
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of the plant alkaloid racemic microthecaline A: first example of a natural product bearing a tricyclic quinoline-serrulatane scaffold
Thirupathi Reddy Penjarlaab,
Maheshwar Kundarapub,
Syed Mohd. Baquerb and
Anupam Bhattacharya *a
*a
aDepartment of Chemistry, Birla Institute of Technology and Science-Pilani (Hyderabad Campus), Hyderabad-500078, India. E-mail: anupam@hyderabad.bits-pilani.ac.in; Fax: +91-40-66303998; Tel: +91-40-66303522
bDepartment of Medicinal Chemistry, GVK Biosciences Pvt. Ltd., Survey No. 125 (Part) & 126, IDA Mallapur, Hyderabad 500076, India
First published on 26th July 2019
Abstract
The first total synthesis of racemic microthecaline A, a quinoline serrulatane alkaloid, isolated from the Australian desert plant Eremophila microtheca is described. The natural product was synthesized in ten steps, starting from ethyl 4-bromo-6-methoxy-8-methylquinoline-3-carboxylate in 8% overall yield.
Introduction
Isolation and characterization of the alkaloid (R)-microthecaline A was reported by Davis and co-workers from the Australian desert plant Eremophila microtheca.1 The molecule is the first example of a natural product bearing the tricyclic quinoline-serrulatane scaffold and displayed moderate activity against 3D7 and Dd2 Plasmodium falciparum strains. Bioactive serrutalane based molecules are well known for their diverse activities including anti-tubercular, anti-inflammatory and inhibition of phagocytosis.2 These properties along with interesting structural features have made serrutalane diterpenoids important targets in synthetic explorations. In a paper reported last year by Rajan Babu and coworkers they disclosed the use of stereoselective hydrovinylation en route to the synthesis of serrulatane diterpenes and several of their diastereomeric analogs.3 This paper attempted to address the installation of stereogenic centers on exocyclic locations adjacent to chiral centers and also developed the synthesis of various serrulatane diterpenoids. Aggarwal et al. in another paper have reported the synthesis of all diastereomers of erogorgiaene, a serrulatane diterpenoid via a lithiation–borylation methodology.4Given our research interest in the area of fused quinoline based heterocycles,5 total synthesis of microthecaline A was undertaken. Key feature of its structure is the presence of a tetra-substituted 5,6-dihydro-4H-benzo[de]quinoline ring. While several reports are available in the literature for the synthesis of the aforementioned ring system, most of them disclose the synthesis in a fused ring format along with other aromatic rings.6 Very few examples reveal a stand-alone synthesis of this scaffold. In a recent paper AlCl3 assisted synthesis of 6-methyl-5,6-dihydro-1H-benzo[de]quinolin-2(4H)-one from 4-(but-3-en-1-yl)quinolin-2(1H)-one was reported by Xu et al.7 Kanai and co-workers have reported synthesis of these rings by reaction between methyl quinolines and styrenes using an in situ generated cobalt hydride catalyst.8 Li et al. in a separate paper have reported the formation of 2-methyl-6-phenyl-5,6-dihydro-4H-benzo[de]quinoline, in their attempt to carry out photo-induced methylation of heteroarenes.9 Ellman and co-workers have reported synthesis of 2,3-dihydro-1H-benzo[kl]acridine bearing the 5,6-dihydro-4H-benzo[de]quinoline unit in their attempt to synthesize unsymmetrical acridines and phenazines.10 In a recent paper by Kuhn and coworkers AlCl3 mediated intramolecular α-alkylation was used for the synthesis of diverse tricyclic scaffolds starting from α,β-unsaturated lactones and lactams.11 Most of the reported compounds represented either 4a,5,7,8,9,10-hexahydrophenanthridin-6(10bH)-one or 7,8,9,10-tetrahydro-6H-benzo[c]chromen-6-one type of ring system, except one example which showed the formation of 6-methyl-5,6-dihydro-1H-benzo[de]quinolin-2(4H)-one compound.
Results and discussion
Retrosynthetic analysis
For microthecaline A synthesis, construction of the 5,6-dihydro-4H-benzo[de]quinoline ring was crucial along with its C-3 appendage and other substituents. Accordingly, a retrosynthetic strategy was envisaged relying mainly on introduction of a pivotal cyclohexane ring by Friedel–Crafts reaction. The initial 5,6-dihydro-4H-benzo[de]quinoline ring system was supposed to be synthesized starting from ethyl 4-bromo-6-methoxy-8-methylquinoline-3-carboxylate and subsequent implementation of Heck coupling and Friedel–Crafts acylation. As per this strategy Wittig reaction followed by asymmetric reduction would generate the required C-6 chiral center with a substituted methyl group. Further systematic two fold implementation of partial reduction and Wittig/Wittig–Horner reaction, respectively would provide the methoxy-substituted penultimate compound. The final step required demethylation to generate the target molecule (Scheme 1).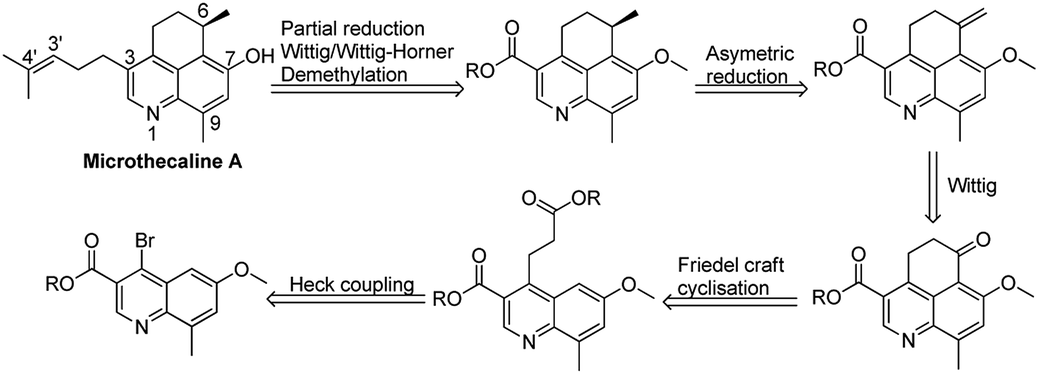 | ||
| Scheme 1 Preliminary retrosynthetic analysis of microthecaline A. | ||
Synthetic strategy
Our synthetic efforts started with commercially available 4-methoxy-2-methylaniline which was converted to ethyl 4-bromo-6-methoxy-8-methylquinoline-3-carboxylate (1) using well established synthetic protocol.12 Compound 1 was subjected to Heck coupling which yielded corresponding α,β-unsaturated ester (2) in 75% yield. On reaction with NiCl2–NaBH4in CH3CN–H2O as solvent system, compound 2 was converted to the corresponding reduced ester (3). Subsequent attempts to carry out Friedel–Crafts acylation on 2 in the presence of CF3SO3H gave the tricyclic cyclohexanone moiety in 60% yield. At this juncture our repeated attempts (by using KOtBu, NaOtBu, NaH, n-BuLi) to generate ethyl 7-methoxy-9-methyl-6-methylene-5,6-dihydro-4H-benzo[de]quinoline-3-carboxylate (4) via Wittig reaction resulted in the formation of only α,β-unsaturated cyclohexanone derivative 5 instead of the expected product (ESI). This forced us to abandon the initially articulated route of asymmetric reduction of the terminal double bond (Scheme 2).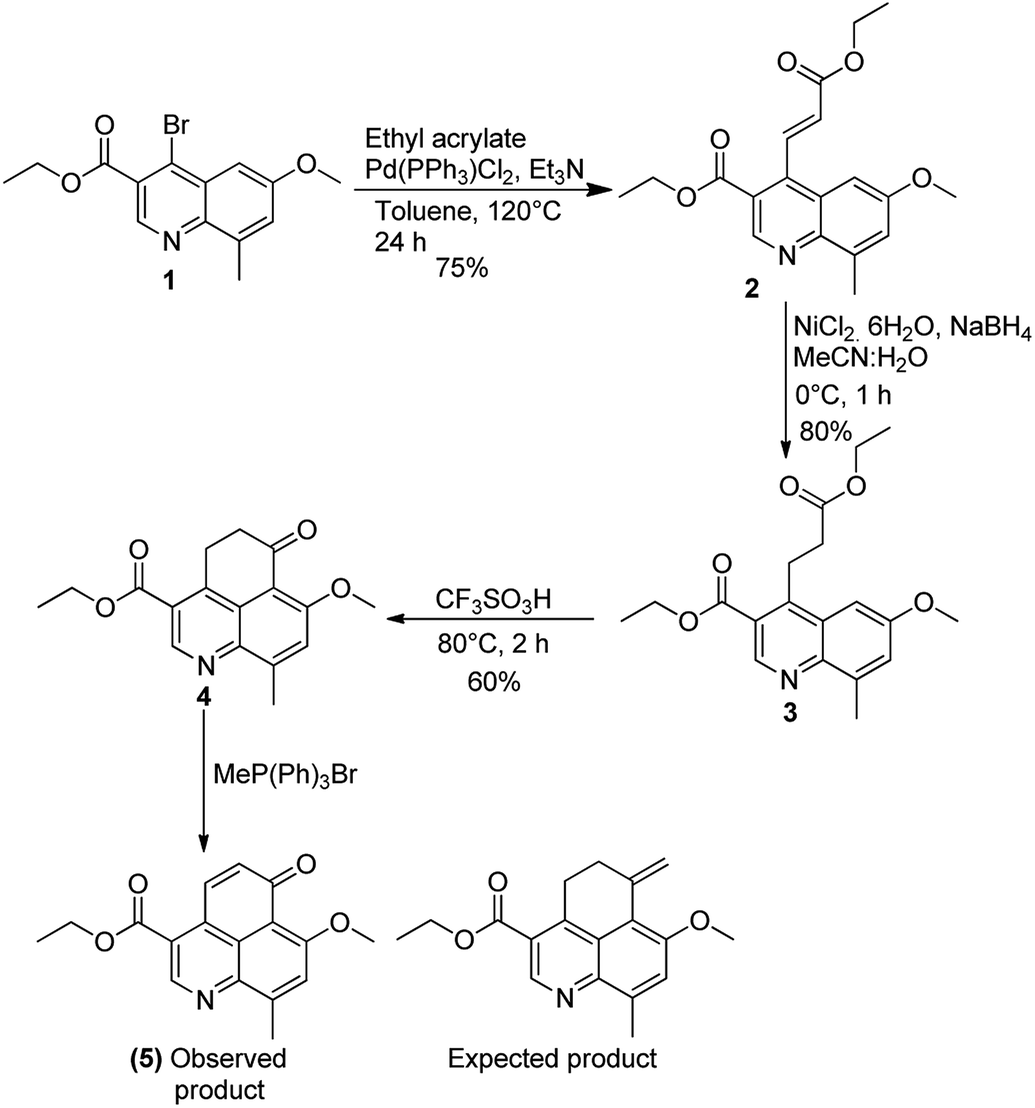 | ||
| Scheme 2 Initial attempt to synthesize the tricyclic core and introduction of the chiral center. | ||
We subsequently carried out a Grignard reaction on the cyclohexanone carbonyl in 4 and the resulting tertiary alcohol (6) was treated with HCl to generate ethyl 7-methoxy-6,9-dimethyl-4H-benzo[de]quinoline-3-carboxylate. The rational here was to attempt asymmetric reduction of the double bond to generate the appropriate chiral center. Several attempts to carry out elimination reaction using HCl, HBr, p-TSA (p-toluene sulfonic acid), PPTS (pyridinium-p-toluenesulfonate), TESiH (triethylsilane) and SiO2did not yield the desired compound ethyl 7-methoxy-6,9-dimethyl-4H-benzo[de]quinoline-3-carboxylate (7) (Scheme 3).
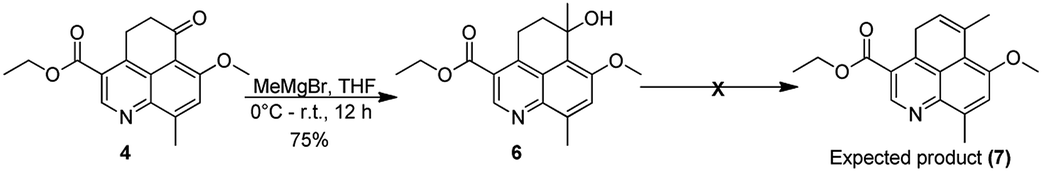 | ||
| Scheme 3 Grignard reaction on the tricyclic core and subsequent elimination attempts. | ||
Our initial setbacks in obtaining the chiral center with appropriate stereochemistry forced us to follow a different approach and focus mainly on the synthesis of the racemic microthecaline A. Our revised method was also initiated from 1, which when subjected to reductive Heck coupling (initial attempt with Pd(OAc)2 and o-tolylphospine led to the formation of reductive Heck product and Heck product in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, while reaction using PdCl2(PPh3)2 as catalyst gave 9:1 ratio of the reductive Heck product and Heck product) yielded compound 8. Subsequent reduction of ketonic carbonyl in 7 using NaBH4 gave the corresponding tertiary alcohol (9) in 91% yield. Subjecting 9 to intramolecular Friedel–Crafts alkylation, by using well known route with FeCl3–AgSbF6 did not give the expected product.13 This observation forced us to defer the intramolecular Friedel–Crafts alkylation step and instead chlorination of compound 9 was attempted with N-chlorosuccinimide in the presence of PPh3. The reaction was successful and gave compound 10 in 85% yield. Further, the available ester on quinoline ring was reduced with DIBAL-H (attempts to terminate the reaction at aldehyde stage failed) and compound 11 thus obtained was oxidized by Dess–Martin periodinane (DMP) to the corresponding aldehyde. It was not isolated and was directly used as a substrate for the Wittig–Horner reaction in the presence of NaH in THF to produce the α,β-unsaturated ester (12) in 70% yield. Subjecting compound 12 to the deferred intramolecular Friedel–Crafts alkylation step using AlCl3 yielded the required tricyclic scaffold (13) in 68% yield. The compound was thoroughly characterized by 1H/13C and NOE experiments (ESI). Subsequent attempts were directed on installing the remaining C3 fragment on the side chain. It was carried out by reduction of the double bond in compound 13 to its corresponding saturated congener 14, which was further reduced to the 1° alcohol (similar to our previous observation, attempts to terminate the reaction at aldehyde stage failed) 15. Oxidation with DMP followed by treatment with the appropriate ylide in the presence of n-BuLi led to the penultimate compound 16. Final step of demethylation was attempted with various reagents such as BBr3, HBr, HCl, NaSEt but was only successful with LiCl in DMF under microwave conditions and gave (±)-microthecaline A as a free base in 65% yield. The product obtained was thoroughly characterized and the spectroscopic data was found to be comparable with the literature values available for microthecaline A (ESI).1 Given the importance of enantiomeric purity to evaluate the biological activity of the molecule, attempts are currently underway in our lab to develop a synthetic route for (R)-microthecaline A, the naturally occurring stereoisomer (Scheme 4).
:1 ratio, while reaction using PdCl2(PPh3)2 as catalyst gave 9:1 ratio of the reductive Heck product and Heck product) yielded compound 8. Subsequent reduction of ketonic carbonyl in 7 using NaBH4 gave the corresponding tertiary alcohol (9) in 91% yield. Subjecting 9 to intramolecular Friedel–Crafts alkylation, by using well known route with FeCl3–AgSbF6 did not give the expected product.13 This observation forced us to defer the intramolecular Friedel–Crafts alkylation step and instead chlorination of compound 9 was attempted with N-chlorosuccinimide in the presence of PPh3. The reaction was successful and gave compound 10 in 85% yield. Further, the available ester on quinoline ring was reduced with DIBAL-H (attempts to terminate the reaction at aldehyde stage failed) and compound 11 thus obtained was oxidized by Dess–Martin periodinane (DMP) to the corresponding aldehyde. It was not isolated and was directly used as a substrate for the Wittig–Horner reaction in the presence of NaH in THF to produce the α,β-unsaturated ester (12) in 70% yield. Subjecting compound 12 to the deferred intramolecular Friedel–Crafts alkylation step using AlCl3 yielded the required tricyclic scaffold (13) in 68% yield. The compound was thoroughly characterized by 1H/13C and NOE experiments (ESI). Subsequent attempts were directed on installing the remaining C3 fragment on the side chain. It was carried out by reduction of the double bond in compound 13 to its corresponding saturated congener 14, which was further reduced to the 1° alcohol (similar to our previous observation, attempts to terminate the reaction at aldehyde stage failed) 15. Oxidation with DMP followed by treatment with the appropriate ylide in the presence of n-BuLi led to the penultimate compound 16. Final step of demethylation was attempted with various reagents such as BBr3, HBr, HCl, NaSEt but was only successful with LiCl in DMF under microwave conditions and gave (±)-microthecaline A as a free base in 65% yield. The product obtained was thoroughly characterized and the spectroscopic data was found to be comparable with the literature values available for microthecaline A (ESI).1 Given the importance of enantiomeric purity to evaluate the biological activity of the molecule, attempts are currently underway in our lab to develop a synthetic route for (R)-microthecaline A, the naturally occurring stereoisomer (Scheme 4).
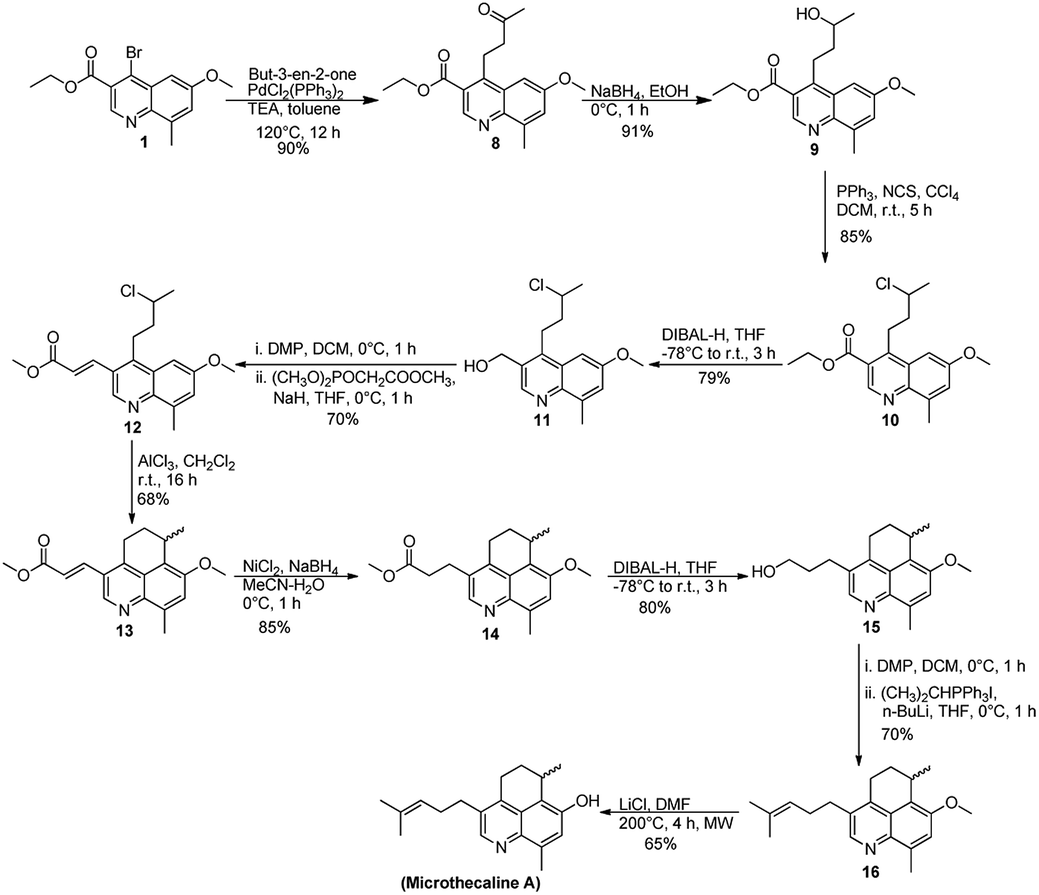 | ||
| Scheme 4 Reactions which led to the final synthesis of (±)-microthecaline A. | ||
Conclusion
In summary, we have developed the first total synthesis of quinoline serrulatane alkaloid (±)-microthecaline A in 8% total yield. Given the generic nature of the reactions incorporated in the synthetic route, we feel that diverse analogues of the aforesaid molecule can be prepared, if required.Experimental
All the compounds and reagents required were purchased from commercial sources and were used without further purification. Solvents were dried and distilled using standard procedures, prior to use. Antonpaar-Monowave microwave synthesizer was used for reactions carried out under microwave conditions. 1H NMR (500 MHz and 400 MHz) and 13C (125 MHz and 100 MHz) spectra were recorded in CDCl3, DMSO-d6 and CD3OD using (CH3)4Si as internal standard. IR spectra were recorded as KBr plates on Shimadzu-IR Affinity instrument. Melting points were recorded on a Buchi-M565 melting point apparatus and are uncorrected.(E)-Ethyl 4-(3-ethoxy-3-oxoprop-1-en-1-yl)-6-methoxy-8-methylquinoline-3-carboxylate (2)
To a stirred and de-gassed solution of 1 (1 g, 3.09 mmol) in dry toluene (20 mL) was added ethylacrylate (460 mg, 4.6 mmol), triethylamine (1.3 mL, 9.2 mmol) and PdCl2(PPh3)2 (217 mg, 0.3 mmol). The reaction mixture was further de-gassed for 2 minutes in a seal-tube and the reaction mixture was then heated at 120 °C for 24 hours. On completion of the reaction as indicated by TLC, the reaction mixture was cooled to room temperature and filtered through Celite pad. The Celite pad was further washed with 50 mL of ethyl acetate (EtOAc). The organic layers were subsequently combined and evaporated to afford crude compound. Column chromatography was performed on the crude compound using silica and 30% EtOAc/hexanes as the mobile phase afforded compound 2 (796 mg, 75%) as a light yellow solid (mp 99–101 °C).1H NMR (400 MHz, CDCl3) δ: 9.2 (s, 1H), 8.33–8.29 (dd, J1 = 16.4 Hz, J2 = 0.8 Hz, 1H), 7.33 (d, 1H), 7.17 (d, 1H), 6.16–6.12 (dd, J1 = 16.4 Hz, J2 = 1.2 Hz, 1H), 4.4–4.36 (q, 2H), 4.36–4.32 (q, 2H), 3.9 (s, 3H), 2.78 (s, 3H), 1.42–1.39 (t, 6H); 13C NMR (100 MHz, CDCl3) δ: 166, 165.6, 158, 146.3, 144.5, 142.8, 142.4, 139.5, 126.3, 125.8, 123.9, 121.5, 103.8, 61.6, 60.9, 55.5, 18.4, 14.2, 14.1; νmax (KBr)/cm−1: 3301, 1610; HRMS-ESI (+) m/z: calculated for C19H22NO5 [M + H]+, 344.1492; found, 344.1473.Ethyl 4-(3-ethoxy-3-oxopropyl)-6-methoxy-8-methylquinoline-3-carboxylate (3)
To a stirred solution of 2 (700 mg, 2.04 mmol) in MeCN:H2O (9:1, 15 mL) at 0 °C was added NiCl2·6H2O (1.45 g, 6.1 mmol), followed by portion-wise addition of NaBH4 (232 mg, 6.1 mmol). The reaction mixture was then allowed to stir at 0 °C for 1 hour. On completion, the reaction mixture was filtered through Celite pad, which was further washed with EtOAc (30 mL). The combined organic layers were then washed with water and saturated brine solution; it was subsequently dried with anhydrous Na2SO4 and evaporated to afford crude compound. Column chromatography of the crude compound using silica and elution with 30% EtOAc/hexanes provided 3 (560 mg, 80%) as a light yellow solid (mp 70–72 °C).1H NMR (400 MHz, CDCl3) δ: 9.18 (s, 1H), 7.25 (s, 2H), 4.48–4.42 (q, 2H), 4.28–4.14 (q, 2H), 3.96 (s, 3H), 3.7–3.66 (t, J = 7.6 Hz, 2H), 2.77 (s, 3H), 2.76–2.72 (t, J = 7.6 Hz, 2H), 1.4–1.42 (t, J = 7.6 Hz, 3H), 1.27–1.23 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 172.8, 166.8, 158, 147.5, 146.8, 144.5, 139.8, 127.4, 122.2, 122, 100.1, 61.5, 60.7, 35.4, 34.3, 24.4, 18.4, 14.2, 14.1; νmax (KBr)/cm−1: 3310, 1625; HRMS-ESI (+) m/z: calculated for C19H24NO5 [M + H]+, 346.1649; found, 346.1638.
Ethyl 7-methoxy-9-methyl-6-oxo-5,6-dihydro-4H-benzo[de]quinoline-3-carboxylate (4)
A mixture of 3 (500 mg, 1.45 mmol) and trifluoromethanesulphonic acid (5 mL) were heated at 80 °C for 2 hours. On completion, the reaction mixture was cooled to room temperature, diluted with ice cold water and quenched with saturated NaHCO3 solution. The resulting aqueous solution was then extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over Na2SO4 evaporated to afford crude compound, which was chromatographed on silica by eluting with 65% EtOAc/hexanes to give 4 (260 mg, 60%) as light yellow solid (mp 148–150 °C). 1H NMR (400 MHz, CDCl3) δ: 9.2 (s, 1H), 7.56 (s, 1H), 4.49–4.44 (q, 2H), 4.19 (s, 3H), 3.82–3.78 (t, J = 7.4 Hz, 2H), 2.88 (s, 3H), 2.9–2.86 (t, J = 7.4 Hz, 2H), 1.47–1.42 (q, 3H); 13C NMR (100 MHz, CDCl3) δ: 196.9, 166.2, 157.4, 147.8, 146.4, 143.8, 142.3, 128.4, 122.9, 118.9, 114.3, 61.6, 56.7, 39.2, 26.9, 19.4, 14.3; νmax (KBr)/cm−1: 3255, 1710; HRMS-ESI (+) m/z: calcd for C17H18NO4 [M + H]+, 300.123; found, 300.122.Ethyl 7-methoxy-9-methyl-6-oxo-6H-benzo[de]quinoline-3-carboxylate (5)
To a stirred suspension of KOtBu (112 mg, 1 mmol) in dry THF (10 mL) was added dropwise methyltriphenylphosphonium bromide (358 mg, 1 mmol) in THF at 0 °C (reaction mixture turned to light red) and stirred for 30 min at same temperature. To this compound 4 (250 mg, 0.83 mmol) dissolved in dry THF was added and the resulting reaction mixture was allowed to stir for 1 hour at 0 °C. The reaction mixture was then diluted with water and extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with brine, dried over Na2SO4 and evaporated to afford the crude compound, which was chromatographed on SiO2 by eluting with 50% EtOAc/hexanes to give ethyl 7-methoxy-9-methyl-6-oxo-6H-benzo[de]quinoline-3-carboxylate (180 mg, 73%) as light yellow solid (mp 148–150 °C)·1H NMR (400 MHz, CDCl3) δ 9.32 (s, 1H), 8.67 (d, J = 10.2 Hz, 1H), 7.56 (s, 1H), 6.89 (d, J = 10.2 Hz, 1H), 4.53 (d, J = 7.1 Hz, 2H), 4.22 (s, 3H), 2.94 (s, 3H), 1.72 (s, 1H), 1.49 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 182.72, 165.82, 163.48, 149.32, 147.31, 142.44, 135.66, 134.20, 133.59, 124.29, 123.14, 118.60, 113.30, 62.12, 56.93, 19.45, 14.29; HRMS-ESI (+) m/z: calcd for C17H16NO4 [M + H]+, 298.1074; found, 298.1065.Ethyl 6-hydroxy-7-methoxy-6,9-dimethyl-5,6-dihydro-4H-benzo[de]quinoline-3-carboxylate (6)
To a stirred solution of 4 (100 mg, 0.33 mmol) in dry THF (5 mL) was added a solution of 1 M methylmagnesium bromide (0.5 mL, 0.5 mmol) in THF at 0 °C and the reaction mixture was allowed to stir at room temperature for 12 hours. The reaction was then quenched with NH4Cl solution, diluted with water and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine solution, dried with Na2SO4 and evaporated to afford the crude compound. Column chromatography on silica by eluting with 30% EtOAc/hexanes to give 6 (80 mg, 75%) as brown gummy solid. 1H NMR (400 MHz, CDCl3) δ: 9.17 (s, 1H), 7.45 (s, 1H), 5.06 (s, 1H), 4.47–4.41 (m, 2H), 4.08 (s, 3H), 3.91–3.83 (m, 1H), 3.17–3.08 (m, 1H), 2.81 (s, 3H), 2.27–2.09 (m, 1H), 1.64 (s, 3H), 1.46–1.42 (t, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.8, 153.1, 148, 147.3, 143.3, 138.2, 125.6, 124.8, 121.4, 118.2, 71.2, 61.4, 56.6, 36.5, 27.8, 25.9, 18.8, 14.4; νmax (KBr)/cm−1: 3315, 1690; HRMS-ESI (+) m/z: calcd for C18H22NO4 [M + H]+, 316.1543; found, 316.1539.Ethyl 6-methoxy-8-methyl-4-(3-oxobutyl)quinoline-3-carboxylate (8)
To a stirred and de-gassed solution of 1 (5 g, 15.4 mmol) in dry toluene was added but-3-en-2-one (2.5 mL, 30.9 mmol) followed by TEA (8.7 mL, 61.2 mmol) and PdCl2(PPh3)2 (1 g, 1.54 mmol). The reaction mixture was further de-gassed for 2 min in a seal-tube and the reaction mixture was then heated at 120 °C for 24 hours. On completion of the reaction as indicated by TLC, the reaction mixture was cooled to room temperature and filtered through Celite pad. The Celite pad was washed with EtOAc (3 × 50 mL). The combined organic layers were evaporated to afford crude compound, which was then chromatographed using SiO2 by eluting with 30% EtOAc/hexanes to give 8 (4.38 g, 90%) as white solid (mp 79–81 °C).1H NMR (400 MHz, CDCl3) δ: 9.13 (s, 1H), 7.31 (d, J = 4 Hz, 1H), 7.2 (d, 1H), 4.46–4.41 (q, 2H), 3.92 (s, 3H), 3.62–3.58 (t, J = 8 Hz, 2H), 2.91–2.87 (t, J = 8 Hz, 2H), 2.77 (s, 3H), 2.21 (s, 3H), 1.45–1.41 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.3, 166.2, 158, 148.4, 146.7, 144.4, 139.2, 127.4, 123.3, 123, 100.1, 61.4, 55.4, 43.6, 29.9, 23.2, 18.4, 14.2; νmax (KBr)/cm−1: 3411, 2972, 1720; HRMS-ESI (+) m/z: calcd for C18H22NO4 [M + H]+, 316.1543; found, 316.1538.Ethyl 4-(3-hydroxybutyl)-6-methoxy-8-methylquinoline-3-carboxylate (9)
To a stirred solution of 8 (2.5 g, 7.93 mmol) in EtOH (25 mL) at 0 °C was added NaBH4 (361 mg, 9.52 mmol) portion wise and the reaction mixture was stirred for 1 hour. On completion, the reaction was quenched with ice cold water, EtOH evaporated and the compound precipitated in water was filtered and dried to get 9 (2.26 g, 91%) as white solid (mp 113–115 °C).1H NMR (400 MHz, CDCl3) δ: 9.14 (s, 1H), 7.34–7.33 (d, J = 2.4 Hz, 1H), 7.3 (d, J = 1 Hz, 1H), 4.47–4.42 (q, J = 7.2 Hz, 2H), 3.94 (s, 3H), 3.9–3.8 (m, 1H), 2.77 (s, 3H), 2.51–2.5 (d, J = 4 Hz, 2H), 1.98–1.83 (m, 2H), 1.45–1.42 (t, J = 7.2 Hz, 3H), 1.25–1.24 (d, J = 6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 167.7, 157.7, 149.7, 146.8, 144.4, 139.6, 127.9, 123.2, 122.7, 100.9, 67.1, 63.5, 55.4, 39.3, 25.1, 23.5, 19.4, 14.2; νmax (KBr)/cm−1: 3310, 2970, 1710; HRMS-ESI (+) m/z: calcd for C18H24NO4 [M + H]+, 318.17; found, 318.1695.Ethyl 4-(3-chlorobutyl)-6-methoxy-8-methylquinoline-3-carboxylate (10)
To a stirred solution of 9 (1 g, 3.15 mmol) in CH2Cl2:CCl4 (1:1, 20 mL) at room temperature was added PPh3 (1.23 g, 4.75 mmol) followed by N-chlorosuccinimide (503 mg, 3.78 mmol) and the reaction mixture was stirred for 5 hours. On completion of the reaction, the solvent was removed under reduced pressure to afford crude compound, which was further chromatographed on SiO2 by eluting with 60% EtOAc/hexanes to give 10 (898 mg, 85%) as white solid (mp 64–66 °C).1H NMR (500 MHz, CDCl3) δ: 9.13 (s, 1H), 7.38–7.37 (d, J = 2.5 Hz, 1H), 7.29 (s, 1H), 4.46–4.44 (q, J = 7 Hz, 2H), 4.3–4.26 (m, 1H), 3.94 (s, 3H), 3.6–3.5 (m, 1H), 2.76 (s, 3H), 2.23–2.19 (m, 1H), 2.1–2 (m, 1H), 1.62–1.6 (d, J = 6.5 Hz, 3H), 1.45–1.42 (t, J = 7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 166.9, 157.9, 148.6, 146.7, 144.5, 139.6, 127.9, 123.6, 122.8, 100.3, 61.3, 59.5, 55.5, 40.3, 26.3, 25.3, 18.4, 14.3; νmax (KBr)/cm−1: 3450, 2960, 1736; HRMS-ESI (+) m/z: calcd for C18H23ClNO3 [M + H]+, 336.1361; found, 336.1351.
(4-(3-Chlorobutyl)-6-methoxy-8-methylquinolin-3-yl)methanol (11)
To a stirred solution of 10 (700 mg, 2.08 mmol) in dry THF (10 mL) at −78 °C dropwise addition of 1 M DIBAL-H (3.13 mL, 3.13 mmol) in hexane was carried out. The reaction mixture was then warmed to room temperature and stirred for 3 hours. On completion, the reaction was quenched with aqueous NH4Cl solution and further extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with brine solution, dried with Na2SO4 and evaporated under reduced pressure. The resulting crude compound was chromatographed on SiO2 by eluting with 50% EtOAc/hexanes to give 11 (480 mg, 79%) as white solid (mp 143–145 °C).1H NMR (500 MHz, CDCl3) δ: 8.64 (s, 1H), 7.26 (s, 1H), 7.23 (s, 1H), 4.87 (s, 2H), 4.25–4.21 (m, 1H), 3.93 (s, 3H), 3.43–3.37 (m, 1H), 3.24–3.18 (m, 1H), 2.75 (s, 3H), 2.13–2.01 (m, 2H), 1.59–1.58 (d, J = 7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 157.6, 147.6, 144.6, 143.6, 139.4, 130.5, 128.1, 121.8, 99.6, 61.5, 59.1, 55.5, 40.4, 25.3, 25.2, 18.5; νmax (KBr)/cm−1: 3231, 1614; HRMS-ESI (+) m/z: calcd for C16H21ClNO2 [M + H]+, 294.1255; found, 294.1247.(E)-Methyl 3-(4-(3-chlorobutyl)-6-methoxy-8-methylquinolin-3-yl)acrylate (12)
To a stirred solution of 11 (450 mg, 1.53 mmol) in CH2Cl2 (20 mL) was added Dess–Martin periodinane (813 mg, 1.91 mmol) portion wise at 0 °C and the reaction mixture was left to stir at same temperature for 1 hour. On completion, the resulting mixture was filtered through Celite pad, which was further washed with CH2Cl2(50 mL). The collected organic layers were then combined and further washed with aqueous NaHCO3, water, brine and dried with anhydrous Na2SO4. The dried organic layer was then evaporated under reduced to afford aldehyde (1.53 mmol) compound as gummy oil, which was directly used for the next step.To a stirred suspension of trimethylphosphonoacetate (0.37 mL, 2.3 mmol) in dry THF (25 mL) at 0 °C was added 60% NaH (92 mg, 2.3 mmol) in small portions. After 10 minutes a solution of aldehyde (1.53 mmol) in dry THF was added and the reaction mixture was left to stir at 0 °C for 1 h. On completion, the reaction was quenched with aqueous NH4Cl solution and extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with brine, dried with Na2SO4 and evaporated to afford crude compound. The resulting crude product was chromatographed on SiO2 by eluting with 30% EtOAc/hexanes to give 12 (373 mg, 70%) as gummy solid. 1H NMR (400 MHz, DMSO-d6) δ: 9 (s, 1H), 8.14–8.1 (d, J = 15.6 Hz, 1H), 7.34 (s, 2H), 6.84–6.8 (d, J = 14.4 Hz, 1H), 4.4 (m, 1H), 3.91 (s, 3H), 3.77 (s, 1H), 3.35–3.39 (m, 2H), 2.67 (s, 3H), 2.02–2.01 (m, 1H), 1.88 (m, 1H), 1.56–1.54 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.9, 157.6, 144.8, 144.5, 143.7, 139.9, 139.4, 127.7, 125.5, 122.6, 120.5, 100.1, 59.5, 55.4, 51.8, 40.2, 25.3, 25.1, 18.3; νmax (KBr)/cm−1: 3416, 2915, 1715; HRMS-ESI (+) m/z: calcd for C19H23ClNO3 [M + H]+, 348.1361; found, 348.1354.
(E)-Methyl 3-(7-methoxy-6,9-dimethyl-5,6-dihydro-4H-benzo[de]quinolin-3-yl)acrylate (13)
To a stirred solution of 12 (400 mg, 1.15 mmol) in dry DCM (20 mL) at 0 °C was added AlCl3 (306 mg, 2.3 mmol) and the reaction mixture was stirred at room temperature for 16 hours. On completion, it was quenched with aqueous NH4Cl solution and extracted with DCM (3 × 75 mL). The combined organic layers were further washed with brine, dried with Na2SO4 and evaporated under reduced pressure to afford the crude compound. Pure product was obtained by performing column chromatography on silica gel and eluting with 25% EtOAc/hexanes to give 13 (276 mg, 68%) as white solid (mp 128–130 °C).1H NMR (500 MHz, DMSO-d6) δ: 9 (s, 1H), 8.06–8.03 (d, J = 16 Hz, 1H), 7.54 (s, 1H), 6.79–6.76 (d, J = 16 Hz, 1H), 3.94 (s, 3H), 3.51 (m, 1H), 3.2–3.1 (m, 2H), 2.7 (s, 3H), 2–1.98 (m, 1H), 1.88–1.83 (m, 1H), 1.16–1.15 (d, J = 7 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ: 166.4, 152.3, 144.6, 143.2, 141.7, 139.2, 135.6, 123.8, 123.5, 123.3, 120, 117.5, 56.2, 51.5, 27.2, 25.4, 20.8, 18.9, 17.9; νmax (KBr)/cm−1: 3410, 2970, 1711; HRMS-ESI (+) m/z: calcd for C19H22NO3 [M + H]+, 312.1594; found, 312.1584.Methyl 3-(7-methoxy-6,9-dimethyl-5,6-dihydro-4H-benzo[de]quinolin-3-yl)propanoate (14)
To a stirred solution of 13 (250 mg, 0.8 mmol) in MeCN:H2O (9:1, 10 mL) at 0 °C was added NiCl2·6H2O (571 mg, 2.41 mmol), followed by portion wise addition of NaBH4 (91 mg, 2.41 mmol). The reaction mixture was then left to stir at same temperature for 1 hour. On completion, the reaction mixture was filtered through Celite pad, which was further washed with EtOAc (40 mL). The combined organic layers were then washed with water, brine, dried with anhydrous Na2SO4 and evaporated under reduced pressure. The resulting crude compound was chromatographed on silica gel by eluting with 30% EtOAc/hexanes to give 14 (213 mg, 85%) as gummy solid.1H NMR (500 MHz, CDCl3) δ: 8.58 (s, 1H), 7.28 (s, 1H), 3.95 (s, 3H), 3.69 (s, 3H), 3.61–3.59 (m, 1H), 3.14–3.11 (t, J = 8 Hz, 2H), 2.69–2.6 (m, 2H), 2.03–1.93 (m, 2H), 1.22–1.2 (d, J = 7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 172.9, 152.2, 147.9, 141.7, 141.3, 135.8, 128.6, 125.1, 123.1, 116, 56.2, 51.8, 34.5, 27.8, 26.1, 25.7, 21.2, 19.2, 18.6; νmax (KBr)/cm−1: 2950, 1714; HRMS-ESI (+) m/z: calcd for C19H24NO3 [M + H]+, 314.1751; found, 314.1747.
3-(7-Methoxy-6,9-dimethyl-5,6-dihydro-4H-benzo[de]quinolin-3-yl)propan-1-ol (15)
Compound 14 (200 mg, 0.63 mmol) converted to 15 (white gummy solid, 145 mg, 80%) by following the same procedure as that of compound 12.1H NMR (500 MHz, CDCl3) δ: 8.59 (s, 1H), 7.27 (s, 1H), 3.95 (s, 3H), 3.74–3.72 (t, J = 6.2 Hz, 2H), 3.61–3.59 (m, 1H), 3.08–2.98 (m, 2H), 2.9–2.87 (t, J = 7.7 Hz, 2H), 2.78 (s, 3H), 2.03–1.86 (m, 4H), 1.22–1.21 (d, J = 7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 152.1, 148.4, 141.3, 141.2, 135.5, 130.1, 125.2, 123.1, 115.8, 62, 56.2, 33.2, 27.9, 26.6, 26, 21.1, 19.2, 18.6; νmax (KBr)/cm−1: 3385, 2910, 1621; HRMS-ESI (+) m/z: calcd for C18H24NO2 [M + H]+, 286.1802; found, 286.1795.7-Methoxy-6,9-dimethyl-3-(4-methylpent-3-en-1-yl)-5,6-dihydro-4H-benzo[de]quinolines (16)
Compound 15 (130 mg, 0.45 mmol) was converted to the corresponding aldehyde (0.45 mmol)by using the same procedure as that for the conversion of compound 10 to its corresponding aldehyde. To a stirred solution of isopropyltriphenylphosphonium iodide (295 mg, 0.68 mmol) in dry THF (10 mL) was added 1.6 M BuLi (0.42 mL, 0.68 mmol) in THF at 0 °C. After 5–10 minutes, a solution of aldehyde (0.45 mmol) in dry THF was introduced to it and the resulting mixture was allowed to stir at 0 °C for 1 hour. On completion, the reaction was quenched with aqueous NH4Cl solution and extracted with EtOAc (3 × 25 mL). The combined organic layers were then washed with brine, dried with anhydrous Na2SO4 and evaporated to afford the crude compound. Chromatographic purification of the crude compound on SiO2 by eluting with 25% EtOAc/hexanes led to the pure product 16 (98 mg, 70%) as a gummy solid. 1H NMR (500 MHz, CD3OD) δ: 8.41 (s, 1H), 7.38 (s, 1H), 5.21–5.18 (q, 1H), 3.94 (s, 3H), 3.59–3.56 (m, 1H), 3.14–3 (m, 2H), 2.83–2.8 (m, 2H), 2.83–2.8 (m, 2H), 2.71 (s, 3H), 2.34–2.31 (t, J = 7.25 Hz, 2H), 2.04–2.0 (m, 1H), 1.88–1.87 (m, 1H), 1.64 (s, 3H), 1.41 (s, 3H), 1.2–1.18 (d, J = 7 Hz, 3H); 13C NMR (125 MHz, CD3OD) δ: 153.8, 149.4, 143.4, 141.9, 136.1, 133.7, 131.8, 126.5, 124.3, 124.2, 117.4, 31.3, 29.8, 28.9, 27.4, 25.9, 22.3, 19.5, 18.9, 17.5; νmax (KBr)/cm−1: 2931, 1620; HRMS-ESI (+) m/z: calcd for C21H28NO [M + H]+, 310.2165; found, 310.2152.6,9-Dimethyl-3-(4-methylpent-3-en-1-yl)-5,6-dihydro-4H-benzo[de]quinolin-7-ol [(±)-Microthecaline A]
To a stirred solution of 16 (70 mg, 0.22 mmol) in DMF (5 mL) was added LiCl (47 mg, 1.1 mmol) in closed vessel and the reaction mixture was then irradiated in a microwave at 200 °C for 4 h. The reaction mixture was then cooled to room temperature and diluted with water. It was further extracted with EtOAc (3 × 20 mL) and the combined organic layers were washed with water, brine, dried with anhydrous Na2SO4. The dried layer was evaporated under reduced pressure to afford the crude compound, which was further chromatographed on silica gel by eluting with 40% EtOAc/hexanes to give the free base of (±)-microthecaline A (43 mg, 65%) as a white solid (mp 78–80 °C). 1H NMR (400 MHz, CD3OD) δ: 8.35 (s, 1H), 7.12 (s, 1H), 5.23–5.19 (t, J = 7.4 Hz, 1H), 3.57–3.54 (m, 1H), 3.14–3.09 (m, 1H), 3.06–3.01 (m, 1H), 2.86–2.79 (m, 2H), 2.63 (s, 3H), 2.37–2.3 (m, 2H), 2.07–2.02 (m, 1H), 1.94–1.88 (m, 1H), 1.65 (s, 3H), 1.43 (s, 3H), 1.23–1.22 (d, J = 4.8 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ: 151.5, 148.3, 143, 141.8, 135.5, 133.6, 131.5, 127.1, 124.2, 121.9, 121.2, 31.3, 29.9, 28.5, 27.4, 25.9, 22.3, 19.3, 18.5, 17.5; νmax (KBr)/cm−1: 2924, 1628; HRMS-ESI (+) m/z: calcd for C20H26NO [M + H]+, 296.2009; found, 296.2001.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors gratefully acknowledge BITS-Pilani Hyderabad campus and GVK Biosciences Pvt. Ltd. for the laboratory facilities and the analytical assistance. TRP and AB are grateful on behalf of Department of Chemistry, BITS-Pilani Hyderabad campus to DST-India for FIST support (SR/FST/CSI-240/2012).Notes and references
- R. Kumar, S. Duffy, V. M. Avery, A. R. Carroll and R. A. Davis, J. Nat. Prod., 2018, 81, 1079–1083 CrossRef CAS PubMed.
- (a) F. Flachsmann, K. Schellhaas, C. E. Moya, R. S. Jacobs and W. Fenical, Bioorg. Med. Chem., 2010, 18, 8324–8333 CrossRef CAS PubMed; (b) H. Correa, A. L. Valenzuela, L. F. Ospina and C. Duque, J. Inflamm., 2009, 6, 5 CrossRef PubMed; (c) W. Zhong, C. Moya, R. S. Jacobs and R. D. Little, J. Org. Chem., 2008, 73, 7011–7016 CrossRef CAS PubMed; (d) C. E. Moya and R. S. Jacobs, Pharmacol. Toxicol., 2006, 143, 436–443 Search PubMed; (e) E. Sansinenea and A. Ortiz, Curr. Org. Synth., 2016, 13, 556–568 CrossRef CAS; (f) N. Karinel, J. Prudhomme, K. G. Le Roch, S. G. Franzblau and A. D. Rodriguez, Bioorg. Med. Chem. Lett, 2016, 26, 854–857 CrossRef PubMed; (g) I. I. Rodriguez and A. D. Rodriguez, J. Nat. Prod., 2003, 66, 855–857 CrossRef CAS PubMed.
- S. Tenneti, S. Biswas, G. A. Cox, D. J. Mans, H. J. Lim and T. V. Rajan Babu, J. Am. Chem. Soc., 2018, 140, 9868–9881 CrossRef CAS PubMed.
- T. G. Elford, S. Nave, R. P. Sonawane and V. K. Aggarwal, J. Am. Chem. Soc., 2011, 133, 16798–16801 CrossRef CAS PubMed.
- (a) M. Akula, Y. Thigulla, C. Davis, M. Jha and A. Bhattacharya, Org. Biomol. Chem., 2015, 13, 2600–2605 RSC; (b) Y. Thigulla, M. Akula, P. Trivedi, B. Ghosh, M. Jha and A. Bhattacharya, Org. Biomol. Chem., 2016, 14, 876–883 RSC; (c) T. R. Penjarla, M. Kundarapu, S. Baquer and A. Bhattacharya, ChemistrySelect, 2018, 3, 5386–5389 CrossRef CAS; (d) M. Akula, J. Padma Sridevi, P. Yogeeswari, D. Sriram and A. Bhattacharya, Monatsh. Chem., 2014, 145, 811–819 CrossRef CAS.
- (a) E. Sobarzo-Sanchez, B. K. Cassels and L. Castedo, Synlett, 2003, 1647–1650 CrossRef CAS; (b) D. Berney and K. Schuh, Helv. Chim. Acta, 1978, 61, 1262–1273 CrossRef CAS.
- D. Xu, F. Kaiser, H. Li, R. M. Reich, H. Guo and F. E. Kunh, Org. Biomol. Chem., 2019, 17, 49–52 RSC.
- S. Yamamoto, S. Matsunaga and M. Kanai, Heterocycles, 2015, 90, 89–96 CrossRef CAS.
- W. Liu, X. Yang, Z. Z. Zhou and C. J. Li, Chem, 2017, 2, 688–702 CAS.
- Y. Lian, J. R. Hummel, R. G. Bergman and J. E. Ellman, J. Am. Chem. Soc., 2013, 135, 12548–12551 CrossRef CAS PubMed.
- D. Xu, F. Kaiser, H. Li, R. M. Reich, H. Guo and F. E. Kuhn, Org. Biomol. Chem., 2019, 17, 49–52 RSC.
- Y. Fukuda, D. E. Kaelin and S. B. Singh, WO 2013003383 A1, Jan 3, 2013.
- L. R. Jeffries and S. P. Cook, Org. Lett., 2014, 16, 2026–2029 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |