
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh(III)-catalyzed C-7 arylation of indolines with arylsilanes via C–H activation†
Haiqing Luo *,
Qi Xie,
Kai Sun,
Jianbo Deng,
Lin Xu,
Kejun Wang and
Xuzhong Luo*
*,
Qi Xie,
Kai Sun,
Jianbo Deng,
Lin Xu,
Kejun Wang and
Xuzhong Luo*
Department of Chemistry & Chemical Engineering, Gannan Normal University, Ganzhou 341000, China. E-mail: luohaiq@sina.com; luoxuzhong@hotmail.com
First published on 10th June 2019
Abstract
Site-selective synthesis of C-7 arylated indolines has been achieved via oxidative arylation of indolines with arylsilanes under Rh(III)-catalyzed C–H activation of indolines by using CuSO4 as a co-oxidant. This transformation has been explored for a wide range of substrates under mild conditions.
The indole and indoline ring systems represent ubiquitous structural motifs as natural alkaloids and biologically active compounds as well as in functional materials.1,2 With the development of C–H bond functionalization,3 a great deal of effort has been devoted to the formation C-2 and C-3 functionalized indoles.4 Nevertheless, only a few methods have been explored for the direct C–H bond functionalization of indoles at the C-7 position,5 in which the unique work for the direct Pd-catalyzed C–H arylation of indoles with arylboronic acids at the C-7 position was developed by the Shi group using a designed di-tert-butylphosphine oxide (TBPO) directing-group.5b In general, the C-7 arylated indolines can be conveniently oxidized to transform into C-7 arylated indoles. So far, transition metals such as Pd,6 Rh,7 Ru,8 Ir,9 etc.,10 have been employed as catalysts for the C-7 C–H bond functionalization of indolines, among which the C–H arylation in the C-7 position of indoline was mainly established by using Pd complexes as catalysts (Scheme 1a).11 In 2016, we also reported the Pd-catalyzed C-7 arylation of indolines with arylsilanes via C–H bond activation.12 Recently, the Punniyamurthy group developed the site-selective C-7-arylation of indolines with arylboronic acids by using low-cost and earth-abundant cobalt(II)-PCy3 as a catalyst.13 Even so, in view of the importance of indole and indoline scaffolds, it is still highly desirable to develop more convenient and efficient approaches for the synthesis of C-7 arylated indolines and C-7 arylated indoles.
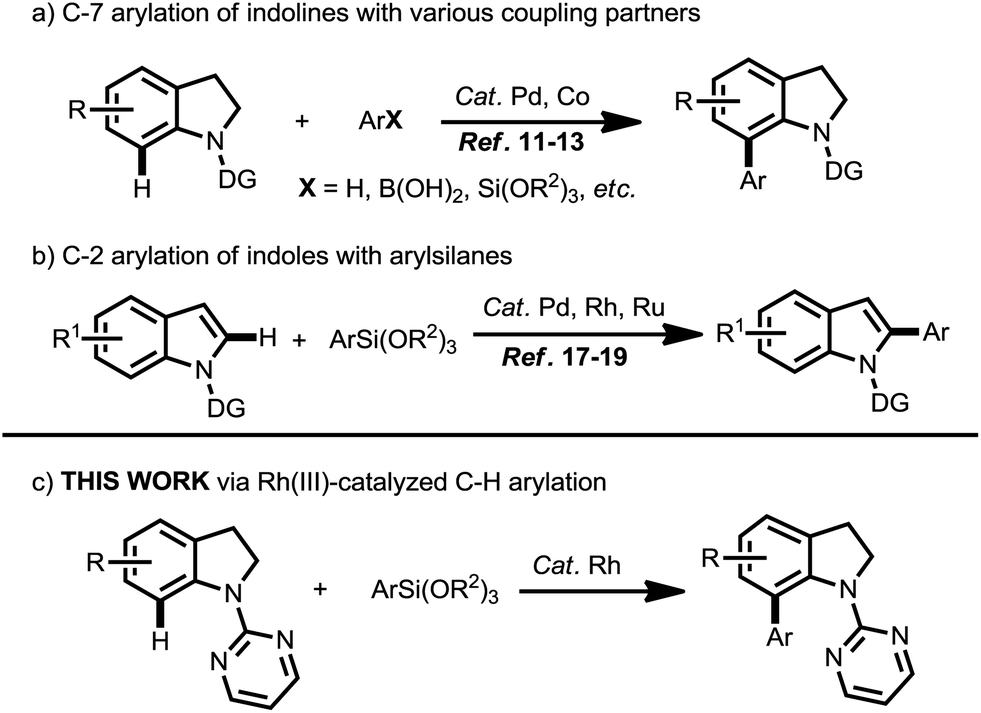 | ||
| Scheme 1 C-2 arylation of indoles with arylsilanes and C-7 arylation of indolines. | ||
Meanwhile, recent years have witnessed tremendous developments in the area of Rh(III)-catalyzed C–H bond functionalization,14 which has allowed effective construction of various C–C bonds. The major advantages of Rh(III) catalysis are (i) high functional-group tolerance, (ii) broad substrate scope, (iii) low catalyst loading, high activity, and high catalytic efficiency.
Since the Hiyama cross-coupling reaction had been first reported in 1988,15 as one of the most useful and reliable synthetic methods for the construction C–C bonds, the Hiyama cross-coupling reaction has drawn increasing attention.16 As attractive organometallic coupling partners with many unique advantages such as low toxicity, high stability and environmental benignity, organosilicon reagents were used as coupling partners for the C–H arylation of indoles and indolines (Scheme 1b).17 Zhang group reported the Pd-catalyzed C-2 arylation of indoles with arylsilanes in acidic medium in 2010. In 2014, Loh group developed the Rh(III) catalyzed C-2 C–H arylation of indoles with arylsilanes in aqueous media.18 Very recently, Szostak group developed the Ru(II) catalyzed arylation of indoles with arylsilanes via C–H activation,19 it is worth noting that the reaction was carried out in water. However, the methods for the synthesis of C-7 arylated indolines and C-7 arylated indoles are very limited by using arylsilanes as the coupling partners. To the best of our knowledge, the Rh(III) catalyzed C-7 arylation of indolines with arylsilanes has not been achieved. Due to our continuous interest in the arylsilanes-based coupling reaction,12,20 we herein would like to report a Rh(III)-catalyzed regioselective C-7 arylation of indolines with arylsilanes via C–H activation (Scheme 1c), the reaction provides a facile access to C-7 arylated indolines.
At the outset of the investigation, various N-protecting indolines (Table 1, 1a–1f) were initially used as coupling partners for the direct C-7 C–H arylation with phenyl-triethoxysilane 2a in the presence of 1 mol% of [Cp*RhCl2]2 in dioxane at 80 °C, 3.0 equiv. of AgF as the activator for C–Si bond as well as the oxidant, and 2.0 equiv of Cu(OAc)2 as the co-oxidant. The choice of the N-protecting group was found to be crucial for this arylation, and only pyrimidyl-protected indoline (1f) afforded desired arylated product 3fa. Then, different fluorine sources such as AgF, CsF, KF, and TBAF were examined (Table 1, entries 3–6). Only AgF was found to be effective, thus affording the desired product 3fa. The reaction can afford the desired product in 72% yield by using no co-oxidant (entry 7). In order to increase the yield, various co-oxidants such as Ag2CO3, Ag2O, Cu(OAc)2 and CuSO4 were utilized (entries 8–10), CuSO4 proved to be the optimal choice. Subsequently, the effect of solvent was then investigated (entries 10–15), and dioxane was found to be suitable for the reaction. To our surprise, this reaction could be performed in aqueous media (dioxane/H2O = 1/10) in 47% yield (Table 1, entry 15). Furthermore, in order to gain the milder reaction condition, the reaction temperature was tried to reduce. The results revealed that the reaction carried out at 60 °C offered a relatively low yield in 87% (Table 1, entry 16).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrates | Co-oxidant | F source | Solvent | Yieldb (%) |
| a Unless otherwise noted, the reaction conditions are as follows: 1 (0.3 mmol), 2a (0.9 mmol), [Cp*Rh(III)Cl2]2 (1 mol%), Co-oxidant (0.6 mmol), F resource (0.9 mmol), solvent (3.0 mL).b Isolated yield after purification by flash column chromatography on silica gel, n.d.p or trace product was determined by TLC.c The reaction temperature was 60 °C. | |||||
| 1 | 1a–1d | Ag2CO3 | AgF | Dioxane | 0 |
| 2 | 1e | Ag2CO3 | AgF | Dioxane | 0 |
| 3 | 1f | Ag2CO3 | AgF | Dioxane | 81 |
| 4 | 1f | Ag2CO3 | CsF | Dioxane | 0 |
| 5 | 1f | Ag2CO3 | KF | Dioxane | 0 |
| 6 | 1f | Ag2CO3 | TBAF | Dioxane | 0 |
| 7 | 1f | — | AgF | Dioxane | 72 |
| 8 | 1f | AgOAc | AgF | Dioxane | 89 |
| 9 | 1f | Cu(OAc)2 | AgF | Dioxane | 85 |
| 10 | 1f | CuSO4 | AgF | Dioxane | 98 |
| 11 | 1f | CuSO4 | AgF | DMF | 62 |
| 12 | 1f | CuSO4 | AgF | DMSO | 0 |
| 13 | 1f | CuSO4 | AgF | iPrOH | 81 |
| 14 | 1f | CuSO4 | AgF | H2O | Messy |
| 15 | 1f | CuSO4 | AgF | Dioxane/H2O = 1/10 | 47 |
| 16c | 1f | CuSO4 | AgF | Dioxane | 87 |

With the optimized reaction conditions in hand, we then proceeded to explore the scope of the direct C-7 C–H arylation of N-(2-pyrimidyl)-indoline with a series of arylsilanes (Table 2). It was found that various phenylsilanes had good compatibility under these reaction conditions. At first, phenyltrimethoxy-silane shows good reactivity affording 67% yield. To investigate the steric effect, the phenyltriethoxysilanes bearing methyl group on different positions of the phenyl ring were employed as the substrates (3fb–3fd). The results indicate that the phenyltriethoxysilane with ortho-methyl substituent was found to afford the desired product for the diminished yield in 33%, which could be due to the steric effect. The direct C–H arylation of N-(2-pyrimidyl)-indoline has shown excellent tolerance to both electron-withdrawing and electron-donating groups as aromatic substituents, including alkyl (3fb–3fe), alkyloxyl (3ff–3fh), aryl (3fi), fluoro (3fj), chloro (3fk), and trifluoromethyl groups (3fl) in good to excellent yields except the ortho-methyl substituent (3fd). In addition, the heterocyclic triethoxy(thiophen-2-yl)silane was also used to explore the possibility of the C–H arylation. To our delight, the reaction also afforded the corresponding product in moderate yield (3fm).


To further evaluate the substrate scope, various substituted N-(2-pyrimidyl)-indolines were used to test the reaction with the phenyltriethoxysilane 2a under the optimized reaction conditions. The results are summarized in Table 3. In general, the (2-pyrimidyl)-indolines bearing electron-donating groups on the aromatic ring such as 5-methyl and 5-alkyloxyl group react smoothly to afford the corresponding arylated products in excellent yields (4ga–4ha). In addition, the reactions with the (2-pyrimidyl)-indolines bearing electron-withdrawing group on the aromatic ring also afford moderate to good yield (4ja–4oa), except the substrate 1n with 6-bromo group showed low activity affording 17% yield, which may be attributed to the steric hindrance of the bromo functionality. Furthermore, the substrates with C-2 or C-3 methyl substituted group on the indoline ring also show excellent compatibility with the reaction condition in 86% (4pa) and 90% yield (4qa), respectively.


To explore the utility of this transformation, we tried to explore direct C-8 C–H arylation with N-(2-pyrimidyl)-tetrahydroquinoline 5 and phenyltrimethoxy-silane 2a under the standard condition, to our delight, the desired C-8 C–H arylated product was obtained in 48% yield (Scheme 2).
 | ||
| Scheme 2 Rh(III)-catalyzed the direct C-8 arylation of N-(2-pyrimidyl)-tetrahydroquinoline 5 with phenyltriethoxysilane 2a. | ||
To further demonstrate the synthetic utility of the method, the transformation of C-7 arylated indoline into C-7 arylated indole was studied.13,21 As shown in Scheme 3, the transformation was begun with the oxidation of C-7 arylated N-(2-pyrimidyl)-indoline 3fa by the use of DDQ (2,3-dicyano-5,6-dichlorobenzoquinone), followed by removing pyrimidyl group in the present of base. Finally, 83% yield of the C-7arylated indole product 7a was successfully obtained.
 | ||
| Scheme 3 Transformation of C-7-arylated indoline 3fa to the corresponding indole 7a. | ||
To gain some preliminary mechanistic insights, kinetic isotope experiments were conducted. The parallel reactions of 1f and deuterio-1f with 2a resulted kH/KD = 0.80 (Scheme 4), which suggested that the C–H cleavage was not the rate-determining step in the catalytic cycle. On the basis of previous literatures, 18,22,23 a plausible mechanism is proposed as shown Scheme 5. The process is likely to be initiated by the coordination of the nitrogen atom of 2-pyrimidyl group of 1f to the rhodium catalyst, leading directly to cyclometalation process via C–H bond activation to afford the five-membered rhodacycle A.24 The Ph–Ag species can be generated via the C–Si activation by the nucleophilic attack of a fluoride ion on silicon,20c,25 followed by the transmetalation with intermediate A to afford the Rh(III) intermediate B, from which reductive elimination would provide C-7 arylated indolines 3fa and regenerate the Rh(I) catalyst, which is reoxidized to Rh(III) species by Ag(I) or Cu(II) salts to complete the catalytic cycle.
 | ||
| Scheme 4 Parallel kinetic isotope experiment. | ||
 | ||
| Scheme 5 Plausible catalytic cycle for Rh(III)-catalyzed the direct C-7 arylation of indolines with phenyltriethoxysilane. | ||
In summary, we have demonstrated a Rh(III)-catalyzed oxidative arylation of indolines with arylsilanes via C–H activation by using CuSO4 as an co-oxidant. This general transformation exhibits excellent reactivity and broad substrate scopes, various functional groups are well tolerated under the mild reaction conditions. This reaction constitutes a complement method for the synthesis the desired C-7 arylated indolines up to excellent yields, which can be conveniently transformed into C-7 arylated indoles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was supported by the National Natural Science Foundation of China (No. 21762002, 21562003) and Undergraduate Training Programs for Innovation and Entrepreneurship of Gannan Normal University (CX170049).Notes and references
- (a) J. S. Mason, I. Morize, P. R. Menard, D. L. Cheney, C. Hulme and R. F. Labaudiniere, J. Med. Chem., 1999, 42, 3251 CrossRef CAS PubMed; (b) K. C. Nicolaou, J. A. Pfefferkorn, A. J. Roecker, G. Cao, S. Barluenga and H. J. Mitchell, J. Am. Chem. Soc., 2000, 122, 9939 CrossRef CAS; (c) J. D. Podoll, Y. Liu, L. Chang, S. Walls, W. Wang and X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15573 CrossRef CAS PubMed.
- (a) U. Pindur and T. Lemster, Curr. Med. Chem., 2001, 8, 1681 CrossRef CAS PubMed; (b) H. Knçlker and K. R. Reddy, Chem. Rev., 2002, 102, 4303 CrossRef; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (d) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (e) M. G. Bell, D. L. Gernert, T. A. Grese, M. D. Belvo, P. S. Borromeo, S. A. Kelley, J. H. Kennedy, S. P. Kolis, P. A. Lander and R. Richey, J. Med. Chem., 2007, 50, 6443 CrossRef CAS PubMed; (f) T. Owa, A. Yokoi, K. Yamazaki, K. Yoshimatsu, T. Yamori and T. Nagasu, J. Med. Chem., 2002, 45, 4913 CrossRef CAS PubMed.
- For selected reviews on C–H bond functionalization, see: (a) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maesa and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (b) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (c) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864 CrossRef CAS PubMed; (d) J. Wencel-Delord, T. Drçge, F. Kiu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (e) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., 2012, 124, 10382 (Angew. Chem., Int. Ed., 2012, 51, 10236) CrossRef; (f) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412 CrossRef CAS PubMed.
- For selected reviews, see: (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (b) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673 CrossRef CAS; (c) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85 CrossRef CAS PubMed; (d) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215 CrossRef PubMed.
- (a) D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068 CrossRef CAS PubMed; (b) Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495 CrossRef CAS PubMed; (c) L. Xu, C. Zhang, Y. He, L. Tan and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 321 CrossRef CAS PubMed.
- (a) L.-Y. Jiao and M. Oestreich, Org. Lett., 2013, 15, 5374 CrossRef CAS PubMed; (b) D. Yang, S. Mao, Y.-R. Gao, D.-D. Guo, S.-H. Guo, B. Li and Y.-Q. Wang, RSC Adv., 2015, 5, 23727 RSC; (c) M. Kim, N. K. Mishra, J. Park, S. Han, Y. Shin, S. Sharma, Y. Lee, E.-K. Lee, J. H. Kwak and I. S. Kim, Chem. Commun., 2014, 50, 14249 RSC.
- (a) X.-F. Yang, X.-H. Hu, C. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 2532 RSC; (b) X.-F. Yang, X.-H. Hu and T.-P. Loh, Org. Lett., 2015, 17, 1481 CrossRef CAS PubMed; (c) S. H. Han, M. Choi, T. Jeong, S. Sharma, N. K. Mishra, J. Park, J. S. Oh, W. J. Kim, J. S. Lee and I. S. Kim, J. Org. Chem., 2015, 80, 11092 CrossRef CAS PubMed; (d) M. Jeon, N. K. Mishra, U. De, S. Sharma, Y. Oh, M. Choi, H. Jo, R. Sachan, H. S. Kim and I. S. Kim, J. Org. Chem., 2016, 81, 9878 CrossRef CAS PubMed; (e) N. K. Mishra, M. Jeon, Y. Oh, H. Jo, J. Park, S. Han, S. Sharma, S. H. Han, Y. H. Jung and I. S. Kim, Org. Chem. Front., 2017, 4, 241 RSC.
- (a) C. S. Yi, S. Y. Yunand and I. A. Guzei, J. Am. Chem. Soc., 2005, 127, 5782 CrossRef CAS PubMed; (b) A. Mishra, T. K. Vats, M. P. Nair, A. Das and I. Deb, J. Org. Chem., 2017, 82, 12406 CrossRef CAS PubMed; (c) P. B. De, S. Banerjee, S. Pradhan and T. Punniyamurthy, Org. Biomol. Chem., 2018, 16, 5889 RSC; (d) C. Pan, A. Abdukader, J. Hang, Y. Cheng and C. Zhu, Chem. –Eur. J., 2014, 20, 3606 CrossRef CAS PubMed.
- (a) S. Pan, T. Wakaki, N. Ryu and T. Shibata, Chem. –Asian J., 2014, 9, 1257 CrossRef CAS PubMed; (b) Y. Kim, J. Park and S. Chang, Org. Lett., 2016, 18, 1892 CrossRef CAS PubMed; (c) Y. Wu, Y. Yang, B. Zhou and Y. Li, J. Org. Chem., 2015, 80, 1946 CrossRef CAS PubMed.
- See review: A. S. Tariq, P. B. De, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2019, 55, 572 RSC , and references cited therein.
- (a) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed; (b) Z. Shi, B. Li, X. Wan, J. Cheng, Z. Fang, B. Cao, C. Qin and Y. Wang, Angew. Chem., Int. Ed., 2007, 46, 5554 CrossRef CAS PubMed; (c) T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 4978 CrossRef CAS PubMed; (d) L. Y. Jiao and M. Oestreich, Eur. J. Org. Chem., 2013, 19, 10845 CrossRef CAS PubMed; (e) L. Y. Jiao, P. Smirnov and M. Oestreich, Org. Lett., 2014, 16, 6020 CrossRef CAS PubMed.
- H. Luo, H. Liu, Z. Zhang, Y. Xiao, S. Wang, X. Luo and K. Wang, RSC Adv., 2016, 6, 39292 RSC.
- P. B. De, S. Pradhan, S. Banerjee and T. Punniyamurthy, Chem. Commun., 2018, 54, 2494 RSC.
- For reviews on Rh(III)-catalyzed C–H activation, see: (a) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS PubMed; (b) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (c) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (d) T. Satoh and M. Miura, Chem. –Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (e) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (f) C. Dutta and J. Choudhury, RSC Adv., 2018, 8, 27881 RSC.
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS.
- (a) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631 CrossRef CAS; (b) S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982 CrossRef CAS PubMed; (c) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (d) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486 CrossRef CAS PubMed.
- Z. J. Liang, B. B. Yao and Y. H. Zhang, Org. Lett., 2010, 12, 3185 CrossRef CAS PubMed.
- M. Z. Lu, P. Lu, Y. H. Xu and T. P. Loh, Org. Lett., 2014, 16, 2614 CrossRef CAS PubMed.
- P. Nareddy, F. Jordan and M. Szostak, Org. Lett., 2018, 20, 341 CrossRef CAS PubMed.
- (a) H. Luo, Z. Zhang, H. Liu and H. Liu, Chin. J. Org. Chem., 2015, 35, 802 CrossRef CAS; (b) H. Luo, H. Liu, X. Chen, K. Wang, X. Luo and K. Wang, Chem. Commun., 2017, 53, 956 RSC; (c) H. Luo and W. Dong, Synth. Commun., 2013, 43, 2733 CrossRef CAS.
- P. Gandeepan, J. Koeller and L. Ackermann, ACS Catal., 2017, 7, 1030 CrossRef CAS.
- K. Cheng, H. Li, Y. Li, X.-S. Zhang, Z.-Q. Lei and Z.-J. Shi, Chem. Sci., 2012, 3, 1645 RSC.
- S. D. Yang, B. J. Li, X. B. Wan and Z. J. Shi, J. Am. Chem. Soc., 2007, 129, 6066 CrossRef CAS PubMed.
- H. Li, J. Jie, S. Wu, X. Yang and H. Xu, Org. Chem. Front., 2017, 4, 250 RSC.
- A. Sugiyama, Y. Ohnishi, M. Nakaoka, Y. Nakao, H. Sato, S. Sakaki, Y. Nakao and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 12975 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04142g |
| This journal is © The Royal Society of Chemistry 2019 |