DOI:
10.1039/C9RA02617G
(Paper)
RSC Adv., 2019,
9, 13646-13657
[Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] compounds with 2-(thiophen-2-yl)pyridines†
Received
8th April 2019
, Accepted 24th April 2019
First published on 2nd May 2019
Abstract
A series of [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] compounds (POP = bis(2-(diphenylphosphino)phenyl)ether, xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) in which the N^S ligand is a 2-(thiophen-2-yl)pyridine (1), 2-(thiophen-2-yl)-6-methylpyridine (2), 2-(5-methylthiophen-2-yl)pyridine (3) or 2-(5-methylthiophen-2-yl)-6-methylpyridine (4) have been prepared and characterized in solution and the solid state. Single crystal structures of [Cu(POP)(1)][PF6], [Cu(xantphos)(1)][PF6], [Cu(xantphos)(2)][PF6], [Cu(POP)(3)][PF6]·CH2Cl2, and [Cu(xantphos)(4)][PF6] confirm chelating N^S and P^P ligands, and distorted tetrahedral copper(I) centres. There is close cation⋯anion association, particularly in [Cu(xantphos)(1)][PF6]. Although the compounds are stable over days in CH2Cl2 solution, they are susceptible to the effects of competing ligands such as chloride ion and MeCN. Analysis of the NMR spectroscopic data of a solution of [Cu(POP)(3)][PF6] with added Cl−, gives a Kd value of 0.14 ± 0.03 mM, indicative of ion-pairing. [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] exhibit quasi-reversible or irreversible Cu+/Cu2+ oxidations. They are blue emitters in solution, and the presence of the 5-methyl group in the thiophene ring in 3 and 4 leads to a red-shift in the emission. The highest photoluminescence quantum yields are for [Cu(POP)(2)][PF6] (30.8%) and [Cu(POP)(4)][PF6] (33.2%), both of which have a 6-methyl-substituted pyridine ring in the N^S ligand. Excited-state lifetimes are <5 ns. On going from solution to powder samples, red-shifts of 133 to 163 nm are observed leading to yellow emitters. The brightest emitter, [Cu(xantphos)(1)][PF6], was tested in a LEC device but showed poor electroluminescence and poor charge transporting characteristics.
Introduction
McMillin was the first to investigate the photoluminescence of the metal-to-ligand charge transfer (MLCT) state of copper(I) complexes containing diimine (2,2′-bipyridine, bpy or 1,10-phenanthroline) and phosphane or chelating bis(phosphane) ligands.1,2 In the last few years, the field has been rejuvenated with [Cu(P^P)(N^N)]+ complexes (N^N = diimine chelates and P^P = bis(phosphane)) being applied in light-emitting electrochemical cells (LECs).3,4 The discovery that [Cu(P^P)(N^N)]+ complexes exhibit thermally activated delayed fluorescence (TADF) leading to indirect harvesting of triplet-state fluorescence has injected additional research vigour into the area.5–7
The combination of hard and soft donors at a metal centre to control electronic, photophysical, redox and reaction chemistry is an old and recurring theme in coordination chemistry.8,9 The combination of both hard and soft donors in the same ligand is also an established theme, illustrated in the motivation for the preparation of the first synthetic macrocyclic complex by a template route.10 A few copper(I) complexes with a P2N2 donor set generated from two P^N ligands containing a pyridine with an appended phosphorus moiety have been reported.11–17 In an effort to broaden the scope of emissive copper(I) coordination compounds, we decided to investigate N^S donor ligands as an alternative hard–soft combination.
Our particular interest lies in the use of 2-(2-thienyl)pyridines as N^S donor ligands. These ligands have only been sporadically investigated and can act as monodentate N- or S-donors, chelating N^S donors or bridging NS donors. Although there is an analogy between the chelating N^S mode and the N^N mode of bpy, there are significant stereoelectronic consequences as the presence of two lone pairs of electrons on sulfur result in a non-planar M–N–C–C–S chelate ring. 2-(2-Thienyl)pyridines have been shown to adopt N^S18–21 chelating and monodentate N-bonding modes22 with copper(II), but no copper(I) complexes with simple 2-(2-thienyl)pyridines having no other chelating capacity (for example, with the N-donor in a bpy or 1,10-phenanthroline) appear to have been reported. As copper(I) is a classical soft metal centre, we considered it likely that the thiophene sulfur might coordinate to the metal. We recently reported a series of [Cu(P^P)(N^S)][PF6] complexes with P^P = bis(2-(diphenylphosphino)phenyl)ether (POP) or 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos) (Scheme 1) and N^S being a 2-(alkylthio)pyridine.23 These investigations were prompted by the fact that several previously reported copper(I) complexes combining heterocyclic thioamide and phosphane ligands are emissive24,25 as is [Cu2(dppdtbpf)2(μ-NCS)2] (dppdtbpf = 1-diphenylphosphino-1′-di-tert-butylphosphinoferrocene).26 Like [Cu(P^P)(N^N)]+ complexes,3 those with a [Cu(P^P)(N^S)]+ composition exhibit partitioning of the characters of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) such that the HOMO has dominant copper character with smaller contributions from the ligands and the LUMO is localized on the N^S ligand.23 By analogy with what is observed with [Cu(P^P)(N^N)]+ complexes,3,27,28 this should, in principle, allow the LUMO energy in [Cu(P^P)(N^S)]+ complexes to be altered by structural variation of the N^S ligand. This in turn permits tuning of their photophysical and electrochemical properties.
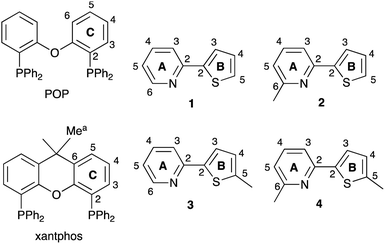 |
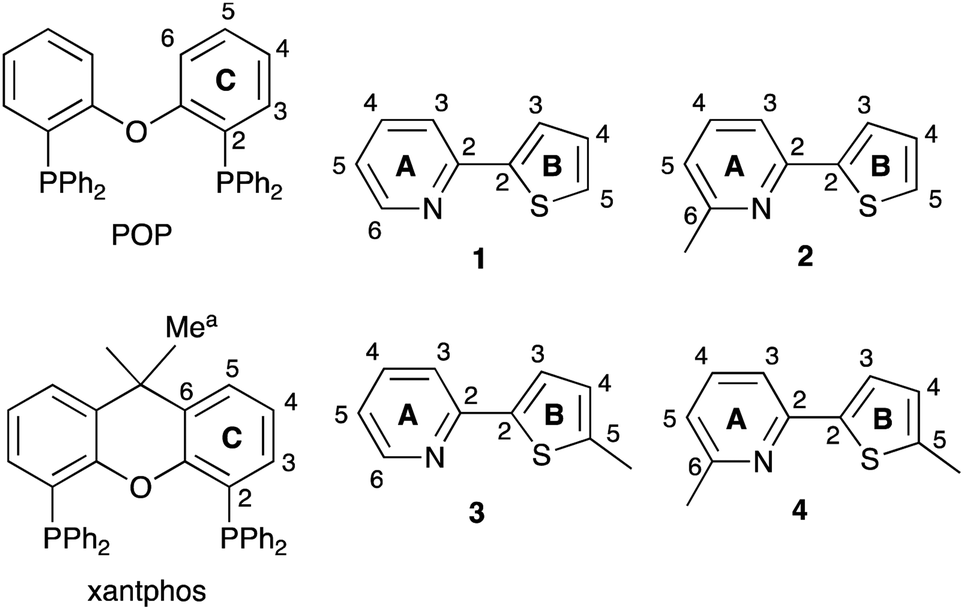
| | Scheme 1 Structures of POP and xantphos and ligands 1–4. Atom labels for NMR spectroscopic assignments are shown; the phenyl rings in POP and xantphos are labelled D. | |
We now report an investigation of [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] complexes in which the N^S ligand is one of the series of 2-(thiophen-2-yl)pyridines shown in Scheme 1.
Experimental
General
1H, 13C{1H} and 31P{1H} NMR spectra were recorded on a Bruker Avance 500 spectrometer at 298 K. 1H and 13C NMR chemical shifts were referenced to the residual solvent peaks with respect to δ (TMS) = 0 ppm and 31P NMR chemical shifts with respect to δ (85% aqueous H3PO4) = 0 ppm.
Solution absorption and emission spectra were measured using a Cary-5000 spectrophotometer and a Shimadzu RF-5301PC spectrofluorometer, respectively. A Shimadzu LCMS-2020 instrument was used to record electrospray ionization (ESI) mass spectra. Quantum yields (CH2Cl2 solution and powder) were measured using a Hamamatsu absolute photoluminescence (PL) quantum yield spectrometer C11347 Quantaurus-QY. Powder emission spectra were measured with a Hamamatsu Compact Fluorescence lifetime Spectrometer C11367 Quantaurus-Tau with an LED light source (λexc = 280 nm). Electrochemical measurements used a CH Instruments 900B potentiostat with [nBu4N][PF6] (0.1 M) as supporting electrolyte and a scan rate of 0.1 V s−1; the solvent was CH2Cl2. The working electrode was glassy carbon, the reference electrode was a leakless Ag+/AgCl (eDAQ ET069-1) and the counter-electrode was a platinum wire. Final potentials were internally referenced with respect to the Fc/Fc+ couple.
[Cu(MeCN)4][PF6] was prepared according to the literature.29 Compound 1 was purchased from Apollo Scientific. Compounds 2 and 3 were prepared by Suzuki-coupling reactions as previously described30 and the NMR spectroscopic data matched those reported.30,31 Other chemicals were purchased from Sigma Aldrich, Acros Organics or Fluorochem and were used as received.
Compound 4
[Pd(PPh3)4] (388 mg, 0.34 mmol) was added to a degassed mixture of 2-bromo-6-methylpyridine (1000 mg, 0.66 mL, 5.8 mmol), 4,4,5,5-tetramethyl-2-(5-methyl-2-thienyl)-1,3,2-dioxaborolane (1300 mg, 1.4 mL, 5.8 mmol), toluene (6 mL), aqueous Na2CO3 (2 M, 6 mL) and EtOH (3 mL). The mixture was heated at reflux under an N2 atmosphere for 5 h. Then the mixture was allowed to cool to room temperature and was diluted with water (20 mL). The aqueous phase was extracted with toluene (60 mL). The combined organic phases were washed with water (20 mL) and brine (20 mL), dried over MgSO4 and the solvent was removed under reduced pressure. The crude product was purified by column chromatography using a Biotage Selekt automated column (CH2Cl2/hexane, 30% → 50% CH2Cl2) to give 4 as a pale yellow oil (740 mg, 3.9 mmol, 67%). 1H (500 MHz, CDCl3, 298 K) δ/ppm: 7.52 (t, J = 7.7 Hz, 1H, HA4), 7.37 (overlapping m, 2H, HA3+B3), 6.95 (d, J = 7.5 Hz, 1H, HA5), 6.74 (m, 1H, HB4), 2.55 (s, 3H, HMe-A6), 2.51 (s, 3H, HMe-B5). 13C{1H} (126 MHz, CDCl3, 298 K) δ/ppm: 158.4 (CA6), 152.4 (CA2), 142.8 (CB2), 142.1 (CB5), 136.8 (CA4), 126.3 (CB4), 124.5 (CB3), 121.1 (CA5), 115.5 (CA3), 24.7 (CMe-A6), 15.8 (CMe-B5). UV-Vis (CH2Cl2, 6.0 × 10−5 mol dm−3) λ/nm 290 sh (ε/dm3 mol−1 cm−1 5000), 316 (8800). ESI-MS m/z 190.00 [M + H]+ (calc. 190.06). Found: 69.41, H 5.75, N 7.48; C46H37CuF6NOP3S requires C 69.80, H 5.86, N 7.40.
Synthesis of copper(I) complexes
All complexes were synthesized according to the following general procedure. POP (1.0 eq.) or xantphos (1.0 eq.) and [Cu(MeCN)4][PF6] (1.0 eq.) were dissolved in CH2Cl2 and the reaction mixture was stirred for 1 h. Then the N^S ligand (1.0 eq.) was added and the reaction mixture was stirred for 1 h. The solvent was then removed under reduced pressure and the residue was washed with Et2O. The crude product was purified by crystallization from CH2Cl2/Et2O by vapour diffusion.
[Cu(POP)(1)][PF6]
POP (367 mg, 0.68 mmol), [Cu(MeCN)4][PF6] (231 mg, 0.62 mmol), 1 (100 mg, 0.62 mmol). [Cu(POP)(1)][PF6] was isolated as a colourless solid (158 mg, 0.45 mmol, 75%). 1H NMR (500 MHz, acetone-d6) δ/ppm 8.39 (ddd, J = 5.3, 1.4, 0.9 Hz, 1H, HA6), 8.14 (dt, J = 8.1, 1.2 Hz, 1H, HA3), 8.08 (td, J = 7.7, 1.6 Hz, 1H, HA4), 7.73 (dd, J = 3.7, 1.1 Hz, 1H, HB5), 7.51 (dd, J = 5.0, 1.1 Hz, 1H, HB3), 7.50–7.43 (m, 6H, HC5+D4), 7.41–7.33 (m, 9H, HA5+D3), 7.25–7.17 (m, 10H, HC6+D2), 7.16–7.11 (m, 3H, HB4+C4), 6.82 (m, 2H, HC3). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 158.7 (t, JPC = 6 Hz, CC1), 152.8 (CA2), 150.3 (CA6), 143.5 (CB2), 140.1 (CA4), 135.2 (CC3), 134.2 (t, JPC = 6 Hz, CD2), 133.5 (CC5), 131.5 (CD4), 130.7 (t, JPC = 20 Hz, CD1), 130.1 (CB4), 130.0 (t, JPC = 5 Hz, CD3), 128.5 (CB3+B5), 126.0 (t, JPC = 2 Hz, CC4), 124.7 (CA5), 123.7 (t, JPC = 17 Hz, CC2), 122.9 (CA3), 121.1 (CC6). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −15.3 (POP), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 762.10 [M–PF6]+ (calc. 762.12), 601.10 [M–PF6–1]+ (base peak, calc. 601.09). Found: C 59.74, H 3.95, N 1.67; C45H35CuF6NOP3S requires C 59.51, H 3.88, N 1.54.
[Cu(xantphos)(1)][PF6]
Xantphos (316 mg, 0.50 mmol), [Cu(MeCN)4][PF6] (185 mg, 0.50 mmol), 1 (80 mg, 0.50 mmol). [Cu(xantphos)(1)][PF6] was isolated as a colourless solid (126 mg, 0.37 mmol, 75%). 1H NMR (500 MHz, acetone-d6) δ/ppm 8.59 (d, J = 5.2 Hz, 1H, HA6), 8.02 (overlapping m, 2H, HA3+A4), 7.85 (d, J = 6.6 Hz, 2H, HC5), 7.65 (d, J = 3.7 Hz, 1H, HB3), 7.44 (t, J = 7.4 Hz, 4H, HD4), 7.39 (m, 1H, HA5), 7.35–7.28 (m, 10H, HC4+D3), 7.27 (d, J = 4.9 Hz, 1H, HB5), 7.23–7.19 (m, 8H, HD2), 7.09 (dd, J = 5.0, 3.9 Hz, 1H, HB4), 6.72 (m, 2H, HC3), 1.74 (s, 6H, HMea). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 155.1 (CC1), 153.1 (CA2), 150.5 (CA6), 143.9 (CB2), 139.4 (CA4), 134.6 (CC6), 134.1 (t, JPC = 8 Hz, CD2), 132.3 (CC3), 131.4 (CD4), 131.1 (t, JPC = 20 Hz, CD1), 130.4 (CB4), 129.9 (t, JPC = 5 Hz, CD3), 129.0 (CC5), 128.2 (CB5), 127.5 (CB3), 126.2 (t, JPC = 2 Hz, CC4), 124.3 (CA5), 122.0 (CA3), 119.9 (CC2), 42.5 (Cxanthphos bridge), 28.9 (CMea). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −14.9 (xantphos), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 802.15 [M–PF6]+ (calc. 802.15), 641.10 [M–PF6–1]+ (base peak, calc. 641.12). Found: C 60.66, H 4.95, N 1.62; C48H39CuF6NOP3S requires C 60.79, H 4.15, N 1.48.
[Cu(POP)(2)][PF6]
POP (215 mg, 0.40 mmol), [Cu(MeCN)4][PF6] (150 mg, 0.40 mmol), 2 (69.9 mg, 0.40 mmol). [Cu(POP)(2)][PF6] was isolated as a colourless solid (154 mg, 0.17 mmol, 42%). 1H NMR (500 MHz, acetone-d6) δ/ppm 7.98 (t, J = 7.6 Hz, 1H, HA4), 7.85 (d, J = 7.6 Hz, 1H, HA3), 7.64 (d, J = 4.9 Hz, 1H, HB3), 7.59 (d, J = 3.7 Hz, 1H, HB5), 7.51–7.44 (m, 6H, HD4+C5), 7.38 (t, J = 7.5 Hz, 8H, HD3), 7.34 (d, J = 7.6 Hz, 1H, HA5), 7.24–7.10 (m, 13H, HD2+C6+C4+B4), 6.85–6.76 (m, 2H, HC3), 2.32 (s, 3H, HMe). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 159.4 (CA6), 158.5 (CC1), 152.9 (CA2), 143.7 (CB2), 139.8 (CA4), 135.2 (CC3), 134.1 (t, JPC = 8 Hz, CD2), 133.5 (CC5), 131.5 (CD4), 130.9 (CD1), 130.7 (CB4), 130.0 (t, JPC = 5 Hz, CD3), 128.0 (CB3), 127.8 (CB5), 126.1 (CC4), 124.3 (CA5), 123.6 (CC2), 121.2 (CC6), 119.9 (CA3), 24.9 (CMe). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −16.0 (POP), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 776.15 [M–PF6]+ (calc. 776.14), 601.10 [M–PF6–2]+ (base peak, calc. 601.09). Found: 59.34, H 4.21, N 1.50; C46H37CuF6NOP3S requires C 59.90, H 4.04, N 1.52.
[Cu(xantphos)(2)][PF6]
Xantphos (231 mg, 0.40 mmol), [Cu(MeCN)4][PF6] (150 mg, 0.40 mmol), 2 (69.9 mg, 0.40 mmol). [Cu(xantphos)(2)][PF6] was isolated as a colourless solid (183 mg, 0.19 mmol, 48%). 1H NMR (500 MHz, acetone-d6) δ/ppm 8.02 (t, J = 7.8 Hz, 1H, HA4), 7.87 (m, 3H, HC5+A3), 7.48–7.38 (m, 6H, HD4+B3+A5), 7.36–7.27 (m, 11H, HD3+C4+B5), 7.16–7.09 (m, 8H, HD2), 7.04 (dd, J = 5.0, 3.7 Hz, 1H, HB4), 6.72–6.65 (m, 2H, HC3), 2.40 (s, 3H, HMe-A6), 1.76 (s, 6H, HMea). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 159.0 (CA6), 155.1 (t, JPC = 6 Hz, CC1), 152.7 (CA2), 143.6 (CB2), 140.0 (CA4), 134.5 (t, JPC = 2 Hz, CC6), 133.8 (t, JPC = 8 Hz, CD2), 132.2 (CC3), 131.5 (CD4), 131.0 (t, JPC = 19 Hz, CD1), 130.5 (CB4), 130.0 (t, JPC = 5 Hz, CD3), 129.1 (CC5), 127.9 (CB3), 127.6 (CB5), 126.3 (t, JPC = 2.5 Hz, CC4), 124.8 (CA5), 120.2 (CA3), 120.1 (CC2), 36.6 (Cxanthphos bridge), 28.9 (CMea), 25.1 (CMe-A6). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −14.2 (xantphos), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 816.15 [M–PF6]+ (calc. 816.17), 641.10 [M–PF6–2]+ (base peak, calc. 641.12). Found: C 61.18, H 4.27, N 1.46; C49H41CuF6NOP3S requires C 61.15, H 4.29, N 1.46.
[Cu(POP)(3)][PF6]
POP (153 mg, 0.29 mmol), [Cu(MeCN)4][PF6] (106 mg, 0.29 mmol), 3 (49.9 mg, 0.29 mmol). [Cu(POP)(3)][PF6] was isolated as a colourless solid (154 mg, 0.17 mmol, 59%). 1H NMR (500 MHz, acetone-d6) δ/ppm 8.53 (d, J = 5.2 Hz, 1H, HA6), 8.10 (d, J = 8.1 Hz, 1H, HA3), 8.09–8.02 (m, 1H, HA4), 7.54 (d, J = 3.7 Hz, 1H, HB3), 7.52–7.44 (m, 6H, HD2+C5), 7.37 (m, 9H, HD3+A5), 7.25–7.18 (m, 10H, HD2+C6), 7.15 (t, J = 7.6 Hz, 2H, HC4), 6.86–6.79 (m, 3H, HC3+B4), 2.19 (s, 3H, HMe). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 158.9 (t, JPC = 6 Hz, CC1), 153.0 (CA2), 150.0 (CA6), 143.4 (CB5), 140.8 (CB2), 140.0 (CA4), 135.2 (CC3), 134.1 (t, JPC = 8 Hz, CD2), 133.5 (CC5), 131.5 (CD4), 130.8 (t, JPC = 19 Hz, CD1), 130.0 (t, JPC = 5 Hz, CD3), 129.6 (CB4), 129.0 (CB3), 126.2 (t, JPC = 2.5 Hz, CC4), 124.3 (CA5), 123.8 (t, JPC = 16 Hz, CC2), 122.7 (CA3), 121.3 (t, JPC = 2 Hz, CC6), 15.4 (CMe). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −14.7 (POP), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 776.10 [M–PF6]+ (calc. 776.14), 601.05 [M–PF6–3]+ (base peak, calc. 601.09). Found: C 56.63, H 4.04, N 1.56; C46H37CuF6NOP3S·CH2Cl2 requires C 56.05, H 3.90, N 1.39.
[Cu(xantphos)(3)][PF6]
Xantphos (330 mg, 0.57 mmol), [Cu(MeCN)4][PF6] (213 mg, 0.57 mmol), 3 (100 mg, 0.57 mmol). [Cu(xantphos)(3)][PF6] was isolated as a colourless solid (237 mg, 0.25 mmol, 43%). 1H NMR (500 MHz, acetone-d6) δ/ppm 8.75 (d, J = 5.2 Hz, 1H, HA6), 8.10–8.03 (m, 2H, HA3+A4), 7.88 (dd, J = 7.8, 1.4 Hz, 2H, HC5), 7.55 (d, J = 3.7 Hz, 1H, HB4), 7.43 (t, J = 7.5 Hz, 4H, HD4), 7.39 (m, 1H, HA5), 7.34–7.28 (m, 10H, HD3+C4), 7.17 (m, 8H, HD2), 6.81 (m, 1H, HB3), 6.69 (m, 2H, HC3), 1.90 (s, 3H, HMe–B5), 1.75 (s, 6H, HMea). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 155.2 (t, JPC = 6 Hz, (CC1), 152.8 (CA2), 150.4 (CA6), 140.0 (CA4), 143.3 (CB5), 140.1 (CB2), 134.8 (t, JPC = 2 Hz, CC6), 134.0 (t, JPC = 8 Hz, CD2), 132.3 (CC3), 131.5 (CD4), 131.0 (t, JPC = 19 Hz, CD1), 130.0 (t, JPC = 5 Hz, CD3), 129.3 (CB4), 129.1 (CC5), 128.9 (CB3), 126.3 (t, JPC = 3 Hz, CC4), 124.4 (CA5), 122.5 (CA3), 120.3 (dd, JPC = 16 Hz, CC2), 36.8 (Cxanthphos bridge), 28.7 (CMea), 14.9 (CMe–B5). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −14.2 xantphos), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 816.15 [M–PF6]+ (calc. 816.17), 641.10 [M–PF6–3]+ (base peak, calc. 641.12). Found: C 60.36, H 4.39, N 1.41; C49H41CuF6NOP3S requires C 61.15, H 4.29, N 1.46.
[Cu(POP)(4)][PF6]
POP (116 mg, 0.22 mmol), [Cu(MeCN)4][PF6] (80.0 mg, 0.22 mmol), 4 (40.7 mg, 0.22 mmol). [Cu(POP)(4)][PF6] was isolated as a colourless solid (134 mg, 0.14 mmol, 67%). 1H NMR (500 MHz, acetone-d6) δ/ppm 8.00 (t, J = 7.8 Hz, 1H, HA4), 7.86 (d, J = 7.9 Hz, 1H, HA3), 7.54–7.45 (m, 6H, HD4+C5), 7.40–7.36 (m, 9H, HD3+B3), 7.34 (d, J = 7.7 Hz, 1H, HA5), 7.28–7.23 (m, 2H, HC6), 7.19–7.09 (m, 10H, HD2+C4), 6.88–6.81 (m, 3H, HC3+B4), 2.39 (d, J = 1.1 Hz, 3H, HMe-B5), 2.34 (s, 3H, HMe-A6). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 159.3 (CA6), 158.7 (t, JPC = 6 Hz, CC1), 153.0 (CA2), 142.9 (CB5), 140.8 (CB2), 140.3 (CA4), 135.1 (CC3), 133.9 (t, JPC = 8 Hz, CD2), 133.3 (CC5), 131.6 (CD4), 130.9 (t, JPC = 19 Hz, CD1), 130.0 (t, JPC = 5 Hz, CD3), 129.7 (CB3), 129.5 (CB4), 126.3 (t, JPC = 2 Hz, CC4), 124.4 (CA5), 123.5 (CC2), 121.2 (t, JPC = 2 Hz, CC6), 120.0 (CA3), 25.2 (CMe-A6), 15.6 (CMe-B5). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −15.5 (POP), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 790.10 [M–PF6]+ (calc. 790.15), 601.05 [M–PF6–4]+ (base peak, calc. 601.09). Found: C 60.57, H 4.11, N 1.78; C47H39CuF6NOP3S requires C 60.29, H 4.20, N 1.50.
[Cu(xantphos)(4)][PF6]
Xantphos (124 mg, 0.22 mmol), [Cu(MeCN)4][PF6] (80.1 mg, 0.22 mmol), 4 (40.7 mg, 0.22 mmol). [Cu(xantphos)(4)][PF6] was isolated as a colourless solid (157 mg, 0.16 mmol, 75%). 1H NMR (500 MHz, acetone-d6) δ/ppm 8.02 (t, J = 7.8 Hz, 1H, HA4), 7.92–7.86 (m, 3H, HC5+A3), 7.44 (t, J = 7.5 Hz, 4H, HD4), 7.39 (d, J = 7.6 Hz, 1H, HA5), 7.35–7.28 (m, 11H, HD3+C4+B3), 7.12 (m, 8H, HD2), 6.76 (m, 1H, HB4), 6.71 (m, 2H, HC3), 2.46 (s, 3H, HMe-A6), 2.08 (s, 3H, HMe-B5), 1.76 (s, 6H, HMea). 13C{1H} NMR (126 MHz, acetone-d6) δ/ppm 158.6 (CA6), 155.1 (t, JPC = 6 Hz, CC1), 152.8 (CA2), 142.9 (CB5), 140.5 (CB2), 140.0 (CA4), 134.8 (t, JPC = 2 Hz, CC6), 133.8 (t, JPC = 8 Hz, CD2), 132.2 (CC3), 131.5 (CD4), 131.0 (t, JPC = 19 Hz, CD1), 130.0 (t, JPC = 5 Hz, CD3), 129.5 (CB4), 129.1 (CC5+B3), 126.5 (t, JPC = 2.5 Hz, CC4), 124.6 (CA5), 120.3 (CA3), 120.1 (dd, JPC = 16 Hz, CC2), 36.6 (Cxantphos bridge), 28.3 (CMea), 25.5 (CMe-A6), 15.2 (CMe-B5). 31P{1H} NMR (202 MHz, acetone-d6, 298 K) δ/ppm −14.4 (xantphos), −144.2 (septet, JPF = 707 Hz, PF6−). ESI-MS positive mode m/z 830.19 [M–PF6]+ (calc. 830.18), 641.10 [M–PF6–4]+ (base peak, calc. 641.12). Found: C 61.83, H 5.13, N 1.48; C50H43CuF6NOP3S requires C 61.51, H 4.44, N 1.43.
Crystallography
Single crystal data were collected on a Bruker APEX-II diffractometer (Bruker AXS GmbH, Karlsruhe, Germany); data reduction, solution and refinement used APEX2,32 SuperFlip33 and CRYSTALS34 or OLEX35 respectively. Structure analysis and ORTEP-style diagrams used Mercury v. 3.8.36,37 Crystallographic data are presented in Table 1.
Table 1 Crystallographic data
| Compound |
[Cu(POP)(1)][PF6] |
[Cu(POP)(3)][PF6]·CH2Cl2 |
[Cu(xantphos)(1)][PF6] |
| Formula |
C45H35CuF6NOP3S |
C47H39Cl2CuF6NOP3S |
C48H39CuF6NOP3S |
| Formula weight |
908.30 |
1007.20 |
948.36 |
| Crystal colour and habit |
Colourless block |
Colourless needle |
Colourless block |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n |
| a, b, c/Å |
10.8352(14), 12.0146(15), 16.754(2) |
10.5738(10), 12.4356(12), 18.8853(17) |
10.5055(13), 21.487(3), 19.094(2) |
| α, β, γ/° |
92.081(3), 98.546(3), 107.937(3) |
71.646(3), 75.679(3), 70.826(3) |
90, 95.118(4), 90 |
| U/Å3 |
2044.2(4) |
2197.3(4) |
4293.0(9) |
| Dc/Mg m−3 |
1.476 |
1.522 |
1.467 |
| Z |
2 |
2 |
4 |
| μ(Cu-Kα)/mm−1 |
2.897 |
3.848 |
2.785 |
| T/K |
123 |
130 |
123 |
| Refln. collected (Rint) |
26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 148 (0.027) 148 (0.027) |
28575 (0.027) |
56802 (0.033) |
| Unique refln. |
7456 |
7961 |
7797 |
| Refln. for refinement |
7376 |
7776 |
7732 |
| Parameters |
523 |
669 |
602 |
| Threshold |
2σ |
2σ |
2σ |
| R1 (R1 all data) |
0.0388 (0.0389) |
0.0383 (0.0390) |
0.0504 (0.0505) |
| wR2 (wR2 all data) |
0.0933 (0.0933) |
0.0922 (0.0927) |
0.0877 (0.0877) |
| Goodness of fit |
0.9868 |
1.050 |
0.9592 |
| CCDC deposition number |
1900493 |
1900489 |
1900490 |
| Compound |
[Cu(xantphos)(2)][PF6] |
[Cu(xantphos)(4)][PF6] |
[Cu(xantphos)(NCMe)2][PF6]·0.5Et2O |
| Formula |
C49H41CuF6NOP3S |
C50H43CuF6NOP3S |
C45H43CuF6N2O1.5P3 |
| Formula weight |
962.39 |
976.36 |
863.42 |
| Crystal colour and habit |
Colourless block |
Colourless plate |
Colourless plate |
| Crystal system |
Orthorhombic |
Triclinic |
Triclinic |
| Space group |
Pbca |
P |
P |
| a, b, c/Å |
19.1120(14), 20.3777(15), 22.2356(16) |
11.5350(5), 12.1924(6), 17.0548(8) |
9.7193(10), 14.8522(17), 17.864(2) |
| α, β, γ/° |
90, 90, 90 |
82.221(3), 71.606(3), 88.977(3) |
110.080(5), 99.380(5), 102.758(5) |
| U/Å3 |
8659.8(11) |
2254.30(19) |
2280.5(4) |
| Dc/Mg m−3 |
1.476 |
1.438 |
1.257 |
| Z |
8 |
2 |
2 |
| μ(Cu-Kα)/mm−1 |
2.769 |
2.668 |
2.188 |
| T/K |
130 |
130 |
130 |
| Refln. collected (Rint) |
38128 (0.033) |
27121 (0.045) |
25739 (0.0412) |
| Unique refln. |
7944 |
8187 |
8116 |
| Refln. for refinement |
7100 |
6150 |
7338 |
| Parameters |
559 |
572 |
554 |
| Threshold |
2σ |
2σ |
2σ |
| R1 (R1 all data) |
0.0338 (0.0382) |
0.0603 (0.0842) |
0.0789 (0.0839) |
| wR2 (wR2 all data) |
0.0868 (0.0903) |
0.1525 (0.1695) |
0.2135 (0.2179) |
| Goodness of fit |
0.9988 |
1.025 |
1.114 |
| CCDC deposition number |
1900494 |
1900492 |
1900491 |
Results and discussion
Synthesis and characterization of compound 4
Ligand 4 (Scheme 1) was prepared by a Suzuki coupling of 2-bromo-6-methylpyridine and 4,4,5,5-tetramethyl-2-(5-methyl-2-thienyl)-1,3,2-dioxaborolane, following a similar procedure reported for the syntheses of 2 and 3.30 Compound 4 was isolated as a pale yellow oil in 67%. The base and highest mass peak in the electrospray mass spectrum appeared at m/z 190.00 and was assigned to [M + H]+. The 1H and 13C NMR spectra (Fig. S1 and S2†) were assigned by 2D methods and were consistent with the structure shown in Scheme 1.
Synthesis and solution characterization of the [Cu(P^P)(N^S)][PF6] complexes
The [Cu(P^P)(N^S)][PF6] complexes were prepared at room temperature by combining POP or xantphos and [Cu(MeCN)4][PF6] in CH2Cl2 followed, after stirring, by addition of the N^S ligand. After purification by crystallization, the complexes were obtained in yields ranging from 42 to 75%. The positive mode electrospray mass spectrum of each [Cu(P^P)(N^S)][PF6] compound (Fig. S3–S10†) exhibited peak envelopes corresponding to the [M–PF6]+ and [Cu(POP)]+ or [Cu(xantphos)]+ ions. In each case, the base peak arose from [Cu(POP)]+ or [Cu(xantphos)]+.
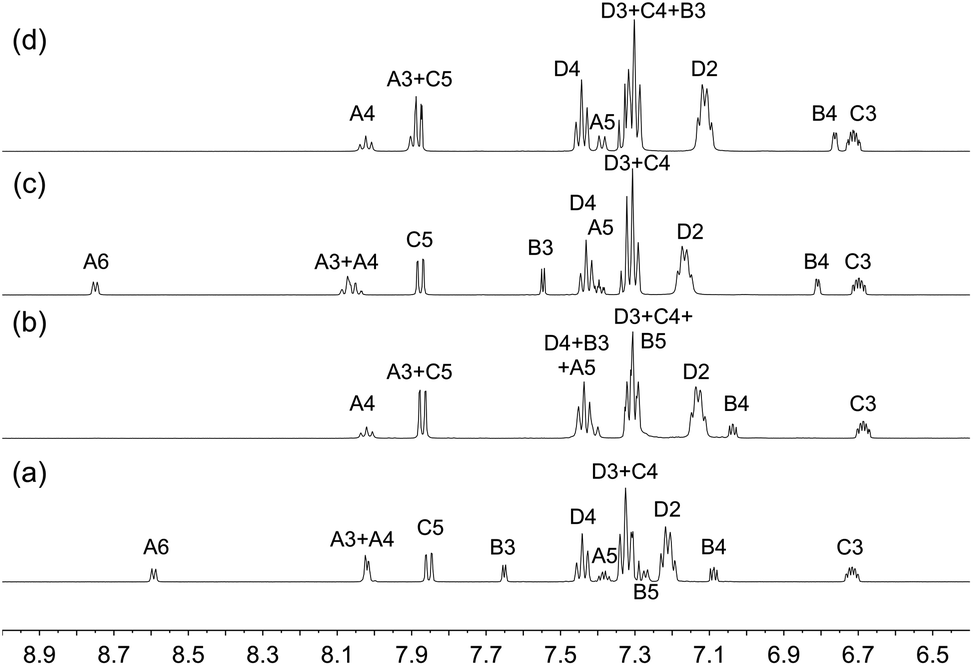
Solution 1H, 13C{1H} and 31P{1H} NMR spectra were recorded at 298 K in acetone-d6. The 31P{1H} NMR spectrum of each compound (Fig. S11†) exhibits a broadened singlet arising from the coordinated POP or xantphos ligand and a septet from the [PF6]− ion. The coordinated sulfur retains a lone pair of electrons and is therefore a stereogenic centre. Inversion at the sulfur atom is a low energy process; in the absence of inversion, the two phosphorus atoms would be inequivalent (see figures in the structural discussion). The observation of only one 31P NMR resonance is consistent with the POP ligand also undergoing dynamic behaviour in solution. The coordinated xantphos ligand is more rigid than POP as can be appreciated from the figures presented in the structural discussion below. The 1H and 13C{1H} NMR spectra were assigned using COSY, NOESY, HMQC and HMBC techniques. The 1H NMR spectra are shown in Fig. S12–S19,† and Fig. 1 compares the aromatic regions of the spectra of the [Cu(xantphos)(N^S)][PF6] compounds. The absence of a signal for proton HA6 in [Cu(xantphos)(2)][PF6] (Fig. 1b) and [Cu(xantphos)(4)][PF6] (Fig. 1d) is consistent with the introduction of the 2-methyl substituent in the pyridine ring in ligands 2 and 4. Similarly, the spectra presented in Fig. 1c and d exhibit no signal for HB5, consistent with the presence of the 5-methyl substituent in the thienyl ring. In [Cu(POP)(4)][PF6], methyl protons HMe-B5 give rise to a doublet (J = 1.1 Hz) in the 1H NMR spectrum and a COSY crosspeak confirms coupling to HB4. This coupling was not resolved in [Cu(POP)(3)][PF6], [Cu(xantphos)(3)][PF6] or [Cu(POP)(4)][PF6]. In contrast to the broadened room temperature 1H NMR spectra of [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] in which N^S = 2-(iso-propylthio)pyridine or 2-(tert-butylthio)pyridine,23 those of the current series of compounds are well-resolved at 298 K. This is consistent with (but does not prove) lower energy barriers to dynamic processes in the compounds containing the 2-(alkylthio)pyridine versus 2-(thiophen-2-yl)pyridine ligands.
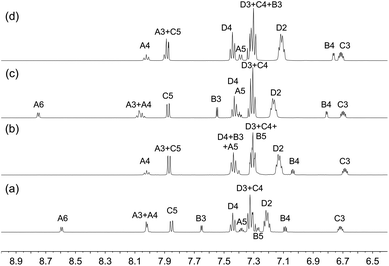 |
| | Fig. 1 Aromatic regions in the 1H NMR spectra (500 MHz, acetone-d6, 298 K) of (a) [Cu(xantphos)(1)][PF6], (b) [Cu(xantphos)(2)][PF6], (c) [Cu(xantphos)(3)][PF6] and (d) [Cu(xantphos)(4)][PF6]. See Scheme 1 for atom labelling. | |
Structural characterizations
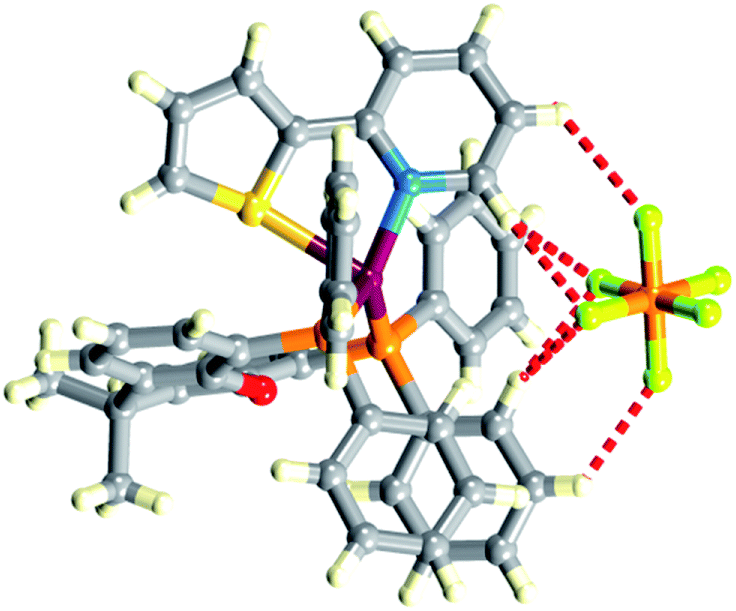
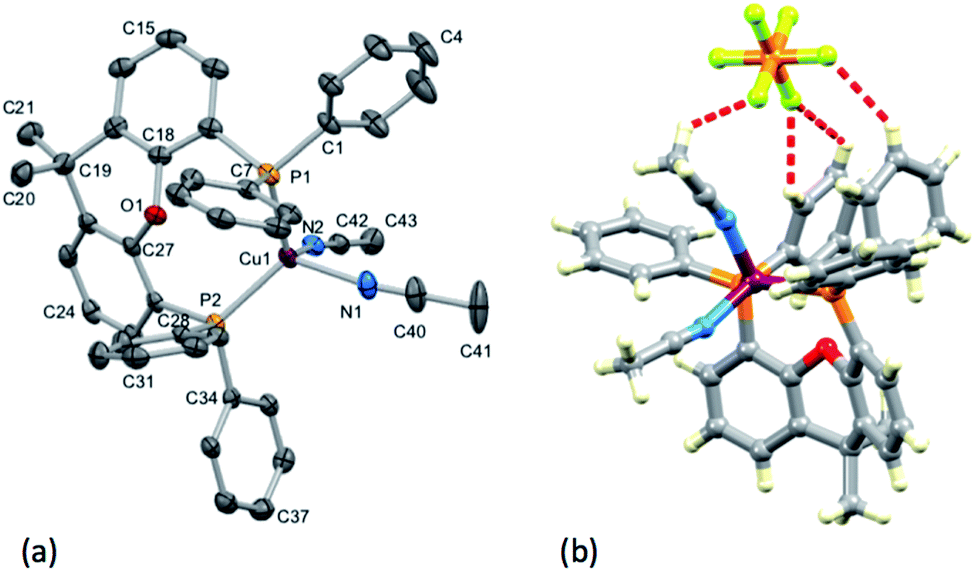
Single crystals of [Cu(POP)(1)][PF6], [Cu(xantphos)(1)][PF6], [Cu(xantphos)(2)][PF6], [Cu(POP)(3)][PF6]·CH2Cl2, and [Cu(xantphos)(4)][PF6] were grown by slow diffusion of Et2O into CH2Cl2 solutions of the compounds. [Cu(POP)(1)][PF6], [Cu(POP)(3)][PF6]·CH2Cl2 and [Cu(xantphos)(4)][PF6] crystallize in the triclinic space group P with rather similar cell dimensions (Table 1). [Cu(xantphos)(1)][PF6] and [Cu(xantphos)(2)][PF6] crystallize in the monoclinic P21/n and orthorhombic Pbca space groups, respectively. ORTEP-style diagrams of the copper(I) complex cations are shown in Fig. S20–24† and selected bond distances and angles are given in the figure captions. In each complex, the Cu atom is in a distorted tetrahedral environment and the bond angles are compared in Table 2. The τ4 parameter defined by Houser and coworkers for 4-coordinate complexes lies in the range 0.76–0.89; for a perfect tetrahedral geometry, τ4 = 1.38 The rather large N–Cu–P angle of 128.30(5)° in [Cu(xantphos)(1)][PF6] is associated with tight cation⋯anion binding. Fig. 2 illustrates that the anion sits in a pocket between the pyridine and phenyl rings with CH⋯F contacts in the range 2.48–2.82 Å; values of the van der Waals radii of H and F are in the range 1.10–1.20 Å and 1.35–1.47 Å, respectively.39 This said, it should be noted that crystal packing inevitably involves extensive C–H⋯F contacts in all the [Cu(P^P)(N^S)][PF6] compounds. Only in [Cu(POP)(3)][PF6]·CH2Cl2 is solvent also involved.
Table 2 Bond angles within the copper(I) coordination sphere of the [Cu(P^P)(N^S)]+ cations. For disordered structures of [Cu(xantphos)(1)][PF6] and [Cu(POP)(3)][PF6]·CH2Cl2, only the major occupancy sites are given
| Cation |
P–Cu–P/° |
N–Cu–S/° |
N–Cu–P/° |
S–Cu–P/° |
| [Cu(POP)(1)]+ |
115.38(2) |
81.34(5) |
110.83(5), 115.14(5) |
110.02(2), 119.44(2) |
| [Cu(POP)(3)]+ |
116.02(2) |
78.2(3) |
116.9(3), 123.4(3) |
100.95(4), 110.40(5) |
| [Cu(xantphos)(1)]+ |
117.89(2) |
80.52(5) |
112.83(5), 128.30(5) |
96.66(2), 103.47(2) |
| [Cu(xantphos)(2)]+ |
127.804(19) |
80.34(5) |
102.04(4), 124.62(4) |
99.701(19), 109.172(19) |
| [Cu(xantphos)(4)]+ |
120.86(4) |
79.40(10) |
111.35(10), 121.48(10) |
96.23(4), 118.75(4) |
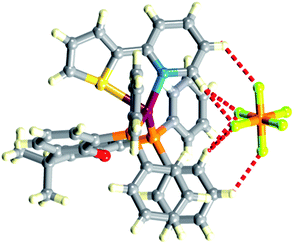 |
| | Fig. 2 Cation⋯anion CH⋯F contacts in [Cu(xantphos)(1)][PF6]. | |
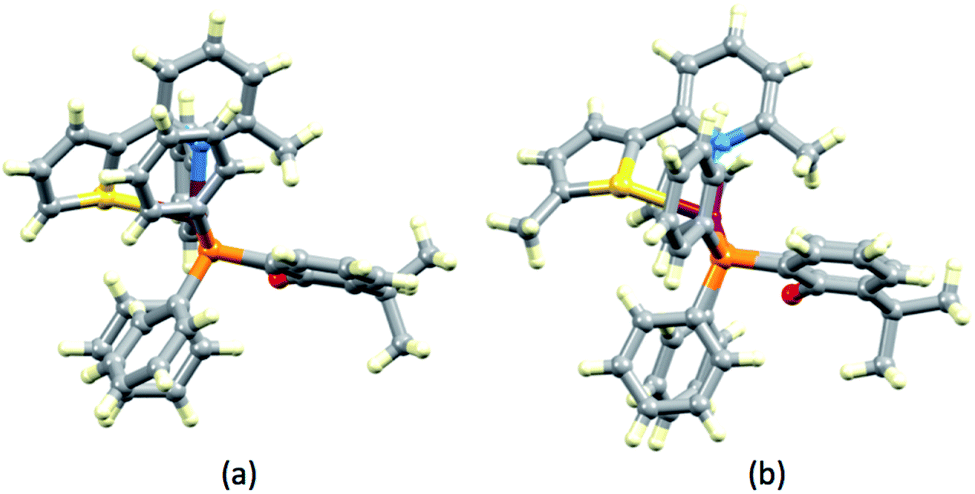
In [Cu(xantphos)(1)][PF6], ligand 1 is disordered and has been modelled over two sites with occupancies of 0.75:0.25. In [Cu(POP)(3)][PF6]·CH2Cl2, ligand 3 is similarly disordered over two sites (0.80:0.20). In both cases, the disorder involves the orientation of the N^S ligand, with the thiophene ring facing towards or away from the O atom of the POP or xantphos ligand. The backbone of xantphos is more rigid than that of POP, and in the complexes containing xantphos, the latter presents a well-defined ‘bowl’. We have previously reported detailed investigations of the orientation preferences of asymmetrical 6-substituted-2,2′-bipyridine (6-Rbpy) ligands in [Cu(xantphos)(6-Rbpy)][PF6].40,41 In [Cu(xantphos)(2)][PF6] and [Cu(xantphos)(4)][PF6], the N^S ligand is oriented with the 6-methyl substituent of the pyridine ring lying over the xanthene ‘bowl’ (Fig. 3). In [Cu(xantphos)(1)][PF6], in which the N^S ligand carries no substituents, the observed ligand disorder indicates that there is little energy difference between the two orientations of the N^S unit in the crystal lattice.
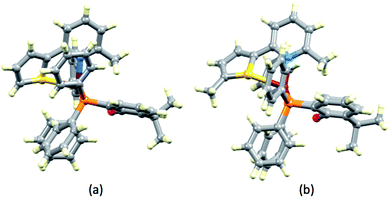 |
| | Fig. 3 Structures of (a) [Cu(xantphos)(2)]+ and (b) [Cu(xantphos)(4)]+ cations showing the positioning of the 6-methyl group of the pyridine ring of N^S ligand over the xanthene ‘bowl’. | |
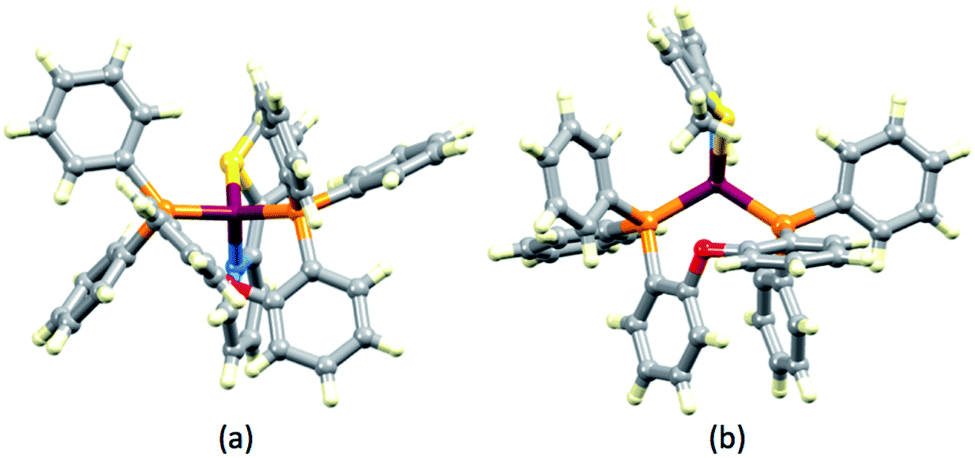
The structures of the [Cu(POP)(1)]+ and [Cu(POP)(3)]+ cations are compared in Fig. 4. The views emphasize the tetrahedral copper(I) centre and the twisted backbones of ligands 1 and 4 which are associated with the presence of the sulfur lone pair. The stereoactive sulfur lone pair results in each of ligands 1–4 in all five [Cu(P^P)(N^S)][PF6] complexes deviating from planarity. However, none crystallizes in a chiral space group and, therefore, both enantiomers of each [Cu(P^P)(N^S)]+ cation are present in the crystal lattice.
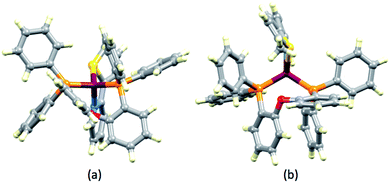 |
| | Fig. 4 Structures of (a) [Cu(POP)(1)]+ and (b) [Cu(POP)(3)]+ cations emphasizing the tetrahedral Cu(I) centre in the former and the stereogenic coordinated S-atoms. | |
We note that in [Cu(POP)(1)][PF6] and [Cu(xantphos)(2)][PF6], close Cu–O contacts are observed (Cu1–O1 = 2.8405(13) Å in [Cu(POP)(1)]+ and 2.8260(13) Å in [Cu(xantphos)(2)]+). The corresponding distances in [Cu(xantphos)(1)][PF6], [Cu(POP)(3)][PF6]·CH2Cl2 and [Cu(xantphos)(4)][PF6] are 3.008(2), 3.019(1) and 3.066(3) Å, respectively. These distances are less than the sum of the van der Waals radii of Cu and O (3.30–3.36 Å using radii for Cu of 1.90 and 1.966 Å from Batsanov42 and Hu43 rather than Bondi's value of 1.4 Å44 or the value of 2.38 Å deduced by Alvarez45).46
Electrochemistry
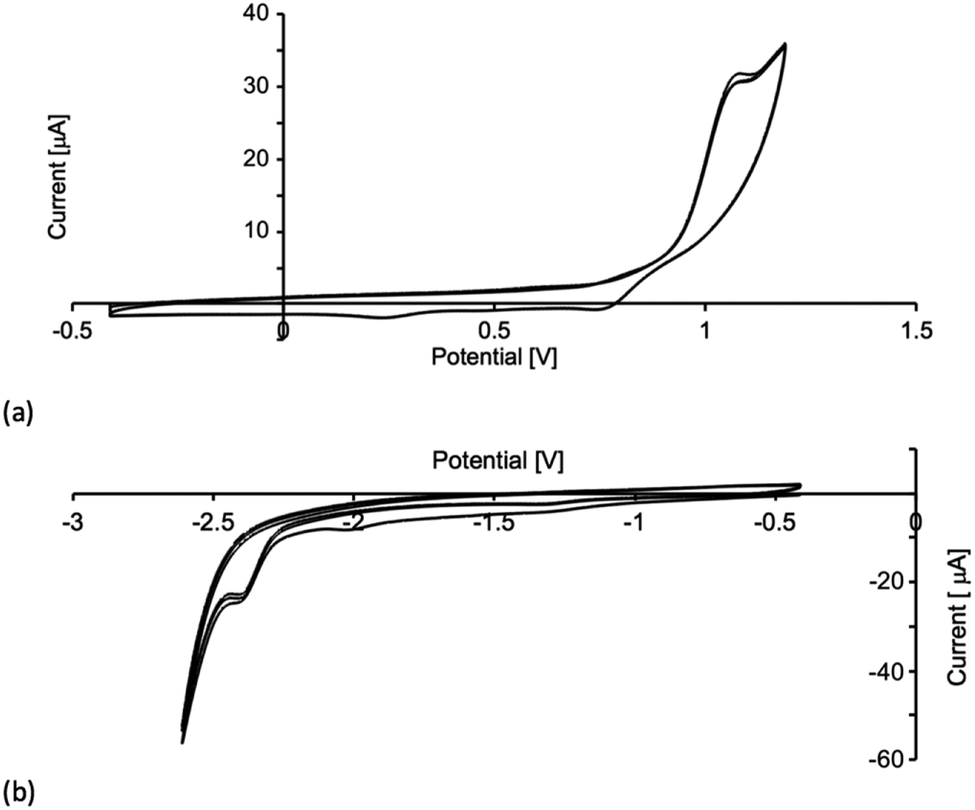
Cyclic voltammograms (CVs) of the [Cu(P^P)(N^S)][PF6] compounds were recorded in CH2Cl2 solution with [nBu4N][PF6] as supporting electrolyte. The observed processes (referenced to Fc/Fc+) are summarized in Table 3 and the CV of [Cu(xantphos)(4)][PF6] is shown in Fig. 5. Each compound undergoes a quasi-reversible (Fig. 5a) or irreversible oxidation assigned to a Cu+/Cu2+ process. If the forward CV scan is taken beyond +1.2 V, a second (irreversible) process is observed due to phosphane oxidation. The Cu+/Cu2+ oxidation potentials in Table 3 compare to values of +0.86 and +0.96 V for [Cu(POP)(N^S)][PF6] and +1.02 and +0.92 V for [Cu(xantphos)(N^S)][PF6] where N^S = 2-(iso-propylthio)pyridine or 2-(tert-butylthio)pyridine.23 For [Cu(POP)(2)][PF6], [Cu(xantphos)(3)][PF6] and [Cu(xantphos)(4)][PF6], irreversible processes are observed at −2.37 or −2.40 V (Fig. 5b) and are assigned to an N^S-based reduction. For the remaining compounds, reduction processes within the solvent accessible window are poorly resolved.
Table 3 Cyclic voltammetry data for [Cu(P^P)(N^S)][PF6] complexes in CH2Cl2 (1–5 × 10−3 M, vs. Fc+/Fc, [nBu4N][PF6] as supporting electrolyte, scan rate = 0.1 V s−1). irrev = irreversible
| Compound |
Oxidative process |
Reductive processes |
| Eox1/2/V |
Epc − Epa/mV |
Ered1/2/V |
| [Cu(POP)(1)][PF6] |
+0.79 |
210 |
|
| [Cu(POP)(2)][PF6] |
+0.89 |
265 |
−2.37 (irrev) |
| [Cu(POP)(3)][PF6] |
+0.84 |
240 |
|
| [Cu(POP)(4)][PF6] |
+1.11 (irrev) |
|
|
| [Cu(xantphos)(1)][PF6] |
+1.06 (irrev) |
|
|
| [Cu(xantphos)(2)][PF6] |
+1.11 (irrev) |
|
|
| [Cu(xantphos)(3)][PF6] |
+0.95 |
245 |
−2.40 (irrev) |
| [Cu(xantphos)(4)][PF6] |
+0.92 |
310 |
−2.40 (irrev) |
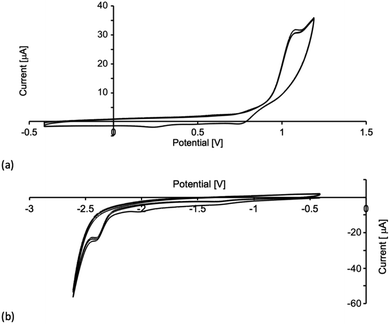 |
| | Fig. 5 Three successive scans in the cyclic voltammogram of [Cu(xantphos)(4)][PF6] in CH2Cl2 (ca. 2 × 10−3 M, vs. Fc+/Fc, [nBu4N][PF6] as supporting electrolyte, scan rate = 0.1 V s−1): (a) oxidative scans to +1.2 V, (b) reductive scans to −2.6 V. | |
Absorption spectra
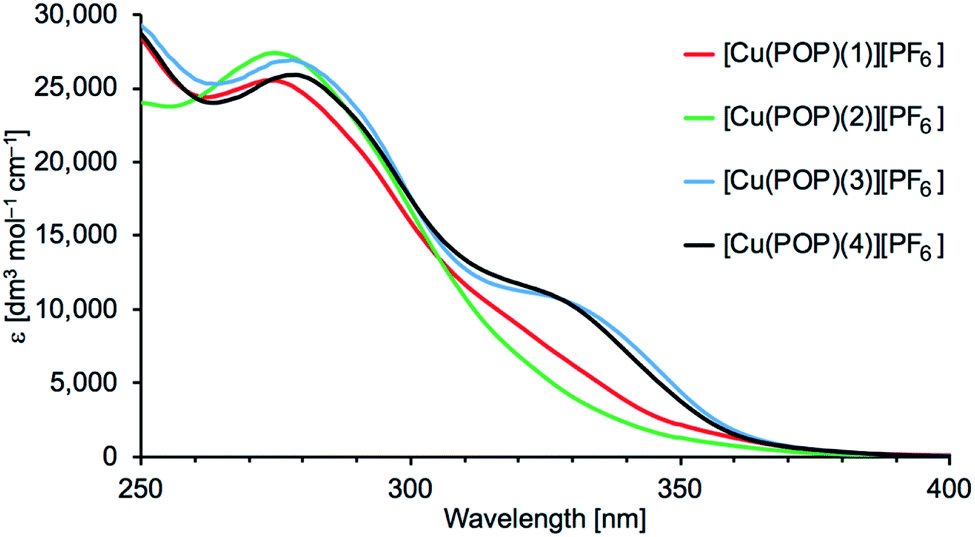
Absorption spectra of the [Cu(P^P)(N^S)][PF6] complexes were recorded in CH2Cl2 solution. Acetone (the solvent used for NMR spectroscopy) is unsuitable because of the cut-off at 330 nm. Fig. 6 and S25† display the absorption spectra of 5 × 10−5 M solutions of [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6], respectively. Values of λmax and εmax are given in Table 4. The intense high-energy absorptions arise from ligand-based, spin-allowed π* ← π and π* ← n transitions. Excitation spectra discussed later (see Emission properties) give further insight into the absorption processes.
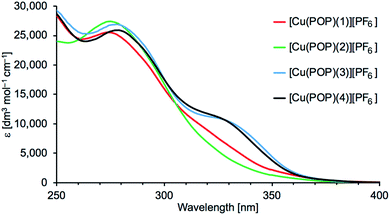 |
| | Fig. 6 Absorption spectra of 5 × 10−5 M solutions of the [Cu(POP)(N^S)][PF6] complexes. | |
Table 4 Solution (CH2Cl2, 5 × 10−5 mol dm−3) absorption maxima for [Cu(P^P)(N^S)][PF6]
| Compound |
λmax/nm (εmax/dm3 mol−1 cm−1) |
| [Cu(POP)(1)][PF6] |
275 (25500) |
| [Cu(POP)(2)][PF6] |
275 (27500) |
| [Cu(POP)(3)][PF6] |
278 (26900), 327 (10400) |
| [Cu(POP)(4)][PF6] |
279 (25900), 327 (10800) |
| [Cu(xantphos)(1)][PF6] |
277 (33500), 326 (11500) |
| [Cu(xantphos)(2)][PF6] |
275 (32900), 326 sh (7700) |
| [Cu(xantphos)(3)][PF6] |
277 (32800), 329 (11300) |
| [Cu(xantphos)(4)][PF6] |
277 (33600), 329 (11300) |
Effects of added chloride ion: absorption spectra and NMR titration
The close cation⋯anion interactions observed in the solid state structures prompted us to investigate whether there were similar interactions in solution. In metal complexes of bpy, the H3 and H3′ protons (analogous to HA3 in ligands 1–4) are relatively acidic. They undergo H/D exchange in [Ru(bpy)3]2+ under basic conditions47 and orthometallation in bpy-containing Ir(III),48 Ni(II), Pd(II) and Pt(II)49 complexes. Strong ion-pairing between the bpy H3 and H3′ protons and chloride ion has been reported for both [Ru(bpy)3]2+ derivatives50 and [Ir(bpy)(ppy)2]+.51
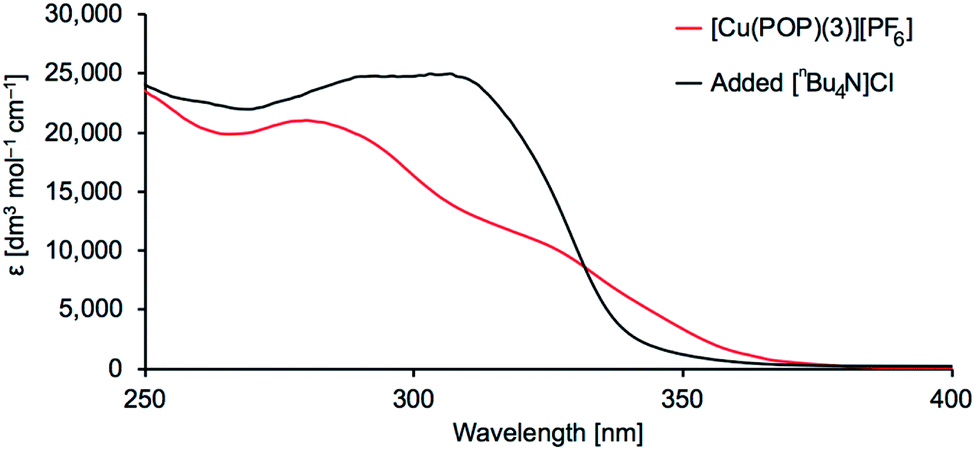
We chose to make a detailed investigation of [Cu(POP)(3)][PF6]. When [nBu4N]Cl was added to a CH2Cl2 solution of [Cu(POP)(3)][PF6], a significant change in the profile of the absorption spectrum was observed (Fig. 7). To confirm the stability of [Cu(POP)(3)][PF6] in CH2Cl2 in the absence of added chloride ion, a 5 × 10−5 M CH2Cl2 solution of [Cu(POP)(3)][PF6] was maintained at room temperature for 3 days. The absorption spectrum remained unchanged (Fig. S26†).
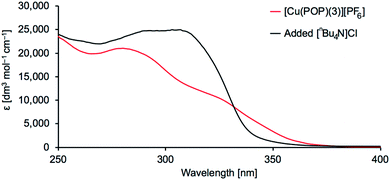 |
| | Fig. 7 Absorption spectra of [Cu(POP)(3)][PF6] in CH2Cl2 (7 × 10−5 M) with and without added chloride ion. | |
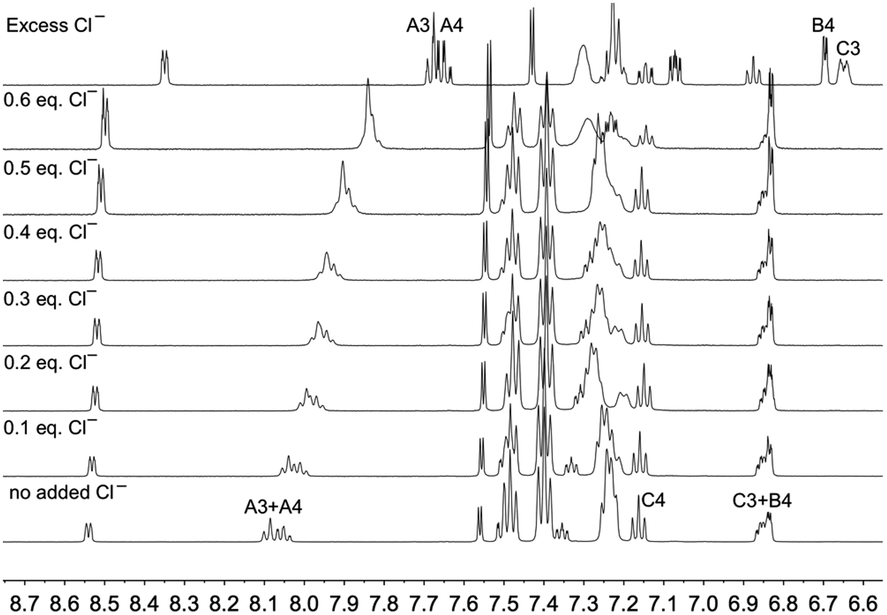
In order to further investigate the effects of chloride ion on [Cu(POP)(3)][PF6], the 1H NMR spectrum of the compound in acetone-d6 in the presence of increasing amounts of [nBu4N]Cl was recorded. Fig. 8 reveals significant changes in the aromatic region, with signals for the pyridine protons HA3 and HA4 undergoing a gradual shift to lower frequencies as the concentration of chloride ion increases, and Fig. 9a shows that the protons most affected are HA3, HA4 and HMe-B6. A Job's plot (Fig. 9b) confirms the formation of an adduct with 1:1 stoichiometry and analysis of the NMR spectroscopic data for the methyl resonance yields a Kd value of 0.14 ± 0.03 mM.52 The magnitude of this Kd is similar to that reported for the ion pair between the 1-methylquinoxalinium ion and iodide in acetone53 and strongly suggests the formation of an ion pair rather than coordination of the chloride to the copper centre to form a five-coordinate complex. No examples are known of structurally characterized five-coordinate {Cu(N^N)(P^P)Cl} or {Cu(N^S)(P^P)Cl} copper(I) complexes (Cambridge Structural Database, CSD v. 5.40,54 searched using Conquest v. 2.0.155).
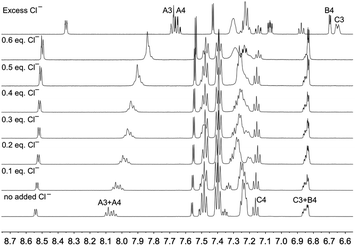 |
| | Fig. 8 Changes in the aromatic region of the 500 MHz 1H NMR spectrum of [Cu(POP)(3)][PF6] (0.01 M in acetone-d6) as a function of added chloride ion ([nBu4N]Cl). See Scheme 1 for atom labelling. | |
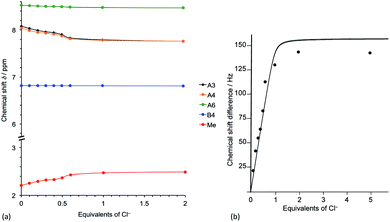 |
| | Fig. 9 (a) Changes in the chemical shifts of protons HA3, HA4, HA6 (pyridine ring) and HB4 and HMe (thiophene ring). Ring protons HA5 and HB3 do not shift and behave like HB4. (b) Job's plot for the methyl resonance of [Cu(POP)(3)][PF6] as function of added chloride ions. The numerical two-parameter fit according to ref. 46, eqn (6) is shown. | |
Crystal structure of [Cu(xantphos)(NCMe)2][PF6]·0.5Et2O
The instability of the [Cu(POP)(N^S)]+ or [Cu(xantphos)(N^S)]+ complexes in the presence of other coordinating ligands was additionally demonstrated by attempted growth of single crystals of [Cu(xantphos)(1)][PF6] from an MeCN solution of the compound with vapour diffusion of Et2O. The integrity of the [Cu(xantphos)(1)][PF6] bulk sample had been confirmed by NMR spectroscopy and mass spectrometry before crystal growth. Colourless plates of [Cu(xantphos)(NCMe)2][PF6]·0.5Et2O were obtained, confirming that ligand 1 had been replaced by the coordinating solvent. The structure of [Cu(xantphos)(NCMe)2][PF6]·0.5Et2O [Cu(xantphos)(NCMe)2][PF6]·CHCl3 was reported in 2013, but suffers from disordering of one MeCN ligand.56 [Cu(xantphos)(NCMe)2][PF6]·0.5Et2O crystallizes in the triclinic space group P, and Fig. 10a depicts the [Cu(xantphos)(NCMe)2]+ cation; selected bond parameters are given in the figure caption. Atom Cu1 is in a distorted tetrahedral environment, and the angle P2–Cu1–P1 of 113.73(4)° is similar to the 115.1° in [Cu(xantphos)(NCMe)2][PF6]·CHCl3 (CSD v. 5.40,54 refcode OGAYIG accessed using Conquest v. 2.0.155), but is smaller than the xantphos chelate angles for [Cu(xantphos)(1)][PF6], [Cu(xantphos)(2)][PF6] and [Cu(xantphos)(4)][PF6] (Table 2). Other structural features of the [Cu(xantphos)(NCMe)2]+ cation are unexceptional, but the close association of [Cu(xantphos)(NCMe)2]+ and [PF6]− ions is worthy of note (Fig. 10b). The C–H⋯F contacts lie between 2.54 and 2.87 Å, a similar range as those in [Cu(xantphos)(1)][PF6] (Fig. 2).
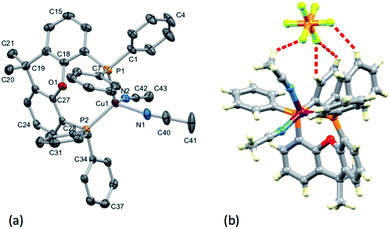 |
| | Fig. 10 (a) ORTEP-style plot of the [Cu(xantphos)(NCMe)2]+ cation in [Cu(xantphos)(NCMe)2][PF6]·0.5Et2O with ellipsoids plotted at 40% probability level. H atoms and solvent molecules are omitted. Selected bond parameters: Cu1–P2 = 2.2593(11), Cu1–P1 = 2.2676(12), Cu1–N2 = 2.032(4), Cu1–N1 = 2.016(4), O1–C27 = 1.391(5), O1–C18 = 1.388(5) Å; P2–Cu1–P1 = 113.73(4), N2–Cu1–P2 = 109.72(11), N2–Cu1–P1 = 114.98(11), N1–Cu1–P2 = 116.10(13), N1–Cu1–P1 = 107.63(14), N1–Cu1–N2 = 93.16(16), C18–O1–C27 = 114.1(3)°. (b) Short C–H⋯F contacts between [Cu(xantphos)(NCMe)2]+ and [PF6]− ions. | |
Emission properties
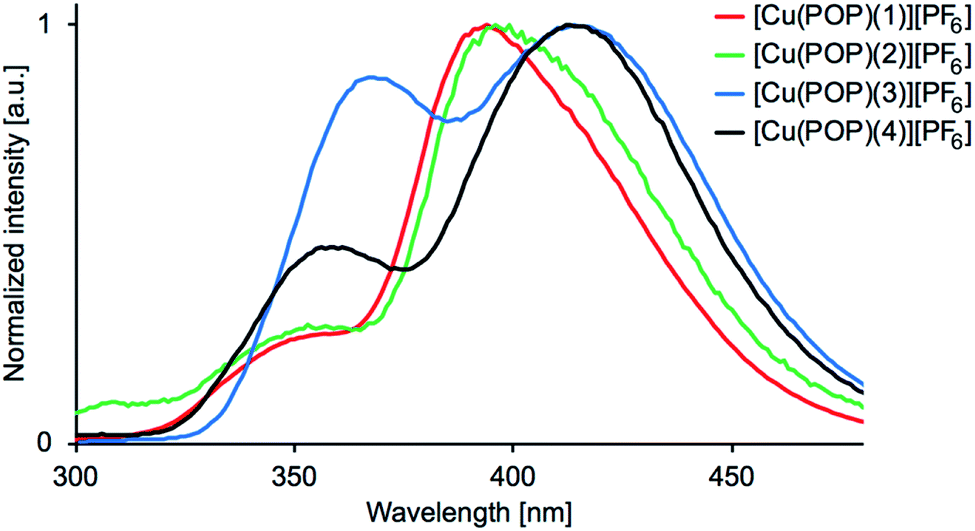
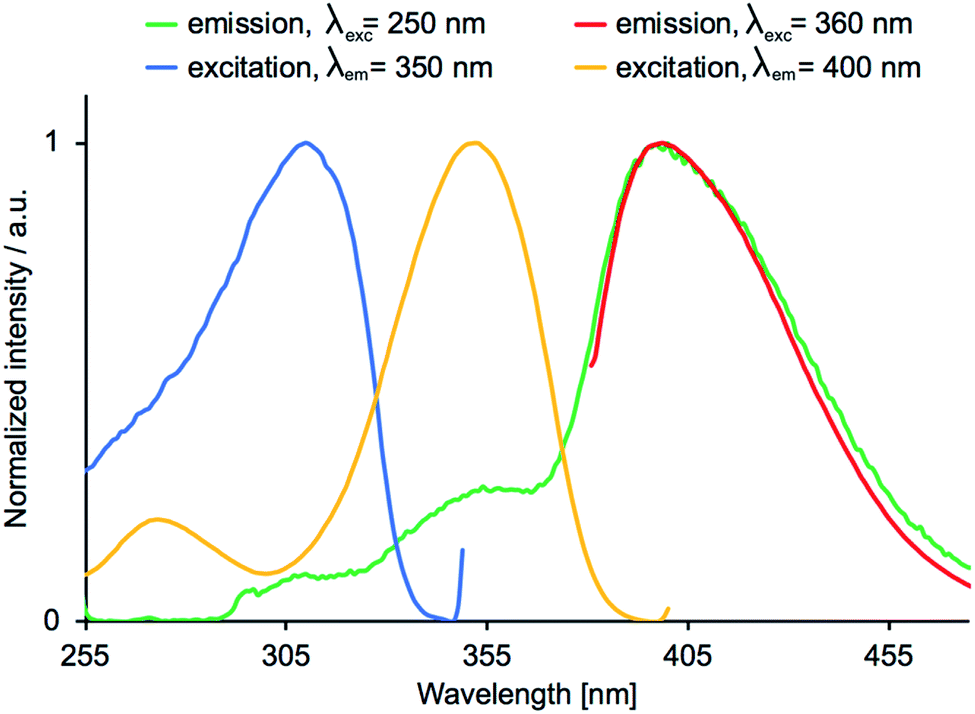
The emission behaviour of the copper(I) compounds was investigated in CH2Cl2 solution and in the solid state. Each [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] compound is a blue emitter and exhibits a dual emission. Fig. 11 and S27† show the solution emission spectra for the POP-and xantphos-containing compounds, respectively. We can eliminate free ligands as being the source of the higher energy emission, since in CH2Cl2 solution, free POP and xantphos emit at 320 and 344 nm, respectively. Moreover, the profile of the excitation band does not correspond to that of free POP or xantphos. Values of λemmax for the complexes are given in Table 5 and in all but [Cu(xantphos)(3)][PF6], the lower energy band is the more dominant. Consistent with the solution absorption spectra (Fig. 6 versus S25), the change from POP to xantphos has little influence on the emission wavelength, as we have previously noted for several series of [Cu(POP)(N^N)][PF6] and [Cu(xantphos)(N^N)][PF6] or [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] compounds.23,28,40,57 A red-shift in the emission is observed as the 5-methyl substituent is introduced into the thienyl ring in ligands 3 and 4 (Table 5). Excitation spectra were recorded for each compound and confirm the origins of the emissions as coming from bands centred at 311–317 nm and 353–380 nm for [Cu(POP)(N^S)][PF6] (Fig. 12), and 302–317 nm and 344–371 nm for [Cu(xantphos)(N^S)][PF6]. We propose that these correspond to coordinated P^P ligand and metal-to-ligand charge transfer (MLCT) bands.
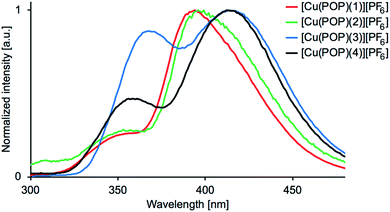 |
| | Fig. 11 Emission spectra for CH2Cl2 solutions of [Cu(POP)(N^S)][PF6] (5.0 × 10−5 M). λexc = 275 nm for 1, 250 nm for 2, 280 nm for 3, 270 nm for 4. | |
Table 5 Solution (CH2Cl2, 5 × 10−5 mol dm−3) and powder emission data for [Cu(P^P)(N^S)][PF6]
| Compound |
Solution |
Powder |
| λemmax/nma |
PLQY/%b |
λemmax/nmc |
PLQY/%d |
| λexc = 250, 275 and 320 or 360 nm. λexc = 360 nm, solutions were deaerated. λexc = 280 nm. λexc = 365 nm. |
| [Cu(POP)(1)][PF6] |
394, 352 |
<1 |
547 |
<1 |
| [Cu(POP)(2)][PF6] |
399, 354 |
30.8 |
555 |
3.1 |
| [Cu(POP)(3)][PF6] |
414, 369 |
1.5 |
569 |
<1 |
| [Cu(POP)(4)][PF6] |
413, 360 |
33.2 |
546 |
<1 |
| [Cu(xantphos)(1)][PF6] |
392, 358 |
4.5 |
555 |
10.8 |
| [Cu(xantphos)(2)][PF6] |
394, 355 |
4.8 |
552 |
4.7 |
| [Cu(xantphos)(3)][PF6] |
418, 367 |
4.5 |
555 |
9.4 |
| [Cu(xantphos)(4)][PF6] |
422, 369 |
17.2 |
560 |
2.9 |
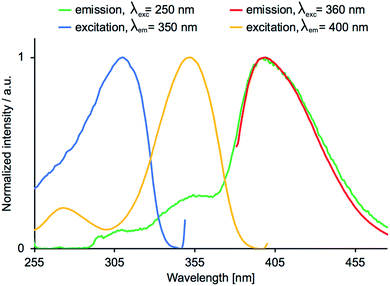 |
| | Fig. 12 Emission (red and green) and excitation (yellow and blue) spectra for a CH2Cl2 solution of [Cu(POP)(2)][PF6]. | |
Photoluminescence quantum yields (PLQY) were measured in deaerated (Ar purge for 15 minutes) solutions and the values (Table 5) were essentially the same for non-deaerated solutions. The highest PLQY was observed for [Cu(POP)(2)][PF6] (30.8%) and [Cu(POP)(4)][PF6] (33.2%), in which the N^S ligands contain a 6-methyl-substituted pyridine ring. The benefits of methyl-substitution in both rings (ligand 4) are also seen in the xantphos-series with the highest PLQY being for [Cu(xantphos)(4)][PF6] (17.2%). However, all excited-state lifetimes in deaerated solutions were <5 ns.
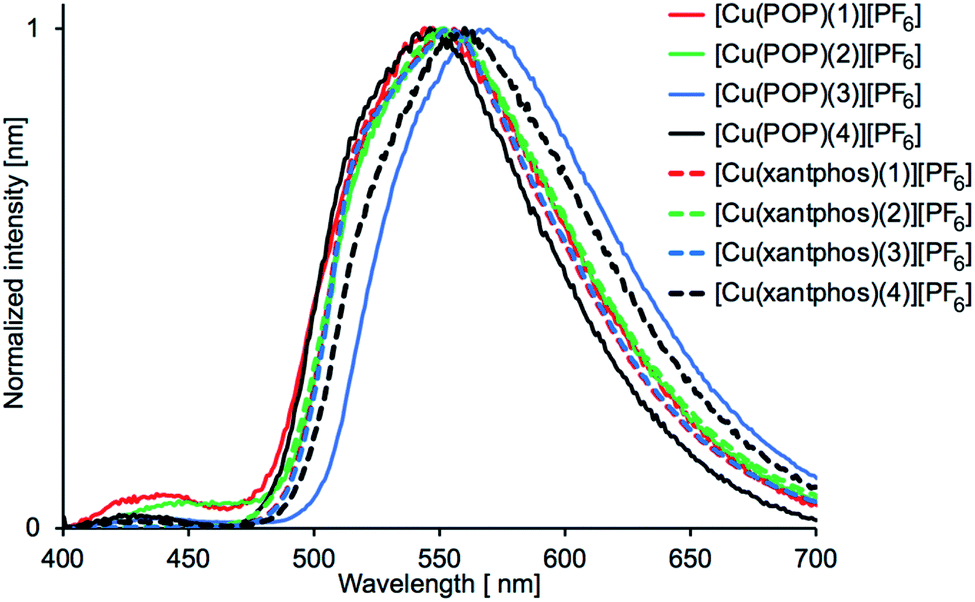
On going from solution to powder samples, values of λemmax undergo a red-shift (Table 5), and only one dominant emission band is observed (Fig. 13). The powder samples are yellow emitters, the brightest being [Cu(xantphos)(1)][PF6]. From the emission profiles in Fig. 13, it is reasonable to assume that the principal solid-state emission band corresponds to the lower energy solution band, and thus red-shifts in the range 133 to 163 nm are observed on going from solution to powder. Free POP and xantphos are weak emitters in the solid state with λemmax = 443 and 455 nm, respectively.56 The low intensity emissions may therefore arise from free ligand, or from coordinated POP or xantphos. With the exception of [Cu(xantphos)(1)][PF6] and [Cu(xantphos)(3)][PF6], the PLQY values for the powder samples of the compounds are lower than in solution and this may be indicative of aggregation-caused quenching (ACQ) in the solid state, although this is typically associated with π-stacking of planar molecular species.58,59
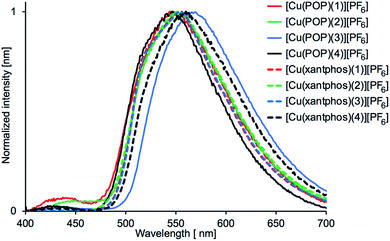 |
| | Fig. 13 Solid-state emission spectra of [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6]. λexc = 365 nm. | |
The most promising solid-state emitter, [Cu(xantphos)(1)][PF6], was also tested in a thin film with ionic liquid but the PL signal was below the detection limit and no PLQY could be measured. In a LEC configuration, [Cu(xantphos)(1)][PF6] exhibited no electroluminescence, even when the LEC was driven under a high bias (18 V). We conclude that, in addition to being poorly emissive, the compound is not a good charge transporter in a device configuration, possible due to tight ion-pairing as observed in the crystal structure (Fig. 2).
Conclusions
We have prepared a series of [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] compounds in which the N^S ligand is a 2-(thiophen-2-yl)pyridine (1–4). Characterization by NMR and absorption spectroscopies in acetone or CH2Cl2 solution shows that the compounds are stable with respect to ligand dissociation. Single crystal structures of [Cu(POP)(1)][PF6], [Cu(xantphos)(1)][PF6], [Cu(xantphos)(2)][PF6], [Cu(POP)(3)][PF6]·CH2Cl2, and [Cu(xantphos)(4)][PF6] confirm the chelating nature of the N^S ligand and the distorted tetrahedral environment of the copper(I) centre. Close cation⋯anion CH⋯F contacts, notably in [Cu(xantphos)(1)][PF6], are observed. This prompted us to investigate the influence of competing ligands. Significant changes are seen in the absorption and 1H NMR spectra of [Cu(POP)(3)][PF6] when chloride ion is added to a CH2Cl2 (or CD2Cl2) solution. When single crystals were grown from an MeCN solution of [Cu(xantphos)(1)][PF6], ligand replacement occurs to give [Cu(xantphos)(MeCN)2][PF6].
The [Cu(POP)(N^S)][PF6] and [Cu(xantphos)(N^S)][PF6] compounds show quasi-reversible (Eox1/2 in the range +0.79 to +0.95 V) or irreversible Cu+Cu2+ oxidations. The compounds are blue emitters in solution, and the introduction of the 5-methyl substituent in the thiophene in ligands 3 and 4 leads to a red-shift in the emission. The highest PLQYs are for [Cu(POP)(2)][PF6] (30.8%) and [Cu(POP)(4)][PF6] (33.2%), in which there is a 6-methyl-substituted pyridine in the N^S ligand. All compounds have very short excited-state lifetimes (<5 ns). On going from solution to powder samples, red-shifts of 133 to 163 nm are observed leading to yellow emitters. The brightest emitter, [Cu(xantphos)(1)][PF6], was tested in a LEC device but showed poor electroluminescence and show poor charge transporting characteristics.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the Swiss National Science Foundation (grant numbers 200020_162631 and 200020_182000) and the University of Basel for financial support. We thank Dr Michele Sessolo and Cristina Momblona (University of Valencia) for fabricating and testing a LEC with [Cu(xantphos)(1)][PF6].
References
- M. T. Buckner and D. R. McMillin, J. Chem. Soc., Chem. Commun., 1978, 759 RSC.
- R. A. Rader, D. R. McMillin, M. T. Buckner, T. G. Matthews, D. J. Casadonte, R. K. Lengel, S. B. Whittaker, L. M. Darmon and F. E. Lytle, J. Am. Chem. Soc., 1981, 103, 5906 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- M. Elie, S. Gaillard and J.-L. Renaud in Light-emitting electrochemical cells: concepts, advances and challenges, ed. R. D. Costa, Springer, Cham, 2017, ch. 11, p. 287 Search PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032 CrossRef CAS PubMed.
- H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508 CrossRef CAS PubMed.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier, H. Yersin and H. Coord, Chem. Rev., 2016, 325, 2 CAS and references therein.
- R. G. Pearson, Coord. Chem. Rev., 1990, 100, 403 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
- M. C. Thompson and D. H. Busch, J. Am. Chem. Soc., 1964, 86, 3651 CrossRef CAS.
- H.-B. Song, Z. Z. Zhang and T. C. Mak, J. Chem. Soc., Dalton Trans., 2002, 1336 RSC.
- B. Nohra, E. Rodriguez-Sanz, C. Lescop and R. Reau, Chem.–Eur. J., 2008, 14, 3391 CrossRef CAS PubMed.
- P. Arce, C. Vera, D. Escudero, J. Guerrero, A. Lappin, A. Oliver, D. H. Jara, G. Ferraudi and L. Lemus, Dalton Trans., 2017, 46, 13432 RSC.
- V. Vreshcha, M. el S. Moussa, B. Nohra, M. Srebro, N. Vanthuyne, C. Roussel, J. Autschbach, J. Crassous, C. Lescop and R. Reau, Angew. Chem., Int. Ed., 2013, 52, 1968 CrossRef PubMed.
- W. Shen, S. Graule, J. Crassous, C. Lescop, H. Gornitzka and R. Reau, Chem. Commun., 2008, 850 RSC.
- S. Graule, M. Rudolph, W. Shen, J. A. G. Williams, C. Lescop, J. Autschbach, J. Crassous and R. Reau, Chem.–Eur. J., 2010, 16, 5976 CrossRef CAS PubMed.
- P. Coburger, J. Schulz, J. Klose, B. Schwarze, M. B. Sarosi and E. Hey-Hawkins, Inorg. Chem., 2017, 56, 292 CrossRef CAS PubMed.
- C. Amari, S. Ianelli, C. Pelizzi, G. Pelizzi and G. Predieri, Inorg. Chim. Acta, 1993, 211, 89 CrossRef CAS.
- M. N. Patel, C. R. Patel and H. N. Joshi, Spectrochim. Acta, Part A, 2012, 97, 66 CrossRef CAS PubMed.
- M. N. Patel, C. R. Patel and H. N. Joshi, Z. Anorg. Allg. Chem., 2012, 638, 1224 CrossRef CAS.
- H. Gökce and S. Bahçeli, Spectrochim. Acta, Part A, 2012, 96, 139 CrossRef PubMed.
- M. Munakata, J. Han, A. Nabei, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and N. Gunjima, Polyhedron, 2006, 25, 3519 CrossRef CAS.
- I. Nohara, A. Prescimone, C. E. Housecroft and E. C. Constable, Inorganics, 2019, 7, 11 CrossRef.
- G. K. Batsala, V. Dokorou, N. Kourkoumelis, M. J. Manos, A. J. Tasiopoulos, T. Mavromoustakos, M. Simčič, S. Golič-Grdadiłnik and S. K. Hadjikakou, Inorg. Chim. Acta, 2012, 382, 146 CrossRef CAS.
- T. S. Lobana, A. K. Sandhu, R. Sultana, A. Castineiras, R. J. Butcher and J. P. Jasinski, RSC Adv., 2014, 4, 30511 RSC.
- M. Trivedi, S. K. Ujjain, G. Singh, A. Kumar, S. K. Dubey and N. P. Rath, J. Organomet. Chem., 2014, 772–773, 202 CrossRef CAS.
- S. Keller, F. Brunner, J. M. Junquera-Hernández, A. Pertegás, M.-G. La-Placa, A. Prescimone, E. C. Constable, H. J. Bolink, E. Ortí and C. E. Housecroft, ChemPlusChem, 2018, 83, 217 CrossRef CAS and references therein.
- M. Alkan-Zambada, S. Keller, L. Martínez-Sarti, A. Prescimone, J. M. Junquera-Hernández, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, J. Mater. Chem. C, 2018, 6, 8460 RSC and references therein.
- G. J. Kubas, Inorg. Synth., 1979, 19, 90 CAS.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971 CrossRef CAS PubMed.
- S. W. Thomas, K. Venkatesan, P. Müller and M. Swager, J. Am. Chem. Soc., 2006, 128, 16641 CrossRef CAS PubMed.
- Bruker Analytical X-ray Systems, Inc., APEX2, Version 2 User Manual, M86-E01078, Madison, WI, 2006 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- L. Wang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 Search PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS and references therein.
- S. Keller, A. Pertegás, G. Longo, L. Martinez, J. Cerdá, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, C. E. Housecroft, E. Ortí and H. J. Bolink, J. Mater. Chem. C, 2016, 4, 3857 RSC.
- F. Brunner, S. Graber, Y. Baumgartner, D. Häussinger, A. Prescimone, E. C. Constable and C. E. Housecroft, Dalton Trans., 2017, 46, 6379 RSC.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS.
- S.-Z. Hu, Z.-H. Zhou and B. E. Robertson, Z. Kristallogr., 2009, 224, 375 CAS.
- A. Bondi, J. Phys. Chem., 1966, 70, 3006 CrossRef CAS.
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC.
- S.-Z. Hu, Z.-H. Zhou, Z.-Z. Xie and B. E. Robertson, Z. Kristallogr., 2014, 229, 517 CAS.
- E. C. Constable and K. R. Seddon, J. Chem. Soc., Chem. Commun., 1982, 34 RSC.
- P. S. Braterman, G. A. Heath, A. J. MacKenzie, B. C. Noble, R. D. Peacock and L. J. Yellowlees, Inorg. Chem., 1984, 23, 3425 CrossRef CAS; A. C. Hazell and R. G. Hazell, Acta Crystallogr., Sect. C., 1984, C40, 806 CrossRef; P. J. Spellane, R. J. Watts and C. J. Curtis, Inorg. Chem., 1983, 22, 4060 CrossRef; G. Nord, A. C. Hazell, R. G. Hazell and O. Farver, Inorg. Chem., 1983, 22, 3429 CrossRef.
- B. Butschke and H. Schwarz, Organometallics, 2010, 29, 6002–6011 CrossRef CAS; B. Butschke, M. Schlangen, D. Schroeder and H. Schwarz, Chem.–Eur. J., 2008, 14, 11050 CrossRef PubMed.
- W. M. Ward, B. H. Farnum, M. Siegler and G. J. Meyer, J. Phys. Chem. A, 2013, 117, 8883 CrossRef CAS PubMed.
- G. E. Schneider, H. J. Bolink, E. C. Constable, C. D. Ertl, C. E. Housecroft, A. Pertegas, J. A. Zampese, A. Kanitz, F. Kessler and S. B. Meier, Dalton Trans., 2014, 43, 1961 RSC.
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1 CrossRef CAS PubMed.
- M. V. Tolstykh, Y. V. Nelyubinab and V. Yu. Kotov, Mendeleev Commun., 2017, 27, 78 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- J. Yuasa, M. Dan and T. Kawai, Dalton Trans., 2013, 42, 16096 RSC.
- S. Keller, A. Prescimone, H. J. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí and C. E. Housecroft, Dalton Trans., 2018, 47, 14263 RSC.
- L. Le Bras, K. Chaitou, S. Aloïse, C. Adamo and A. Perrier, Phys. Chem. Chem. Phys., 2019, 21, 46 RSC and references therein.
- X. Ma, R. Sun, J. Cheng, J. Liu, F. Gou, H. Xiang and X. Zhou, J. Chem. Educ., 2016, 93, 345 CrossRef CAS and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2: NMR spectra of 4; Fig. S3–S10: electrospray mass spectra; Fig. S11–S19. 31P{1H} and 1H NMR spectra of complexes; Fig. S20–24: structural figures; Fig. S25 and S26: absorption spectra; Fig. S27: solution emission spectra of [Cu(xantphos)(N^S)][PF6]. CCDC: 1900489–1900494. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra02617g. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Daniel Häussinger
a,
Daniel Häussinger