DOI:
10.1039/C9RA02296A
(Paper)
RSC Adv., 2019,
9, 17631-17638
Green synthesis of benzonitrile using ionic liquid with multiple roles as the recycling agent†
Received
26th March 2019
, Accepted 18th May 2019
First published on 5th June 2019
Abstract
Preparation of benzonitrile from benzaldehyde and hydroxylamine hydrochloride is one of the most advantageous approaches. Nevertheless, it suffers from various constraints such as longer reaction time, corrosion and recovery of hydrochloric acid, the use of metal salt catalysts and their separation. For these reasons, a novel green benzonitrile synthetic route was proposed with ionic liquid as the recycling agent in this study. The results indicated that hydroxylamine 1-sulfobutyl pyridine hydrosulfate salt ((NH2OH)2·[HSO3-b-Py]·HSO4) was an expert alternative to hydroxylamine hydrochloride. Meanwhile, the ionic liquid [HSO3-b-Py]·HSO4 exhibited the multiple roles of co-solvent, catalysis and phase separation, thus the use of metal salt catalyst was eliminated, and no additional catalyst was needed. Hence, the separation process was greatly simplified. When the molar ratio of benzaldehyde to (NH2OH)2·[HSO3-b-Py]·HSO4 was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5, the volume ratio of paraxylene to [HSO3-b-Py]·HSO4 was 2:1, the benzaldehyde conversion and benzonitrile yield were both 100% at 120 °C in 2 h. Even better, the ionic liquid could be recovered easily by phase separation, and recycled directly after reaction. Additionally, this novel route is applicable to the green synthesis of a variety of aromatic, heteroaromatic and aliphatic nitriles with excellent yields.
:1.5, the volume ratio of paraxylene to [HSO3-b-Py]·HSO4 was 2:1, the benzaldehyde conversion and benzonitrile yield were both 100% at 120 °C in 2 h. Even better, the ionic liquid could be recovered easily by phase separation, and recycled directly after reaction. Additionally, this novel route is applicable to the green synthesis of a variety of aromatic, heteroaromatic and aliphatic nitriles with excellent yields.
Introduction
Benzonitrile is a versatile chemical that plays key roles in the synthesis of benzoic acid, benzylamine, benzamide, pesticides and dyes.1 It is also a prominent intermediate for the production of benzoguanamine,2 which is a widely used advanced coating. Classical methods for benzonitrile synthesis are the cyanation of benzene halides,3,4 toluene halides and ammonia reaction,5 ammoxidation of toluene, ammonia and air,6–8 benzoic acid and urea reaction,9 and the cyanidation of benzaldehyde.10–13 Among them, preparation of benzonitrile from benzaldehyde and hydroxylamine hydrochloride (NH2OH·HCl) is one of the most interesting and advantageous approaches due to the constant carbon number in the reactant and final product, mild reaction conditions, low production cost and prospect for industrial-scale application.14
The transformation of benzaldehyde and NH2OH·HCl to benzonitrile can be grouped into two-step and one-pot methods.15 For the two-step reaction, it is carried out to form benzaldoxime, and then the isolated benzaldoxime is dehydrated to benzonitrile.15,16 Both of the two steps can be catalyzed by acid.17,18 As for the one-pot route, it includes oximation of benzaldehyde to benzaldoxime, followed by the dehydration of in situ generated benzaldoxime to benzonitrile.19 Obviously, the one-pot reaction helps to reduce the energy consumption, solvent waste and reaction time, which in turn, substantially avoids the isolation of benzaldoxime. It is a valuable strategic way in terms of economic and environmental aspects.14 Owing to these advantageous features, several studies for the one-pot synthesis of benzonitrile have been conducted in recent years.
Enthaler et al.20 presented a direct conversion of benzaldehyde to benzonitrile catalyzed by Zn(CH3COO)2 (5 mol%). When benzaldehyde was 0.72 mmol, n(benzaldehyde):n(NH2OH·HCl) = 1:1.2, toluene was 2 mL, reaction temperature was 100 °C, reaction time was 24 h, the benzonitrile yield was 79.0%. Zhuang et al.21 studied SnCl2·2H2O (10 mol%) catalyzed transformation of benzaldehyde to benzonitrile. When benzaldehyde was 1 mmol, n(benzaldehyde):n(NH2OH·HCl) = 1:1.1, CH3CN was 0.5 mol, NaHCO3 was 1 mmol, the benzonitrile yield was 80.0% at 80 °C within 24 h. Leggio et al.22 investigated the catalytic properties of TiCl4. When benzaldehyde was 1 mmol, n(benzaldehyde):n(NH2OH·HCl) = 1:2, pyridine was 2 mL, TiCl4 was 2.5 mmol, the benzonitrile yield was 85.0% at 40 °C in 3 h. Wang et al.19 reviewed the synthesis of benzonitrile catalyzed by NaHSO4·H2O. Under the optimized conditions of molar ratio of n(benzaldehyde):n(NH2OH·HCl) = 1:1.4, and reaction at 135 °C for 4.5 h in the presence of tetrabutylammonium bromide as phase transfer catalyst and DMF as solvent, benzonitrile was obtained in excellent yield of 92.8%. In addition to homogeneous catalysts, heterogeneous catalysts were also reported. Fe3O4-CTAB NPs (1.8 mol%) was applied as a competent catalyst for one-pot synthesis of benzonitrile.14 The combination of benzaldehyde (0.5 mmol), NH2OH·HCl (0.75 mmol), Fe3O4-CTAB NPs (1.8 mol%), and DMF (5 mL) at 80–90 °C for 1 h were found to be the optimized reaction conditions for the desired transformation, with the benzonitrile yield being 97.0%. A green chitosan supported magnetic ionic liquid (CSMIL) was prepared for direct conversion of benzaldehyde to benzonitrile.23 When benzaldehyde was 1 mmol, NH2OH·HCl was 1.2 mmol, dry-CSMIL was 15 mg, MeSO2Cl was 1.2 mmol, the benzonitrile yield was 93.0% (70 °C, 1.5 h). It can be seen that benzonitrile could be efficiently synthesized from benzaldehyde and NH2OH·HCl. However, it suffers from various constrains such as longer reaction time, use of metal salt catalysts and inconvenient post treatment.24,25 Furthermore, the using of NH2OH·HCl will inevitably lead to problems of equipment corrosion and environmental pollution because it releases HCl.
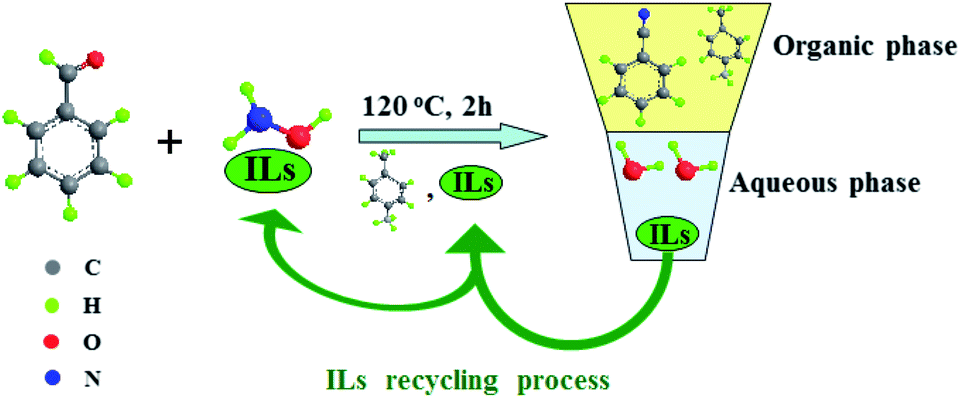
Recently, a series of ionic liquids (ILs) were used as alternatives to conventional inorganic acids in hydroxylamine stabilization, leading to the formation of several eco-friendly hydroxylamine ionic liquid salts ((NH2OH)2·ILs) that exhibited better reactivity in one-step clean synthesis of caprolactam, phenol and aniline in comparison with NH2OH·HCl and (NH2OH)2·H2SO4.26–28 In continuation of our work for application of these (NH2OH)2·ILs, a green and high efficient reaction route was developed for direct synthesis of benzonitrile from benzaldehyde and (NH2OH)2·ILs with ILs as the co-solvent, catalyst, phase separation and recycling agent, as shown in Scheme 1. Compared with the existing method with NH2OH·HCl as raw material, this novel route has the following advantages: (1) it is eco-friendly since no metal salt catalyst was added and no inorganic acid was released. (2) When the reaction was completed, the reaction mass was separated into organic phase and aqueous phase automatically. The organic phase could be analyzed by gas chromatography and the ionic liquid in aqueous phase could be easily recovered. (3) The recovered ionic liquid not only can be recycled as the co-solvent and catalyst, but also as the hydroxylamine stabilizer for preparation of (NH2OH)2·ILs. Thus, the recovery of the ionic liquid with multiple roles was fully realized. Moreover, the one-pot clean synthesis of benzonitrile was achieved.
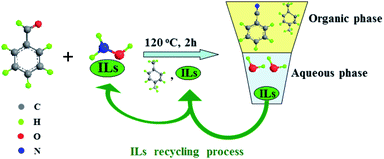 |
| | Scheme 1 Green synthesis of benzonitrile using ionic liquid with multiple roles as the recycling agent. The roles of ionic liquid: hydroxylamine stabilizer; co-solvent and catalyst; phase separation; recycled as the co-solvent and catalyst; recycled as the hydroxylamine stabilizer. | |
Experimental
Materials and methods
All the chemicals (AR grade) were commercially available and used without further purification. FTIR spectra were recorded on a Bruker Vector 22 FTIR spectrometer in the 4000–400 cm−1 range using liquid film or KBr tablet. The 1H NMR and 13C NMR spectra were recorded on a Bruker AMX FT 400 MHz NMR spectrometer using D2O as solvent, and the chemical shifts were expressed in ppm.
Preparation of the (NH2OH)2·ILs
The (NH2OH)2·ILs were prepared according to the method described in the literature.26,27 The synthesis of hydroxylamine 1-sulfobutyl pyridine hydrosulfate salt is given here as an example. 1-Sulfobutyl pyridine hydrosulfate ([HSO3-b-Py]·HSO4, 32.0 mmol) was added into a 250 mL three-necked flask equipped with a dropping funnel and a stirrer. The flask was then placed in a low-temperature reaction bath, and the temperature was continuously kept under 2 °C. The aqueous solution of hydroxylamine (80.0 mmol) was added dropwise to the three-necked flask that contained the [HSO3-b-Py]HSO4 while stirring. When the neutralization process was completed, a clear solution was obtained. The solution was then evaporated to dryness under reduced pressure to obtain the white hydroxylamine 1-sulfobutyl pyridine hydrosulfate salt ((NH2OH)2·[HSO3-b-Py]·HSO4). The preparation method of hydroxylamine N,N,N-trimethyl-N-sulfobutyl hydrosulfate salt ((NH2OH)2·[HSO3-b-N(CH3)3]·HSO4) and hydroxylamine 1-sulfobutyl-3-methyl imidazole hydrosulfate salt ((NH2OH)2·[HSO3-b-mim]·HSO4) were similar to that of the (NH2OH)2·[HSO3-b-Py]·HSO4, the only difference was to replace the [HSO3-b-Py]·HSO4 with [HSO3-b-N(CH3)3]·HSO4 or [HSO3-b-mim]·HSO4.27,28
One-pot synthesis of benzonitrile from benzaldehyde and (NH2OH)2·ILs
The reaction was performed in a 100 mL three-necked flask equipped with a stirrer and a reflux condenser. Typically, benzaldehyde (14.4 mmol), (NH2OH)2·[HSO3-b-Py]·HSO4 (21.6 mmol), [HSO3-b-Py]·HSO4 (16 mL) and the organic solvent (32 mL) were charged into the flask, and the reaction mixture was heated in an oil bath at 90–120 °C and kept for 0.5–2 h. Along with the reaction, the organic components (1 mL) were taken every five minutes and analyzed with an Agilent 7890B gas chromatograph connected with a DB-WAX capillary column and a flame ionisation detector (FID). The analysis conditions were as follows: N2 as carrier, FID temperature 320 °C, the injection port temperature 250 °C, and the program-controlled column temperature as follows: initial temperature of 90 °C for 1 min, increasing to 225 °C at a rate of 15 °C min−1 and holding for 1 min. The benzaldehyde conversion and benzonitrile yield were determined by the area normalization method.
Recycling of the ILs
For recycling of the ILs, the one-step synthesis of benzonitrile from benzaldehyde and (NH2OH)2·[HSO3-b-Py]·HSO4 was carried out under optimal reaction conditions. After the reaction, the resulting mixture was cooled and separated into organic phase and aqueous phase automatically. The organic phase was analyzed by gas chromatography, and the leaving ionic liquid in aqueous phase was easily recovered by distilled with a rotary evaporator under vacuum to remove the water. The recovered ILs could be recycled as the co-solvent and catalyst for one-pot synthesis of benzonitrile. Furthermore, it also could be used again as the hydroxylamine stabilizer to synthesis the (NH2OH)2·[HSO3-b-Py]·HSO4.
Results and discussion
Co-solvent and catalysis role of the ionic liquid
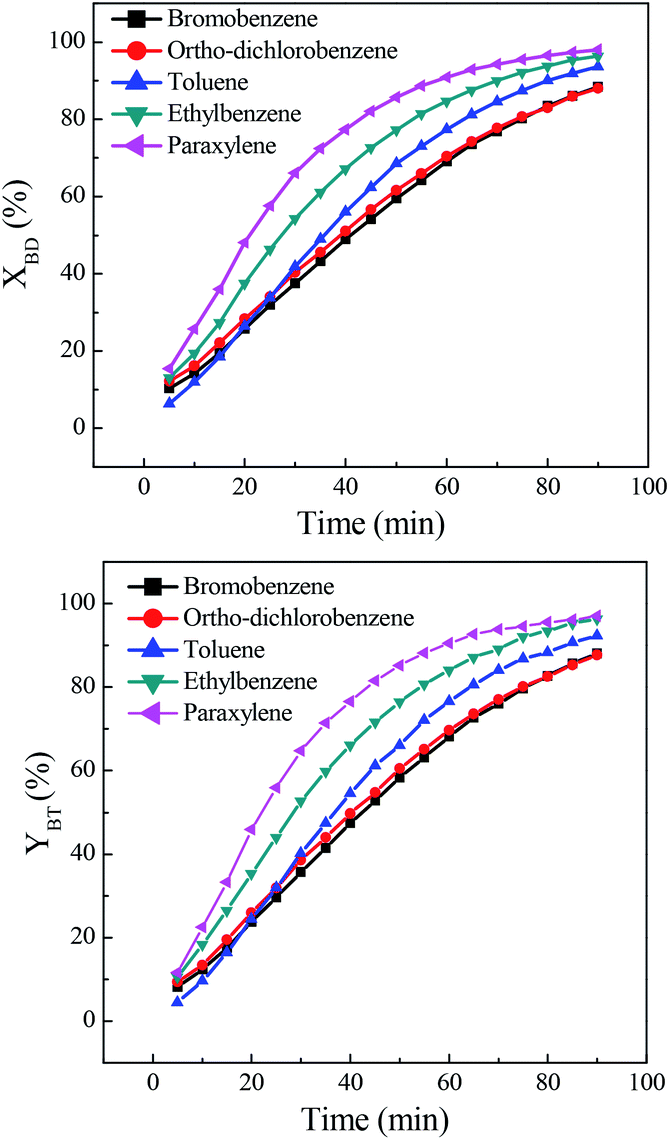
The effect of solvent on direct synthesis of benzonitrile was presented in Fig. 1. It could be seen that solvent strongly affected the results. When bromobenzene and ortho-dichlorobenzene were used as solvents, the benzaldehyde conversion and benzonitrile yield were relatively low. While toluene, ethylbenzene and paraxylene were used, the benzaldehyde conversion and benzonitrile yield were very high, especially the latter two solvents. The conversion and yield were both improved to over 96.0%. The solvent effect of aryl halide was inferior to that of aromatic hydrocarbon. The probably reason is that, the present reaction includes oximation of benzaldehyde to benzaldoxime, followed by the dehydration of the in situ generated benzaldoxime to benzonitrile. Being a multistep process, the outcome of the reaction depends on the reactivity of the substrate towards both oximation and dehydration step. As for the first step, it is believed that benzaldoxime is produced through the nucleophilic attack of nitrogen electron pairs in the NH2OH to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbon in the benzaldehyde.29,30 Free NH2OH,31 resulted from the decomposition of hydroxylamine salt, is the nucleophile necessary for conversion of benzaldehyde to the corresponding benzaldoxime. It was evident that, the aromatic hydrocarbon solvent, such as toluene, ethylbenzene and paraxylene, demonstrated an obvious promotion on the decomposition of hydroxylamine salt to form free NH2OH,32,33 since high benzaldehyde conversions were obtained in these solvents. This solvent promoted decomposition role was probably due to the formation of hydrogen bond between the nitrogen electron pairs in the hydroxylamine salts and hydrogen in the solvent.34 Among them, paraxylene was a better choice for direct synthesis of benzonitrile.
O carbon in the benzaldehyde.29,30 Free NH2OH,31 resulted from the decomposition of hydroxylamine salt, is the nucleophile necessary for conversion of benzaldehyde to the corresponding benzaldoxime. It was evident that, the aromatic hydrocarbon solvent, such as toluene, ethylbenzene and paraxylene, demonstrated an obvious promotion on the decomposition of hydroxylamine salt to form free NH2OH,32,33 since high benzaldehyde conversions were obtained in these solvents. This solvent promoted decomposition role was probably due to the formation of hydrogen bond between the nitrogen electron pairs in the hydroxylamine salts and hydrogen in the solvent.34 Among them, paraxylene was a better choice for direct synthesis of benzonitrile.
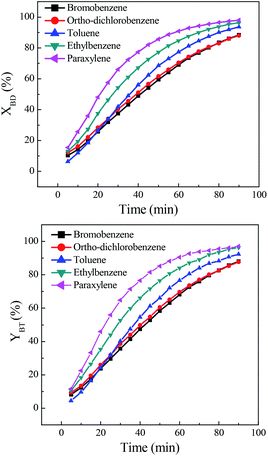 |
| | Fig. 1 Effect of solvent on direct synthesis of benzonitrile (BT). Reaction conditions: benzaldehyde (BD) 14.4 mmol, n(BD):n((NH2OH)2·[HSO3-b-Py]·HSO4) = 1:1.5, solvent 32 mL, [HSO3-b-Py]·HSO4 16 mL, temperature 100 °C. | |
Besides paraxylene, a certain amount of [HSO3-b-Py]·HSO4 was also added. To clarify their roles, a comparative study on paraxylene alone, [HSO3-b-Py]·HSO4 alone and different ratio of paraxylene to [HSO3-b-Py]·HSO4 was made and the results were listed in Table 1. When paraxylene was added alone, a large number of white particles, which was the undissolved (NH2OH)2·[HSO3-b-Py]·HSO4, was dispersed in the reaction mass. It indicated that pure paraxylene could not effectively dissolve the (NH2OH)2·[HSO3-b-Py]·HSO4, which then inhibited the reaction between benzaldehyde and hydroxylamine, resulting in a low benzaldehyde conversion of 54.8%. Moreover, most of the product was benzaldoxime, the benzonitrile yield was only 2.12%, indicating that paraxylene only acted as solvent and had no catalytic effect. When [HSO3-b-Py]·HSO4 was added, the reaction turned to liquid–liquid two phase. It could be concluded that the addition of [HSO3-b-Py]·HSO4 was favorable for the dissolution of (NH2OH)2·[HSO3-b-Py]·HSO4. Moreover, [HSO3-b-Py]·HSO4 being a Brønsted acid coordinated with the carbonyl oxygen of benzaldehyde thus increasing its electrophilicity and facilitating its reaction with hydroxylamine. Therefore, the [HSO3-b-Py]·HSO4 prompted the benzaldehyde oximation. The benzaldehyde conversion was greatly increased to over 95%. Furthermore, when the [HSO3-b-Py]·HSO4 was added alone, the reaction was homogeneous. The benzaldehyde conversion and benzonitrile yield were 100% and 96.3% respectively, which were much higher than those in pure paraxylene. The results demonstrated that [HSO3-b-Py]·HSO4 not only can be used as solvent to promote the benzaldehyde oximation, but also as catalyst to promote the benzaldoxime dehydration.35–37
Table 1 The amount of paraxylene and [HSO3-b-Py]·HSO4 on direct synthesis of benzonitrilea
| No |
Paraxylene (mL) |
[HSO3-b-Py]·HSO4 (mL) |
XBD (%) |
YBT (%) |
| Reaction conditions: benzaldehyde 3.6 mmol, n(BD):n(NH2OH) = 1:3, temperature 100 °C, time 90 min. NH2OH·HCl. (NH2OH)2·H2SO4. |
| 1 |
12 |
0 |
54.8 |
2.12 |
| 2 |
10 |
2 |
95.0 |
92.0 |
| 3 |
8 |
4 |
98.0 |
97.0 |
| 4 |
6 |
6 |
98.8 |
96.9 |
| 5 |
4 |
8 |
100 |
96.7 |
| 6 |
0 |
12 |
100 |
96.3 |
| 7b |
12 |
0 |
3.71 |
2.43 |
| 8c |
12 |
0 |
3.34 |
6.29 |
Considering that when the [HSO3-b-Py]·HSO4 alone was applied as both the solvent and catalyst, the reaction liquid was too viscous to separate and recover the ionic liquid. For this reason, a mixture of paraxylene and [HSO3-b-Py]·HSO4 were employed as solvent for the synthesis of benzonitrile. It can be seen that the benzonitrile yield was related to their ratio. With the increase of the amount of [HSO3-b-Py]·HSO4, the benzonitrile yield improved first, then remained almost unchanged. The benzonitrile yield reached a maximum value of 97.0% when the volume ratio of paraxylene and [HSO3-b-Py]·HSO4 was 2:1. With NH2OH·HCl and (NH2OH)2·H2SO4 as raw materials and paraxylene as a solvent, the benzaldehyde conversion and benzonitrile yield were extremely low, which verified the co-solvent and catalytic role of the [HSO3-b-Py]·HSO4.
Influence of reaction parameters on direct synthesis of benzonitrile
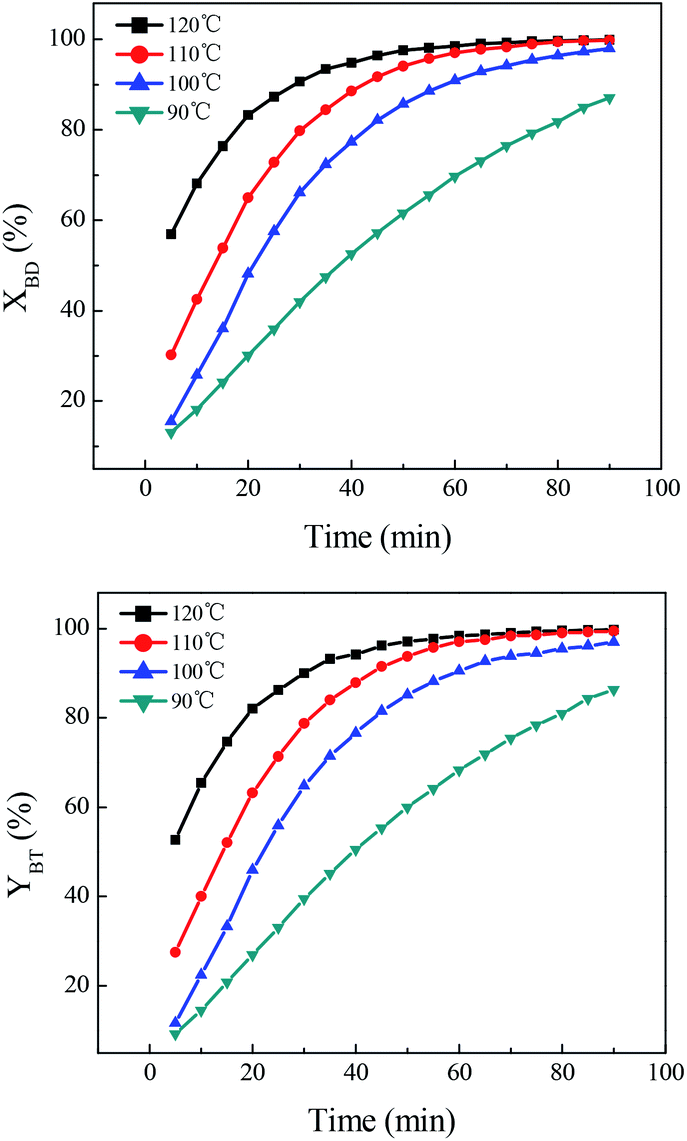
(1) Temperature. The influence of reaction temperature on direct synthesis of benzonitrile was depicted in Fig. 2. It could be seen that the benzaldehyde conversion and benzonitrile yield were relatively low when the temperature was 90 °C. Meanwhile, the reaction rate was slower since it took longer time to achieve the same conversion and yield. However, the benzaldehyde conversion and benzonitrile yield increased significantly with rising temperature from 90 to 120 °C. The reaction rate also increased rapidly, indicating that higher reaction temperature could enhance the reaction rate, favoring the synthesis of benzonitrile. The highest conversion and yield obtained (120 °C) were 99.9% and 99.7% respectively.
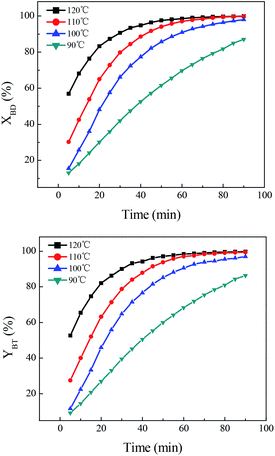 |
| | Fig. 2 Effect of temperature on direct synthesis of benzonitrile. Reaction conditions: benzaldehyde 14.4 mmol, n(BD):n((NH2OH)2·[HSO3-b-Py]·HSO4) = 1:1.5, paraxylene 32 mL, [HSO3-b-Py]·HSO4 16 mL. | |
(2) Time. The influence of reaction time was also investigated to see whether the benzaldehyde conversion and benzonitrile yield could reach 100% by prolonging the time. As shown in Fig. 3, the benzaldehyde conversion and benzonitrile yield depended greatly on time at the early stage of reaction. They increased rapidly with the extension of time, and reached a value of 98.6% and 98.4% respectively when the time was 1 h. Then the benzaldehyde conversion and benzonitrile yield increased slightly when further prolonging the time. When the reaction time was 2 h, they both reached a value of 100%.
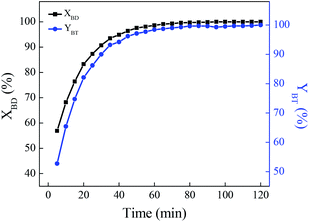 |
| | Fig. 3 Effect of time on direct synthesis of benzonitrile. Reaction conditions: benzaldehyde 14.4 mmol, n(BD):n((NH2OH)2·[HSO3-b-Py]·HSO4) = 1:1.5, paraxylene 32 mL, [HSO3-b-Py]·HSO4 16 mL, temperature 120 °C. | |
Reactivity comparison of various hydroxylamine salts
A comparison between the (NH2OH)2·[HSO3-b-Py]·HSO4 and other (NH2OH)2·ILs, as well as the traditional hydroxylamine inorganic acid salts was summarized in Table 2. It can be seen that (NH2OH)2·[HSO3-b-mim]·HSO4 and (NH2OH)2·[HSO3-b-Py]·HSO4 demonstrated better reactivity than (NH2OH)2·[HSO3-b-N(CH3)3]·HSO4. The benzonitrile yield was around 99.9% and 100%, respectively. No other by-products were generated.
Table 2 Effect of different hydroxylamine salts on direct synthesis of benzonitrilea
| No |
Hydroxylamine salt |
XBD (%) |
YBT (%) |
| Reaction conditions: benzaldehyde 3.6 mmol, n(BD):n(NH2OH) = 1:3, paraxylene 8 mL, IL 4 mL, temperature 120 °C, time 2 h. IL is [HSO3-b-N(CH3)3]·HSO4. IL is [HSO3-b-mim]·HSO4. IL is [HSO3-b-Py]·HSO4. |
| 1 |
(NH2OH)2·[HSO3-b-N(CH3)3]·HSO4b |
84.3 |
84.3 |
| 2 |
(NH2OH)2[HSO3-b-mim]·HSO4c |
99.9 |
99.9 |
| 3 |
(NH2OH)2·[HSO3-b-Py]·HSO4d |
100 |
100 |
| 4 |
NH2OH·HCld |
99.7 |
99.7 |
| 5 |
(NH2OH)2·H2SO4d |
94.3 |
94.3 |
Ionic liquid was reported to be the catalyst for the dehydration of benzaldehyde oxime.35–37 The dehydration process could be promoted by the protonation of hydroxyl group in the benzaldehyde oxime through the H+ provided by the ionic liquid, converted this moiety to a better leaving group and thus facilitated the formation of benzonitrile.36 In order to explore the relationship between the catalytic performance and properties of the ionic liquids, which were combined in the (NH2OH)2·ILs, their acid strengths were measured according to the method reported previously.38 The results were shown in Table 3 and Fig. S1,† from which we can see that the acid strength order is as follows: [HSO3-b-N(CH3)3]·HSO4 < [HSO3-b-mim]·HSO4 ≈ [HSO3-b-Py]·HSO4, which was in accordance with their catalytic activity. Thus it could be deduced that the catalytic performance of ionic liquids depended greatly on their acid strength. The ionic liquid with greater acid intensity had the stronger ability to give proton and higher catalytic activity. Moreover, the reactivity of (NH2OH)2·[HSO3-b-Py]·HSO4 was better than that of (NH2OH)2·H2SO4 and NH2OH·HCl. Hence, (NH2OH)2·[HSO3-b-Py]·HSO4 was regarded to be a better alternative to traditional hydroxylamine salts.
Table 3 Calculation and comparison of H0 values of different ionic liquidsa
| No |
IL |
Amax |
I (%) |
HI (%) |
H0 |
| Indicator: 4-nitroaniline (5 mg L−1, pKa = +0.99), ILs (5 mmol L−1). |
| 1 |
Blank |
0.53 |
100 |
|
|
| 2 |
[HSO3-b-N(CH3)3]·HSO4 |
0.44 |
83.0 |
17.0 |
1.68 |
| 3 |
[HSO3-b-mim]·HSO4 |
0.38 |
71.7 |
28.3 |
1.39 |
| 4 |
[HSO3-b-Py]·HSO4 |
0.37 |
69.8 |
30.2 |
1.35 |
Plausible mechanism for the present reaction
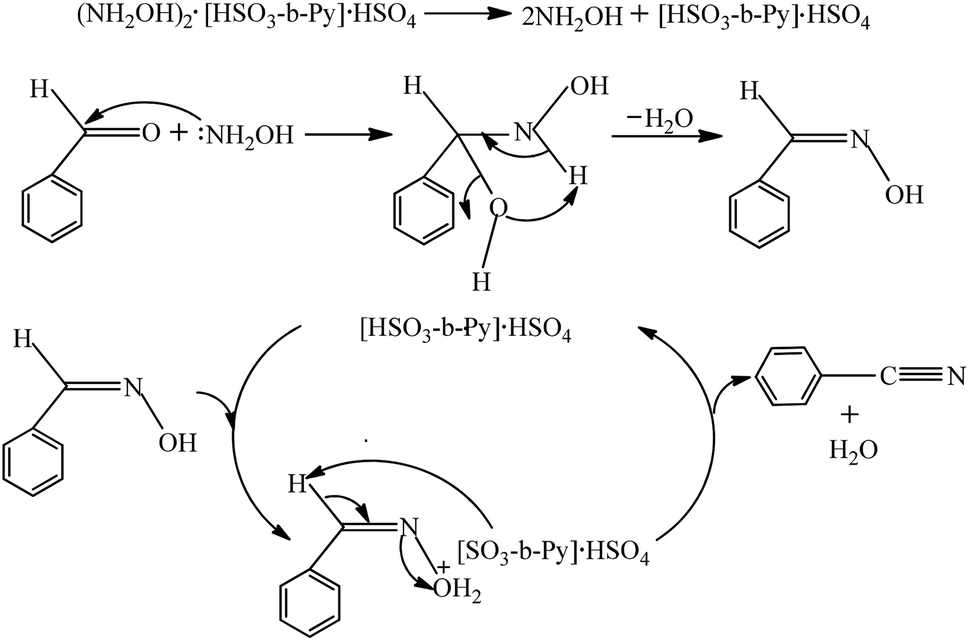
Based on the reported reaction mechanism for aldehyde and hydroxylamine to produce aldehyde oxime,30,31 followed by benzaldoxime dehydration to benzonitrile,35,36 a plausible mechanism for the present reaction was proposed and depicted in Scheme 2. Firstly, free NH2OH and [HSO3-b-Py]·HSO4 were released by the decomposition of (NH2OH)2·[HSO3-b-Py]·HSO4. Then a nucleophilic attack of NH2OH to benzaldehyde occurred, followed by a dehydration step to form the benzaldehyde oxime. At last, the hydroxyl group in the benzaldehyde oxime was protonated by the H+ offered by the [HSO3-b-Py]·HSO4 to form the intermediate, which then converted to benzonitrile through a dehydration step, along with the regeneration of [HSO3-b-Py]·HSO4 to complete the catalytic cycle.
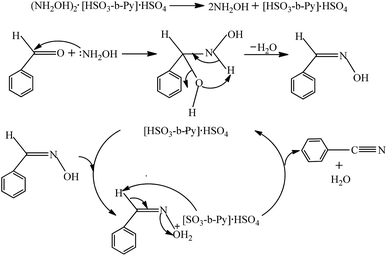 |
| | Scheme 2 Plausible mechanism for synthesis of benzonitrile from benzaldehyde and (NH2OH)2·[HSO3-b-Py]·HSO4. | |
The recovery and recycling of the ionic liquid
(1) Recycling of the ionic liquid as co-solvent and catalyst. To investigate the feasibility of the recovery and recycling of the ionic liquid, direct synthesis of benzonitrile from benzaldehyde and (NH2OH)2·[HSO3-b-Py]·HSO4 was carried out under the above mentioned optimal reaction conditions. After completion of the reaction, the resulting mixture was cooled and separated into organic phase and aqueous phase automatically. The organic phase was analyzed by gas chromatography, and the leaving ionic liquid in aqueous phase was distilled with a rotary evaporator under vacuum to recover the [HSO3-b-Py]·HSO4. The recovered [HSO3-b-Py]·HSO4 was further used as co-solvent and catalyst for the next round. It can be seen that the benzaldehyde conversion and benzonitrile yield were still as high as 96.7% after five times of recycling (Table 4), indicating that [HSO3-b-Py]·HSO4 was stable enough to be recycled.
Table 4 Recycling of the [HSO3-b-Py]·HSO4 as co-solvent and catalysta
| Recycle times |
XBD (%) |
YBT (%) |
| Reaction conditions: benzaldehyde 3.6 mmol, n(BD):n((NH2OH)2·[HSO3-b-Py]·HSO4) = 1:1.5, paraxylene 8 mL, [HSO3-b-Py]·HSO4 4 mL, temperature 120 °C, time 2 h. |
| 1 |
100 |
100 |
| 2 |
100 |
100 |
| 3 |
99.5 |
99.5 |
| 4 |
98.9 |
98.9 |
| 5 |
96.7 |
96.7 |
After the [HSO3-b-Py]·HSO4 was recycled for five times, it was recovered, collected and characterized. The FTIR spectra of the recovered and fresh [HSO3-b-Py]·HSO4 were similar (Fig. 4). In addition, the 1H NMR and 13C NMR spectra (Fig. S2–S5†) of the recovered [HSO3-b-Py]·HSO4 was consistent with that of the fresh [HSO3-b-Py]·HSO4. These results demonstrated that the [HSO3-b-Py]·HSO4 was structurally stable even after recycling five times.
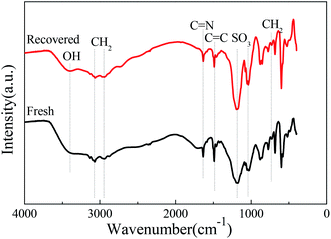 |
| | Fig. 4 FTIR spectra of the recovered and fresh [HSO3-b-Py]·HSO4. | |
(2) Recycling of the ionic liquid as hydroxylamine stabilizer. [HSO3-b-Py]·HSO4 was recovered when it was recycled for five times, and then reused as the hydroxylamine stabilizer. The (NH2OH)2·[HSO3-b-Py]·HSO4 was prepared again. The FTIR spectra of (NH2OH)2·[HSO3-b-Py]·HSO4 obtained from the fresh and recovered [HSO3-b-Py]·HSO4 was presented in Fig. 5. It can be seen that they were almost identical. The peaks at 3120 and 2725 cm−1 were assigned to the NH3+ stretching mode. The peaks at 2041 and 1540 cm−1 were attributed to the characteristic peaks of primary ammonium salt and the NH3+ deformation frequencies.26,27 Moreover, the 1H NMR and 13C NMR spectra (Fig. S6–S9†) of the (NH2OH)2·[HSO3-b-Py]·HSO4 obtained from the recovered [HSO3-b-Py]·HSO4 was consistent with the (NH2OH)2·[HSO3-b-Py]·HSO4 obtained from the fresh one, indicating that the recovered [HSO3-b-Py]·HSO4 could be used again as the hydroxylamine stabilizer even after it was recycled as co-solvent and catalyst for five times.
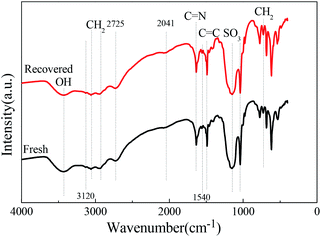 |
| | Fig. 5 FTIR spectra of the (NH2OH)2·[HSO3-b-Py]·HSO4 obtained from the recovered and fresh [HSO3-b-Py]·HSO4. | |
Next, the (NH2OH)2·[HSO3-b-Py]·HSO4 obtained from the recovered [HSO3-b-Py]·HSO4 was used in direct synthesis of benzonitrile under the optimal reaction conditions. The benzaldehyde conversion and benzonitrile yield were 100% and 98.2%, respectively. The results demonstrated that the reactivity of the (NH2OH)2·[HSO3-b-Py]·HSO4 obtained from the recovered and fresh [HSO3-b-Py]·HSO4 were much the same.
Reactivity of different aldehydes
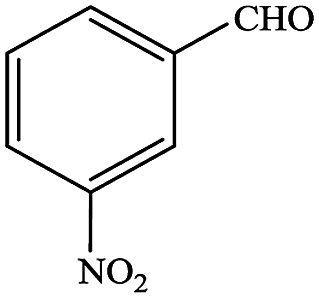
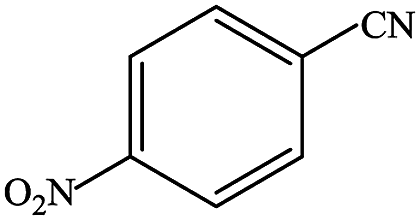
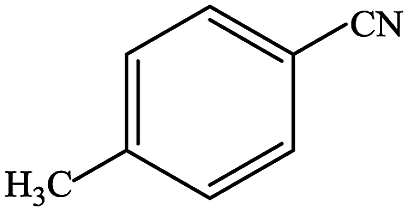
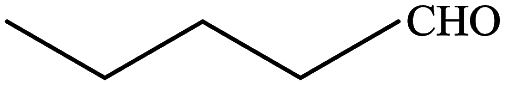
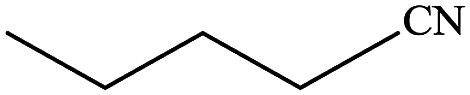
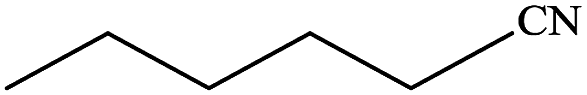
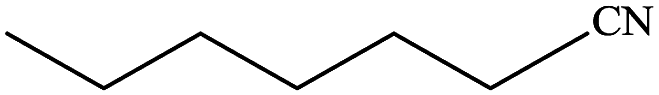
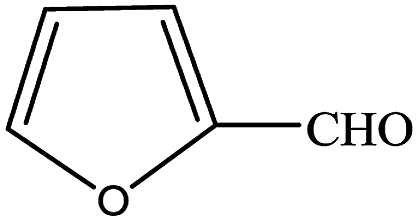
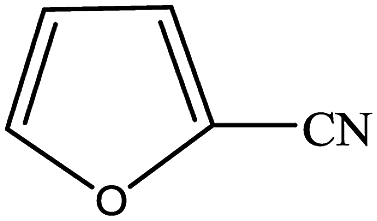
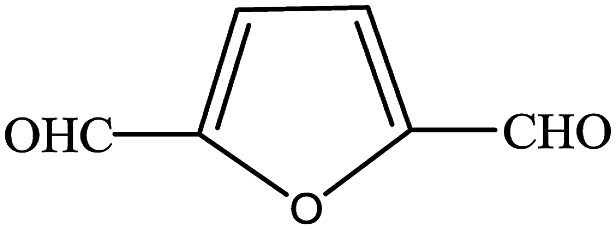
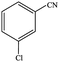
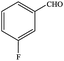
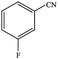
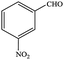
In subsequent efforts, various aldehydes including structurally diverse aryl aldehydes, heterocyclic aldehydes, aliphatic aldehydes, even dialdehyde were chosen as potential substrates to produce their corresponding nitriles (Table 5). For substituted aromatic aldehydes, the substituents and their positions could affect their reactivity. For example, the conversion of the aldehyde with electron withdrawing group was increased along with the enhancement of the electron attract ability (line 1–3, Table 5). Moreover, the results of the para-substituted aldehydes were better than those of the meta-substituted aldehydes (line 4–5, 6–7, Table 5). For aliphatic aldehydes, the yield of aliphatic nitrile decreased with the increase of the chain length of the aliphatic aldehyde. The conversions of the heterocyclic aldehydes were nearly 100%. As evident from Table 5, stronger electron withdrawing groups (like nitro) have greater influence on the carbonyl group of aromatic aldehydes leading to an increase in its electrophilicity thus increasing the yield of nitrile. This effect is more pronounced for an electron withdrawing substituent in the para position compared to its meta position. Moreover, for aliphatic aldehydes, increasing chain length decreases the electrophilicity of the aldehydic group due to increase in positive inductive effect of the aliphatic chain thus decreasing the overall yield of aliphatic nitriles for higher aliphatic chain aldehydes.
Table 5 Reactivity of different aldehydesa
| No |
Aldehyde |
Nitrile |
X (%) |
Y (%) |
| Reaction conditions: aldehyde 3.6 mmol, n(aldehyde):n((NH2OH)2·[HSO3-b-Py]·HSO4) = 1:1.5, paraxylene 8 mL, [HSO3-b-Py]·HSO4 4 mL, temperature 120 °C, time 2 h. |
| 1 |
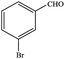 |
 |
80.6 |
80.6 |
| 2 |
 |
 |
86.7 |
86.7 |
| 3 |
 |
 |
94.4 |
94.4 |
| 4 |
 |
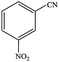 |
99.7 |
99.7 |
| 5 |
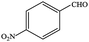 |
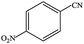 |
99.9 |
99.9 |
| 6 |
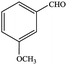 |
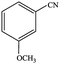 |
77.1 |
77.1 |
| 7 |
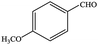 |
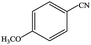 |
99.8 |
99.8 |
| 8 |
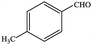 |
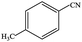 |
91.5 |
91.5 |
| 9 |
 |
 |
100 |
100 |
| 10 |
 |
 |
100 |
100 |
| 11 |
 |
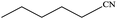 |
100 |
98.7 |
| 12 |
 |
 |
85.4 |
83.1 |
| 13 |
 |
 |
41.0 |
38.7 |
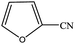
| 14 |
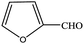 |
 |
99.4 |
99.4 |
| 15 |
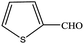 |
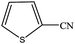 |
100 |
98.1 |
| 16 |
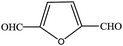 |
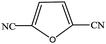 |
99.9 |
99.9 |
Conclusions
In summary, a convenient and facile method for direct conversion of aldehydes into nitriles using ionic liquid as the recycling agent has been established. The optimum reaction condition was at 120 °C for 2 h. The suitable molar ratio of benzaldehyde to (NH2OH)2·[HSO3-b-Py]·HSO4 was 1:1.5, and the suitable volume ratio of paraxylene to [HSO3-b-Py]·HSO4 was 2:1. Under the optimal reaction conditions, the benzaldehyde conversion and benzonitrile yield were both 100%. Ionic liquid exhibited the multiple roles of co-solvent, catalysis and phase separation. It could be recovered easily, and recycled directly after reaction. Additionally, this novel route is applicable to green synthesis of a variety of aromatic, heteroaromatic and aliphatic nitriles with excellent yields.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors are grateful for the financial support of the National Natural Science Foundation of China (No. 21236001, 21878069, 21576069 and 201802031), National Natural Science Foundation of Hebei Province (No. B2016202335, B2015202369 and B2017202133).
Notes and references
- B. M. Xu, H. Y. Ruan, Y. Zhou, W. Xiong and K. Chen, Chem. World, 2012, 5, 316–320 Search PubMed.
- L. P. OuYang, X. Q. You, Y. Y. Huang, A. Q. Zhang and G. Y. Xie, Appl. Chem. Ind., 2017, 46, 1692–1694 Search PubMed.
- C. H. Yang and J. M. Williams, Org. Lett., 2004, 6, 2837–2840 CrossRef CAS PubMed.
- T. Schareina, A. Zape and M. Beller, Chem. Commun., 2004, 12, 1388–1389 RSC.
- S. Iida, R. Ohmura and H. Togo, Tetrahedron, 2009, 65, 6257–6262 CrossRef CAS.
- H. F. Lu, Y. Zhou, H. F. Huang, H. Y. Liu and Y. F. Chen, Chin. J. Catal., 2005, 26, 101–105 CAS.
- E. Rombi, I. Ferino, R. Monaci, C. Picciau, V. Solinas and R. Buzzoni, Appl. Catal., A, 2004, 266, 73–79 CrossRef CAS.
- K. V. R. Chary, K. R. Reddy, T. Bhaskar and V. S. Guggilla, Green Chem., 2002, 4, 206–209 RSC.
- H. Chen, C. Y. Lu and X. H. Liu, J. Huaihai Inst. Technol., Nat. Sci. Ed., 1997, 6, 51–53 Search PubMed.
- K. Murugesan, T. Senthamarai, M. Sohail, M. Sharif, N. V. Kalevarua and R. V. Jagadeesh, Green Chem., 2018, 20, 266–273 RSC.
- B. Mitra, G. C. Pariyar, R. Singha and P. Ghosh, Tetrahedron Lett., 2017, 58, 2298–2301 CrossRef CAS.
- C. J. Fang, M. C. Li, X. Q. Hu, W. M. Mo, B. X. Hu, N. Sun, L. Q. Jin and Z. L. Shen, RSC Adv., 2017, 7, 1484–1489 RSC.
- V. P. Gozum and R. C. Mebane, Green Chem. Lett. Rev., 2013, 6, 149–150 CrossRef CAS.
- P. Ghosh, B. Saha, G. C. Pariyar, A. Tamang and R. Subba, Tetrahedron Lett., 2016, 57, 3618–3621 CrossRef CAS.
- S. L. Guan and W. B. Huang, Chem. World, 1993, 9, 418–423 Search PubMed.
- J. M. DeMott Jr and C. J. Kelley, J. Chem. Educ., 2001, 78, 780 CrossRef.
- S. C. Xu, Organic Chemistry, Higher Education Press, Beijing, 2nd edn, 1993, p.284 Search PubMed.
- S.-I. Fukuzawa, Y. Yamaishi, H. Furuya, K. Terao and F. Iwasaki, Tetrahedron Lett., 1997, 38, 7203–7206 CrossRef CAS.
- Z. Wang and J. Luo, Fine Chem. Intermed., 2013, 43, 14–18 CAS.
- S. Enthaler, M. Weidauer and F. Schröder, Tetrahedron Lett., 2012, 53, 882–885 CrossRef CAS.
- Y. J. Zhuang, J. Liu and Y. B. Kang, Tetrahedron Lett., 2016, 57, 5700–5702 CrossRef CAS.
- A. Leggio, E. L. Belsito, S. Gallo and A. Liguori, Tetrahedron Lett., 2017, 58, 1512–1514 CrossRef CAS.
- A. Khalafi-Nezhad and S. Mohammadi, RSC Adv., 2014, 4, 13782–13787 RSC.
- S. H. Khezri, N. Azimi, M. Mohammed-Vali, B. Eftekhari-Sis, M. M. Hashemi, M. H. Baniasadi and F. Teimouri, ARKIVOC, 2007, 15, 162–170 Search PubMed.
- L. V.-B. Hoelz, B. T. Gonçalves, J. C. Barros and J. F. Mendes da Silva, Molecules, 2010, 15, 94–99 Search PubMed.
- Z. H. Li, Q. S. Yang, X. D. Qi, Y. Y. Xu, D. S. Zhang, Y. J. Wang and X. Q. Zhao, Chem. Commun., 2015, 51, 1930–1932 RSC.
- Z. H. Li, Q. S. Yang, L. Y. Gao, Y. Y. Xu, D. S. Zhang, S. F. Wang, X. Q. Zhao and Y. J. Wang, RSC Adv., 2016, 6, 83619–83625 RSC.
- Z. H. Li, X. D. Qi, L. Y. Gao, Y. Y. Xu, D. S. Zhang, S. F. Wang, X. Q. Zhao and Y. J. Wang, Chem. Lett., 2017, 46, 289–292 CrossRef CAS.
- H. J. Zang, M. L. Wang, B. W. Cheng and J. Song, Ultrason. Sonochem., 2009, 16, 301–303 CrossRef CAS PubMed.
- A. Elmakssoudi, K. Abdelouahdi, M. Zahouily, J. Clark and A. Solhy, Catal. Commun., 2012, 29, 53–57 CrossRef CAS.
- D. Lorenzo, A. Romero and A. Santos, Ind. Eng. Chem. Res., 2016, 55, 6586–6594 CrossRef CAS.
- M. Matsumoto, M. Tanimura, T. Akimoto, N. Watanabe and H. K. Ijuin, Tetrahedron Lett., 2008, 49, 4170–4173 CrossRef CAS.
- O. Phillips, J. M. Schwartz and P. A. Kohl, Polym. Degrad. Stab., 2016, 125, 129–139 CrossRef CAS.
- J. Wang, X. B. Zhang, F. M. Wang and W. F. Cai, Chem. Ind. Eng. Prog., 2016, 35, 1309–1313 Search PubMed.
- D. Saha, A. Saha and B. C. Ranu, Tetrahedron Lett., 2009, 50, 6088–6091 CrossRef CAS.
- D. Abolghasem, K. Amir, B. Mehdi and T.-H. Niloofar, Chin. J. Chem., 2011, 29, 978–982 CrossRef.
- J. L. Zhang, X. R. Chen, Y. P. Yu, K. L. Xu and J. Huang, Chem. Res. Chin. Univ., 2011, 27, 712–714 CAS.
- X. L. Zhang, H. L. An, H. Q. Zhang, X. Q. Zhao and Y. J. Wang, Ind. Eng. Chem. Res., 2014, 53, 16707–16714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and the characterization data. See DOI: 10.1039/c9ra02296a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a