
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural exploration of rhodium catalysts and their kinetic studies for efficient parahydrogen-induced polarization by side arm hydrogenation†
Marino Itodaa,
Yuki Naganawa *ab,
Makoto Itoa,
Hiroshi Nonakaa and
Shinsuke Sando*ac
*ab,
Makoto Itoa,
Hiroshi Nonakaa and
Shinsuke Sando*ac
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ssando@chembio.t.u-tokyo.ac.jp
bInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: yuki.naganawa@aist.go.jp
cDepartment of Bioengineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 10th June 2019
Abstract
Parahydrogen-induced polarization (PHIP) is a rapid and cost-effective hyperpolarization technique using transition metal-catalysed hydrogenation with parahydrogen. We examined rhodium catalysts and their kinetic studies, rarely considered in the research of current PHIP. It emerged that rhodium complexes with electron-donating bisphosphine ligands, with a dicyclohexylphosphino group, appear to be more effective than conventional rhodium catalysts.
Introduction
Nuclear magnetic resonance (NMR) spectroscopy is one of the attractive techniques for non-invasive biological analysis. However, in vivo analysis at a molecular level by NMR spectroscopy is often problematic because of the intrinsic low sensitivity. Hyperpolarization techniques have been developed to enhance the intensity of NMR signals of target nuclei dramatically. Theoretically in the case of 13C nuclei, hyperpolarization should enhance intensities of NMR signals up to 105 times more than in the thermal equilibrium.1 Among various protocols, the dissolution dynamic nuclear polarization (d-DNP) method has become a mainstream technique to achieve high polarization levels, although several unsolved issues do remain, such as the requirement for expensive equipment and prolonged operating times of several hours.2Meanwhile, parahydrogen-induced polarization (PHIP),3 in addition to signal amplification by reversible exchange (SABRE),4 has drawn increasing attention as an easy and cost-effective hyperpolarization technique. PHIP utilizes transition metal-catalysed hydrogenation of carbon–carbon unsaturated bonds in precursor molecules with parahydrogen, the spin isomer of molecular hydrogen with antiparallel nuclear spins (Scheme 1). The rapid decay of hyperpolarization generally proceeds through spin–lattice relaxation, and thus it is preferable that the polarization is transferred from 1H to other heteronuclei such as 13C and 15N with larger T1, by field cycling or using radio frequency pulse, where the hyperpolarized spin state can be retained for longer periods of time.5 Therefore, the first hydrogenation with parahydrogen is regarded as the dominant step of PHIP to realize sufficient polarization levels. Hyperpolarization levels are positively correlated with the reaction rate of the hydrogenation reaction; therefore, this key step should be completed as soon as possible with highly active transition metal catalysts.
 | ||
| Scheme 1 Experimental concept of PHIP. | ||
Regarding the choice of metal catalysts, homogeneous rhodium complexes have been applied to hydrogenation in PHIP because the pairwise addition to unsaturated bonds must take place to retain the initial spin character of parahydrogen.6,7 Commercially available [Rh(nbd)(dppb)]BF4 (1) (nbd = 2,5-norbornadiene, dppb = 1,4-bis(diphenylphosphino)butane) has been employed in typical PHIP experiments, although the catalytic activity of this conventional cationic rhodium complex 1 (see Fig. 1a) is sometimes inadequate. For example, Chekmenev and co-workers8 reported PHIP experiments of [1-13C]vinyl acetate (VA) with 1, wherein high hydrogen pressure was required to complete the hydrogenation within a few seconds. In this case, hydrogenation of 80 mM VA solution in methanol-d4 at 40 °C under standard pressure (1 bar) was not completed after 100 s.8a
 | ||
| Fig. 1 (a) Structural exploration of conventional rhodium complex 1. (b) Rhodium complexes 2–5 selected based on the screening of ligands. | ||
In the field of organometallic chemistry, the development of various rhodium catalysts for hydrogenation has thus far been devoted to the achievement of high reactivity and efficiency. In contrast, such careful investigations on rhodium catalysts have rarely been considered for PHIP experiments,9 especially for PHIP by side arm hydrogenation (PHIP-SAH).10 PHIP-SAH is a promising method which can hyperpolarize carboxylic acid such as pyruvic acid, an attractive substrate for hyperpolarized magnetic resonance imaging (MRI).11 We therefore focused our attention and efforts on the structural exploration of [Rh(nbd)(PP)]BF4 (PP = bisphosphine ligand) and kinetic studies to improve the efficiency of PHIP experiments (see Fig. 1a).
Experimental
1 Synthesis
1H NMR (CDCl3, 400 Hz) δ = 5.54 (s, 4H), 4.23 (s, 2H), 2.03–1.70 (m, 20H), 1.39–1.02 (m, 30H); 13C NMR (CDCl3, 125 MHz) δ = 85.4, 71.7, 55.7, 35.4–35.7 (m), 29.5, 29.0, 27.1 (t, J = 4.7 Hz), 26.7 (t, J = 4.0 Hz), 26.4, 25.9, 21.1 (t, J = 16 Hz); 31P NMR (CDCl3, 162 Hz) δ = 70.2 (d, J = 153 Hz); HRMS(ESI): m/z calc. for C33H56P2Rh+ [M–BF4−]+ = 617.2907, found = 617.2900.
1H NMR (CDCl3, 400 Hz) δ = 5.18 (s, 4H), 4.14 (s, 2H), 2.20–1.14 (m, 52H); 13C NMR (CDCl3, 100 MHz) δ = 80.2, 70.7, 54.4, 36.9 (t, J = 11 Hz), 30.4, 28.7, 27.5 (t, J = 5.7 Hz), 26.8 (t, J = 4.8 Hz), 26.0, 22.5, 17.8 (t, J = 13.8 Hz); 31P NMR (CDCl3, 162 Hz) δ = 16.3 (d, J = 148 Hz); HRMS(ESI): m/z calc. for C34H58P2Rh+ [M–BF4−]+ 631.3063, found 631.3050.
1H NMR (CDCl3, 400 Hz) δ = 4.93 (s, 4H), 4.09 (s, 2H), 2.15 (m, 4H), 2.03–1.60 (m, 30H), 1.46–1.18 (m, 20H); 13C NMR (CDCl3, 100 MHz) δ = 76.7–77.3 (m), 69.8, 53.7, 36.7 (t, J = 11 Hz), 31.0, 28.7, 27.6 (t, J = 5.7 Hz), 26.9 (t, J = 4.3 Hz), 26.1, 24.2, 17.1 (t, J = 11 Hz); 31P NMR (CDCl3, 162 Hz) δ = 27.7 (d, J = 153 Hz); HRMS(ESI): m/z calc. for C35H60P2Rh+ [M–BF4−]+ = 645.3220, found 645.3208.
1H NMR (CDCl3, 400 Hz) δ = 5.06 (s, 4H), 4.47 (s, 4H), 4.29 (s, 4H), 4.15 (s, 2H), 2.33 (m, 4H), 2.18–1.06 (m, 42H); 13C NMR (CDCl3, 100 MHz) δ = 75.4, 73.5, 72.0, 69.6, 53.7, 37.1 (t, J = 10 Hz), 30.0, 30.0, 27.6 (t, J = 6.2 Hz), 26.7 (t, J = 4.8 Hz), 26.0; 31P NMR (CDCl3, 162 Hz) δ = 29.8 (d, J = 162 Hz); HRMS(ESI): m/z calc. for C41H60FeP2Rh [M–BF4−]+ 773.2575, found 773.2551.
2 Ligand screening
3 Hydrogenation kinetics
4 Hyperpolarization experiments
5 Hyperpolarization using flow system
Results and discussion
Initial ligand screening based on catalytic activity
We commenced our evaluation of various rhodium catalysts in the hydrogenation of VA, which is a minimal model molecule usually applied to PHIP-SAH.10a All experiments were conducted in methanol or methanol-d4 because most of the ligands were insoluble in water. We examined the hydrogenation of VA in the presence of rhodium complexes prepared in situ from [Rh(nbd)]2BF4 with various phosphine ligands (Scheme S1†). The reaction was carried out using a solution of VA in methanol-d4 (150 mM) with 5 mM of rhodium catalyst, under constant bubbling of hydrogen (1 atm, 20 mL min−1) into an NMR tube for 30 s at 50 °C. The conversion rate was then determined by 1H NMR analysis. Screening of various phosphine ligands revealed that highly electron-donating bisphosphine ligands with a dicyclohexylphosphino group (PCy2 group) were particularly promising in terms of their catalytic activity (Table 1, entries 7–9 and 11).| Entry | Ligand | Conversion of VAb [%] |
|---|---|---|
| a Hydrogen (20 mL min−1, 1 atm) was bubbled into a solution of VA in methanol-d4 (150 mM) with 5 mM of rhodium catalyst in an NMR tube for 30 s at 50 °C.b The conversion rate was determined by 1H NMR analysis. n.d. = not detected. | ||
| 1 | 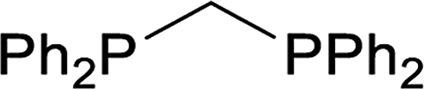 |
Trace |
| 2 | 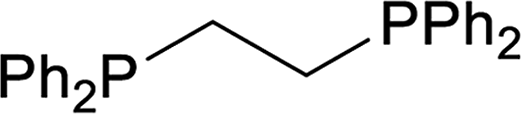 |
16 |
| 3 | 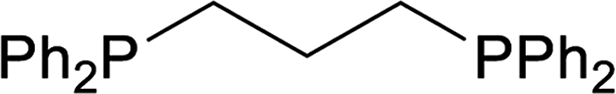 |
24 |
| 4 | 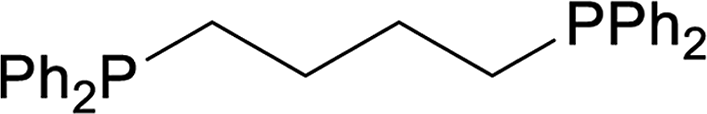 |
33 |
| 5 |  |
39 |
| 6 |  |
23 |
| 7 | 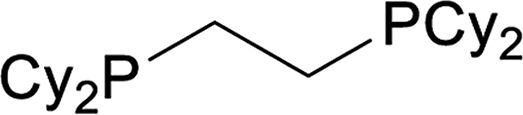 |
71 |
| 8 |  |
69 |
| 9 |  |
73 |
| 10 |  |
25 |
| 11 |  |
81 |
| 12 |  |
29 |
| 13 | 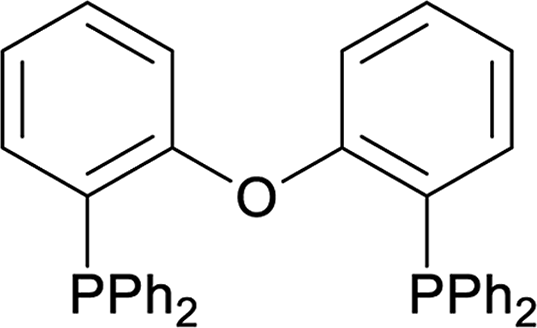 |
45 |
| 14 |  |
5 |
| 15 |  |
n.d. |
| 16 | PPh3 (2 eq.) | 19 |
| 17 | PCy3 (2 eq.) | 5 |
Hyperpolarization experiment of VA
Based on the screening of ligands, we next prepared rhodium complexes [Rh(nbd)(dcpe)]BF4 (2), [Rh(nbd)(dcpp)]BF4 (3), [Rh(nbd)(dcpb)]BF4 (4) and [Rh(nbd)(dcpf)]BF4 (5) (see Fig. 1b). Rhodium complexes of bisphosphine ligands with a dicyclohexylphosphino group have been reported as catalysts for hydrogenation; however, to our knowledge, they have not been widely employed for PHIP experiments.13With these promising rhodium catalysts in hand, we conducted the PHIP-SAH experiment of VA according to procedure described by Chekmenev and co-workers.8a We performed our hyperpolarization experiment by bubbling parahydrogen (1 atm, 20 mL min−1) for 10 s into a methanol-d4 solution of 13C natural abundance VA (50 mM) and rhodium catalysts 1–5 (5 mM) in NMR tubes at 50 °C under the Earth magnetic field. Then, the sample was quickly inserted in 0.2 μT field and pulled out slowly (ca. 4 s) to transfer the hyperpolarization from nascent protons to 13C with longer T1 (ALTADENA conditions) (Fig. 2a and Scheme S2†). Our PHIP-SAH experiments of VA using catalysts 1–5 resulted in the successful observation of hyperpolarized 13C NMR signals of ethyl acetate (EA). The polarization level % P13C at the carbonyl carbon of EA reached about 1.5%, which was comparable in the case of conventional catalyst 1 and modified catalysts 2–5 (Fig. 2b). In contrast, the conversion rate of the hydrogenation with catalysts 2–5 was uniformly higher than that with 1 (Fig. 2c).14 As a result, the signal-to-noise (S/N) ratios of the 13C NMR signals obtained when using catalysts 2–5 were enhanced; they were three to four times higher than when using conventional catalysis 1 (Fig. 2d–f).
 | ||
| Fig. 2 (a) Conditions of the PHIP experiment of VA. (b) Polarization level % P13C at carbonyl carbon of EA. (c) Conversion rate of VA to EA. (d) 13C NMR spectrum of natural abundant EA (ca. 40 mM) in thermal equilibrium state (30° pulse, 1 scan). (e) 13C NMR spectra (30° pulse, 1 scan) of EA produced upon PHIP (initial concentration of VA 50 mM) with catalyst 1 or 4. S/N means signal to noise ratio with standard deviation (n = 3). (f) Stacked 13C NMR spectra of the hyperpolarized EA. PHIP experiments were conducted under the following conditions: 20 mL min−1 parahydrogen (1 atm) was bubbled for 10 s into 700 μL of methanol-d4 containing 5 mM catalyst and 50 mM VA, followed by polarization transfer. | ||
Hydrogenation kinetics
To gain further insight into the origin of the better catalytic activities of 2–5, we investigated the kinetics of the hydrogenation. In the initial stage of hydrogenation, the removal of the coordinated nbd ligand of 1–5 must be carried out to generate active species under reductive conditions (Step 1 in Scheme 2). | ||
| Scheme 2 Two steps of rhodium-catalysed hydrogenation. | ||
Thus, we first determined the activation rate of 1–5 under hydrogen (1 atm) by UV-Vis analysis of the coordinated nbd ligands, with the maximum absorption wavelength at 470–480 nm (Fig. 3a). Subsequently, the removal rate of a nbd ligand of modified catalysts 2–5 was determined to be 4.0–5.7 × 10−3 mM s−1,14 whereas that of 1 was 1.2 × 10−3 mM s−1 (Fig. 3b).
 | ||
| Fig. 3 Measurement of activation rates. (a) UV-vis spectra of 0.5 mM of Rh-dcpb complex with and without nbd. (b) Determination of activation rates. Activation rates were determined by monitoring absorption around 470–480 nm of 0.5 mM catalyst solution at 1 atm of hydrogen. | ||
Next, we checked the turnover frequency (TOF) of the completely activated rhodium catalysts in the hydrogenation of VA with bubbling of hydrogen (1 atm) (Step 2 in Scheme 2). The TOF of catalysts 2–5 was determined to be 1.8–2.0 times higher than that of 1 (Fig. 4).14 Overall, these two kinetic studies revealed that both catalyst activation (Step 1) and hydrogenation (Step 2) proceeded rapidly in the case of catalysts 2–5, resulting in higher performance in the PHIP experiments.
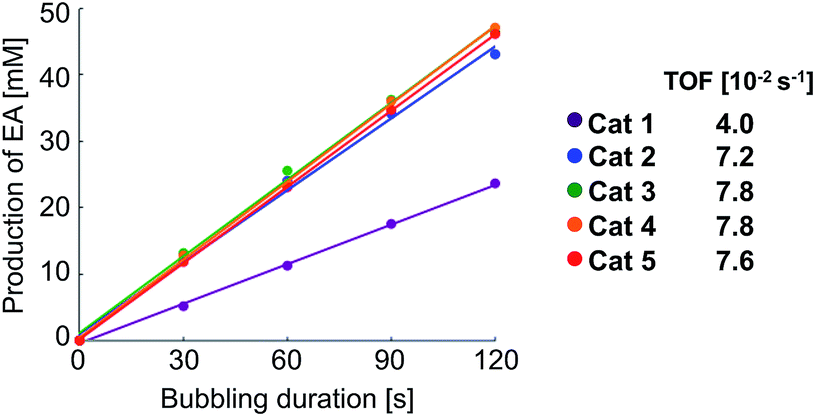 | ||
| Fig. 4 TOF of Rh catalysts 1–5 in hydrogenation for vinyl acetate (VA). Determination of TOF for VA (cat 1–5). 5 mM of catalyst solution in methanol-d4 containing 1 M of VA was set into an NMR tube, and bubbled with normal hydrogen (20 mL min–1). | ||
Possible reaction mechanism
To further consider the reasons for the high catalytic activity of 2–5, details of differences in each of the possible reaction mechanism are illustrated in Fig. 5. In the general pathway, with cationic rhodium catalysts such as 1, it is well known that hydrogenation takes place via the initial coordination of substrates before the oxidative addition of hydrogen, which is irreversible15 (‘Unsaturated route’, left in Fig. 5).16 In contrast, Imamoto and co-workers propose that cationic rhodium complexes [RhS2(PP)]BF4 with an electron-donating bisphosphine ligand bearing alkyl groups are first converted to the corresponding dihydride complexes [RhH2S2(PP)]BF4, rather than the coordination of substrates, due to the stabilizing effect on the high oxidation state of a rhodium centre (‘Hydride route’, right in Fig. 5).17 It has been also examined that the reversible addition of hydrogen and ortho–para interconversion occur in the hydrogenation with catalyst 1 and the formation of rhodium hydride species is more favoured with the more basic phosphine ligands.18 In addition, an isolation of the related stable cationic rhodium hydride complex of a bisphosphine ligand with a diisopropylphosphino group was reported.19 Thus, it may be possible to reason that hydrogenation with our modified catalysts 2–5 might proceed mainly via the Hydride route.6,20,21 It should be also noted that the Hydride route has been thought to be less preferable for PHIP.6,22 In our attempt to determine the plausible mechanism, the generation of the corresponding dihydride species after H2 bubbling with both catalysts 1 and 4 was observed (Fig. S1†). Therefore, further investigations, e.g., identification of the rate-determining step, are required to conclude which pathway would be more plausible for hydrogenation with catalysts 2–5.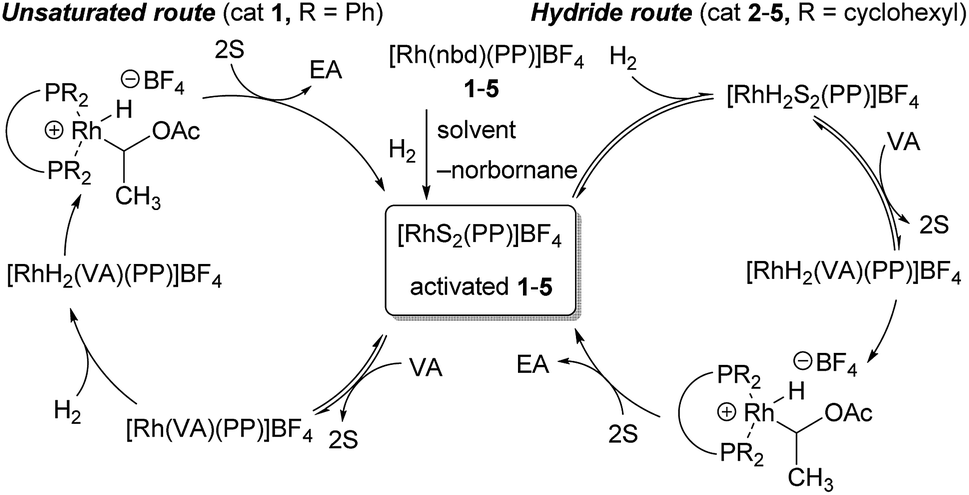 | ||
| Fig. 5 Two different catalytic cycles of hydrogenation. nbd = 2,5-norbornadiene, S = solvent, VA = vinyl acetate, EA = ethyl acetate, PP = bisphosphine ligand. | ||
Hyperpolarization experiment of PA
With high-performance catalysts in hand, we then moved to examine the hydrogenation of propargyl esters, which have also been employed for PHIP-SAH experiments (Fig. 6).9,20 Fig. 6b shows the conversion rate of the hydrogenation of a model compound, propargyl acetate (PA), to allyl acetate (AA). As in the case of VA, the yields of AA with 2–5 were uniformly higher than that with 1. In reactions with 2–5, double hydrogenation occurred to afford n-propyl acetate (n-PA), suggesting the stronger reductive ability of these rhodium catalysts. Thus, we demonstrated PHIP experiments of PA and observed the same or slightly higher level of hyperpolarization at the carbonyl carbon (Fig. 6c). Consequently, PHIP with catalyst 4 resulted in 13C NMR signals that were about four times higher than with catalyst 1 (Fig. 6d and e). | ||
| Fig. 6 (a) Conditions of PHIP experiment of PA. (b) Conversion rate of PA to AA and n-PA. (c) Polarization level % P13C at carbonyl carbon of AA and n-PA. (d) 13C NMR spectrum of natural abundant AA (ca. 35 mM) in thermal equilibrium state (30° pulse, 1 scan). (e) 13C NMR spectra (30° pulse, 1 scan) of AA and n-PA produced upon PHIP (initial concentration of PA 50 mM) with catalyst 1 or 4. S/N means signal to noise ratio with standard deviation (n = 3). PHIP experiments were conducted under the following conditions: 20 mL min−1 parahydrogen (1 atm) was bubbled for 10 s into 700 μL of methanol-d4 containing 2.5 mM catalyst and 50 mM PA, followed by polarization transfer. | ||
Hyperpolarization using flow system
Finally, we tried to apply these rhodium catalysts to flow hydrogenation systems23 to achieve continuous acquisition of hyperpolarized samples in high concentration. We conducted PHIP-SAH of 1 M VA in methanol using a flow reactor in a combined phase of 100 mL min−1 parahydrogen and 3 mL min−1 substrate in methanol, as illustrated in Fig. 7. When using one of the modified catalysts 4, the PHIP experiment yielded a high concentration of EA (ca. 300 mM) with a polarization level of ∼0.7%. Because of the slightly longer operation time, in comparison with the batch reaction system, the polarization level % P13C at the carbonyl carbon of EA was slightly lower. Further improvements of a flow reactor, specifically in terms of a shorter reaction pathway, would be helpful in producing large amounts of product with higher polarization efficiency. | ||
| Fig. 7 (a) Illustration of flow hydrogenation system used in this study. (b) Reaction conditions for PHIP of VA in (a). | ||
Conclusions
In conclusion, we investigated structural modification of the conventional rhodium catalyst 1 for PHIP experiments based on a screening approach of organometallic chemistry. We found that the modified catalyst candidates 2–5 with electron-donating bisphosphine ligands, with a dicyclohexylphosphino group, resulted in faster hydrogenation, with the same polarization level, under our experimental conditions (50 °C, parahydrogen 1 atm, continuous parahydrogen bubbling), thus leading to higher 13C NMR signals. To further demonstrate the utility of these catalysts, we used them in PHIP-SAH experiments of VA in liquid–gas two-phase flow systems. Here, we recorded the production of hyperpolarized EA in significantly high concentration (ca. 300 mM). It should be noted that in this study we evaluated and optimized ligands based on the catalytic hydrogenation activity. Further screening study focusing on hyperpolarization efficiency, especially retention of para spin during the catalytic reaction, would allow finding better catalysts for PHIP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CREST (JPMJCR13L4), Japan Science and Technology Agency. We thank Dr F. Reineri of the University of Torino for experimental support and helpful discussions. We also thank Prof. Dr K. Nozaki and Dr S. Kusumoto of the University of Tokyo for their kind donation of phosphine ligands and carrying out NMR measurement.Notes and references
- (a) A. Viale and S. Aime, Curr. Opin. Chem. Biol., 2010, 14, 90 CrossRef CAS PubMed; (b) A. Viale, F. Reineri, D. Santelia, E. Cerutti, S. Ellena, R. Gobetto and S. Aime, Q. J. Nucl. Med. Mol. Imaging, 2009, 53, 604 CAS.
- A. Bornet, R. Melzi, A. J. P. Linde, P. Hautle, B. van den Brandt, S. Jannin and G. Bodenhausen, J. Phys. Chem. Lett., 2013, 4, 111 CrossRef CAS PubMed.
- (a) M. G. Pravica and D. P. Weitekamp, Chem. Phys. Lett., 1988, 145, 255 CrossRef CAS; (b) C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541 CrossRef CAS; (c) T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft and R. Eisenberg, J. Am. Chem. Soc., 1987, 109, 8089 CrossRef CAS; (d) C. R. Bowers and D. P. Weitekamp, Chem. Phys. Lett., 1986, 57, 2645 CAS.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708 CrossRef CAS.
- Recent efforts for designing long T1 structures, see for example: (a) Y. Imakura, H. Nonaka, Y. Takakusagi, K. Ichikawa, N. R. Maptue, A. M. Funk, C. Khemtong and S. Sando, Chem.–Asian J., 2018, 13, 280 CrossRef CAS PubMed; (b) H. Nonaka, M. Hirano, Y. Imakura, Y. Takakusagi, K. Ichikawa and S. Sando, Sci. Rep., 2017, 7, 40104 CrossRef CAS PubMed; (c) H. Nonaka, R. Hata, T. Doura, T. Nishihara, K. Kumagai, M. Akakabe, M. Tsuda, K. Ichikawa and S. Sando, Nat. Commun., 2013, 4, 2411 CrossRef PubMed; (d) T. Doura, R. Hata, H. Nonaka, K. Ichikawa and S. Sando, Angew. Chem., Int. Ed., 2012, 51, 10114 CrossRef CAS PubMed.
- (a) E. Cavallari, C. Carrera and F. Reineri, Isr. J. Chem., 2017, 57, 833 CrossRef CAS; (b) J. B. Hçvener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Glçggler, K. V. Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier, A. Jerschow, F. Reineri, T. Theis, R. V. Shchepin, S. Wagner, P. Bhattacharya, N. M. Zacharias and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2018, 57, 11140 CrossRef PubMed.
- PHIP using heterogeneous catalysts were also reported, see for example: (a) I. V. Koptyug, K. V. Kovtunov, S. R. Burt, M. S. Anwar, C. Hilty, S.-I. Han, A. Pines and R. Z. Sagdeev, J. Am. Chem. Soc., 2007, 129, 5580 CrossRef CAS PubMed; (b) S. Glçggler, A. M. Grunfeld, Y. N. Ertas, J. McCormick, S. Wagner, P. P. M. Schleker and L.-S. Bouchard, Angew. Chem., Int. Ed., 2015, 54, 2452 CrossRef PubMed; (c) E. W. Zhao, H. Zheng, R. Zhou, H. E. Hagelin-Weaver and C. R. Bowers, Angew. Chem., Int. Ed., 2015, 54, 14270 CrossRef CAS PubMed.
- (a) R. V. Shchepin, D. A. Barskiy, A. M. Coffey, I. V. Manzanera Esteve and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2016, 55, 6071 CrossRef CAS PubMed; (b) K. V. Kovtunov, D. A. Barsliy, R. V. Shchepin, A. M. Coffey, K. W. Waddell, I. V. Koptyug and E. Y. Chekmenev, Anal. Chem., 2014, 86, 6192 CrossRef CAS PubMed.
- (a) M. Roth, Sensitivity enhancement in NMR by using parahydrogen induced polarization, PhD thesis, Johannes Gutenberg-Universität Mainz, 2010; (b) R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, J. Phys. Chem. Lett., 2012, 3, 3281 CrossRef CAS PubMed.
- (a) F. Reineri, T. Boi and S. Aime, Nat. Commun., 2015, 6, 5858 CrossRef CAS PubMed; (b) N. V. Chukanov, O. G. Salnikov, R. V. Shchepin, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, ACS Omega, 2018, 3, 6673 CrossRef CAS PubMed; (c) O. G. Salnikov, R. V. Shchepin, N. V. Chukanov, L. Jaigirdar, W. Pham, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, J. Phys. Chem. C, 2018, 122, 24740 CrossRef CAS.
- K. Golman, R. I. Zandt and M. Thaning, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11270 CrossRef CAS PubMed.
- D. Heller, S. Barns, W. Baumann and R. Selke, Chem. Ber., 1996, 129, 85 CrossRef CAS.
- (a) H.-J. Drexler, W. Baumann, A. Spannenberg, C. Fisher and D. Heller, J. Organomet. Chem., 2001, 621, 89 CrossRef CAS; (b) H. G. Nedden, WO Pat., 2010001173A1, 2010.
- Activities among catalysts 2–5 could not be differentiated with sufficient statistical significances. Therefore, precise comparison among catalysts 2–5 is not discussed in this research..
- F. Reineri, S. Aime, R. Gobetto and C. Nervi, J. Chem. Phys., 2014, 140, 094307 CrossRef CAS PubMed.
- R. A. Sánchez-Delgado and M. Rosales, Coord. Chem. Rev., 2000, 196, 249 CrossRef.
- (a) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183 CrossRef CAS; (b) I. D. Gridnev, Y. Yamanoi, N. Higashi, H. Tsuruta, M. Yasutake and T. Imamoto, Adv. Synth. Catal., 2001, 343, 118 CrossRef CAS; (c) T. Imamoto, K. Tamura, Z. Zhang, Y. Horiuchi, M. Sugiya, K. Yoshida, A. Yanagisawa and I. D. Gridnev, J. Am. Chem. Soc., 2012, 134, 1754 CrossRef CAS PubMed; (d) I. D. Gridnev and T. Imamoto, Acc. Chem. Res., 2004, 37, 633 CrossRef CAS PubMed; (e) I. D. Gridnev and T. Imamoto, Chem. Commun., 2009, 7447 RSC; (f) T. Imamoto, Chem. Rec., 2016, 16, 2659 CrossRef PubMed.
- J. M. Brown, L. R. Canning, A. J. Downs and A. M. Forster, J. Organomet. Chem., 1983, 255, 103 CrossRef CAS.
- K. Tani, T. Yamagata, Y. Tatsuno, T. Saito, Y. Yamagata and N. Yasuoka, J. Chem. Soc., Chem. Commun., 1986, 494 RSC.
- E. Cavallari, C. Carrera, S. Aime and F. Reineri, Chem.–Eur. J., 2017, 23, 1200 CrossRef CAS PubMed.
- PHIP experiments with the conventional neutral rhodium complex such as RhCl(PPh3)3 (Wilkinson's catalyst) showed poor polarization levels because the reversible addition of parahydrogen was included, see: R. U. Kirss and R. Eisenberg, J. Organomet. Chem., 1989, 359, C22 CrossRef CAS.
- Under high pressure PHIP in a batch system, relatively lower % P13C values were observed for cat 4 (data not shown) than for cat 1. This may be attributed to ortho–para conversion in the ‘Hydride route’..
- Examples of PHIP experiments using flow systems, see: (a) L.-S. Bouchard, S. R. Burt, M. S. Anwar, K. V. Kovtunov, I. V. Koptyug and A. Pines, Science, 2008, 319, 442 CrossRef CAS PubMed; (b) V.-V. Telkki, V. V. Zhivonitko, S. Ahola, K. V. Kovtunov, J. Jokisaari and I. V. Koptyug, Angew. Chem., Int. Ed., 2010, 49, 8363 CrossRef CAS PubMed; (c) K. V. Kovtunov, V. V. Zhivonitko, I. V. Skovpin, D. A. Barskiy, O. G. Salnikov and I. V. Koptyug, J. Phys. Chem. C, 2013, 117, 22887 CrossRef CAS; (d) D. A. Barskiy, O. G. Salnikov, K. V. Kovtunov and I. V. Koptyug, J. Phys. Chem. A, 2015, 119, 996 CrossRef CAS PubMed; (e) O. G. Salnikov, K. V. Kovtunov and I. V. Koptyug, Sci. Rep., 2015, 5, 13930 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02580d |
| This journal is © The Royal Society of Chemistry 2019 |