
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, functionalization, and isolation of planar-chiral pillar[5]arenes with bulky substituents using a chiral derivatization agent†
Talal F. Al-Azemi *,
Mickey Vinodh,
Fatemeh H. Alipour and
Abdirahman A. Mohamod
*,
Mickey Vinodh,
Fatemeh H. Alipour and
Abdirahman A. Mohamod
Chemistry Department, Kuwait University, P. O. Box 5969, Safat 13060, Kuwait. E-mail: t.alazemi@ku.edu.kw; Fax: +965-2481-6482; Tel: +965-2498-554
First published on 26th July 2019
Abstract
Bulky perneopentyloxy-pillar[5]arene (Pillar-1) was synthesized and its conformational mobility was investigated using variable-temperature 1H NMR spectroscopy. The host–guest interactions between Pillar-1 and n-octyltrimethylammonium hexafluorophosphate (OMA) were investigated, and the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex was revealed via 1H NMR. Planar-chiral isomers were synthesized via the reaction of a hydroxy-functionalized pillar[5]arene with chiral derivatization agent (S)-(+)-MTPA-Cl. The (Sp, R)-and (Rp, R)-forms of the pillar[5]arene diastereomers were isolated by HPLC, and their structures were analyzed by 19F NMR. HPLC measurements indicated that racemization did not take place at 40 °C for 72 h.
:1 complex was revealed via 1H NMR. Planar-chiral isomers were synthesized via the reaction of a hydroxy-functionalized pillar[5]arene with chiral derivatization agent (S)-(+)-MTPA-Cl. The (Sp, R)-and (Rp, R)-forms of the pillar[5]arene diastereomers were isolated by HPLC, and their structures were analyzed by 19F NMR. HPLC measurements indicated that racemization did not take place at 40 °C for 72 h.
Introduction
Examples of macrocyclic host molecules are crown ethers,1 calixarenes,2 cucurbiturils,3 cyclodextrins,4 cyclophanenes5 and their structurally similar scaffolds.6 Pillararenes, a relatively new class of macrocyclic compounds, have garnered considerable attention recently due to their interesting conformational, physicochemical, and host–guest properties.7 Pillararenes and their derivatives are good hosts for a variety of organic compounds such as viologens,8a alkanediamines,8b dinitrobenzene,8c azobenzene derivatives,8d and neutral molecules.8e–g Functionalized pillararenes provide a useful platform for the construction of various interesting supramolecular systems and functionalization enables tuning of certain physical properties such as solubility, optical response, and crystallinity. Functionalized pillar[5]arenes bearing bromo,9a amino,9b alkyne,9c and hydroxy groups10–12 have been synthesized using different methods such as controlled de-O-methylation,9a selective oxidation–reduction,10 or by co-cyclization with a functional monomer.11,12Recently, planar-chiral pillar[5]arene enantiomers with bulky percyclohexylmethyl-substituents were successfully isolated using chiral column HPLC. The bulky substituents on both rims play an important role in inhibiting the rotational motion round the units.7d Notably, planar-chiral molecules have been widely used as chiral auxiliaries and ligands for transition metal-catalyzed asymmetric reactions. In addition, planar-chiral molecules have many potential applications as scaffolds for chiral guest receptors and architectures for chiral supramolecular assemblies and polymers.13 Appropriate structural modifications of the receptors are always necessary in order to tune the guest encapsulation and reactivity. Although chiral-planar pillar[5]arenes have been isolated by restricting the rotation around the units with bulky groups of cyclohexylmethyl groups, the synthesis and isolation of functionalized chiral-planar pillar[5]arenes with different bulky groups has not been reported. Herein, we report the synthesis of pillar[5]arenes with bulky perneopentyl groups, as well as their variable-temperature 1H NMR measurements, and guest-binding properties. The derivatization, isolation, and characterization of chiral-planar functionalized pillar[5]arenes with (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride ((S)-(+)-MTPA-Cl) are also reported.
Results and discussion
Synthesis
1,4-Bis(neopentyloxy)benzene was synthesized via the reaction of 1-iodo-2,2-dimethylpropane with hydroquinone in DMF in the presence of K2CO3 at 120 °C in a sure seal tube. Despite the 10 bulky groups on both rims, perneopentyloxy-pillar[5]arene (Pillar-1) was synthesized by cyclization of 1,4-bis(neopentyloxy)benzene with paraformaldehyde in the presence of BF3·OEt2, according to the literature procedure shown in Scheme 1.7a In contrast, bulky percyclohexylmethyl pillar[5]arene was synthesized by the reaction of perhydroxypillar[5]arene with (bromomethyl)cyclohexane.7d Suitable crystals for X-ray single crystal diffraction analysis were grown by slow diffusion of hexane into a solution of Pillar-1 in dichloroethane. In the solid state, Pillar-1 exhibited stacking in the edge-to-face style and the unit cell was quite large with six pillar[5]arenes and 14 dichloroethane molecules (see ESI†). | ||
| Scheme 1 Synthesis of perneopentyloxy-pillar[5]arene (Pillar-1), and X-ray crystal structure. | ||
In the 1H NMR spectrum of Pillar-1, the methylene protons (H2) split into two doublets with a 1:1 ratio due to their locations in the inner and outer spaces of the electron-rich cyclic structure, which were shielded and deshielded, respectively; this indicates no or slow mobility on the NMR time scale at 25 °C. To investigate the conformational mobility of Pillar-1, variable-temperature 1H NMR spectroscopy was performed in toluene-d8 (Fig. 1). No changes were observed in the two doublets corresponding to the methylene protons (H2) even at 90 °C due to the rigidity of the bulky t-butyl groups. Further variable-temperature 1H NMR experiments conducted with pillar[5]arene with bulky isobutyl substituents on the benzene rings showed that the protons coalesced into one peak upon heating above 75 °C, indicating that increasing the branching of the alkoxy groups on the pillar[5]arene ring hindered the conformational mobility.7b
 | ||
| Fig. 1 Variable-temperature 1H NMR spectra (600 MHz, toluene-d8) of Pillar-1. | ||
Binding studies
The host–guest interactions between Pillar-1 and n-octyltrimethyl ammonium hexafluorophosphate (OMA) were investigated using a 1H NMR titration method, as OMA was previously reported to be a good guest species for pillar[5]arenes.7d,14 The 1H NMR spectra of mixtures of OMA (12 mM) and varying concentrations of Pillar-1 in CDCl3 at 25 °C confirmed host–guest complexation, as evidenced by the upfield shifts of the resonances corresponding to the trimethyl protons (Ha) of OMA. The presence of signals corresponding to complexed (2.35 ppm) and uncomplexed guest OMA (3.18 ppm) suggested a slow exchange on the NMR time scale (see ESI†). Fig. 2 shows the 1H NMR spectra of Pillar-1 and OMA before and after complexation, along with the corresponding peak assignments, which were based on the host and guest 1H NMR spectra before complexation and 2D NMR spectra of the complex. After complexation, methyl and methylene protons Ha-f shifted upfield significantly, indicating that these protons were located in the shielding region of the cyclic pillar structure. The calculated upfield chemical shift changes of methylene protons Hb, Hc, and methyl protons Ha of OMA were −3.25, −3.74, and 0.79 ppm, respectively. In contrast, methylene protons Hg-h and methyl protons Hi were located in the deshielding region of the pillararene cavity, as evidenced by the downfield shift of methyl protons Hj from 0.88 to 1.03 ppm in the 1H NMR spectrum of the complex. These results indicated that the methylene protons were threaded through the cavity of the cyclic host. | ||
| Fig. 2 1H NMR spectra (600 MHz, CDCl3, 25 °C) of (a) 12 mM OMA, (b) 12 mM OMA and 14 mM Pillar-1, and (c) Pillar-1. | ||
The relative position of the protons and through-space 1H–1H interactions due to the inclusion complex formed between Pillar-1 and OMA were investigated using 2D ROESY NMR spectroscopy. The strong correlation between methylene hydrogen Hc and trimethyl group protons Ha of OMA with the methylene hydrogens of Pillar-1 provided further evidence for the formation of the inclusion complex (Pillar-1⊃OMA) (see ESI†).
The stoichiometry of the host–guest complex was established using Job's plots, which was formed using the mole fraction of the guest (Xguest) and the difference between the uncomplexed and observed chemical shift change of the trimethyl protons on OMA in 1H NMR multiplied by the mole fraction (Xguest). The Job's plot of Pillar-1 with OMA in CDCl3 showed a maximum at a mole fraction of 0.5 (Fig. 3a), indicating a 1:1 host-to-guest stoichiometric ratio. Further evidence of the 1:1 complex was obtained via ESI-MS, where the relevant molecular peak at m/z = 1483.28 corresponding to [(Pillar-1 + OMA)-PF6]+ was observed; these results were also in agreement with the results obtained from the 1H NMR titration experiments (Fig. 3b). The association constant for complexation (Ka) was determined to be 2.9 × 103 M−1 using the integration of the relative peak area of the slowly exchanged protons on the trimethyl group of OMA on the complexed and uncomplexed species. The association constant calculated for Pillar-1 was significantly higher than that of the bulky percyclohexylmethyl-pillar[5]arene.7d
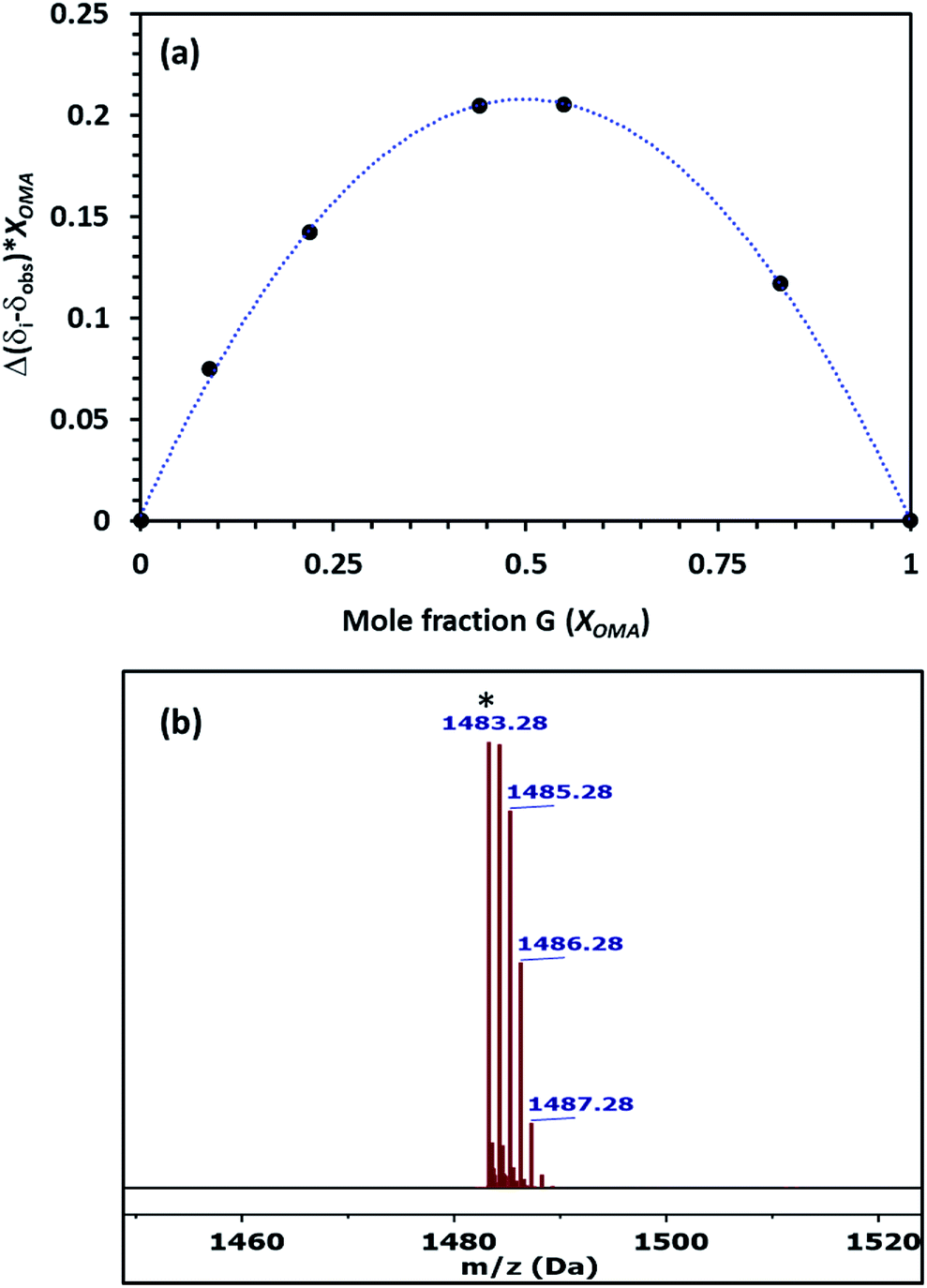 | ||
| Fig. 3 (a) Job's plot of Pillar-1 with OMA determined from 1H NMR titration in CDCl3 at 25 °C. (b) ES-MS spectrum of [(Pillar-1⊃OMA)-PF6]+. | ||
The pillar[5]arenes are racemic and comprised of a 1:1 mixture of the planar chiral (Sp) and (Rp) conformers, as evidenced from their XRD structures. The introduction of bulky groups on pillar[5]arenes is known to restrict the rotation of the units and result in the separation of enantiopure isomers, as demonstrated previously with a pillar[5]arene with bulky cyclohexylmethyl groups. However, attempts to separate enantiopure isomers of Pillar-1 by chiral column HPLC were unsuccessful. Therefore, using our previously reported strategy15 we attempted co-cyclization with a benzoxy-containing monomer, which enabled derivatization with a chiral agent ((S)-(+)-Mosher's acid chloride) after complete removal of the benzyl group by catalytic hydrogenation, as shown in Scheme 2.16
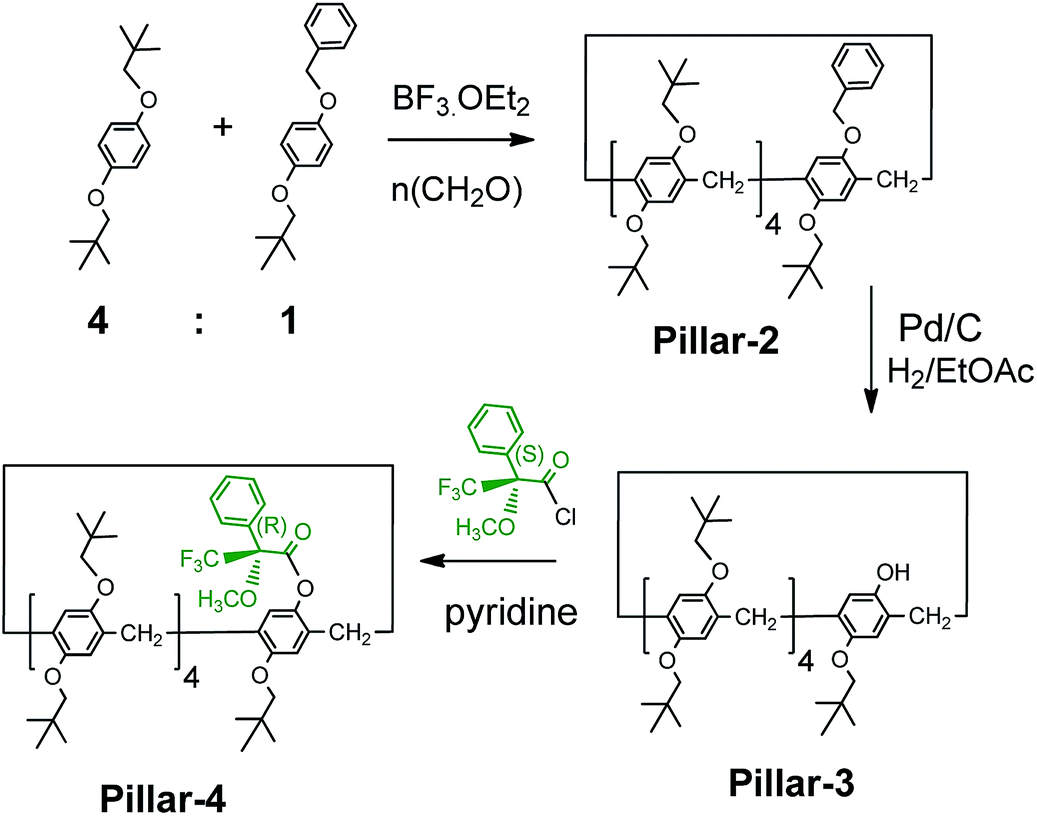 | ||
| Scheme 2 Synthesis and derivatization of monohydroxy-pillar[5]arene with (S)-(+)-Mosher's acid chloride. | ||
TLC analysis of synthesized Pillar-4 showed two spots with equal intensities, which were isolated by silica gel column chromatography. Fig. 4a shows the HPLC chromatograms of the crude reaction mixture and isolated (R)-(+)-Mosher's ester derivatives. Even though pillar[5]arene has eight possible stereoisomeric conformers, two peaks with equal areas were observed. Both fractions exhibited a peak at m/z 1479.9164 and 149.9141 corresponding to [M + Na]+ in their HRMS spectra. Fig. 4b shows the 19F NMR spectra before and after separation by silica gel column chromatography. The 19F NMR spectrum of the crude reaction mixture showed two signals with a 1:1 ratio corresponding to the trifluoromethyl (CF3) group, which indicated the presence of two stereoisomers. The CF3 group in the compound from the first fraction resonated at −70.02 ppm, while than that of the second fraction appeared at −70.87 ppm.
 | ||
| Fig. 4 (a) HPLC traces of Pillar-4 before and after separation. Hexane/CHCl3 = 95/5 (vol%) was used as the eluent. (b) 19F NMR spectra. | ||
Significant differences in the 1H NMR spectra of the isolated fractions were also observed (Fig. 1). The 1H NMR of the first fraction revealed a noticeable upfield shift in the resonance corresponding to one of the bridge methylene protons at 2.81 ppm and methine proton at 6.31 ppm of the aromatic pillar frame; in the second fraction, these protons appeared at 3.35 ppm and 6.68 ppm, respectively. Furthermore, the substituent methylene hydrogens (O–C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2–tBu) of the compound isolated in the first fraction resonated at a higher field compared to those of the second fraction. The shielding effect experienced by the protons in the compound isolated in the first fraction was due to their location in the shielding region of the phenyl group of the Mosher fragment, as evidenced by 1H–1H ROSEY NMR (see ESI†). In contrast, the protons corresponding to the Mosher methoxy group (assigned by 1H–13C HSQC) on the compound isolated in the first fraction resonated at 3.85 ppm, while those from the second fraction resonated at 3.56 ppm. To further investigate the rotational motion in the diastereomers of Pillar-4, HPLC measurements were carried out. No new peaks were detected in either fraction, indicating that the diastereomers did not racemize.
2–tBu) of the compound isolated in the first fraction resonated at a higher field compared to those of the second fraction. The shielding effect experienced by the protons in the compound isolated in the first fraction was due to their location in the shielding region of the phenyl group of the Mosher fragment, as evidenced by 1H–1H ROSEY NMR (see ESI†). In contrast, the protons corresponding to the Mosher methoxy group (assigned by 1H–13C HSQC) on the compound isolated in the first fraction resonated at 3.85 ppm, while those from the second fraction resonated at 3.56 ppm. To further investigate the rotational motion in the diastereomers of Pillar-4, HPLC measurements were carried out. No new peaks were detected in either fraction, indicating that the diastereomers did not racemize.
Experimental
Materials and methods
Nuclear magnetic resonance (NMR) spectroscopy was carried out on Bruker Avance II 600 MHz (Germany), Bruker DPX 400 (Germany), or Agilent 400 MHz (UK) spectrometers. Electron impact ionization (EI) mass spectrometry was performed on a Thermo Scientific DFS High Resolution GC/MS (Germany) mass spectrometer. Electro-spray ionization was carried out in high resolution mode using a Waters Xevo G2-S Qtof (Germany) LC MS/MS mass spectrometer. Single crystal data analysis was carried out on an R-AXIS RAPID II, Rigaku, (Japan) diffractometer. The data were collected at −123 °C (Oxford Cryosystems, UK). High performance liquid chromatography (HPLC) was carried out on a Waters 1525 (USA) HPLC system equipped with a UV-Vis detector (Waters 2487 dual λ absorbance detector). The details were as follows: Waters Spherisorb® 5 μm NH2; solvent polarity: hexane:chloroform (95:5, v/v); run time: 12 min; flow rate: 1 mL per min. Circular dichroism (CD) spectra were recorded on a Jasco J-815 CD (Germany) spectrometer. Ultraviolet-visible (UV-vis) spectra were recorded on a Varian Cary 5 Agilent (USA) spectrophotometer. Flash column chromatography was performed on silica gel (silica gel 60, 230–400 mesh ASTM, EMD Millipore, Merck KGaA, Germany). DMF, dichloroethane, and ethyl acetate were distilled prior to use in the hydrogenation reaction. All other reagents and solvents were of reagent grade purity and were used as received from commercial suppliers without further purification.
Synthesis of n-octyltrimethylammonium hexafluorophosphate (OMA)15c
Chlorooctane (2 mL; 11.76 mmol) and Trimethylamine (4.2 M solution in ethanol; 2.8 mL; 11.76 mmol) were taken in 10 mL round bottom flask and the reaction mixture was refluxed overnight. Ethanol was removed under reduced pressure, then the residue dissolved in deionized water. A solution of ammonium hexafluorophosphate (2.5 g; 15.3 mmol) in 1 mL deionized water, was added. White precipitate of OMA was formed instantly. The precipitate was collected by filtration, washed with water three times and dried at vacuum oven. Yield, 3.5 g white solid (11 mmol); 95%. 1H NMR (600 MHz, CDCl3) δ: 0.90 (t, J = 7.2, J = 7.2, 3H), 1.31 (m, 6H), 1.37 (m, 4H), 1.75 (m, 2H), 3.15 (s, 9H), 3.28 (m, 2H). 13C NMR (150 MHz, CDCl3), δ: 14.2, 22.8, 23.3, 26.2, 29.2, 31.8, 53.4, 67.6. EI-MS: 172.2 [M − PF6].Synthesis of 4-(benzyloxy)phenol
Hydroquinone (6.60 g; 60 mmol) was dissolved in acetone (120 mL) at room temperature and potassium carbonate (12.40 g; 90 mmol) was added. The solution was refluxed overnight after adding benzyl bromide (10.36 mL, 40 mmol). The solvent was removed by evaporation under reduced pressure and the crude product was extracted with dichloromethane. 4-(Benzyloxy)phenol was then purified by column chromatography using a mixture of dichloromethane and hexane (60:40, v/v). Yield = 5.6 g, 70%. 1H NMR (400 MHz, CDCl3) δ: 5.01 (s, 2H), 6.77 (m, 2H), 6.87 (m, 2H), 7.38 (m, 5H); 13C NMR (150 MHz, CDCl3), δ: 71.0, 116.3, 116.3, 127.7, 128.1, 128.8, 137.4, 149.9, 153.2. HRMS: calc.: 200.0832 (for C13H12O2; [M]+); found: 200.0833.
Synthesis of 1-(benzyloxy)-4-(neopentyloxy)benzene
In a sealed tube, 4-(benzyloxy)phenol (4.00 g; 20 mmol) was dissolved in DMF (20 mL) at room temperature and potassium carbonate (4.14 g; 30 mmol) was added with stirring. This solution was stirred for about 30 min and then 1-iodo-2,2-dimethylpropane (2.65 mL, 20 mmol) was added. The sealed tube was heated at 125 °C for 24 h. The crude product precipitated upon addition of water (200 mL) to the reaction mixture. The precipitate was collected, washed with water (2 × 50 mL), and dried over anhydrous Na2SO4. The product was purified by column chromatography using dichloromethane/hexane (50:50, v/v). Yield = 3.5 g, 65%. 1H NMR (600 MHz, CDCl3) δ: 1.08 (s, 9H), 3.56 (s, 2H), 5.03 (s, 2H), 6.85 (m, 2H), 6.91 (m, 2H), 7.32 (m, 1H), 7.39 (m, 2H), 7.44 (m, 2H); 13C NMR (150 MHz, CDCl3), δ: 26.8, 32.1, 70.9, 78.8, 115.6, 116.0, 127.6, 128.0, 128.7, 137.6, 153.0, 154.3. HRMS: calc.: 270.1614 (for C18H22O2; [M]+); found: 270.1614.
Pillar-1
Paraformaldehyde (0.93 g, 30 mmol) was added to a solution of 1,4-bis(neopentyloxy)benzene (2.50 g, 10 mmol) in dry dichloroethane (30 mL) under a nitrogen atmosphere. Boron trifluoride diethyl etherate (BF3·OEt2), (1.25 mL, 10 mmol) was then added to the solution, and the mixture was stirred at room temperature for 30 min. MeOH (100 mL) was poured into the reaction mixture, the solution was concentrated, and the resulting residue was dissolved in CH2Cl2 (100 mL). The solution was then washed with aqueous NaHCO3 (2 × 50 mL) and H2O (50 mL). The organic layer was dried over anhydrous Na2SO4, concentrated under vacuum, and subjected to silica gel chromatography (hexane/CH2Cl2; 40:60, v/v) to give Pillar-1 as a white solid (680 mg, 26%). 1H NMR (600 MHz, CDCl3) δ: 1.15 (S, 90H), 3.43 (d, J = 8.4, 10H), 3.80 (d, J = 8.4, 10H), 3.90 (s, 10H), 6.97 (s, 10H); 13C NMR (150 MHz, CDCl3), δ: 27.1, 29.0, 32.1, 79.8, 115.5, 128.6, 150.4. HRMS: calc.: 1311.9742 (for C85H131O10; [M + H]+); found: 1311.9762.
Pillar-2
Paraformaldehyde (0.93 g, 30 mmol) was added to a solution of 1,4-bis(neopentyloxy)benzene (2.0 g, 8 mmol) and 1-(benzyloxy)-4-(neopentyloxy)benzene (0.54 g, 2 mmol) in dry dichloroethane (30 mL) under a nitrogen atmosphere. BF3·OEt2 (1.25 mL, 10 mmol) was then added to the solution, and the mixture was heated to 35 °C for 30 min. MeOH (50 mL) was poured into the reaction mixture, the solution was concentrated, and the resulting residue was dissolved in CH2Cl2 (100 mL). The solution was then washed with aqueous NaHCO3 (2 × 50 mL) and H2O (50 mL). The organic layer was dried over Na2SO4, concentrated under vacuum, and subjected to silica gel chromatography (hexane/CH2Cl2; 85:15 v/v) to give Pillar-2 as a white solid (320 mg, 12%). 1H NMR (600 MHz, CDCl3) δ: 0.95 (S, 9H), 1.12 (m, 72H), 3.05 (d, J = 8.4, 1H), 3.13 (d, J = 9.0, 1H), 3.41 (m, 8H), 3.75 (m, 8H), 3.86 (m, 10H), 4.90 (d, J = 11.4, 1H), 5.13 (d, J = 11.4), 6.84 (s, 1H), 6.87 (s, 1H), 6.89 (s, 1H), 6.92 (s, 1H), 6.94 (m, 5H), 7.07 (s, 1H), 7.32 (m, 3H), 7.48 (m, 2H); 13C NMR (150 MHz, CDCl3), δ: 14.3, 22.9, 27.2, 27.2, 27.2, 27.3, 27.3, 27.3, 27.4, 28.3, 28.5, 29.2, 29.5, 30.6, 31.8, 32.1, 32.3, 32.3, 32.3, 70.6, 78.8, 79.4, 79.9, 79.9, 79.9, 80.0, 80.4, 114.9, 115.3, 115.3, 115.5, 115.6, 115.6, 115.8, 115.9, 116.0, 116.0, 127.5, 127.8, 127.9, 128.4, 128.5, 128.7, 128.8, 128.8, 128.9, 129.1, 129.1, 129.2, 138.3, 150.1, 150.2, 150.3, 150.4, 150.5, 150.6, 150.7, 150.7, 150.8, 150.9. HRMS: calc.: 1353.9249 (for C87H126O10Na; [M + Na]+); found: 1353.9218.
Pillar-3
50 mg of Pd/C (5%) was added to a solution of Pillar-2 (0.200 g; 0.15 mmol) in 30 mL of anhydrous ethyl acetate. Air was evacuated from the apparatus used for the catalytic hydrogenation and it was filled with H2. After the theoretical quantity of hydrogen was absorbed, the Pd/C catalyst was filtered off and carefully washed with ethyl acetate. The combined filtrate was evaporated to give Pillar-3 as a white solid (0.180 g, 97%). 1H NMR (600 MHz, CDCl3) δ: 0.99 (s, 9H), 1.11 (m, 72H), 3.35 (m, 1H), 3.46 (m, 6H), 3.50 (m, 1H), 3.65 (m, 8H), 3.77 (m, 5H), 3.93 (m, 5H), 4.13 (d, J = 7.2, 1H), 4.16 (d, J = 7.2, 1H), 6.76 (m, 3H), 6.84 (s, 1H), 6.89 (s, 1H), 6.91 (s, 1H), 6.94 (s, 1H), 6.99 (s, 1H), 7.00 (s, 1H), 7.01 (s, 1H); 13C NMR (150 MHz, CDCl3), δ: 22.9, 27.1, 27.2, 27.2, 27.3, 27.3, 27.3, 27.4, 28.6, 29.0, 29.7, 29.9, 30.0, 31.4, 32.2, 32.2, 32.3, 32.3, 32.3, 32.4, 32.4, 79.5, 79.8, 79.9, 80.0, 80.1, 80.2, 80.3, 115.2, 115.5, 115.6, 115.7, 115.7, 115.7, 115.8, 116.0, 118.8, 125.8, 127.3, 128.8, 128.9, 129.0, 129.2, 129.8, 130.0, 147.2, 150.3, 150.5, 150.6, 150.6, 150.7, 150.9, 150.9. HRMS: calc.: 1263.8779 (for C80H120O10Na; [M + Na]+); found: 1263.8757.Derivatization reaction
Pillar-3 (100 mg, 0.08 mmol) was dissolved in 1 mL distilled pyridine, and 20 mg of dimethylaminopyridine (DMAP) was added and stirred at 0 °C for 15 min. (S)-(+)-α-methoxy-α-trifluoromethylphenylacetyl chloride (50 μL, 0.267 mmol) was added and the mixture was stirred for 3 h at room temperature. The reaction mixture was poured into 20 mL aqueous HCl (4 M). The resulting white precipitate was filtered, and washed with distilled water (2 × 50 mL). TLC analysis showed two distinct spots, which corresponded to the diastereomers. The two diastereomers were separated by column chromatography using hexane/dichloromethane (85:15, v/v). Overall yield = 101 mg, 87%.
First fraction
Yield = 49 mg (42%). 1H NMR (600 MHz, CDCl3) δ: 1.01 (s, 9H), 1.09 (m, 63H), 1.22 (s, 9H), 2.84 (d, J = 18.0, 1H) 3.24 (m, 1H), 3.31 (m, 1H), 3.38 (m, 7H), 3.79 (m, 21H), 6.32 (s, 1H), 6.92 (m, 8H), 7.16 (s, 1H), 7.41 (m, 3H), 7.72 (m, 2H); 13C NMR (150 MHz, CDCl3), δ: 22.9, 27.2, 27.3, 27.5, 28.9, 29.1, 29.1, 29.2, 29.3, 31.8, 32.2, 32.2, 32.3, 32.3, 32.4, 56.9, 79.6, 79.6, 79.9, 80.0, 80.0, 80.1, 114.9, 115.0, 115.5, 115.5, 115.8, 115.8, 115.9, 116.0, 124.3, 127.3, 127.4, 128.0, 128.7, 128.8, 129.0, 129.1, 129.1, 129.2, 129.3, 130.1, 131.8, 140.2, 150.2, 150.4, 150.6, 150.6, 150.7, 150.8, 150.8, 150.9, 155.4, 166.4. HRMS: calc.: 1479.9177 (for C90H127O12F3Na; [M + Na]+); found: 1479.9164.Second fraction
Yield = 52 mg (45%).1H NMR (600 MHz, CDCl3) δ: 0.96 (s, 9H), 1.08 (m, 72H), 3.34 (m, 10H), 3.53 (s, 3H), 3.62 (m, 18H), 6.65 (s, 1H), 6.90 (m, 8H), 7.05 (s, 1H), 7.43 (m, 3H), 7.64 (m, 2H); 13C NMR (150 MHz, CDCl3), δ: 27.3, 27.3, 27.3, 27.3, 28.8, 29.1, 29.3, 29.5, 29.9, 32.2, 32.3, 32.3, 55.6, 79.8, 79.8, 79.9, 80.0, 80.0, 80.1, 80.2, 115.0, 115.4, 115.4, 115.5, 115.7, 115.7, 115.9, 116.0, 116.0, 124.1, 127.5, 127.7, 128.4, 128.6, 128.9, 129.0, 129.0, 129.0, 129.3, 129.3, 129.3, 129.3, 130.1, 131.7, 132.0, 140.4, 150.2, 150.3, 150.6, 150.6, 150.7, 150.8, 151.0, 151.1, 155.6, 166.5. HRMS: calc.: 1479.9177 (for C90H127O12F3Na; [M + Na]+); found: 1479.9141.Preparation of single crystals for X-ray diffraction
Crystals suitable for single crystal X-ray diffraction of Pillar-1 were grown by diffusion using dichloroethane and hexane. The single crystal data were collected on a diffractometer (R-AXIS RAPID, Rigaku, Japan) using Rigaku's Crystal clear software package at −123 °C. The structure was solved and refined using the Bruker SHELXTL Software Package (structure solution program: SHELXS-97; refinement program: SHELXL-97). CCDC 1561385.†1H NMR titrations
A 0.5 mL sample of OMA was prepared at 12.0 mM in chloroform-d. All titration experiments were carried out in an NMR tube at 298 K, and 1H-NMR spectra were recorded upon successive addition of the appropriate amount of the host (Pillar-1). The association constant was calculated from the complexed and uncomplexed trimethyl groups of OMA at 2.35 and 3.18 ppm, respectively at 80% complexation.Conclusions
In conclusion, we successfully synthesized a pillar[5]arene with bulky neopentyl groups on both rims. Variable-temperature 1H NMR spectroscopy experiments showed that the bulky substituents inhibited the rotational mobility. Complexation between Pillar-1 and OMA was studied by 1H-NMR and ES-MS, and the formation of a 1:1 inclusion complex Pillar-1⊃OMA was confirmed. Monohydroxy-functionalized pillar[5]arene Pillar-3 was synthesized by co-cyclization of hydroquinone derivatives bearing monobenzyl substituents followed by catalytic hydrogenation over palladium in charcoal in anhydrous ethyl acetate. The pure diastereomeric pair was separated by column chromatography after derivatization with (S)-(+)-Mosher's acid chloride to produce Pillar-3. The diastereomeric purity was confirmed by chiral-column HPLC and 19F NMR analysis. No racemization was observed at 40 °C for 72 h. Further studies and modification of the planar-chiral functionalized pillar[5]arene host in terms of their use as chiral guest receptors and building blocks for chiral supramolecular architectures are underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support of the University of Kuwait, received through research grant no. SC03/16, and the facilities of ANALAB and SAF (grant no. GS01/01, GS03/01, GS01/03, GS01/05, and GS03/08) are gratefully acknowledged.Notes and references
- (a) K. E. Krakowiak, J. S. Bradshaw and D. J. Zamecka-Krakowiak, Chem. Rev., 1989, 89, 929–972 CrossRef CAS; (b) J. S. Bradshaw and R. M. Izatt, Acc. Chem. Res., 1997, 30, 338–345 CrossRef CAS; (c) G. W. Gokel, W. M. Levy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- (a) A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed; (b) C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry, Cambridge, 2nd edn, 2000 Search PubMed; (c) C. D. Gutsche, Calixarenes, An Introduction, Royal Society of Chemistry, Cambridge, 2008 Search PubMed; (d) E. Botana, E. Da Silva, J. Benet-Buchholz, P. Ballester and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 198–201 CrossRef CAS PubMed.
- (a) K. Kim, Chem. Soc. Rev., 2002, 31, 96–107 RSC; (b) A. E. Kaifer, W. Li, S. Silvi and V. Sindelar, Chem. Commun., 2012, 48, 6693 RSC.
- (a) A. Harada, Acc. Chem. Res., 2001, 34, 456–467 CrossRef CAS; (b) L. Zhu, D. Zhang, D. Qu, Q. Wang, X. Ma and H. Tian, Chem. Commun., 2010, 46, 2587–2589 RSC; (c) K.-R. Wang, D.-S. Guo, B.-P. Jiang and Y. Liu, Chem. Commun., 2012, 48, 3644–3646 RSC.
- (a) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed; (b) W. Maes and W. Dehaen, Chem. Soc. Rev., 2008, 37, 2393–2402 RSC; (c) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed.
- (a) F. Diederich, Cyclophanes, The Royal Society of Chemistry, Cambridge, 1991 Search PubMed; (b) J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183 CrossRef CAS PubMed; (c) H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406 CrossRef CAS PubMed.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T. A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) C. Han, F. Ma, Z. Zhang, B. Xia, Y. Yu and F. Huang, Org. Lett., 2010, 12, 4360–4363 CrossRef CAS PubMed; (c) Z. Li, J. Yang, J. He, Z. Abliz and F. Huang, Org. Lett., 2014, 16, 2065–2069 Search PubMed; (d) T. Ogoshi, K. Masaki, R. Shiga, K. Kitajima and T.-a. Yamagishi, Org. Lett., 2011, 13, 1264–1266 CrossRef CAS PubMed; (e) T. Ogoshi, Y. Nishida, T.-a. Yamagishi and Y. Nakamoto, Macromolecules, 2010, 43, 3145–3147 CrossRef CAS; (f) H. Li, Y. Yang, F. Xu, T. Liang, H. Wena and W. Tian, Chem. Commun., 2019, 55, 271–285 RSC; (g) W. Feng, M. Jin, K. Yang, Y. Pei and Z. Pei, Chem. Commun., 2018, 54, 13626–13640 RSC.
- (a) N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed; (b) N. Strutt, D. Fairen-Jimenez, J. Iehl, M. Lalonde, R. Snurr, O. K. Farha, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17436–17439 CrossRef CAS PubMed; (c) T. Ogoshi, K. Kida and T. Yamagishi, J. Am. Chem. Soc., 2012, 134, 20146–20150 CrossRef CAS PubMed; (d) C. Li, S. Chen, J. Li, K. Han, M. Xu, B. Hu, Y. Yu and X. Jia, Chem. Commun., 2011, 47, 11294–11296 RSC; (e) C. Li, K. Han, J. Li, H. Zhang, J. Ma, X. Shu, Z. Chen, L. Weng and X. Jia, Org. Lett., 2012, 14, 42–45 CrossRef CAS PubMed; (f) T. Ogoshi, K. Demachi, K. Kitajima and T.-A. Yamagishi, Chem. Commun., 2011, 47, 10290–10292 RSC; (g) T. Ogoshi, K. Masaki, R. Shiga, K. Kitajima and T. Yamagishi, Org. Lett., 2011, 13, 1264–1266 CrossRef CAS PubMed.
- (a) T. Ogoshi, T. Aoki, K. Kitajima, S. Fujinama, T. A. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76, 328–331 CrossRef CAS PubMed; (b) Y. Chen, M. He, B. Li, L. Wang, H. Meier and D. Cao, RSC Adv., 2013, 3, 21405–21408 RSC; (c) J. Han, X. Hou, C. Ke, H. Zhang, N. L. Strutt, C. L. Stern and J. F. Stoddart, Org. Lett., 2015, 17, 3260–3263 CrossRef CAS PubMed.
- (a) C. Han, Z. Zhang, G. Yu and F. Huang, Chem. Commun., 2012, 48, 9876–9878 RSC; (b) T. Ogoshi, D. Yamafuji, D. Kotera, T. Aoki, S. Fujinami and T.-A. Yamagishi, J. Org. Chem., 2012, 77, 11146–11152 CrossRef CAS PubMed; (c) M. Pan and M. Xue, Eur. J. Org. Chem., 2013, 4787–4793 CrossRef CAS.
- (a) N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668 CrossRef CAS PubMed; (b) N. L. Strutt, D. Fairen-Jimenez, J. Iehl, M. B. Lalonde, R. Q. Snurr, O. K. Farha, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17436 CrossRef CAS PubMed.
- (a) T. Ogoshi, R. Shiga, M. Hashizume and T. Yamagishi, Chem. Commun., 2011, 47, 6927–6929 RSC; (b) W. B. Hu, W.-J. Hu, X.-L. Zhao, Y. A. Liu, J. S. Li, B. Jiang and K. Wen, J. Org. Chem., 2016, 81, 3877–3881 CrossRef CAS PubMed; (c) G. Yu, Y. Ma, C. Han, Y. Yao, G. Tang, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2013, 135, 10310–10313 CrossRef CAS PubMed.
- (a) S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744–5893 CrossRef CAS PubMed; (b) S. Arae and M. Ogasawara, Tetrahedron Lett., 2015, 56, 1751–1761 CrossRef CAS; (c) R. Deng, Y. Huang, X. Ma, G. Li, R. Zhu, B. Wang, Y. Kang and Z. Gu, J. Am. Chem. Soc., 2014, 136, 4472–4475 CrossRef CAS PubMed; (d) Y. Morisaki, M. Gon, T. Sasamori, N. Tokitoh and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 3350–3353 CrossRef CAS PubMed; (e) X. Liu, Y. Ma, W. Duan, F. He, L. Zhao and C. Song, J. Org. Chem., 2011, 76, 1953–1956 CrossRef CAS PubMed; (f) R. Fiesel, J. Huber, U. Apel, V. Enkelmann, R. Hentschke and U. Scherf, Macromol. Chem. Phys., 1997, 198, 2623–2650 CrossRef CAS.
- (a) T. Ogoshi, R. Shiga, T.-a. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76, 618–622 CrossRef CAS PubMed; (b) Z. Zhang, B. Xia, C. Han, Y. Yu and F. Huang, Org. Lett., 2010, 12, 3285–3287 CrossRef CAS PubMed.
- (a) T. F. Al-Azemi, M. Vinodh, F. H. Alipour and A. A. Mohamod, J. Org. Chem., 2017, 82, 10945–10952 CrossRef CAS PubMed; (b) T. F. Al-Azemi, A. A. Mohamod, M. Vinodh and F. H. Alipour, Org. Chem. Front., 2018, 5, 10–18 RSC; (c) T. F. Al-Azemi, M. Vinodh, F. H. Alipour and A. A. Mohamod, Org. Biomol. Chem., 2018, 16, 7513–7517 RSC; (d) P. Demay-Drouhard, K. Du, K. Samanta, X. Wan, W. Yang, R. Srinivasan, A. C.-H. Sue and H. Zuilhof, Org. Lett., 2019, 21(11), 3976–3980 CrossRef CAS PubMed.
- T. F. Al-Azemi, M. Vinodh, F. H. Alipour and A. A. Mohamod, Org. Chem. Front., 2019, 6, 603–610 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1561385. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra03135a |
| This journal is © The Royal Society of Chemistry 2019 |