. Mater., 2009, 164, 1517–1522 CrossRef CAS PubMed.
- F. Ferrarini, L. R. Bonetto, J. S. Crespo and M. Giovanela, Water Sci. Technol., 2016, 73(9), 2132–2141 CrossRef CAS PubMed.
- Z. Qi, H. Lan, T. P. Joshi, R. Liu, H. Liu and J. Qua, RSC Adv., 2016, 6, 66990–67001 RSC.
- M. Y. Nassar, I. S. Ahmed and H. S. Hendy, J. Mol. Liq., 2018, 271, 844–856 CrossRef CAS.
- C. Jiang, X. Wang, D. Qin, W. Da, B. Hou, C. Hao and J. Wu, J. Hazard. Mater., 2019, 369, 50–61 CrossRef CAS PubMed.
- K. Z. Elwakeel, J. Hazard. Mater., 2009, 167, 383–392 CrossRef CAS PubMed.
- S. Chatterjee, S. Lim and S. H. Woo, Chem. Eng. J., 2010, 160, 27–32 CrossRef CAS.
- X. L. Peng, F. Xu, W. Z. Zhang, J. Y. Wang, C. Zeng, M. J. Niu and E. Chmielewska, Colloids Surf., A, 2014, 443, 27–36 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
- M. Y. Nassar, I. S. Ahmed and M. A. Raya, J. Mol. Liq., 2019, 282, 251–263 CrossRef CAS.
- H. Su, X. Wang, Y. Sun, D. Xu, L. Li, C. Liu, S. Zeng and D. Sun, Bull. Korean Chem. Soc., 2017, 38, 1155–1162 CrossRef CAS.
- X. Liang, S. Zhu, Y. Zhong, J. Zhu, P. Yuan, H. He and J. Zhang, Appl. Catal., B, 2010, 19, 151–159 CrossRef.
- W. Wolski, E. Wolska and J. Kaczmarek, Phys. Status Solidi A, 1993, 51, 51–54 CrossRef.
- B. P. Ladgaonkar and A. S. Vaingankar, Mater. Chem. Phys., 1998, 56, 280–283 CrossRef CAS.
- A. Q. Wang, D. Souza and T. Diane, J. Mater. Chem., 2006, 16, 481–488 RSC.
- W. B. White and B. A. D. Anoelis, Spectrochim. Acta, Part A, 1967, 23, 985–995 CrossRef CAS.
- M. Y. Nassar and M. Khatab, RSC Adv., 2016, 6, 79688–79705 RSC.
- M. Y. Nassar, T. Y. Mohamed, I. S. Ahmed and I. Samir, J. Mol. Liq., 2017, 225, 730–740 CrossRef CAS.
- M. Abboud, S. Youssef, J. Podlecki, R. Habchi, G. Germanos and A. Foucaran, Mater. Sci. Semicond. Process., 2015, 39, 641–648 CrossRef CAS.
- J. Tang, M. Myers, K. A. Bosnick and L. E. Brus, J. Phys. Chem. B, 2003, 107, 7501–7506 CrossRef CAS.
- S. O. P. W. S. Phanichphant, J. Electroceram., 2006, 16, 363–368 CrossRef.
- D. Padalia, U. C. Johri and M. G. H. Zaidi, Mater. Chem. Phys., 2016, 169, 89–95 CrossRef CAS.
- B. Bhushan, Z. Wang, J. Tol, N. S. Dalal, A. Basumallick, N. Y. Vasanthacharya, S. Kumar and D. Das, J. Am. Ceram. Soc., 2012, 6, 1985–1992 CrossRef.
- L. Zhao, H. Yang, X. Zhao, L. Yu, Y. Cui and S. Feng, Mater. Lett., 2006, 60, 1–6 CrossRef CAS.
- M. Thommes and K. A. Cychosz, Adsorption, 2014, 20, 233–250 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- M. W. Ruckman, J. Chen, S. L. Qiu, P. Kuiper and M. Strongin, Phys. Rev. Lett., 1991, 67, 2533–2566 CrossRef CAS PubMed.
- D. Channei, B. Inceesungvorn, N. Wetchakun and S. Phanichphant, Int. J. Photoenergy, 2013, 2013, 1–9 CrossRef.
- T. L. Barr, C. G. Fries, F. Cariati, J. C. J. Bart and N. Giordano, J. Chem. Soc., Dalton Trans., 1983, 1825–1829 RSC.
- T. Varnashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef.
- M. Muhler, R. Schlogl and G. Ertl, J. Catal., 1992, 138, 413–444 CrossRef CAS.
- T. M. Petrova, L. Fachikov and J. Hristov, Int. J. Chem. Biol. Eng., 2011, 3, 134–152 Search PubMed.
- S. Han, K. Liu, L. Hu, F. Teng, P. Yu and Y. Zhu, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- F. Helfferich, Ion Exchange, McGraw-Hill Book, 1962, vol. 138, p. 133 Search PubMed.
- M. Khosravi, B. Yahyaei and S. Azizian, J. Dispersion Sci. Technol., 2014, 35, 1135–1142 CrossRef CAS.
- M. Chauhan, B. Sharma, R. Kumar, G. R. Chaudhary, A. A. Hassan and S. Kumar, Environ. Res., 2019, 168, 85–95 CrossRef CAS PubMed.
- A. Debnath, K. Deb, K. K. Chattopadhyay and B. Saha, Desalin. Water Treat., 2016, 57, 13549–13560 CrossRef CAS.
- M. Y. Nassar, A. S. Amin, I. S. Ahmed and S. Abdallah, J. Taiwan Inst. Chem. Eng., 2016, 64, 79–88 CrossRef CAS.
- M. A. Behnajady, S. Yavari and N. Modirshahla, Chem. Ind. Chem. Eng. Q., 2014, 20, 97–107 CrossRef CAS.
- M. Y. Nassar, M. M. Moustafa and M. M. Taha, RSC Adv., 2016, 6, 42180–42195 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03252e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Suman Singhc,
R. J. Choudharyd and
S. K. Mehta
b,
Suman Singhc,
R. J. Choudharyd and
S. K. Mehta