
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrospinning of pyrazole-isothiazole derivatives: nanofibers from small molecules†
Silvia Locarnoa,
Aitziber Eleta-Lopez b,
Maria Giovanna Lupoc,
Maria Luisa Gelmia,
Francesca Clerici*a and
Alexander M. Bittner*bd
b,
Maria Giovanna Lupoc,
Maria Luisa Gelmia,
Francesca Clerici*a and
Alexander M. Bittner*bd
aDepartment of Pharmaceutical Sciences, General and Organic Chemistry Section “A. Marchesini”, University of Milan, Via Venezian 21, 20133 Milano, Italy. E-mail: francesca.clerici@unimi.it
bCIC nanoGUNE, Tolosa Hiribidea 76, 20018 Donostia-San Sebastián, Spain
cDepartment of Pharmaceutical and Pharmacological Sciences, Via Marzolo 5, 35131 Padua, Italy
dIkerbasque, Basque Foundation for Science, Ma Díaz de Haro 3, E-48013 Bilbao, Spain
First published on 2nd July 2019
Abstract
We investigate the electrospinning of small molecules, specifically designed peptide derivatives of the pyrazole-isothiazole scaffold. Such non-natural peptides enhance the spectrum of fundamental materials used for electrospinning. Unlike standard electrospun materials, our peptides are not polymeric, but able to aggregate in solution and especially during processing. They contain donor/acceptor groups that can form hydrogen bonds, and groups that are able to generate π-stacking interactions, which are known as important requirements for assembly processes. The pyrazole-isothiazole derivatives were synthesized by means of a 1,3-dipolar cycloaddition reaction, which is completely regioselective, affording only one isomer. We demonstrate that our compounds can be electrospun from fluoroalcohol solution into solid, quasi-endless micro- and nanofibers. The electrospinnability varies substantially, depending on the amino acids linked to the scaffold. Some compounds provide only short fibers, while Fmoc-glycyl-(N-benzyl)-pyrazole-isothiazole-tert-butyl carboxylate-1,1-dioxide forms continuous, homogenous, and bead-free fibers (droplet-like beads are a common problem in electrospinning). We analyzed the compounds and the fibers with various spectroscopic techniques (MS, IR and Raman). Electrospinning does not change chemical composition and configuration, suggesting the monomeric form of the compounds even in the fibers. Interestingly, we found that the stereochemistry of the scaffold can affect the ability of the peptide to be electrospun.
Introduction
Electrospinning is a simple and versatile technique used for the fabrication of continuous micro and nanofibers, mainly from polymer solutions and polymer melts.1 The approach is versatile, inexpensive, scalable and reliable. Nanofibers possess several interesting physicochemical properties such as an extremely high specific surface (surface-to-volume ratio) that make them very appealing for a broad spectrum of applications, from energy generation and storage, water treatment and environmental remediation, to healthcare and biomedical engineering.2 Many synthetic polymers, additional solutes and solvents are compatible with successful spinning, but also some biomolecules, as documented in several recent reviews.3–7 The process is based on applying a high voltage (kV range) to a liquid, which forms a jet that can be collected in dry form on a grounded substrate. Polymers have the great advantage of good “spinnability”, i.e. the parameter space to form fibers is rather large, and of high versatility. However, they have various degrees of biocompatibility, and the incorporation of additional functionalities (chemical or physical) can be hampered by complex post-modifications, or by inhomogeneous distribution.8,9 The presence of more than one compound also complicates the analysis. For example, a quantification of the assembly is hampered by the very definition of polymers or poly-aggregates, which is usually based on the (average) number of monomers in a polymer chain or in an aggregate, and on chain flexibility.10 On the other hand non-polymeric molecules can usually not be electrospun because only polymer solutions or melts are sufficiently viscous and provide the required degree of molecular entanglement.11 However, high molar mass polymers are not essential for production of uniform electrospun fibers, because the presence of sufficient intermolecular interactions can effectively provide the required chain entanglement.12,13 Currently, only few papers discussing electrospinning of small molecular systems have been reported in the literature. Examples are phospholipid amphiphiles,14,15 tetraphenylporphyrin compounds,16 and cyclodextrin.17,18 In spite of their small size, these molecules can assemble to nanofibers by electrospinning, based solely on noncovalent interactions.Our previous results are based on the use of simple amino acids such as Fmoc-Gly,19 and of dipeptides (Fmoc-Phe-Gly and Phe-Phe)19–21 for nanofiber preparation. They prompted us to test short peptide sequences, containing different amino acids. The modularity of the amino acids and their ability to drive self-assembly and self-organization leads to a high versatility in functions.22,23 Furthermore, by tailoring the functional groups on the side chains and at the N- and C-terminus, one can enormously expand the molecular variability. Notwithstanding these appealing features, natural peptides suffer from a main drawback when they are used for biological applications, namely their well-known low stability to proteases. The insertion of unnatural amino acids in the peptide sequences is a very established tool to overcome this problem. Starting from this consideration, and continuing our research on the preparation of non-coded amino acids and on their use to obtain peptidomimetics24–27 for different applications ranging from biomedical application to nanomaterials, we investigated the electrospinnability of short peptide chains. In this work we used simple systems formed by natural amino acids coupled with an unnatural amino acid, i.e. a bicyclic pyrazole-isothiazole system (Fig. 1) that resembles a β-amino acid (carboxylic group at C-6 and NH of pyrazole ring). Scaffold A (Fig. 1) is characterized by (i) a rigid planar geometry, (ii) the BnNH at C-3 as well as the sulfonyl group, working respectively as hydrogen bond donor and acceptor, (iii) important groups for π–π interactions (scaffold A itself and the benzyl substituent). All these features could influence not only the conformation of the resulting molecules, but also induce specific orientation of the groups involved in hydrogen bonding and π–π interactions.
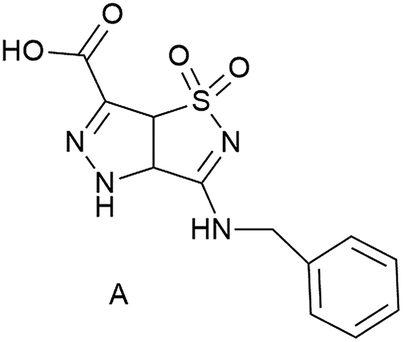 | ||
| Fig. 1 Pyrazole-isothiazole scaffold. | ||
In order to better understand the forces that govern the assembly in electrospun fibers, we started from the racemic scaffold A and designed three different short peptide sequences (1, 2, 3 Fig. 2).
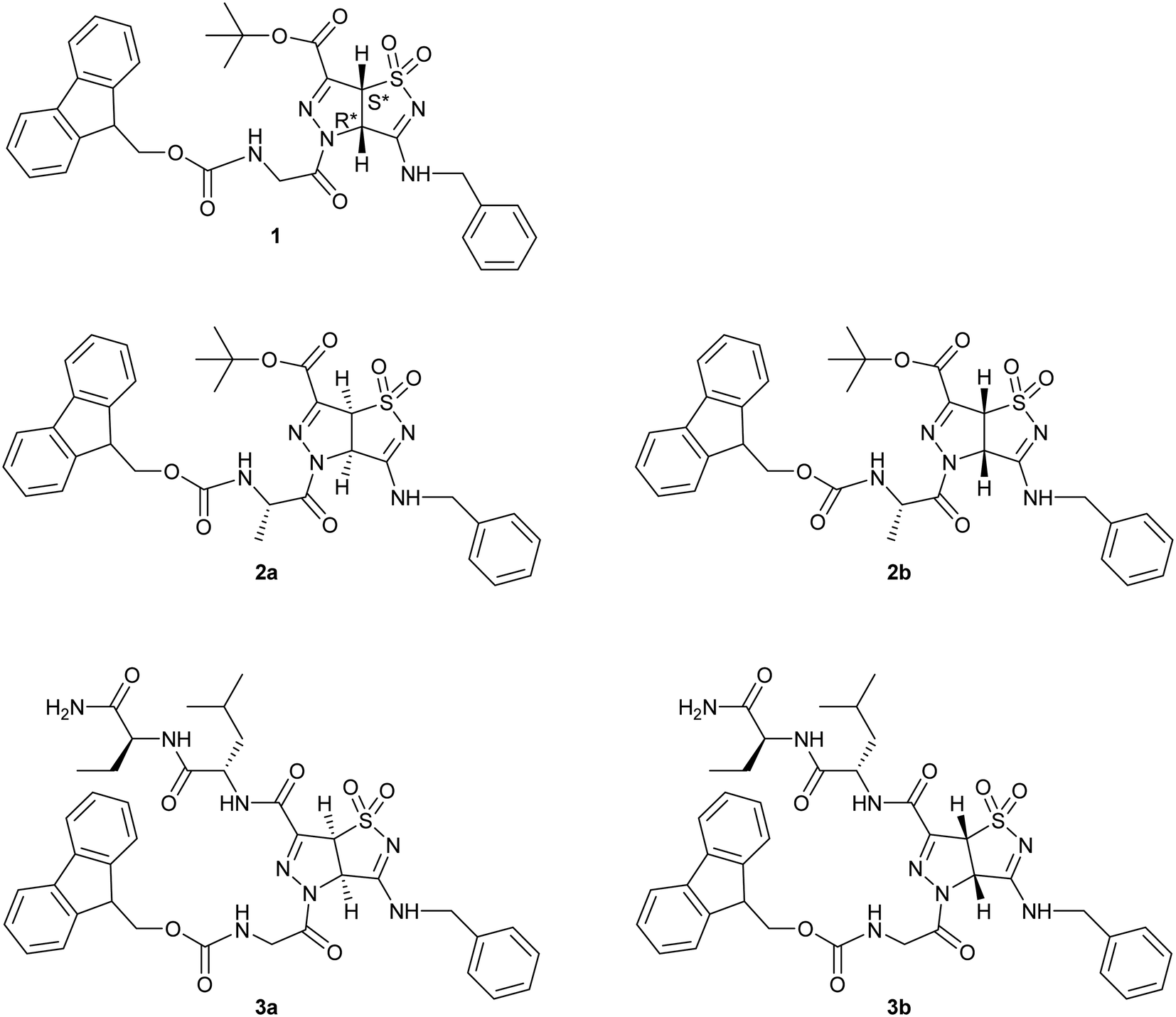 | ||
| Fig. 2 Compounds 1, 2, 3 designed for the study. All the stereoisomers are shown. | ||
Compound 1 is characterized by the presence of a Fmoc-Gly moiety linked through an amidic bond to the heterocyclic scaffold. As Gly is an achiral amino acid, compound 1 exists as a pair of enantiomers. Compound 2 presents Fmoc-Ala, generating two different diastereomers (2a and 2b), based on the two enantiomers of the pyrazole-isothiazole scaffold. Finally, compound 3 is characterized by the insertion of a dipeptide (Leu-Val) and a Fmoc-Gly moiety at the C- and N-terminus, respectively. As in the previous case, two different diastereomers (3a and 3b) are formed.
Our aim was to exploit the use of the above peptides to develop electrospun nanofibers. We point out that the use of the chiral scaffold A is of particular interest because its stereochemistry can influence the electrospinnability of the resulting diastereomeric compounds to a surprisingly large degree, and even suppress it completely for one isomer (under the same conditions). Spectroscopic analyses were performed both on the powder educts and on the fibers, by various techniques (MS, IR and Raman). We found that electrospinning does not change chemical composition and configuration, suggesting the presence of the monomeric form of the compounds even in the fibers.
Thus, our work enhances the spectrum of fundamental materials that can be used for the preparation of nanofibers, mostly for biomedical applications. For this reason, we tested the cytotoxicity of the best compound of the series, demonstrating that neither the powder educts nor the fibers cause cell damage.
Results and discussion
Chemistry
The synthetic strategy adopted for the preparation of compounds 1, 2 and 3 is shown in Scheme 1. The 3-benzylamino isothiazole 1,1-dioxide 4 is produced with a synthetic strategy already developed in our group.28 Then, a cycloaddition reaction was performed with t-butyl diazoacetate 5, taking advantage of the reactivity of the C-4 C-5 double bond toward 1,3-dipoles. The Δ2-pyrazoline derivative 6 (80% yield) was obtained through tautomerization of the primary cycloaddition product, the Δ1-pyrazoline. The reaction is completely regioselective, affording only one regioisomer (ESI, Scheme ESI1†). For the following coupling steps we used amino acids protected at the N-termini with a Fmoc group since the Fmoc and the t-butyl groups are orthogonal and the t-butyl group is easily removed under mild acidic conditions (ESI, Scheme ESI2†). After several attempts, the best coupling conditions were found (EDC, HOBT in DCM in the presence of DIPEA as base) and were applied to the reaction of 6 with Fmoc-Gly and Fmoc-Ala (see ESI† for details). Compound 1 was obtained in satisfactory yield (84%) as inseparable mixture of enantiomers and completely characterized (see ESI†). Compound 2 was obtained as a pair of diastereomers that were separated by flash chromatography, affording compounds 2a and 2b in 44% and 37% yield, respectively (see ESI† for characterization). For the preparation of 3, compound 1 was deprotected (TFA/DCM 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, overnight, 25 °C, quantitative yield) and then coupled with dipeptide H-Leu-Val-NH2, according to the previously described coupling procedure. The diastereomers 3a and 3b were obtained and separated by flash chromatography (see ESI† for characterization).
:1, overnight, 25 °C, quantitative yield) and then coupled with dipeptide H-Leu-Val-NH2, according to the previously described coupling procedure. The diastereomers 3a and 3b were obtained and separated by flash chromatography (see ESI† for characterization).
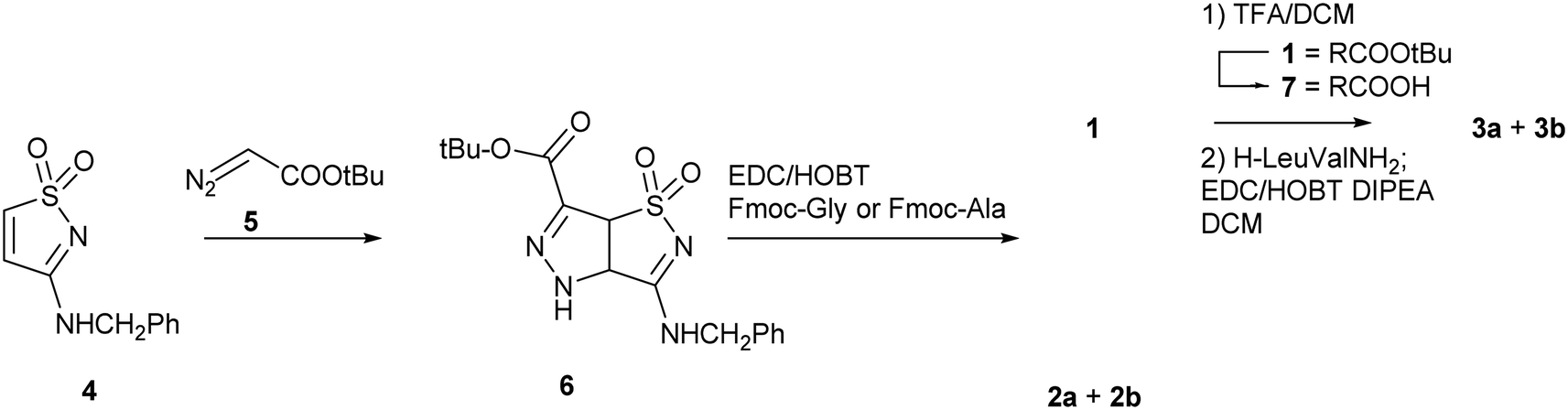 | ||
| Scheme 1 Synthetic strategy for the preparation of compounds 1, 2 and 3. | ||
We performed a range of NMR analyses (1H, 13C, HMBC, HMQC, NOESY) on compounds 1, 2a, 2b, 3a, and 3b. Several important signals in the spectra of the two pairs of diastereoisomers 2a/2b and 3a/3b overlap significantly. Nevertheless, we found spatial proximity between the two ends of the peptide chains for compounds 3a and 3b, suggesting a compact structure. The diastereomer 3a shows NOEs between the two peptidyl arms of the bicyclic system, in particular between the glycine amide group and the leucine α-carbon, and between the aromatic moiety of the Fmoc group and the methyl group of the valine side chain. We additionally detected weak NOE between the leucine NH and the glycinyl methylene group. On the other hand, compound 3b showed strong NOEs between the side chain at the C-terminus and the heterocyclic ring, in particular between the methyl group of the leucine side chain and H-3a and H-6a (see ESI, Fig. ESI1†). However, notwithstanding several attempts, we were unable to obtain crystals that are suitable for X-ray analysis. Therefore, the absolute configuration of the diastereomers 2a/2b and 3a/3b is arbitrarily assigned.
Fig. 3 shows optical (Fig. 3a and b) and scanning electron microscope (SEM, Fig. 3c–f) micrographs of electrospun fibers of compound 1. The fibers appear continuous, fairly homogenous, and without any beads. This is a very satisfactory result, taking into account that compound 1 is a small molecule and not a polymer. The average fiber diameters (762 ± 167 nm) and heights (630 ± 84 nm) were evaluated with ImageJ software. We imaged the fiber cross-section by SEM with the sample stage tilted to 75°, after cutting the fibers with a sharp blade. Fig. 3f shows that compound 1 gives fully filled fibers, not tubes, different from Phe-Phe.21 The nearly circular cross-section means that fiber heights are practically identical with their diameters.
 | ||
| Fig. 3 (a and b) Optical micrographs of compound 1 electrospun nanofibers (15 cm, 15 kV, 0.5 ml h−1); (c and d) SEM micrographs of compound 1 nanofibers; (e) also observed under tilt of 75°; (f) cross section of the fiber. SEM was recorded in vacuum (voltage 2–10 kV), optical images in air. | ||
We imaged details of the fiber surface by AFM. Fig. 4a shows a topography overview, which fits very well to the optical and SEM data (see statistics in ESI, Fig. ESI2†). The AFM heights of the thicker fibers span the range of 400 to 800 nm, while some smaller fibers gave 70 to 200 nm. However, the larger statistical sampling from SEM shows that the diameter distribution is monomodal, with a maximum around 700 nm (see ESI for detailed statistics, Fig. ESI2†).
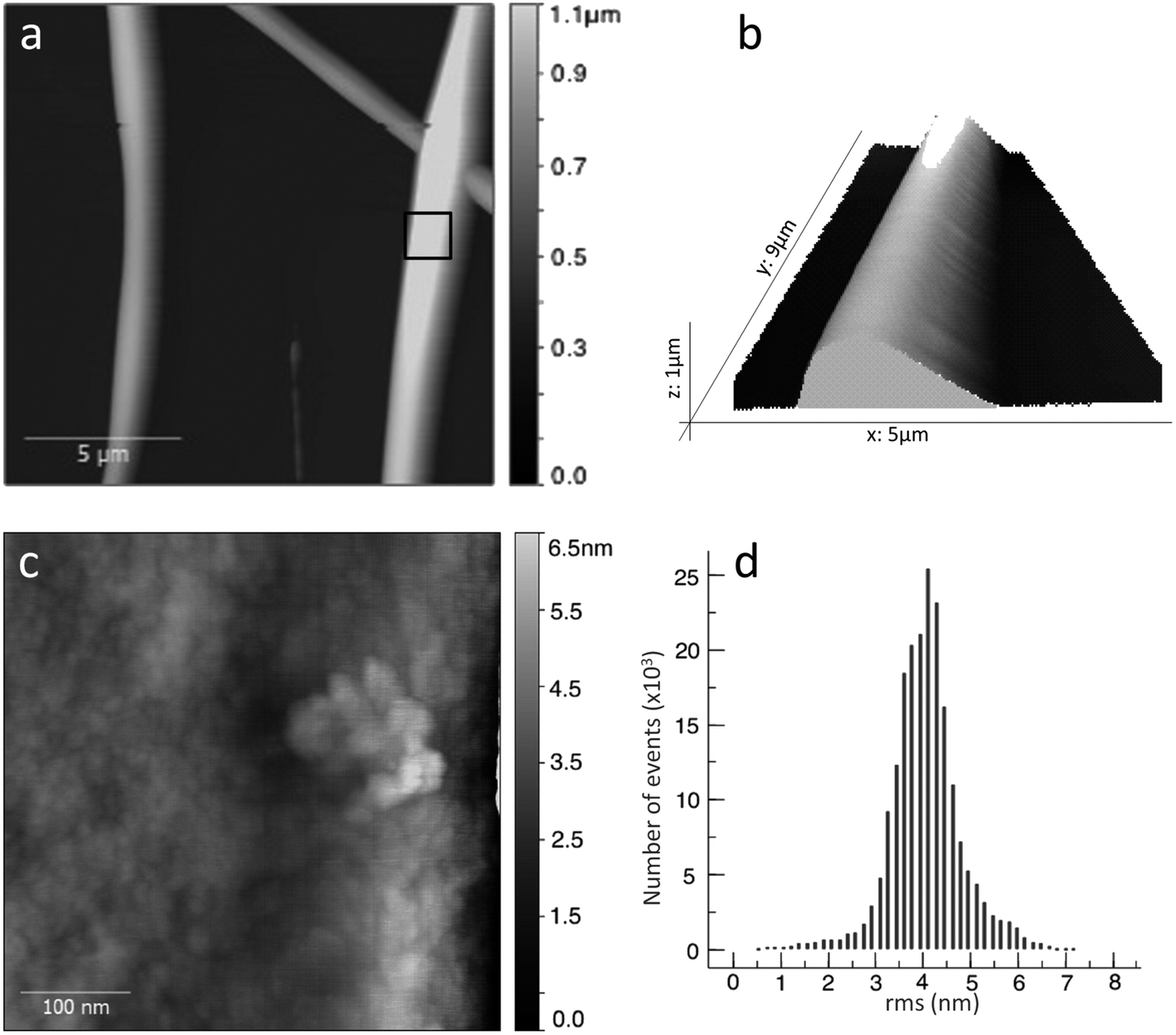 | ||
| Fig. 4 AC mode (tapping) AFM scans of compound 1 electrospun nanofibers (15 cm, 15 kV, 0.5 ml h−1). (a) topography image of compound 1 nanofibers on a silicon wafer substrate, overview; (b) 3D rendering of a fiber detail; (c) zoom into area indicated in (a); (d) height distribution for (c). | ||
AFM can provide much finer details, as we demonstrate in Fig. 4c and d. AFM scans of fibers always suffer from the high curvature, hence we predominantly scanned the very top part of the fibers, where the curvature is minimal, and flattened the images by fitting to a polynomial background. Our fibers exhibit a grainy structure with typical feature sizes that can vary from 7–20 nm. The grains are very compact and do not show any ordered or symmetric structure. The local roughness of these surface is 0.8 ± 0.3 nm, which is exemplified by the height distribution in Fig. 4d (we tested also other smaller and larger areas with similar results, see ESI†). This very low value explains that SEM, even at high resolution, shows extremely smooth fibers. Some protrusions, e.g. on the right hand side of Fig. 4c, show height differences of 2 nm, which correlate with the molecular size, but due to the absence of ordering, features such as the islands found by Lepere et al.30 on amyloid fibers do not exist here. Indeed, X-ray diffraction suggests an amorphous structure (see ESI, Fig. ESI5†).
When we consider the continuous and homogenous nature of the fibers, obtained in absence of a polymer, questions of molecular arrangements that are analogous to entanglements in polymers arise. Entanglement is usually quantified based on bulk properties of polymer melts, which can be correlated with the degree of polymerization. The number of chemical bonds in an entanglement loop can be as small as 75, and for the conformationally restricted “virtual bonds”10 even 20. A single molecule of compound 1 (conformationally restricted by the presence of amides and the bicyclic system) provides 9 virtual bonds, which is not sufficient for substantial entanglement. Hence, in analogy to smaller peptides, we decided to investigate chemical interactions in more detail. We probed chemical nature and conformations of compound 1 by vibrational infrared (IR) and Raman spectroscopy in the solid state (Fig. 5; see ESI† for additional information). Most IR peaks found in the fiber fit very well to those in the powder. The only significant difference is a redshift of the 2940 cm−1 band (powder) to 2910 cm−1 (fiber). This band should stem from some of the three CH groups, and the shift would mean that the bicyclic system in the fibers interacts more closely with neighboring molecules. Another difference is that the vibration at 1075 cm−1 is clearly less intense, suggesting a different orientation for the phenyl group. Overall, this comparison demonstrates an excellent correspondence of all functional group vibrations, and hence that powder and fiber are chemically nearly identical in composition and configuration. This is surprising, given the application of high voltage and organic solvent, and by no means general (compare electrospray ionization processes at similar voltages). We have confirmed the result by mass spectrometry of the fibers, where the presence of mass (m/z) 666.25 [M + Na]+ and 642.00 [M − H]− prove the monomeric form of the compound.
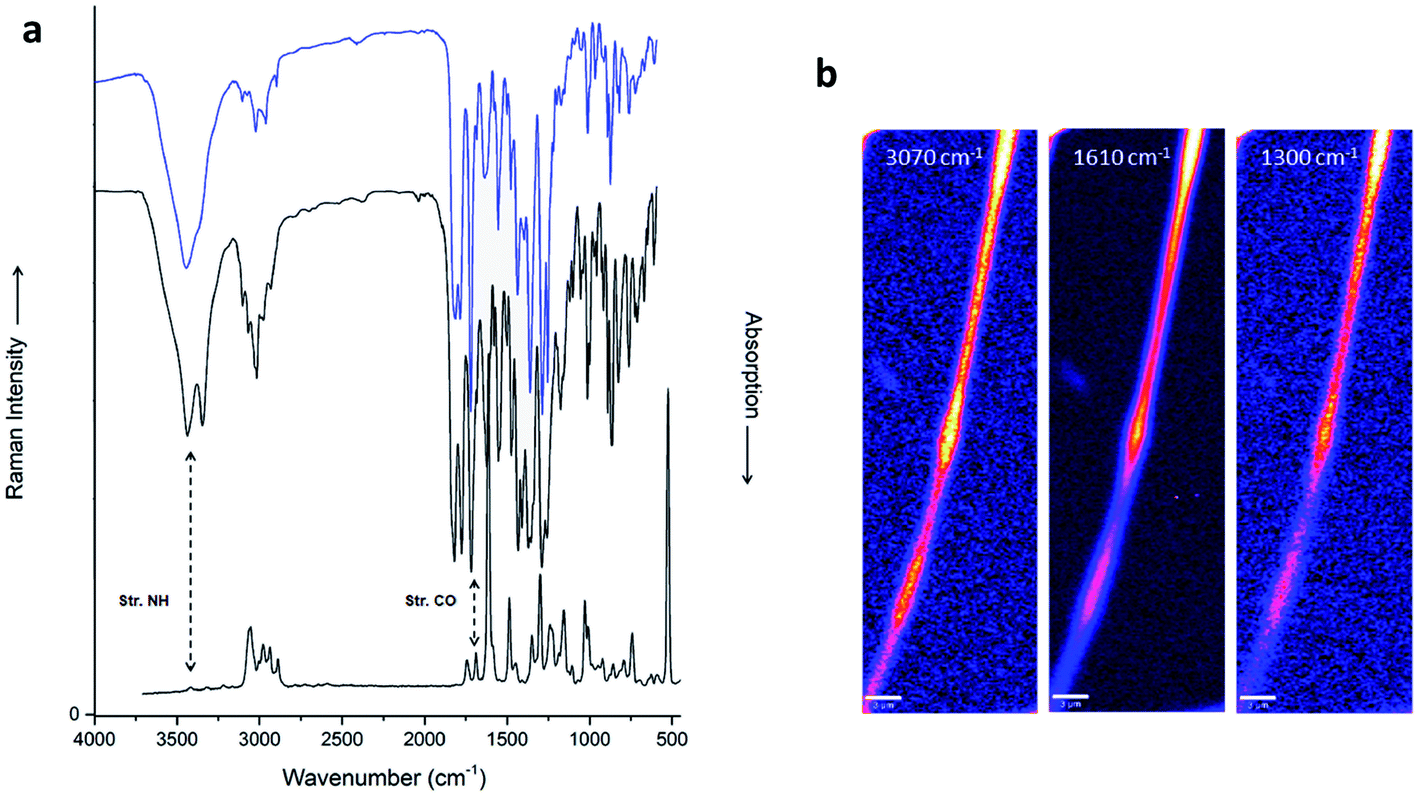 | ||
| Fig. 5 (a) Raman (bottom) and IR spectra (top) of compound 1, scaled and offset for clarity. The IR spectra refer to powder (black) and fibers (blue). (b) Raman hyperspectral micrographs of an electrospun compound 1 fiber. Each pixel contains a complete Raman spectrum; the images are produced by selecting bands at 3070 cm−1, 1610 cm−1, and 1300 cm−1. Note that each band has a different signal/noise ratio. The intensities are shown in false color. The scalebar is 3 μm. The three beads (on top, in the middle, and close to the bottom of the image), each about 9 μm long and 1.5 to 2 μm wide, show enhanced intensity because more material is present in the Raman focus. | ||
The Raman spectra are generally well comparable with the IR spectra, but some characteristic differences show up. They are usually due to selection rules, which often favor symmetric vibrations for Raman. In the electrospun fibers of compound 1 shown in Fig. 5a, the N–H stretching band, expected at ≈3400 cm−1, is absent in Raman, and the “amide A” band (NH stretching in the secondary amides) is barely detectable. The presence of amides manifests best in the “amide I” (mainly C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) at ≈1690 cm−1, which is one of the strongest absorptions in IR. The strong Raman signal at 1610 cm−1, originating from in plane C–H bending vibrations on the fluorenyl, is much weaker in IR. Other bands in the Raman spectra correspond well to the IR bands: 3070 cm−1 and 2900 cm−1 for C–H stretches in the aromatic and aliphatic groups, respectively; 1480 cm−1 for the CH bendings; 1300 cm−1 for amide III (“stretch-open”); 1025 cm−1 for the “ring breathing” modes in the phenyl group. In contrast to IR spectra, Raman scattering can readily be collected from submicron spots. By scanning the Raman laser spot pixelwise, we obtained hyperspectral data sets from the fibers. They can be presented for selected Raman bands, which we show here for C–H stretching (3070 cm−1) and in plane bending (1610 cm−1) in the aromatic rings, and for amide III (Fig. 5b and ESI6† for additional information). The intensities scale exactly with the local fiber diameter, i.e. they are highest at the beads, and lowest at constrictions. This is observed for all Raman bands, and thus proves a very homogeneous structure, and the absence of agglomerates of modified molecules, contaminants, or solvent (HFIP would show very strong C–F stretching Raman bands, and thus be visible20).
O stretching) at ≈1690 cm−1, which is one of the strongest absorptions in IR. The strong Raman signal at 1610 cm−1, originating from in plane C–H bending vibrations on the fluorenyl, is much weaker in IR. Other bands in the Raman spectra correspond well to the IR bands: 3070 cm−1 and 2900 cm−1 for C–H stretches in the aromatic and aliphatic groups, respectively; 1480 cm−1 for the CH bendings; 1300 cm−1 for amide III (“stretch-open”); 1025 cm−1 for the “ring breathing” modes in the phenyl group. In contrast to IR spectra, Raman scattering can readily be collected from submicron spots. By scanning the Raman laser spot pixelwise, we obtained hyperspectral data sets from the fibers. They can be presented for selected Raman bands, which we show here for C–H stretching (3070 cm−1) and in plane bending (1610 cm−1) in the aromatic rings, and for amide III (Fig. 5b and ESI6† for additional information). The intensities scale exactly with the local fiber diameter, i.e. they are highest at the beads, and lowest at constrictions. This is observed for all Raman bands, and thus proves a very homogeneous structure, and the absence of agglomerates of modified molecules, contaminants, or solvent (HFIP would show very strong C–F stretching Raman bands, and thus be visible20).
Altogether, we find no evidence for increased hydrogen bonding in the fibers (different from Phe-Phe,21 for example). Given that the molecules are not charged, only dipoles and π-stacking of the aromatic systems should provide the required intermolecular interactions, which are responsible for the assembly of compound 1. Contrary to what might be expected from the presence of a large electrical field, the spinning process does not involve orienting the molecules, even though they have suitably high dipole moments, while it obviously allows for sufficient time for self-assembly. We furthermore found an amorphous structure, hence no long-range ordering (see ESI for X-ray diffraction, Fig. ESI5†).
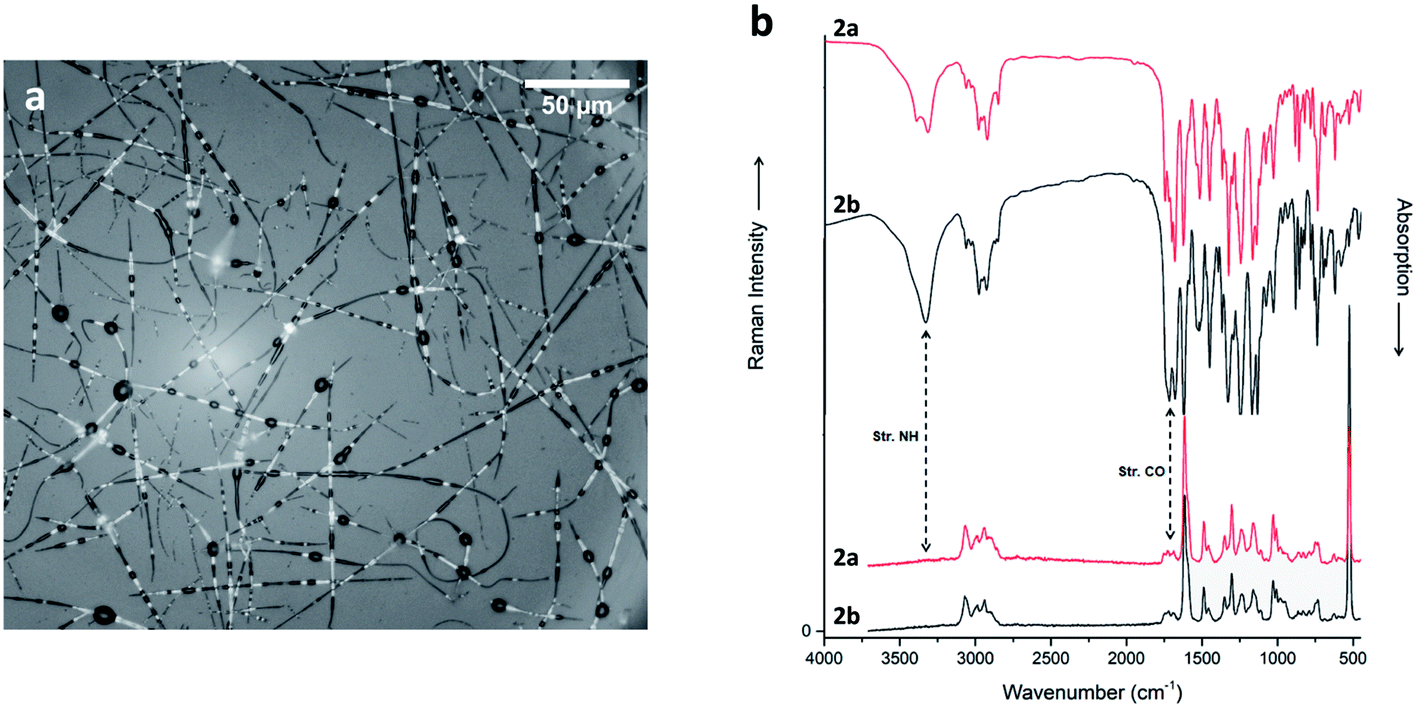 | ||
| Fig. 6 (a) Optical micrographs of compound 2a electrospun nanofibers (15 cm, 15 kV, 0.5 ml h−1), recorded in air in reflection (the strong contrast in the fibers results from various distances from the Si wafer substrate). (b) Raman (bottom) and IR spectra (top) of compounds 2a (black) and 2b (red) powder. Spectra are scaled and offset for clarity. | ||
The two diastereomers of compounds 3 show more subtle differences in behaviour at our selected electrospinning conditions. While both are well soluble in HFIP, compound 3a cannot be spun. Its diastereomer 3b gave only short and discontinuous nanofibers, which, however, are quite homogenous (Fig. 7a; see ESI for detailed statistics in Fig. ESI2†), with the same diameter range as fibers of compound 1. The fibers are, as the original substance, amorphous (ESI, Fig. ESI5†).
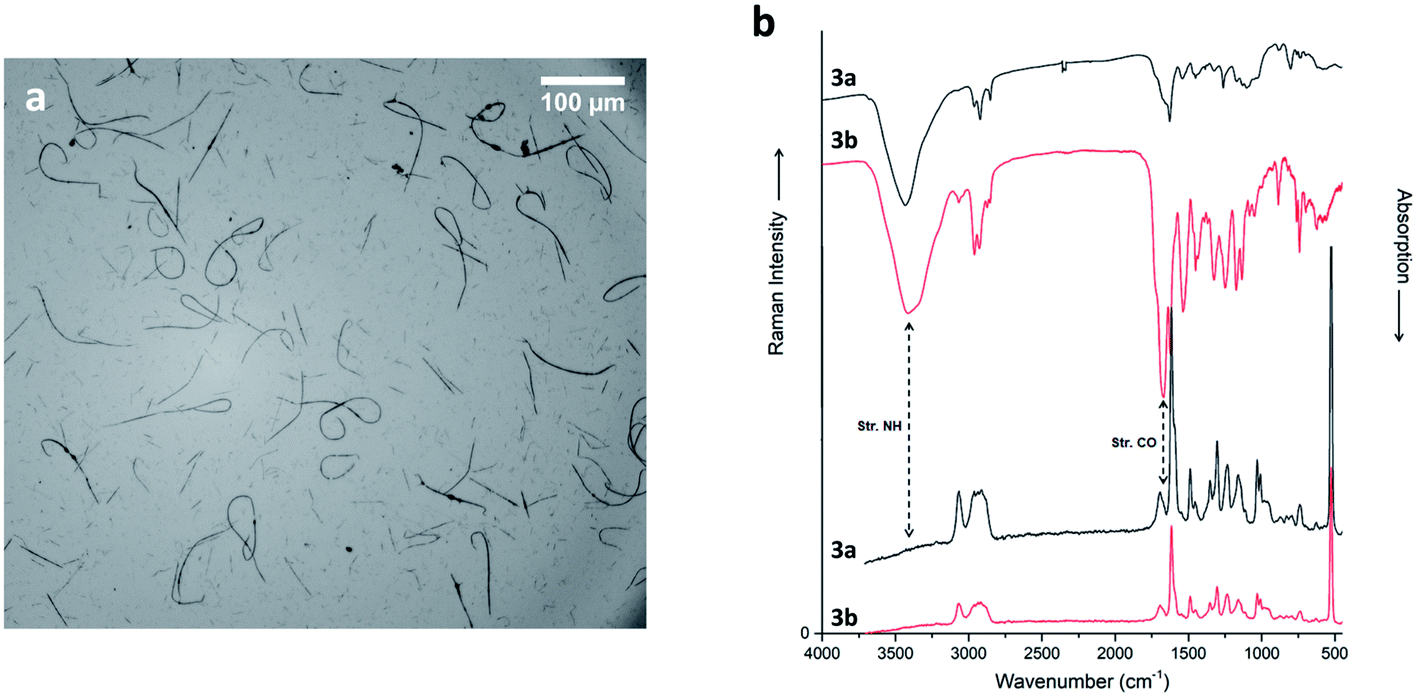 | ||
| Fig. 7 (a) Optical micrograph of compound 3b electrospun nanofibers (15 cm, 15 kV, 0.5 ml h−1), recorded in air. (b) Raman (bottom) and IR spectra (top) of compounds 3a (black) and 3b (red) powder. Spectra are scaled and offset for clarity. | ||
Concerning IR spectra (not Raman), substantial differences between the two diastereoisomers (Fig. 7b; see ESI† for additional information) are apparent. These changes manifest exclusively in the intensity of the peaks, but not in any shifts. This means, presumably, that the different configurations of 3a and 3b do not cause a very different intermolecular interaction (or crystal packing), but that some chemical groups are oriented in a different direction, such that vibrating dipoles (partially) cancel out. This fits nicely to the similar solubility in HFIP, and to the observed electrospinnability. In detail, the C–H stretching band at 2950 cm−1 is much stronger in 3b than in 3a, suggesting a different orientation of some aliphatic groups. Here we see a possible alternative to electrostatic, hydrogen bond and π-stacking interactions. Moreover, the amide I stretching band at 1670 cm−1 is highly intense only in 3b. The explanation could be a “closed” end-to-end structure for both diastereomers, but probably in 3a CH2 and CONH groups point outwards.
Cytotoxicity tests
Finally, the very interesting results obtained in particular with compound 1, prompted us to test the cytotoxicity of both powder and fibers with a view towards bio-applications. We employed a human colorectal adenocarcinoma cell line (Caco-2) in standard medium, with various concentrations of compound 1 (0 to 200 μM) or the corresponding fibers. We measured cell viability by a sulforhodamine B (SRB) assay,31,32 48 h after incubation. The effects of the compounds vs. control were analyzed by a two-tailed Student's t test for unpaired data. From these results it is clear that neither the powder nor the fibers induce significant cell toxicity (Fig. ESI7†). Although our viability test is rather simple, the tolerated high concentrations are a valuable first approach.Conclusion
In conclusion, the pyrazole-isothiazole derivatives 1, 2a, 2b, 3a, and 3b have been designed and synthesized, and their electrospinnability was tested. With exception of compound 2b, all compounds can be electrospun from HFIP solutions to micro- and nanowires, without the need of a polymer matrix. The best results were obtained with compound 1, which gave continuous, fairly homogenous, bead-free fibers. This is a very satisfactory result, taking into account that compound 1 is a small molecule and not a polymer. In fact even polymers can be more problematic in terms of beading (typical for PEO) and diameter inhomogeneity.Interestingly, the diastereomers (2a vs. 2b; 3a vs. 3b) showed different behavior: 2a gave long, although inhomogneous fibers, while 2b was not soluble in HFIP and cannot be electrospun at all under the same conditions. Diastereomers 3a and 3b are both well soluble in HFIP. At the electrospinning parameters set in our experiments, compound 3a cannot be electrospun, while its diastereomer 3b gives short and discontinuous nanofibers, which, however, are quite homogenous, with the same diameter range as fibers of compound 1. Apparently the different stereochemistry of the two enantiomeric bicyclic systems, combined with natural amino acids or short peptides, exerts very substantial influence on the electrospinnability of the two diastereomers. Our hypothesis, basing on vibrational spectroscopy, is that intermolecular hydrogen bonds are less important than π-stacking interactions for the fiber formation, pointing out the key role of aromatic systems for electrospinning.
With our work we disclose the electrospinnability of several small molecules. We enhance the spectrum of fundamental materials that can be used for the synthesis of nanofibers. This opens the way to generate nanofibers from new classes of compounds with the typical (large) application range of electrospun fibers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Mrs D. Nava for performing NMR experiments and Prof. S. Pieraccini for very helpful discussions. We are grateful for financial support from the Basque Government (Elkartek-ng 17). We acknowledge the Erasmus + Traineeship programme of the European Union for the contribution to the mobility of S. L.References
- D. B. Khadka and D. T. Haynie, Nanomedicine, 2012, 8, 1242–1262 CrossRef CAS PubMed.
- Kenry and C. T. Lim, Prog. Polym. Sci., 2017, 70, 1–17 CrossRef CAS.
- N. Bhardwaj and S. C. Kundu, Biotechnol. Adv., 2010, 28, 325–347 CrossRef CAS PubMed.
- B.-M. Min, G. Lee, S. H. Kim, Y. S. Nam, T. S. Lee and W. H. Park, Biomaterials, 2003, 25, 1289–1297 CrossRef.
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- S. A. Sell, P. S. Wolfe, K. Garg, J. M. McCool, I. A. Rodriguez and G. L. Bowlin, Polymers, 2010, 2, 522–553 CrossRef CAS.
- J. F. Almine, D. V. Bax, S. M. Mithieux, L. Nivison-Smith, J. Rnjak, A. Waterhouse, S. G. Wise and A. S. Weiss, Chem. Soc. Rev., 2010, 39, 3371–3379 RSC.
- N. Reddy, R. Reddy and Q. Jiang, Trends Biotechnol., 2015, 33, 362–369 CrossRef CAS PubMed.
- A. Balaji, S. K. Jaganathan, M. V. Vellayappan, A. A. John, A. P. Subramanian, M. SelvaKumar, H. Mohandas, M. Sundar Raj and E. Supriyanto, RSC Adv., 2015, 5, 69660–69679 RSC.
- S. Wu, J. Polym. Sci., Part B: Polym. Phys., 1989, 27, 723–741 CrossRef CAS.
- S. L. Shenoy, W. D. Bates, H. L. Frisch and G. E. Wnek, Polymer, 2005, 46, 3372–3384 CrossRef CAS.
- D. Fang, Y. Liu, S. Jiang, J. Nie and G. Ma, Carbohydr. Polym., 2011, 85, 276–279 CrossRef CAS.
- Z. Li and C. Wang, Effects of Working Parameters on Electrospinning, Springer, Berlin, Heidelberg, 2013, DOI:10.1007/978-3-642-36427-3_2.
- M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science (Washington, DC, U. S.), 2006, 311, 353–355 CrossRef CAS PubMed.
- S. T. Hemp, A. G. Hudson, M. H. Allen Jr, S. S. Pole, R. B. Moore and T. E. Long, Soft Matter, 2014, 10, 3970–3977 RSC.
- W. Nuansing, E. Georgilis, T. V. A. G. de Oliveira, G. Charalambidis, A. Eleta, A. G. Coutsolelos, A. Mitraki and A. M. Bittner, Part. Part. Syst. Charact., 2014, 31, 88–93 CrossRef CAS.
- A. Celebioglu and T. Uyar, Chem. Commun., 2010, 46, 6903–6905 RSC.
- A. Celebioglu and T. Uyar, J. Colloid Interface Sci., 2013, 404, 1–7 CrossRef CAS PubMed.
- W. Nuansing, A. Rebollo, J. M. Mercero, J. Zuniga and A. M. Bittner, J. Raman Spectrosc., 2012, 43, 1397–1406 CrossRef CAS.
- W. Nuansing, D. Frauchiger, F. Huth, A. Rebollo, R. Hillenbrand and A. M. Bittner, Faraday Discuss., 2013, 166, 209–221 RSC.
- G. Singh, A. M. Bittner, S. Loscher, N. Malinowski and K. Kern, Adv. Mater. (Weinheim, Ger.), 2008, 20, 2332–2336 CrossRef CAS.
- F. Clerici, E. Erba, M. L. Gelmi and S. Pellegrino, Tetrahedron Lett., 2016, 57, 5540–5550 CrossRef CAS.
- A. Bonetti, S. Pellegrino, P. Das, S. Yuran, R. Bucci, N. Ferri, F. Meneghetti, C. Castellano, M. Reches and M. L. Gelmi, Org. Lett., 2015, 17, 4468–4471 CrossRef CAS PubMed.
- A. Ruffoni, A. Contini, R. Soave, L. Lo Presti, I. Esposto, I. Maffucci, D. Nava, S. Pellegrino, M. L. Gelmi and F. Clerici, RSC Adv., 2015, 5, 32643–32656 RSC.
- A. Bonetti, F. Clerici, F. Foschi, D. Nava, S. Pellegrino, M. Penso, R. Soave and M. L. Gelmi, Eur. J. Org. Chem., 2014, 3203–3209 CrossRef CAS.
- A. Ruffoni, M. V. Cavanna, S. Argentiere, S. Locarno, S. Pellegrino, M. L. Gelmi and F. Clerici, RSC Adv., 2016, 6, 90754–90759 RSC.
- R. Bucci, P. Das, F. Iannuzzi, M. Feligioni, R. Gandolfi, M. L. Gelmi, M. Reches and S. Pellegrino, Org. Biomol. Chem., 2017, 15, 6773–6779 RSC.
- F. Clerici, M. L. Gelmi, C. Monzani, D. Pocar and A. Sala, J. Heterocycl. Chem., 2006, 43, 1045–1049 CrossRef CAS.
- T. Driessen, R. Jeurissen, H. Wijshoff, F. Toschi and D. Lohse, Phys. Fluids, 2013, 25, 062109 CrossRef.
- M. Lepere, C. Chevallard, G. Brezesinski, M. Goldmann and P. Guenoun, Angew. Chem., Int. Ed., 2009, 48, 5005–5009 CrossRef CAS PubMed.
- Y. P. Keepers, P. E. Pizao, G. J. Peters, J. Van Ark-Otte, B. Winograd and H. M. Pinedo, Eur. J. Cancer, 1991, 27, 897–900 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02486g |
| This journal is © The Royal Society of Chemistry 2019 |