
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterization of phosphoethanolamine-modified lipid A from probiotic Escherichia coli strain Nissle 1917†
Sung-Hyun Jo‡
 a,
Han-Gyu Park‡a,
Won-Suk Songb,
Seong-Min Kima,
Eun-Jung Kimc,
Yung-Hun Yangd,
Jae-Seok Kime,
Byung-Gee Kimb and
Yun-Gon Kim*a
a,
Han-Gyu Park‡a,
Won-Suk Songb,
Seong-Min Kima,
Eun-Jung Kimc,
Yung-Hun Yangd,
Jae-Seok Kime,
Byung-Gee Kimb and
Yun-Gon Kim*a
aDepartment of Chemical Engineering, Soongsil University, 369 Sangdo-Ro, Seoul 06978, Korea. E-mail: ygkim@ssu.ac.kr; Tel: +82-2-828-7099
bSchool of Chemical and Biological Engineering, Seoul National University, Seoul 08826, Korea
cInstitute of Molecular Biology and Genetics, Seoul National University, Seoul 08826, Korea
dDepartment of Biological Engineering, Konkuk University, Seoul 05029, Korea
eDepartment of Laboratory Medicine, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, Seoul 05355, Korea
First published on 25th June 2019
Abstract
Gut microbiota, a complex microbial community inhabiting human or animal intestines recently regarded as an endocrine organ, has a significant impact on human health. Probiotics can modulate gut microbiota and the gut environment by releasing a range of bioactive compounds. Escherichia coli (E. coli) strain Nissle 1917 (EcN), a Gram-negative bacterial strain, has been used to treat gastrointestinal (GI) disorders (i.e., inflammatory bowel disease, diarrhea, ulcerative colitis, and so on). However, endotoxicity of lipopolysaccharide (LPS), a major component of the cell wall of Gram-negative bacteria in the gut, is known to have a strong influence on gut inflammation and maintenance of gut homeostasis. Therefore, characterizing the chemical structure of lipid A which determines the toxicity of LPS is needed to understand nonpathogenic colonization and commensalism properties of EcN in the gut more precisely. In the present study, MALDI multiple-stage mass spectrometry analysis of lipid A extracted from EcN demonstrates that hexaacylated lipid A (m/z 1919.19) contains a glucosamine disaccharide backbone, a myristate, a laurate, four 3-hydroxylmyristates, two phosphates, and phosphoethanolamine (PEA). PEA modification of lipid A is known to contribute to cationic antimicrobial peptide (CAMP) resistance of Gram-negative bacteria. To confirm the role of PEA in CAMP resistance of EcN, minimum inhibitory concentrations (MICs) of polymyxin B and colistin were determined using a wild-type strain and a mutant strain with deletion of eptA gene encoding PEA transferase. Our results confirmed that MICs of polymyxin B and colistin for the wild-type were twice as high as those for the mutant. These results indicate that EcN can more efficiently colonize the intestine through PEA-mediated tolerance despite the presence of CAMPs in human gut such as human defensins. Thus, EcN can be used to help treat and prevent many GI disorders.
1. Introduction
Gut microbiota, a microbial community inhabiting human or animal intestines recently regarded as an endocrine organ, has a significant impact on human health.1,2 Gut bacteria can ferment indigestible nutrients and produce biomolecules (i.e., antioxidants, vitamins, short chain fatty acids, etc.) known to be beneficial to health. Thus, it has a beneficial effect on the human body such as development of the immune system and prevention of colonization by pathogens.3–6 On the other hand, dysbiosis of gut microbiota is known to cause human diseases such as inflammatory bowel disease, diarrhea, and ulcerative colitis.7 In this context, probiotics as beneficial microbes are attracting interest to treat and prevent intestinal diseases.8Escherichia coli strain Nissle 1917 (EcN), a Gram-negative bacterial strain, has been registered as a microbial drug called Mutaflor (Ardeypharm GmbH, Herdecke, Germany) to treat many gastrointestinal (GI) disorders including inflammatory bowel disease (IBD), diarrhea, ulcerative colitis, and so on.9,10 This strain is not pathogenic. It does not have any a major virulence factor such as toxin, mannose-resistant hemagglutination, etc.10–12 It is known to have immunomodulatory and anti-inflammatory effects as well as fitness factors (i.e., microcin M, siderophores) through many studies.10,11 However, it has been reported that lipopolysaccharide (LPS) of intestinal Gram-negative bacteria such as Bacteroides and Prevotella spp. can trigger IBD.13
LPS, the molecule embedded in outer-membrane of Gram-negative bacteria, is composed of three major components: (1) O-polysaccharide (or O-antigen), (2) core-oligosaccharide, and (3) lipid A.15 LPS can cause immune response by activating Toll-like receptor 4 (TLR4). In severe cases, it can cause septic shock. Thus, it is known as an endotoxin.14–16 It is known that the toxicity of LPS is affected by its chemical structure and that lipid A can strongly affect the activation of TLR4.15 Thus, the identification of chemical structure of lipid A is important. The chemical structure (i.e., acylation pattern, phosphorylation pattern, modification of phosphoethanolamine (PEA), modification of 4-amino-4-deoxy-L-arabinose, and so on) of lipid A, the lipophilic anchor part of LPS in outer-membrane of Gram-negative bacteria, differs depending on species. Change of its chemical structure also affects its immunogenicity.14,17,18 Chemical structure of LPS of EcN and serum sensitive properties according to its structure (especially O-antigen region) have been studied.12 However, previous studies did not elucidate the precise chemical structure of lipid A such as acylation pattern or phosphorylation pattern. Therefore, analysis of the chemical structure of lipid A is needed to understand the immunogenicity of LPS from EcN.
Here, we characterized the exact chemical structure of lipid A from EcN, which is not yet elucidated such as acylation and phosphorylation pattern and PEA modification of lipid A, using MALDI-based mass spectrometry systems. Several studies have performed biomolecule qualitative analysis with laser desorption/ionization (LDI)-based mass spectrometry.19–22 However, the average molecular weight of lipid A from E. coli is generally around m/z 1700–2000, so we collected the MALDI spectra using the common matrix and organic solvent.23–25 Thus, MALDI-TOF MS was applied to validate the extraction of lipid A and to analyze the full mass of lipid A from EcN. Structure of PEA-modified lipid A was characterized using MALDI-QIT-TOF MS based multiple stages of fragmentation (up to MS3) to analyze chemical structure more precisely. PEA modification of lipid A is known to increase the resistance to cationic antimicrobial peptide (CAMP) by diminishing net charge of the outer membrane of Gram-negative bacteria.26–29 To confirm this, a mutant was constructed by deleting eptA gene encoding PEA transferase. The effect of PEA modification in EcN on the tolerance to CAMPs was verified by determining minimum inhibitory concentrations (MIC) for polymyxin B and colistin (Fig. 1). Our results indicated that PEA modification of lipid A in EcN may help gut colonization and survival by providing tolerance to CAMPs.
 | ||
| Fig. 1 The characterization of the lipid A molecule of Gram-negative probiotic strain Escherichia coli strain Nissle 1917 and cationic antimicrobial peptide tolerance via phosphoethanolamine modification of lipid A. | ||
2. Materials and methods
2.1. Bacterial growth condition and preparation of lipid A sample
In this study, we used Escherichia coli strain Nissle 1917 kindly provided by Professor Byung-Gee Kim (Seoul National University, Korea) and EcN mutant (ΔeptA). Bacteria were plated onto Luria–Bertani (LB) agar (Becton, Dickinson and Company, Franklin lakes, NJ, USA) and incubated at 37 °C for 24 h. A single colony was obtained and inoculated into 50 mL of LB broth (Becton, Dickinson and Company, Franklin lakes, NJ, USA) and then grown at 37 °C for 16 h in a shaking incubator at 250 rpm. Then 1.5 mL of cell suspension was used to obtain cell pellet by centrifuging at 8000 rpm for 2 min at 4 °C.LPS extraction from the cell pellet was performed using LPS extraction kit (Intron Biotechnology Inc., Sungnam, Korea). Briefly, 1 mL of lysis buffer was added to cell pellet and vigorously vortexed to lyse cells completely. After adding 200 μL of chloroform to the lysate, the mixture was vortexed for 20 s and incubated at room temperature for 5 min. The mixture was then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C. Subsequently, 400 μL of the supernatant was transferred to a new microtube. After adding 800 μL of purification buffer, the mixture was incubated at −20 °C for 10 min. LPS pellet was obtained by centrifugation at 13000 rpm for 15 min at 4 °C. The extracted LPS was washed with 1 mL of 70% ethanol and completely dried using speed-vacuum.
000 rpm for 10 min at 4 °C. Subsequently, 400 μL of the supernatant was transferred to a new microtube. After adding 800 μL of purification buffer, the mixture was incubated at −20 °C for 10 min. LPS pellet was obtained by centrifugation at 13000 rpm for 15 min at 4 °C. The extracted LPS was washed with 1 mL of 70% ethanol and completely dried using speed-vacuum.
Hydrolysis with 1% acetic acid was performed to extract lipid A from LPS. After adding 200 μL of 1% acetic acid to the dried LPS pellet, LPS was hydrolyzed at 100 °C for 1 h. After adding 400 μL of chloroform and 200 μL of methanol to hydrolyzed LPS, the mixture was centrifuged at 8000 rpm for 15 min at 15 °C. Finally, the chloroform layer (bottom layer) was transferred to a new tube and dried by nitrogen gas.
2.2. MALDI-TOF MS and MALDI-QIT-TOF MSn analysis
Dried lipid A samples were dissolved with 10 μL of chloroform. Then 1 μL of lipid A sample and 1 μL of 50 mg mL−1 super-DHB (in 70% acetonitrile/water) (Sigma-Aldrich, St. Louis, MO, USA) were loaded onto a MSP 96 target polished steel MALDI plate and dried at room temperature. Lipid A samples were analyzed using Bruker Daltonics Microflex LRF MALDI-TOF MS instrument and FlexControl 3.0 software (Bruker, Bremen, Germany). Operating conditions were as follows: negative-ion and reflectron mode, detector gain = 6.9, laser frequency = 60.0 Hz, and laser power = 87%. MS/MS analysis of lipid A was performed using an Axima Resonance MALDI-quadrupole ion trap-TOF instrument (Shimadzu, Manchester, UK). Fragment ions were analyzed by collision-induced dissociation (CID) of parent ions and argon gas was used as a collision gas. Operating conditions were as follows: negative ion and reflectron mode, intro endcap = −5.5 kV, extr endcap = 10.0 kV, flight tube = 10.0 kV, reflectron center = −0.2 kV, reflectron back = −0.2 kV, detector = 2.2 kV, and laser intensity: 60–100 power. Data acquisition and processing were performed using Launchpad 2.9.3 software (Kratos Analytical, Manchester, UK).2.3. Homology search of eptA gene and construction of eptA mutant
Homology search of phosphoethanolamine transferase encoding gene (eptA) of Escherichia coli strain Nissle 1917 was performed using BLASTN and BLASTX programs (the National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/BLAST/).11,12Construction of eptA mutant of EcN was performed using Lambda red-mediated recombineering method.30 Briefly, plasmid pKD46 containing the gene expressing Lambda red-recombinase was introduced to EcN using electroporation followed by plating onto LB agar plate containing ampicillin and culturing at 30 °C for 16 h. Next, kanamycin resistance gene cassette from pKD4 plasmid was amplified by polymerase chain reaction (PCR) using Herculase II Fusion DNA polymerase (Agilent, CA, USA). Primers used here are as follows: forward primer 5′-CAGAATCAATCCCTGCAATAACAGCGTATCGTCTTCAACAATCAGAATTTTGTGTAGGCTGGAGCTGCTT-3′, reverse primer 5′-TAATTTTGCTTTGCGAGCATATGCGCACTTTGTTCGATGGAAACACCGTGTCCTCCTTAGTTCCTATTCC-3′. Amplified gene template was cleaved using DpnI (NEB, MA, USA) as the restriction endonuclease and purified using Expin PCR SV kit (Geneall Biotechnology, Korea). The purified kanamycin resistance gene cassette was further introduced by electroporation using EcN with pKD46 plasmid. The eptA deletion mutant of EcN was confirmed by DNA sequencing using a 3730XL DNA Analyzer (Applied Biosystems, CA, USA). Primers used here are as follows: forward primer 5′-GGCAGAGTGTATTTTTTCTG-3′, reverse primer 5′-GCGAAAGAGGTTATGTGGTC-3′.
2.4. Minimum inhibitory concentration (MIC) test of cationic antimicrobial peptides
To confirm the role of PEA modification in lipid A from EcN, we conducted MIC test for polymyxin B and colistin known as cationic antimicrobial peptides using broth microdilution method.31 Briefly, 100 μL of two-fold serial dilutions of antibiotics (concentration range of 16 to 0.031 mg mL−1) in LB broth were dispensed into 96-well microtiter plates. Bacteria cultured for 16 h in 3 mL of LB broth were then diluted with LB broth to a concentration of 1 × 106 cfu mL−1 and 50 μL was added to each well. MICs were determined as the lowest concentration of antibiotic that did not allow visible bacterial growth after incubation at 37 °C for 24 h.3. Results and discussion
3.1. MS analysis of lipid A from Escherichia coli strain Nissle 1917 by MALDI-TOF MS
To obtain lipid A from Escherichia coli strain Nissle 1917, we extracted LPS using phenol/water based LPS extraction method. Lipid A was collected by hydrolysis with 1% acetic acid.32 To elucidate the chemical structure of lipid A, MALDI-TOF MS-based preliminary analysis was performed because MALDI-TOF MS could only provide the spectrum containing structural information by inducing fragmentation of relatively unstable chemical bonds through “in-source decay”.17 In this study, several quasi-molecular ions such as [M–H]−, [M + Na–2H]− and [M + K–2H]− can be detected in MALDI MS analysis on negative ion mode. However, in this study, only one [M + Na–2H]− ion was detected and the rest were all detected as [M–H]−. Therefore, unless otherwise stated, peaks correspond to [M–H]− ion.17,33 Based on preceding studies of lipid A analysis, the major peak at m/z 1796.2 was predicted to represent hexaacylated lipid A structure comprising of glucosamine (GlcN) disaccharide backbone, six acyl groups placed asymmetrically, and phosphate groups at the C-1 and C-4′ position.17,24,34 A peak at m/z 1919.0 was expected to be a structure that might contain phosphoethanolamine (PEA) modification (Fig. 2).35,36 In the mass spectrum, a peak at m/z 1585.5 indicates the elimination of PEA and C14:0 acyl chain from m/z 1919.0 while a peak at m/z 1280.8 indicates elimination of C14:0 (3-OH) acyl chain and phosphate group from m/z 1585.5.17 A peak at m/z 1716.1 is the result of cleavage of a phosphate group at the C-1 position because fragmentation by electron transfer is more likely to occur in the phosphate group at the C-1 position than the phosphate group at the C-4′ position.24,37 A peak at m/z 1510.2 is the result of quasi-molecular ion [M + Na–2H]− cleaved a C14:0 acyl chain from m/z 1796.2.24,37 In addition, peak ‘clusters’ differing by 14 m/z units around each main peak was detected in the mass spectrum. Such cluster might be due to the heterogeneity of acyl chain length.38 By combining this structural information, we could confirm that lipid A contains a C14:0 acyl chain, a C14:0 (3-OH) acyl chain, and a phosphate group.17,18 Therefore, tandem mass spectrometric analysis (MSn) via MALDI-QIT TOF MS was performed to obtain additional structural information.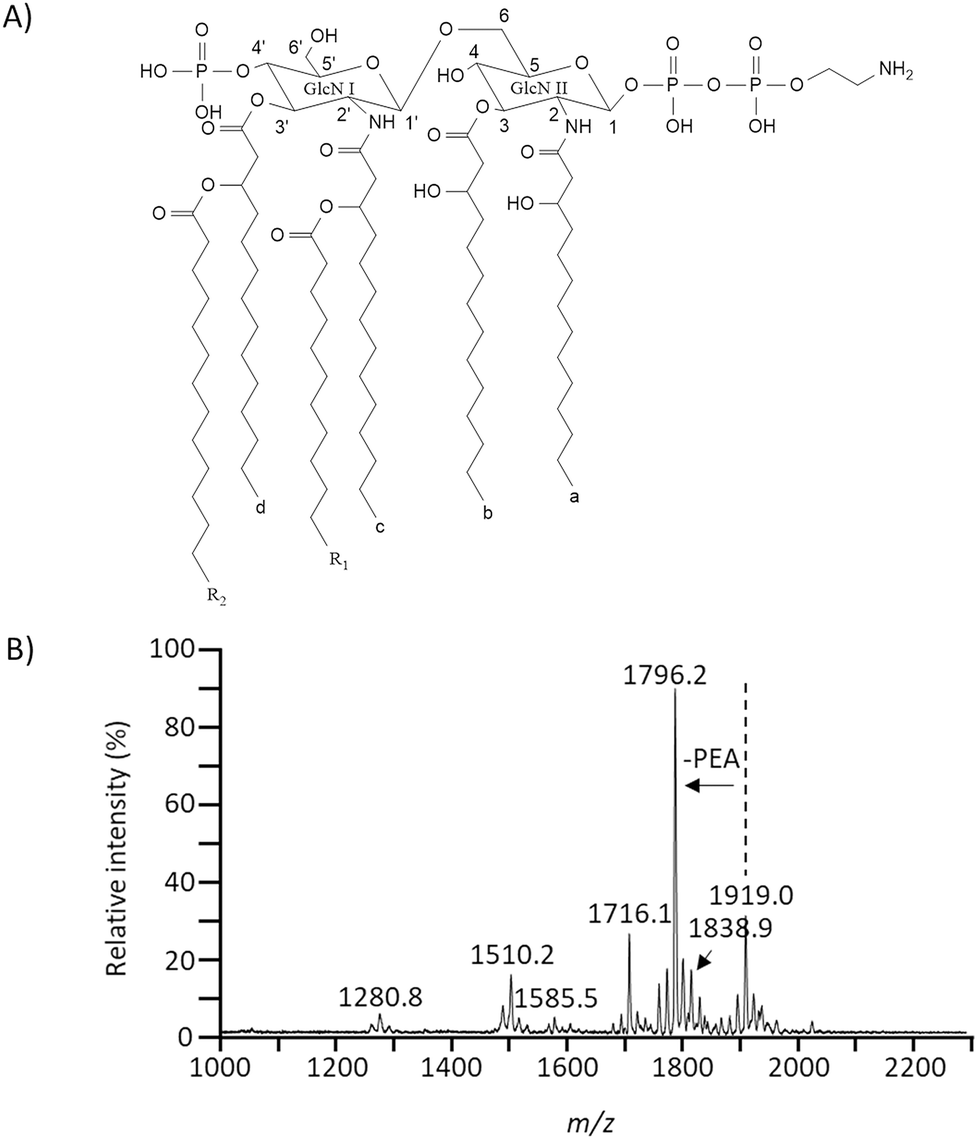 | ||
| Fig. 2 The proposed chemical structure of lipid A from Escherichia coli strain Nissle 1917 (A), negative-ion MALDI-TOF MS spectra of lipid A (B). | ||
3.2. MSn analysis of lipid A from Escherichia coli strain Nissle 1917 by MALDI-QIT TOF MS
MALDI-TOF QIT MS-based MSn analysis is suitable for analysis of chemical structure because it can select and fragment ions corresponding to the desired peak using ion trap and collision-induced dissociation (CID).39 First, fragment ions (fragmented by “in-source decay”) not detected in the analysis using MALDI-TOF MS were detected in MS analysis using MALDI-TOF-QIT MS. Structures corresponding to peaks are shown in Fig. 3. Detected peaks at m/z 1488.43 and 1244.57 were predicted to show cleaved structure of PEA, the phosphate group and C14:0 acyl chain, and PEA, the phosphate group, C14:0 acyl chain and C14:0 (3-OH), respectively. Peaks at m/z 1036.42 and 1018.40 were expected to be due to C14:0 (3-OH) acyl chain removed from the structure corresponding to peak m/z 1244.57 by charge-driven and charge-remote processes, respectively. To explain this, in the fragmentation of lipid A by CID, the acyl chain is removed in the form of neutral ketone derivative by ‘charge-driven process’ and in the form of free fatty acid by ‘charge-remote process’. These two processes are competitive.40,41 The m/z difference of the fragment ion is 18 in the spectrum.41 | ||
| Fig. 3 Negative-ion MALDI-QIT TOF MS mass spectra of lipid A and the lipid A structures corresponding to each ion peak were drawn as neutral molecules. | ||
To characterize these expected structures, MS2 analysis of m/z 1919.85 corresponding to one of parent ions was performed. Through MS2 analysis of m/z 1919.85, it was confirmed that the modification of PEA existed in lipid A based on the mass difference (m/z Δ123) (Fig. 4). MS2 analysis of the major peak at m/z 1796.87 was also performed to obtain further chemical structural information of lipid A (Fig. 5). As a result, a fragment peak at m/z 710.56 was identified as the Y1-type fragment ion. This peak indicated the presence of ester-linked primary acyl chain (C14:0 (3-OH) ‘b’) at the C-3 position and amide-linked primary acyl chain (C14:0 (3-OH) ‘a’) at the C-2 position as well as phosphate group at the C-1 position (Fig. S1†). In addition, because deacylation of lipid A in the analysis by CID generally occurs in ester bond which is relatively labile compared to amide bond, the presence of ester-linked C14:0 (3-OH) acyl chain without a secondary acyl chain and the presence of secondary acyl chain C14:0 liked primary acyl chain were confirmed through fragment peaks at m/z 1552.41 and 1324.12, respectively (Fig. 5).17 A peak at m/z 1244.10 was the result of cleavage of the ‘b’ acyl chain, C14:0 acyl chain, and the phosphate group. The secondary acyl chain C14:0 was found to bind to ester-linked C14:0 (3-OH) acyl chain to form an acyloxyacyl chain because the mass difference between the peak at m/z 1017.89 and 1244.10 had a specific mass difference (m/z 226) that could not be generated by fragmentation of C14:0, C14:0 (3-OH), and C12:0 acyl chain.
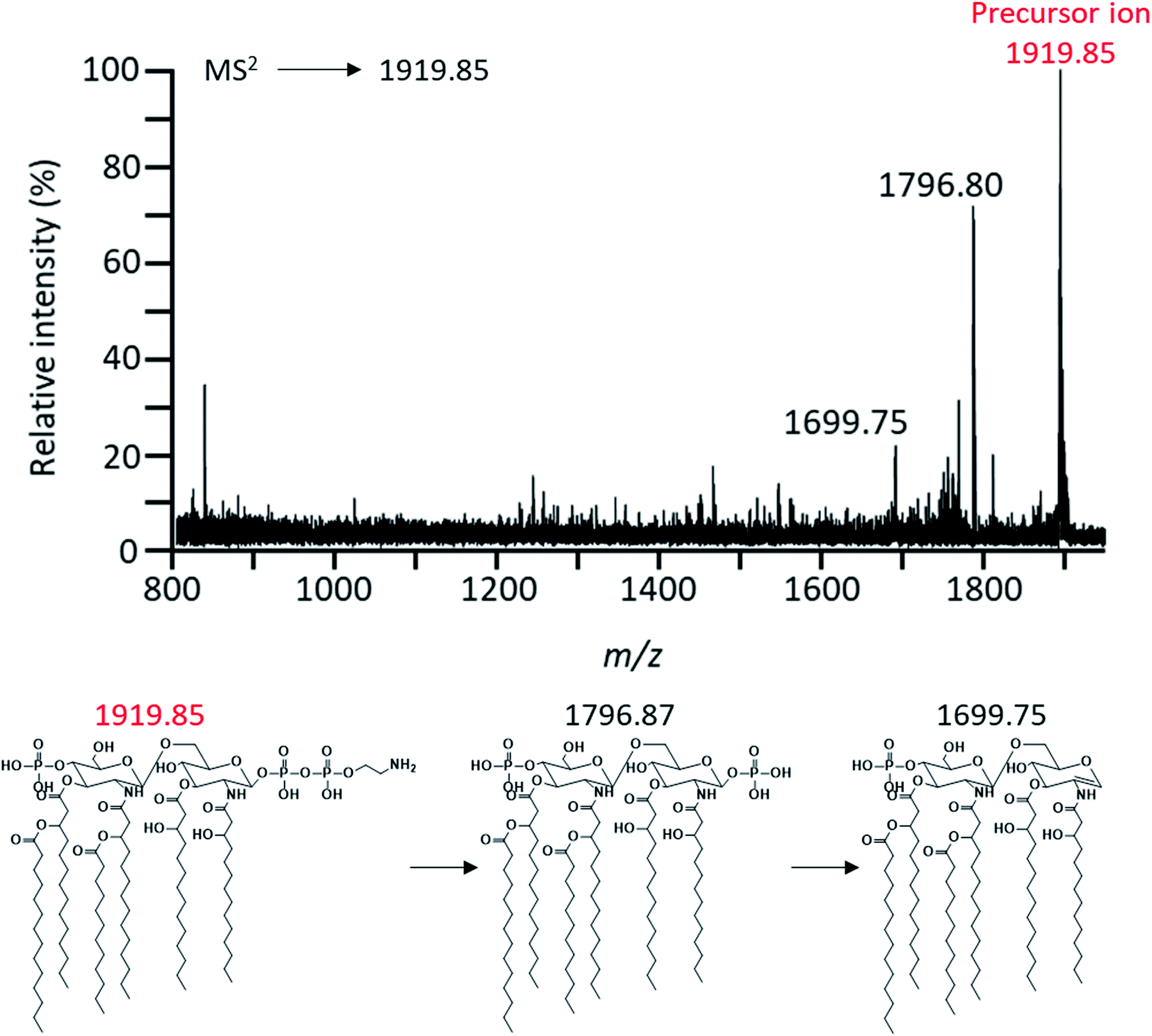 | ||
| Fig. 4 Negative-ion MALDI-QIT TOF MS2 spectrum of parent peak at m/z 1919.85 and fragment structures. Precursor ion was indicated by the mass to charge ratio of the red letter. | ||
 | ||
| Fig. 5 Negative-ion MALDI-QIT TOF MS2 spectrum of parent peak at m/z 1796.87 and fragment structures. Precursor ion was indicated by the mass to charge ratio of the red letter. | ||
MS3 analysis of m/z 1454.37 was performed to determine the position of acyl chain in lipid A and confirm the presence of the secondary acyl chain (C12:0 ‘R1’) and the phosphate group at the C-4′ position (Fig. 6). The fragment ion at m/z 929.16 detected by this analysis was due to cleavage of the ‘R1’ acyl chain, ‘R2’ acyl chain (C14:0) and two phosphate groups. And then, the fragment ion at m/z 650.06 was B1-type fragment ion. The presence of phosphate group at the C-4′ and the acyl chain ‘R1’ was found to bind to amide-linked primary acyl chain (C14:0 (3-OH) ‘c’) on GlcN II due to the parent ion which was cleaved primary acyl chain (C14:0 (3-OH) ‘d’) at the C-3′ position by ‘charge remote process’ already.17,42 In this context, the hydroxyl group at the C-3′ position of this fragment ion was the result of the ‘charge-driven process’ of the acyloxyacyl chain (‘d’ and ‘R2’ acyl chain). This information of the chemical structure of lipid A was also confirmed by tandem mass analysis in other pathways (Fig. S2–4†).
 | ||
| Fig. 6 Negative-ion MALDI-QIT TOF MS3 spectrum of parent peak at m/z 1454.37 and fragment structures. Precursor ion was indicated by the mass to charge ratio of the red letter. | ||
Taken together, we could confirm the lipid A structure from EcN as shown in Fig. 2A. Due to this lipid A structure EcN, we can deduce that LPS from EcN has immunomodulatory effect by up-regulating pro-inflammatory cytokine and IL-10, an anti-inflammatory cytokine.43 Properties of EcN as probiotics are expected to be due to difference in the distinct chemical structure of lipid A from EcN, unlike other known pathogenic strains of E. coli. Through MALDI multiple-stage mass spectrometry analysis, we demonstrated that the chemical structure of lipid A from EcN comprised of glucosamine disaccharide backbone, four 3-hydroxylmyristates, a myristate, a laurate, two phosphate groups, and phosphoethanolamine. Phosphoethanolamine modification of lipid A is mediated by PEA transferase. It is known to contribute to resistance to cationic antimicrobial peptides (CAMPs) such as polymyxin B, colistin, α-defensin, β-defensin, and human cathelicidin (LL-37) by diminishing the negative net charge of the outer membrane.26–29 Therefore, we performed the minimum inhibitory concentration (MIC) test for polymyxin B and colistin belonging to CAMPs to confirm the difference in resistance of EcN wild-type and ΔeptA mutant to CAMPs.
3.3. Phosphoethanolamine modification of lipid A from Escherichia coli strain Nissle 1917 is mediated by eptA gene with effect on tolerance to polymyxin B and colistin
Through the result of the chemical structure analysis of lipid A, we demonstrated the presence of PEA modification in lipid A from EcN (Fig. 2A). To confirm the effect of PEA modification in lipid A on antibiotic resistance of EcN, a comparative experiment was performed by deleting a gene encoding PEA transferase. In order to find the gene sequence encoding phosphoethanolamine transferase in EcN, a homology search was conducted through BLASTN using eptA gene sequence of E. coli K12 as a reference.44 As a result, we found that the sequence shared 92% homologies with the EcN genome. After confirming that this sequence might encode PEA transferase via BLASTX, we constructed a mutant with deletion of eptA gene. Lipid A from the mutant strain was analyzed using MALDI-TOF MS in order to confirm that the gene encoding PEA transferase was deleted. As a result of MALDI-TOF MS analysis, peaks at m/z 1919.0 and 1838.9 corresponding to PEA modified lipid A were not detected, unlike the MS spectrum of wild-type strain (the m/z 1838.9 corresponded to the structure cleaved a phosphate group from m/z 1919.0) (Fig. 2B and 7A). These results suggested that the eptA gene encoding lipid A PEA transferase was successfully deleted from EcN. | ||
| Fig. 7 Negative-ion MALDI-TOF MS mass spectra of lipid from the eptA mutant (A), the result of minimum inhibitory concentration test (B). | ||
The difference in resistance of wild-type and ΔeptA mutant to polymyxin B and colistin belonging to CAMPs was determined through minimum inhibitory concentration (MIC) test.45–47 Although the wild-type strain did not have any inherent antibiotics resistance, we confirmed that the wild-type strain had two-fold higher MIC value (p-value ≤ 0.01, n = 32) than the mutant strain for CAMPs (Fig. 7B).48,49 This result suggests that PEA modification of lipid A in EcN may contribute to resistance to CAMPs and that PEA modification of lipid A in the intestine may help them survive against endogenous human CAMPs (innate immunity).27,47 However, antibiotic is still the most potent factor that can cause an imbalance in intestinal microorganisms for maintaining a symbiotic relationship. It can reduce intestinal microbial diversity and cause relative overgrowth of certain species. Therefore, basic researches are needed to determine how beneficial microorganisms and pathobionts respond to antibiotics and what mechanisms can cause proliferation and development of these organisms in the host environment changed by antibiotics. We expect that such studies will contribute to the development of new therapies for infectious diseases.
4. Conclusion
In this study, we analyzed and demonstrated the chemical structure of lipid A from probiotic bacteria Escherichia coli strain Nissle 1917 (EcN), one of intestinal microorganisms, using MALDI-TOF MS and MALDI-TOF QIT MS. Our results confirmed that lipid A from EcN comprised of glucosamine disaccharide backbone, four 3-hydroxylmyristates, a myristate, a laurate, two phosphate groups, and phosphoethanolamine (PEA). Analysis of lipid A structure of EcN was also helpful for explaining the inherent immunomodulatory properties of EcN. To determine the effect of PEA modification specifically substituted in lipid A form of EcN on resistance to CAMPs, MIC test was performed for polymyxin B and colistin. Our results confirmed that the wild-type strain had two-fold higher MIC values than the mutant.Our results also revealed that PEA modification of lipid A from EcN could diminish the net negative charge of the outer membrane and thus provide tolerance to CAMPs. Therefore, it is expected that this study will be able to improve our understanding of nonpathogenic colonization and commensalism characteristics of EcN as probiotic bacteria in the intestine. It will also be helpful to study the commensalism of intestinal Gram-negative bacteria.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2015M1A5A1037196, NRF-2017M3A9B6062989, NRF-2018R1D1A1B07048185, NRF-2017M3A9E4077234).References
- J. M. Evans, L. S. Morris and J. R. Marchesi, J. Endocrinol., 2013, 218, R37–R47 CAS
.
- G. Clarke, R. M. Stilling, P. J. Kennedy, C. Stanton, J. F. Cryan and T. G. Dinan, Mol. Endocrinol., 2014, 28, 1221–1238 CrossRef PubMed
- F. Sommer and F. Bäckhed, Nat. Rev. Microbiol., 2013, 11, 227–238 CrossRef CAS PubMed
- S. Krishnan, N. Alden and K. Lee, Curr. Opin. Biotechnol., 2015, 36, 137–145 CrossRef CAS PubMed
- W. R. Wikoff, A. T. Anfora, J. Liu, P. G. Schultz, S. A. Lesley, E. C. Peters and G. Siuzdak, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3698–3703 CrossRef CAS PubMed
- L. G. Hersoug, P. Møller and S. Loft, Obes. Rev., 2016, 17, 297–312 CrossRef CAS PubMed
- Y. Belkaid and T. W. Hand, Cell, 2014, 157, 121–141 CrossRef CAS PubMed
- C. Pagnini, R. Saeed, G. Bamias, K. O. Arseneau, T. T. Pizarro and F. Cominelli, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 454–459 CrossRef CAS PubMed
- B. Arribas, M. E. Rodríguez-Cabezas, D. Camuesco, M. Comalada, E. Bailón, P. Utrilla, A. Nieto, A. Concha, A. Zarzuelo and J. Gálvez, Br. J. Pharmacol., 2009, 157, 1024–1033 CrossRef CAS PubMed
- U. Sonnenborn and J. Schulze, Microb. Ecol. Health Dis., 2009, 21, 122–158 CrossRef CAS
- L. Grozdanov, C. Raasch, J. Schulze, U. Sonnenborn, G. Gottschalk, J. Hacker and U. Dobrindt, J. Bacteriol., 2004, 186, 5432–5441 CrossRef CAS PubMed
- L. Grozdanov, U. Zähringer, G. Blum-Oehler, L. Brade, A. Henne, Y. A. Knirel, U. Schombel, J. Schulze, U. Sonnenborn, G. Gottschalk, J. Hacker, E. T. Rietschel and U. Dobrindt, J. Bacteriol., 2002, 184, 5912–5925 CrossRef CAS PubMed
- G. Loh and M. Blaut, Gut Microbes, 2012, 3, 544–555 CrossRef PubMed
- M. L. DeMarco and R. J. Woods, Mol. Immunol., 2011, 49, 124–133 CrossRef CAS PubMed
- A. Aderem and R. J. Ulevitch, Nature, 2000, 406, 782–787 CrossRef CAS PubMed
- R. J. Ulevitch and P. S. Tobias, Curr. Opin. Immunol., 1999, 11, 19–22 CrossRef CAS PubMed
- P. K. Sarkar, P. K. Prajapati, V. J. Shukla, B. Ravishankar and A. K. Choudhary, Mass Spectrom. Rev., 2009, 47, 987–992 CAS
- C. R. H. Raetz, C. M. Reynolds, M. S. Trent and R. E. Bishop, Annu. Rev. Biochem., 2008, 295–329 Search PubMed
- L. Huang, J. Wan, X. Wei, Y. Liu, J. Huang, X. Sun, R. Zhang, D. D. Gurav, V. Vedarethinam, Y. Li, R. Chen and K. Qian, Nat. Commun., 2017, 8, 220 CrossRef PubMed
- V. Vedarethinam, L. Huang, W. Xu, R. Zhang, D. D. Gurav, X. Sun, J. Yang, R. Chen and K. Qian, Small, 2019, 15, 1803051 CrossRef PubMed
- R. A. Picca, C. D. Calvano, N. Cioffi and F. Palmisano, Nanomaterials, 2017, 7, 75 CrossRef PubMed
- X. Sun, L. Huang, R. Zhang, W. Xu, J. Huang, D. D. Gurav, V. Vedarethinam, R. Chen, J. Lou, Q. Wang, J. Wan and K. Qian, ACS Cent. Sci., 2018, 4, 223–229 CrossRef CAS PubMed
- M. Kurogochin and S. I. Nishimura, Anal. Chem., 2004, 76, 6097–6101 CrossRef PubMed
- C. S. Lee, Y. G. Kim, H. S. Joo and B. G. Kim, J. Mass Spectrom., 2004, 39, 514–525 CrossRef CAS PubMed
- T. W. Cullen, J. P. O'Brien, D. R. Hendrixson, D. K. Giles, R. I. Hobb, S. A. Thompson, J. S. Brodbelt and M. S. Trent, Infect. Immun., 2013, 81, 430–440 CrossRef CAS PubMed
- J. Huang, Y. Zhu, M. L. Han, M. Li, J. Song, T. Velkov, C. Li and J. Li, Int. J. Antimicrob. Agents, 2018, 51, 586–593 CrossRef CAS PubMed
- A. Peschel and H. G. Sahl, Nat. Rev. Microbiol., 2006, 4, 529–536 CrossRef CAS PubMed
- M. P. Trombley, D. M. B. Post, S. D. Rinker, L. M. Reinders, K. R. Fortney, B. W. Zwickl, D. M. Janowicz, F. M. Baye, B. P. Katz, S. M. Spinola and M. E. Bauer, PLoS One, 2015, 10, 1–20 CrossRef PubMed
- J. S. Gunn, J. Endotoxin Res., 2001, 7, 57–62 CrossRef CAS PubMed
- A. D. Kirill and L. W. Barry, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640–6645 CrossRef PubMed
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed
- E. Thye Yin, C. Galanos, S. Kinsky, R. A. Bradshaw, S. Wessler, O. Lüderitz and M. E. Sarmiento, Biochim. Biophys. Acta, Gen. Subj., 1972, 261, 284–289 CrossRef
- R. Zenobi and R. Knochenmuss, Mass Spectrom. Rev., 1998, 17, 337–366 CrossRef CAS
- D. Karibian, A. Brunelle, L. Aussel and M. Caroff, Rapid Commun. Mass Spectrom., 1999, 13, 2252–2259 CrossRef CAS PubMed
- Z. Zhou, S. Lin, R. J. Cotter and C. R. H. Raetz, Biochemistry, 1999, 274, 18503–18514 CAS
- S. H. Kim, W. Jia, V. R. Parreira, R. E. Bishop and C. L. Gyles, Microbiology, 2006, 152, 657–666 CrossRef CAS PubMed
- H. G. Park, G. Sathiyanarayanan, C. H. Hwang, D. H. Ann, J. H. Kim, G. Bang, K. S. Jang, H. W. Ryu, Y. K. Lee, Y. H. Yang and Y. G. Kim, Sci. Rep., 2017, 7, 2168 CrossRef PubMed
- F. Renzi, U. Zähringer, C. E. Chandler, R. K. Ernst, G. R. Cornelis and S. J. Ittig, Infect. Immun., 2015, 84, 550–561 CrossRef PubMed
- C. Koy, S. Mikkat, E. Raptakis, C. Sutton, M. Resch, K. Tanaka and M. O. Glocker, Proteomics, 2003, 3, 851–858 CrossRef CAS PubMed
- A. Kussak and A. Weintraub, Anal. Biochem., 2002, 307, 131–137 CrossRef CAS PubMed
- J. T. Fong-Fu Hsu, J. Am. Soc. Mass Spectrom., 2000, 11, 892–899 CrossRef
- L. Sturiale, A. Palmigiano, A. Silipo, Y. A. Knirel, A. P. Anisimov, R. Lanzetta, M. Parrilli, A. Molinaro and D. Garozzo, J. Mass Spectrom., 2011, 46, 1135–1142 CrossRef CAS PubMed
- A.-K. Güttsches, S. Löseke, U. Zähringer, U. Sonnenborn, C. Enders, S. Gatermann and A. Bufe, Innate Immun., 2012, 18, 204–216 CrossRef PubMed
- G. Klein, S. Müller-Loennies, B. Lindner, N. Kobylak, H. Brade and S. Raina, J. Biol. Chem., 2013, 288, 8111–8127 CrossRef CAS PubMed
- G. Wang, Pharmaceuticals, 2014, 7, 545–594 CrossRef CAS PubMed
- N. Frimodt-Moller, A. Poudyal, M. Vaara, J. Apajalahti, T. Vaara, H. He, R. L. Nation, J. Fox, O. Siikanen and J. Li, Antimicrob. Agents Chemother., 2010, 54, 3341–3346 CrossRef PubMed
- A. R. P. Tierney and P. N. Rather, Future Microbiol., 2019, 14, 533–552 CrossRef PubMed
- E. Matuschek, J. Åhman, C. Webster and G. Kahlmeter, Clin. Microbiol. Infect., 2018, 24, 865–870 CrossRef CAS PubMed
- M. Tyers and G. D. Wright, Nat. Rev. Microbiol., 2019, 17, 141–155 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02375e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |