
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilization of two conformers via intra- or inter-molecular hydrogen bonds in a dinuclear vanadium(V) complex with a pendant Schiff base: theoretical insight†
Snehasish Thakura,
Michael G. B. Drewb,
Antonio Franconetti c,
Antonio Frontera*c and
Shouvik Chattopadhyay*a
c,
Antonio Frontera*c and
Shouvik Chattopadhyay*a
aDepartment of Chemistry, Inorganic Section, Jadavpur University, Kolkata 700 032, India. E-mail: shouvik.chem@gmail.com
bSchool of Chemistry, The University of Reading, P.O. Box 224, Whiteknights, Reading RG6 6AD, UK
cDepartament de Química, Universitat de les Illes Balears, Crta. De Valldemossa km 7.5, 07122 Palma, Baleares, Spain
First published on 6th November 2019
Abstract
A dinuclear vanadium(V) complex, (μ-O)2[V(O)(L)]2, [where HL = 2-methoxy-6-((2-(2-hydroxyethylamino)ethylimino)methyl)phenol] has been synthesized and characterized by spectral and elemental analysis. A single crystal X-ray diffraction study confirms it structure. Two different conformations, stabilized via either intra- or inter-dinuclear hydrogen bonding interactions, co-exist in the solid-state structure. The energies of these intra- or inter-dinuclear hydrogen bonding interactions have been estimated by Density functional theory (DFT) calculations. A ‘Non-covalent interaction’ (NCI) plot has also been used to characterize these interactions.
1. Introduction
The ability of vanadium complexes to catalyze oxidations and oxo transfer reactions has provoked the interest of synthetic inorganic chemists for the preparation of various vanadium complexes.1 Many of these complexes have different nuclearity and coordination environments.2 Steric bulk of the substituent on the benzene ring of a ligand may prevent dimerization of the complex and may therefore be used to manipulate the coordination geometry and/or number of vanadium atoms in the final structure by preventing dimerization.3 Usually, final structures of metal complexes have been argued to be dependent on many factors e.g. metal–ligand ratio, solvent used, reaction temperature, and last but not the least, weak forces, such as hydrogen bonding and π⋯π stacking interactions.4 These non-covalent interactions are responsible for determining the three-dimensional structures of chemical and biological systems as a consequence of their specificity and directionality.5 While covalent bonds, once formed, are stable under usual conditions and can only be broken by providing sufficient energy, hydrogen bonds are formed reversibly and their strength depends on the chemical environment, such as the solvent or temperature.6 Hydrogen bonding is indeed the most significant directional inter-molecular interaction which has successfully aroused the interest of solid state chemists for years.7–14 Both intra- and inter-molecular hydrogen bonding interactions can be responsible for the generation of different conformations of a single substance. For example, two different secondary structures (α-helices and β-sheets) of biological macromolecules (e.g. proteins) may result from the competition between these two types of interactions. When short-range N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C inter-molecular hydrogen bonds are favored (energetically) over long-range N–H⋯OC intra-molecular hydrogen bonds, dimer (β-sheet) formation is preferred over self-assembled amino acid monomers (α-helix).8 Numerous hydrogen bonded architectures of different coordination complexes also exist in literature.9–15 Rochon et al. synthesized a coordination complex of manganese, where coexisting intra- and inter-molecular hydrogen bonds result in infinite triple chains and in two equivalent interpenetrating infinite 2D sheets, respectively.9 Hydrogen bonding interaction involving pendant side arms in the complex moiety is also a matter of interest for coordination chemists.10–15 Usually pendant arms are bonded directly to the donor atoms so that they can affect the nature of its metal ion coordination.10 But interesting structural motifs arise when they are uncoordinated to the metal centre and remain as dangling groups. They may consist of flexible components i.e. alkyl chain containing functional groups like amines, alcohols, amides and carboxyl or aromatic groups with other potential donors.11 There are quite a few instances involving macrocyclic ligands bearing first row transition metal complexes where the unbound side arms play a pivotal role in establishing intra- and inter-molecular hydrogen bonding interactions.12,13 Aneetha et al. discussed the hydrogen bonding interaction in a copper(II) complex of a macrocyclic tetraaza ligand, where protons of pendant –CH2–CH2–OH side arm establish hydrogen bonds with non coordinated perchlorate anions in the moiety,12 whereas Nagata et al. described intra-molecular hydrogen bonding interaction of a pendant –CH2–CO–NH2 group within the molecule.13 Literature shows numerous 3d series transition metal coordination complexes,14 including vanadium,15 having uncoordinated hydroxyethyl (–CH2–CH2–OH) moiety involved in intra- or inter-molecular hydrogen bonding interactions. However, computational techniques have scarcely been implemented to identify the energy involved in those kind of hydrogen bonding interactions. Construction of two different hydrogen bonded frameworks exploiting the pendant side arm of the ligand moiety in any vanadium complex has also not been reported prior to the present work. In contrast, the recent vanadium(V) investigations reported in the literature are focused on elucidating the behavior of hydrophobic complexes in terms of their interaction with erythrocytes,16 interface model systems, their impact in anti-cancer activities17 and enzyme inhibitors.18 Moreover, vanadium(V) complexes derived from hydrazone compounds have been used as antimicrobial complexes19 and also as catalysts for the oxidation of cyclohexane20 and the oxidative bromination of salicylaldehyde,21 which were rationalized using DFT and TDDFT calculations.
C inter-molecular hydrogen bonds are favored (energetically) over long-range N–H⋯OC intra-molecular hydrogen bonds, dimer (β-sheet) formation is preferred over self-assembled amino acid monomers (α-helix).8 Numerous hydrogen bonded architectures of different coordination complexes also exist in literature.9–15 Rochon et al. synthesized a coordination complex of manganese, where coexisting intra- and inter-molecular hydrogen bonds result in infinite triple chains and in two equivalent interpenetrating infinite 2D sheets, respectively.9 Hydrogen bonding interaction involving pendant side arms in the complex moiety is also a matter of interest for coordination chemists.10–15 Usually pendant arms are bonded directly to the donor atoms so that they can affect the nature of its metal ion coordination.10 But interesting structural motifs arise when they are uncoordinated to the metal centre and remain as dangling groups. They may consist of flexible components i.e. alkyl chain containing functional groups like amines, alcohols, amides and carboxyl or aromatic groups with other potential donors.11 There are quite a few instances involving macrocyclic ligands bearing first row transition metal complexes where the unbound side arms play a pivotal role in establishing intra- and inter-molecular hydrogen bonding interactions.12,13 Aneetha et al. discussed the hydrogen bonding interaction in a copper(II) complex of a macrocyclic tetraaza ligand, where protons of pendant –CH2–CH2–OH side arm establish hydrogen bonds with non coordinated perchlorate anions in the moiety,12 whereas Nagata et al. described intra-molecular hydrogen bonding interaction of a pendant –CH2–CO–NH2 group within the molecule.13 Literature shows numerous 3d series transition metal coordination complexes,14 including vanadium,15 having uncoordinated hydroxyethyl (–CH2–CH2–OH) moiety involved in intra- or inter-molecular hydrogen bonding interactions. However, computational techniques have scarcely been implemented to identify the energy involved in those kind of hydrogen bonding interactions. Construction of two different hydrogen bonded frameworks exploiting the pendant side arm of the ligand moiety in any vanadium complex has also not been reported prior to the present work. In contrast, the recent vanadium(V) investigations reported in the literature are focused on elucidating the behavior of hydrophobic complexes in terms of their interaction with erythrocytes,16 interface model systems, their impact in anti-cancer activities17 and enzyme inhibitors.18 Moreover, vanadium(V) complexes derived from hydrazone compounds have been used as antimicrobial complexes19 and also as catalysts for the oxidation of cyclohexane20 and the oxidative bromination of salicylaldehyde,21 which were rationalized using DFT and TDDFT calculations.
In this work, a potentially pentadentate Schiff base ligand is used to form a dinuclear vanadium(V) complex, in which the Schiff base behaves as a tridentate ligand keeping its remaining binding sites pendant. Two different conformers of the complex have been stabilized by intra- or inter-molecular hydrogen bonds in its solid state structure. In one conformer, the vanadyl oxygen atoms are involved in intra-dinuclear hydrogen bonding with the pendant hydroxyl group of the Schiff base. The other conformer forms 1D chain via inter-dinuclear hydrogen bonding interactions among vanadyl oxygen atoms and the pendant hydroxyl group of the Schiff base. DFT calculations have been utilized to estimate the energies involved in these interactions. NCI plot index computational tool further assists relevant theoretical findings.
2. Experimental section
VOSO4·5H2O was purchased from Loba Chemie Pvt. Ltd. and was of reagent grade. All other starting materials were commercially available, reagent grade, and used as purchased from Sigma-Aldrich without further purification.2.1 Preparation of (μ-O)2[V(O)(L)]2
The potentially pentadentate ligand, HL, has been prepared by refluxing 3-methoxysalicylaldehyde (152 mg, 1 mmol) with N-(2-hydroxyethyl)ethylenediamine (0.10 mL, 1 mmol) in CH3CN (10 mL) for ca. 1 h. The ligand has not been separated from the reaction mixture and has directly been used for the preparation of the complex. A 20 mL CH3CN solution of VOSO4·5H2O (253 mg, 1 mmol) has been added thereafter to the 15 mL CH3CN solution of the ligand and the resulting solution has then been refluxed for ca. 3 h. It was cooled to room temperature, subsequently filtered and kept for crystallization. Single crystals, appropriate for X-ray diffraction, were obtained after a week on slow evaporation of the filtrate (CH3CN solution) in open atmosphere.Yield: 227 mg (71%), based on vanadium; anal. calc. for C24H34N4O10V2 (FW = 640.43): C, 45.01; H, 5.35; N, 8.75%; found: C, 44.9; H, 5.1; N, 8.9%; FT-IR (KBr, cm−1): 3393 (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) O–H), 3148 (N–H), 1634 (CN), 934 (VO). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN): 388 (4.7 × 103).
O–H), 3148 (N–H), 1634 (CN), 934 (VO). UV-vis, λmax (nm), [εmax (L mol−1 cm−1)] (CH3CN): 388 (4.7 × 103).
1H NMR (DMSO-d6, 400 MHz) (ppm) δ: 8.89 (singlet, 2H, imine H, –CHN), 6.72–7.14 (multiplet, 6H, aromatic C–H), 5.55 (broad singlet, 2H, hydroxyl –OH), 4.87 (multiplet, 2H, –NH), 3.13–4.03 (multiplet, 14H, methylene CH2).
13C NMR (DMSO-d6, 300 MHz) (ppm) δ: 48.619 [–C(43)], 55.968 [–C(41)], 57.952–58.820 [–C(40), –C(44), –C(341)], 116.794, 116.948 [–C(35), –C(36)], 120.828 [–C(34)], 125.542 [–C(37)], 150.209 [–C(32)], 155.999 [–C(33)], 168.772 [–C(38)].
Crystal data: formula = C24H34N4O10V2, formula weight = 640.43, temperature (K) = 150, crystal system = triclinic, space group = P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 6.8424(6) Å, b = 9.4438(8) Å, c = 21.2231(3) Å, α = 102.009(7)°, β = 98.937(7)°, γ = 90.685(7)°, Z = 2, dcalc (g cm−3) = 1.607, μ (mm−1) = 0.771, F(000) = 664, total reflections = 9447, unique reflections = 7403, observed data [I > 2σ(I)] = 5321, no. of parameters = 365, R(int) = 0.035, R1, wR2 (all data) = 0.1002, 0.1432, R1, wR2 [I > 2σ(I)] = 0.0638, 0.1281.
, a = 6.8424(6) Å, b = 9.4438(8) Å, c = 21.2231(3) Å, α = 102.009(7)°, β = 98.937(7)°, γ = 90.685(7)°, Z = 2, dcalc (g cm−3) = 1.607, μ (mm−1) = 0.771, F(000) = 664, total reflections = 9447, unique reflections = 7403, observed data [I > 2σ(I)] = 5321, no. of parameters = 365, R(int) = 0.035, R1, wR2 (all data) = 0.1002, 0.1432, R1, wR2 [I > 2σ(I)] = 0.0638, 0.1281.
2.2 Physical measurements
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a PerkinElmer 240C elemental analyzer. IR spectrum in KBr (4500–500 cm−1) was recorded with a PerkinElmer Spectrum Two spectrophotometer. Electronic spectrum in CH3CN was recorded on a Shimadzu UV-1700 UV-vis spectrophotometer. The magnetic susceptibility measurement was performed with an EG and PAR vibrating sample magnetometer, model 155 at room temperature (300 K) in a 5000 G magnetic field, and diamagnetic corrections were performed using Pascal's constants. 1H NMR spectrum in DMSO-d6 solvent was recorded in a JEOL 400 MHz NMR instrument (Fig. S4, ESI†).13C NMR spectrum of the complex in DMSO-d6 solvent was recorded in a BRUKER 300 MHz NMR Spectrometer (Fig. S5, ESI†).2.3 X-ray crystallography
A suitable single crystal of the complex was picked and mounted on a glass fiber, and diffraction intensities were measured with an Oxford Diffraction X-Calibur diffractometer, equipped with Mo Kα radiation (λ = 0.71073 Å, 50 kV, 40 mA) at temperature 150 K. Data collection and reduction were performed with the Oxford Diffraction CrysAlis system. Structure of the complex was solved by direct method and refined by full-matrix least squares on F2, using the SHELX-2016/6 package.22 Absorption correction type is empirical with ABSPACK program of Oxford Diffraction Ltd. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms attached to nitrogen and oxygen atoms were located by difference Fourier maps. All other hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. Programs used: SHELX-2016/6,22 PLATON,23 WINGX,24 ORTEP,25 and MERCURY.262.4 Theoretical methods
The energy of the complex included in this study was calculated at the M06-2X/def2-TZVP level of theory using the crystallographic coordinates. For calculation, the GAUSSIAN-09 program has been used.27 This level of theory and methodology has been successfully used by us to study similar interactions.28 The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.29 The NCI plot30 isosurfaces have been used to characterize non-covalent interactions. They correspond to both favorable and unfavorable interactions, as differentiated by the sign of the second density Hessian eigenvalue and defined by the isosurface color. The color scheme is a red-yellow-green-blue scale with red for ρcut+ (repulsive) and blue for ρcut− (attractive).2.5 Hirshfeld surface analysis
Hirshfeld surfaces,31 mapped with dnorm, shape index and curvedness along with 2D fingerprint32 plots were calculated with the help of Crystal Explorer33 software using CIF of the complex. Bond lengths to hydrogen atoms were set to standard values. A typical Hirshfeld surface is represented customarily by a set of surface points. Two parameters are essential to provide information about relevant contact distances from each point; di represents the distance from the surface to the nearest atom interior to the surface, while de represents the distance from the surface to the nearest atom exterior to the surface. The normalized contact distance (dnorm) based on de and di is given bywhere rvdwi and rvdwe are the van der Waals radii of the atoms. dnorm was negative or positive depending on inter-molecular contacts which were shorter or longer than the van der Waals separations. It also demonstrated a surface with a red-white-blue color scheme, where bright red spots highlighted shorter contacts, white areas represented contacts around the van der Waals separation, and blue regions were the areas where close contacts were missing. Hirshfeld surface is characteristic for a given crystal structure and set of spherical atomic electron densities,34 and it was this property which indicated the probability of achieving additional insight into the inter-molecular interaction of molecular crystals.

3. Results and discussions
3.1 Synthesis
3-methoxysalicylaldehyde and N-(2-hydroxyethyl)ethylenediamine have been refluxed in CH3CN medium to synthesize a potentially pentadentate Schiff base ligand, HL, following a standard literature procedure.35 A solution of VOSO4·5H2O in CH3CN has then been added to the synthesized ligand solution to prepare a dinuclear vanadium(V) complex, (μ-O)2[V(O)(L)]2. HL has been found to coordinate vanadium(V) in a tridentate fashion in the complex. Vanadium(IV) in the precursor salt is oxidized to vanadium(V) through oxidation by atmospheric air with associated reduction of atmospheric oxygen to water. The effort to synthesize the complex in inert atmosphere (N2 environment) did not succeed. This supported the involvement of molecular oxygen in the oxidation of vanadium(IV). Formation of the complex has been shown in Scheme 1.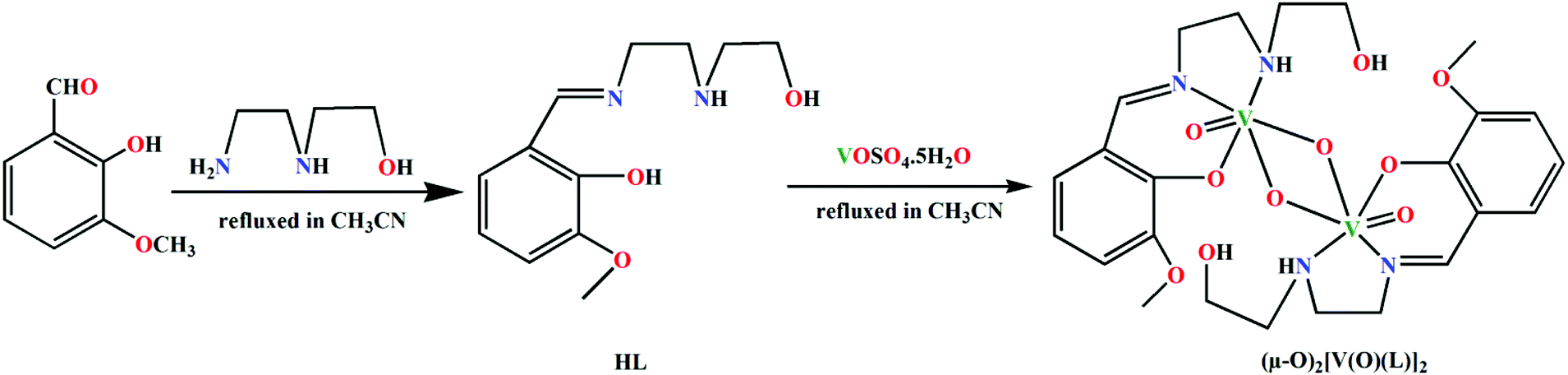 | ||
| Scheme 1 Synthesis of the ligand HL and the complex (μ-O)2[V(O)(L)]2. | ||
3.2 Description of structure of (μ-O)2[V(O)(L)]2
The complex crystallizes in triclinic space group P. The asymmetric unit of the complex contains two independent subunits, named ‘A’ and ‘B’. An outlook of the complex consisting of both subunits with selective atom numbering scheme is demonstrated in Fig. 1. In each subunit, one half of the molecule, constituting the asymmetric part of the structure, is related to the other half by the center of symmetry. Vanadium(V) centers are hexa-coordinated in both subunits. In the V2O2 core of each subunit, one vanadium(V)–oxygen distance is shorter [V(1)–O(1) = 1.675(2) Å in subunit ‘A’; V(2)–O(3) = 1.691(2) Å in subunit ‘B’] compared to the other vanadium(V)–oxygen distance [V(1)–O(1a) = 2.272(2) Å (a = 1 − x, 2 − y, 1 − z) in subunit ‘A’; V(2)–O(3b) = 2.337(2) Å (b = −x, 1 − y, −z) in subunit ‘B’]. The short vanadium–oxygen distances correspond to a VO double bond. The vanadium(V) centers, V(1) and V(1a) in subunit ‘A’ are separated by a distance of 3.0663(7) Å, whereas V(2) and V(2b) in subunit ‘B’ are 3.1437(7) Å apart from each other. These values correspond to the values found in analogous dinuclear vanadium(V) complexes.36 In both subunits, vanadium(V) centers show a distorted octahedral geometry. The slightly distorted octahedra about vanadium(V) centers are constituted by iminic nitrogen atoms, amine nitrogen atoms, phenoxy oxygen atoms, terminal oxo groups [N(19), N(22), O(11), and O(2) for subunit ‘A’; N(39), N(42), O(31) and O(4) for subunit ‘B’] and remaining two bridging oxygen atoms in each subunit [O(1) and O(1a) in subunit ‘A’, O(3) and O(3b) in subunit ‘B’], related by their respective center of symmetries. Thus, the coordination sphere of the vanadium(V) centers in each subunit is comprised of six unique heteroatom types: vanadyl oxo atoms, two bridging oxo atoms at short and long distances, a phenoxy oxygen atom and imine and amine nitrogen atoms. The O(2)–V(1)–O(1) angle of 106.41(1)° in subunit ‘A’ and the analogous O(3)–V(2)–O(4) angle of 107.51(1)° in subunit ‘B’ (Table S1, ESI†) indicate significant double bond characters of the bonds V(1)–O(1) and V(1)–O(2) in subunit ‘A’ and V(2)–O(3) and V(2)–O(4) in subunit ‘B’, respectively.37 The chelating angles, N(19)–V(1)–N(22) and N(19)–V(1)–O(11) of 77.84(10)° and 84.66(10)°, respectively in subunit ‘A’ and N(39)–V(2)–N(42) and N(39)–V(2)–O(31) of 78.27(10)° and 83.29(10)°, respectively in subunit ‘B’, indicate the distortion of the octahedral geometry around the vanadium(V) centers.38 The dimerization of the monomeric VO2L units in both subunits is motivated by the electron deficiency and oxophilicity of vanadium(V) centers.39
 | ||
| Fig. 1 Perspective view of the subunits ‘A’ and ‘B’ of the complex with selective atom numbering scheme. Hydrogen atoms have not been shown. All the symmetry related atoms have not been labeled for clarity. Symmetry transformation: a = 1 − x, 2 − y, 1 − z; b = −x, 1 − y, −z. Selected bond lengths (Å): V(1)–O(1) = 1.675(2); V(1)–O(2) = 1.626(2); V(1)–O(11) = 1.912(2); V(1)–N(19) = 2.162(3); V(1)–N(22) = 2.158(3); V(1)–O(1a) = 2.272(2) [for subunit ‘A’]. V(2)–N(42) = 2.182(3); V(2)–O(3b) = 2.337(2); V(2)–O(31) = 1.898(2); V(2)–O(3) = 1.691(2); V(2)–O(4) = 1.610(2); V(2)–N(39) = 2.133(3) [for subunit ‘B’]. | ||
This complex has a five membered puckered chelate ring in each subunit of ‘A’ and ‘B’. The ring shape is usually assigned explicitly by the Cremer–Pople puckering parameters.40 Puckering parameters are defined by the actual variations in ring shape, which are variations in the distances of the atom from a mean plane, and that the bond lengths and covalent bond angles vary to a more limited extent with less effect on the overall shape. The deviation (q) is customarily expressed in angstrom units. For six-membered rings, there are two additional parameters, θ and φ. The θ parameter suggests the type of shape, such as chair, half chair, boat, or skew, and the φ parameter denotes where the deviations from the mean plane are greatest.41
The saturated five membered chelate rings, V(1)–N(19)–C(20)–C(21)–N(22) in subunit ‘A’ and V(2)–N(39)–C(40)–C(41)–N(42) in subunit ‘B’ assume a half chair and an envelope conformation respectively, having the puckering parameters q = 0.443(4) Å; φ = 271.3(3)° for subunit ‘A’ and q = 0.409(4) Å; φ = 90.1(4)° for subunit ‘B’. It is noteworthy that the hydroxyl group of the pendant hydroxyethyl (–CH2–CH2–OH) side arm of the ligand remains uncoordinated. It makes the ligand a tridentate donor for this complex (the ligand is actually a potentially pentadentate one). Moreover, from the structural point of view, it is evident that in subunit ‘A’, the unbound hydroxyl groups are extended away from the vanadium(V) centers, while in subunit ‘B’, they are pointed inwards the metal centers. This differential orientation of the uncoordinated hydroxyl groups of the pendant –CH2–CH2–OH arms may be the reason of their involvement in inter- and intra-molecular hydrogen bonding interactions in subunit ‘A’ and subunit ‘B’, respectively.
In order to check the substituent effect of ligand on the final structure of the complex, we have synthesized a ligand, HL1 = 2-methoxy-6-((2-(2-hydroxyethylamino)ethylimino)methyl)phenol, which is structurally similar to HL. The only difference being the presence of bromine atom in HL1. Using this ligand, we eventually got success in synthesizing a complex, (μ-O)2[V(O)(L1)]2, structurally similar to the reported complex (μ-O)2[V(O)(L)]2. The complex (μ-O)2[V(O)(L1)]2 has been prepared in an identical procedure (Scheme 2) to that of (μ-O)2[V(O)(L)]2. It is also a μ2-oxo bridged dimer similar to our investigated complex. Unlike (μ-O)2[V(O)(L)]2, which crystallizes in triclinic space group P, (μ-O)2[V(O)(L1)]2 crystallizes in orthorhombic space group Pbca. Perspective view of (μ-O)2[V(O)(L1)]2 with relative atom numbering scheme is shown in Fig. 2, while crystallographic data, selected bond lengths and bond angle values are given in Tables S2–S4 of ESI,† respectively. There are several other examples42 of similar vanadium systems where the other substituents in the benzene ring of the ligand consequently lead to the same μ2-oxo bridged dinuclear complexes described here. Referring to these facts, it can be concluded that variation of substituents on the ligand has practically minimal or no effect in the final structure of vanadium complexes. Table 1 shows few examples of similar vanadium complexes having different substituents in the benzene ring of similar type of ligands.
 | ||
| Scheme 2 Synthetic route to the ligand HL1 and the complex (μ-O)2[V(O)(L1)]2. | ||
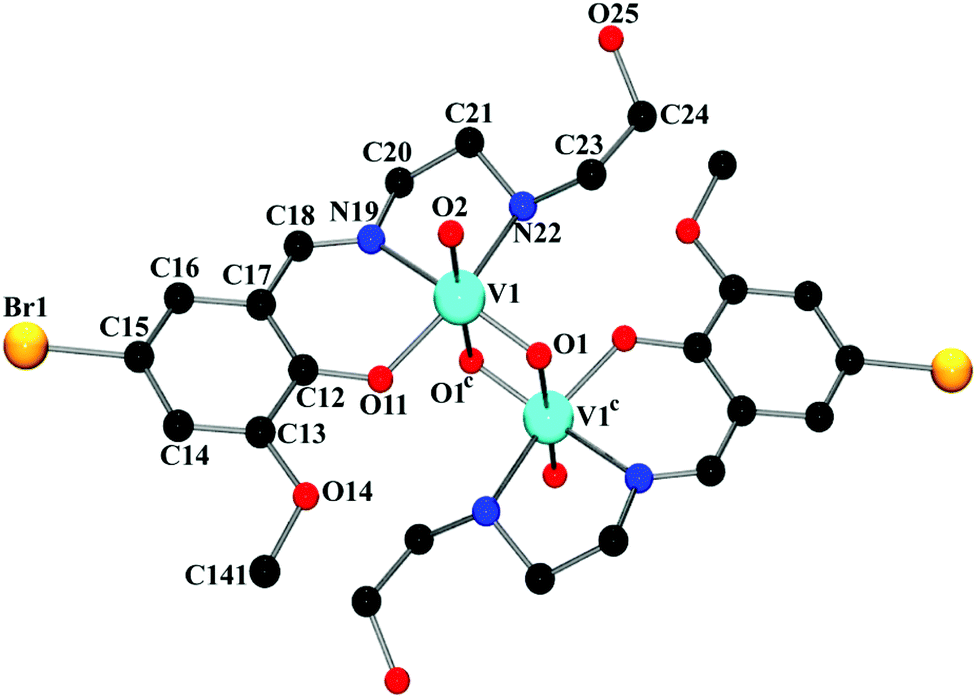 | ||
| Fig. 2 Perspective view of (μ-O)2[V(O)(L1)]2 with selective atom numbering scheme. Hydrogen atoms have not been shown. All the symmetry related atoms have not been labeled for clarity. Symmetry transformation: c = 1 − x, 1 − y, 1 − z. | ||
| Complexes | Crystal system | Space group | Structure | References |
|---|---|---|---|---|
| a HL2 = 2-((2-(2-hydroxyethylamino)ethylimino)methyl)phenol. HL3 = 2-ethoxy-6-((2-(2-hydroxyethylamino)ethylimino)methyl)phenol. HL4 = 4-chloro-2-((2-(2-hydroxyethylamino)ethylimino)methyl)phenol. HL5 = 4-fluoro-2-((2-(2-hydroxyethylamino)ethylimino)methyl)phenol. HL6 = 2-bromo-4-chloro-6-((2-(2-hydroxyethylamino)ethylimino)methyl)phenol. HL7 = 2-((2-(2-hydroxyethylamino)ethylimino)methyl)-6-methylphenol. HL7 = 2-((2-(2-hydroxyethylamino)ethylimino)phenylmethyl)-5-methoxyphenol. | ||||
| (μ-O)2[V(O)(L2)]2 | Monoclinic | P21/c |  |
42a |
| (μ-O)2[V(O)(L3)]2 | Monoclinic | P21/n |  |
42b |
| (μ-O)2[V(O)(L4)]2 | Monoclinic | P21/c |  |
42b |
| (μ-O)2[V(O)(L5)]2 | Monoclinic | P21/c |  |
42c |
| (μ-O)2[V(O)(L6)]2 | Monoclinic | P21/n |  |
42d |
| (μ-O)2[V(O)(L7)]2·bpy | Triclinic | P |
 |
42e |
| (μ-O)2[V(O)(L8)]2 | Triclinic | P |
 |
42f |
3.3 Theoretical studies
The theoretical study is dedicated to analyze the energy responsible to the extensive hydrogen bonding in the complex. Moreover, the co-existence of two different conformations (pendant arms in Fig. 3) in the solid state structure has also been investigated, which provokes the formation of either intra- or inter-molecular hydrogen bonds, as further described below.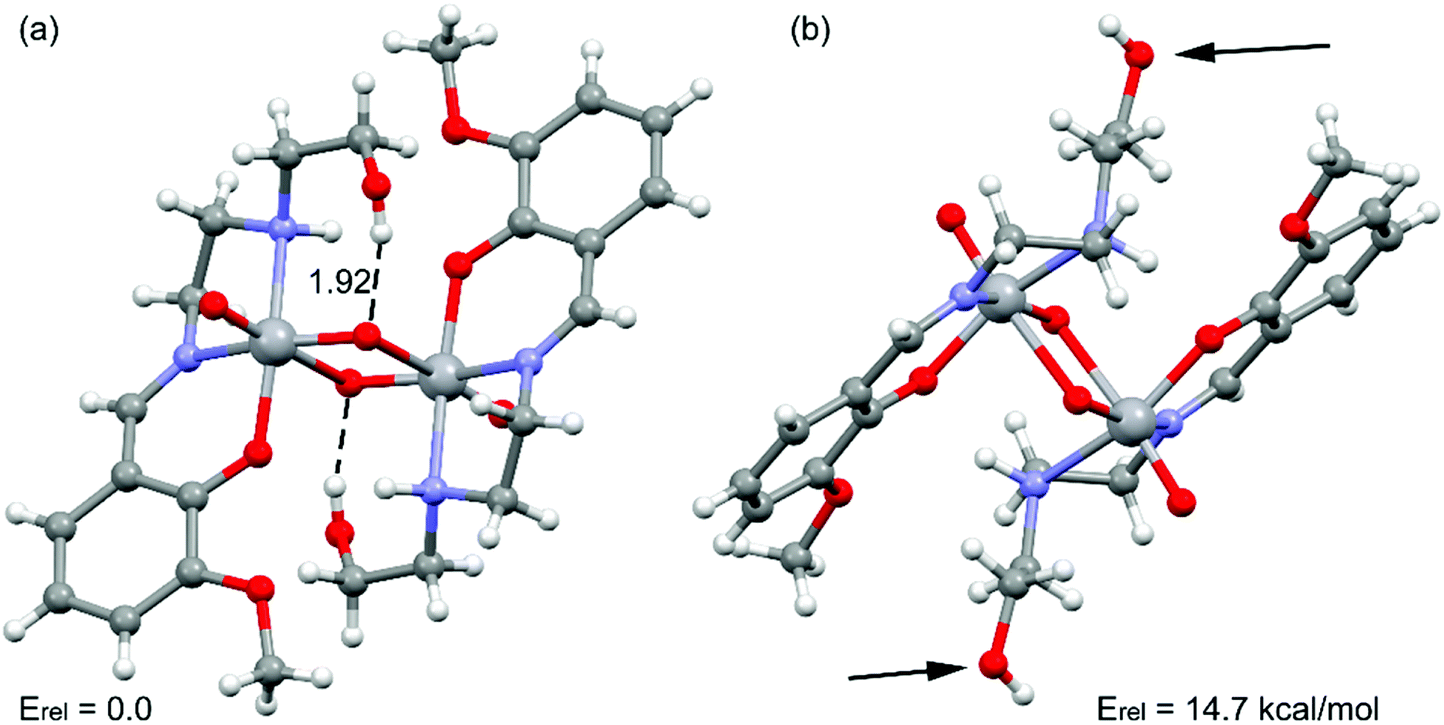 | ||
| Fig. 3 Partial view of the solid state structure of the complex. Distance in Å. The relative energy of both conformations is also indicated. | ||
Emphasis has been put on the analysis of the two different conformation adopted by the –CH2–CH2–OH pendant arm in the complex. As shown in Fig. 3, two different conformers coexist in the same crystal structure. In one case, the hydroxyl groups point to the oxygen atoms of the VO2+ cation, forming intra-molecular hydrogen bonds. In the other case, the hydroxyl groups are not establishing intra-molecular hydrogen bonds (small arrows in Fig. 3, right). The conformer with intra-molecular hydrogen bonds is 14.7 kcal mol−1 more stable than the other, suggesting that each hydrogen bond stabilizes the conformation in approximately 7.35 kcal mol−1. The intra-molecular O–H⋯O distance has been found to be 1.92 Å. This is similar to the intra-molecular O–H⋯O distances found in structurally similar vanadium(V) complexes (Table 2).15a,42a,c,d The existence of the other conformer is due to its crucial participation in the formation of a supramolecular 1D chain in the solid state governed by inter-molecular instead of intra-molecular O–H⋯O hydrogen bonds as shown in Fig. 4a. The interaction energy of one dimer extracted from the 1D chain is large and negative (ΔE = −26.9 kcal mol−1, Fig. 4b), which clearly explains why this conformation (14.7 kcal mol−1 less favorable) is also present in the solid state. The NCI plot (Fig. 4c) shows the existence of small blue isosurfaces characterizing the hydrogen bonding interactions as strong forces, in agreement with the large binding energy. Moreover, it also reveals the existence of weak van der Waals interactions (CH⋯HC) between the hydrogen atoms of the ethylene groups (blue dashed line in Fig. 4b) that also contribute to the stabilization of the supramolecular assembly.
 | ||
| Fig. 4 (a) 1D supramolecular chain observed in the solid state structure of the complex. Non-polar hydrogen atoms have been omitted for clarity. (b) Dimer retrieved from the crystal structure of the complex. Distances in Å. (c) NCI surface of the hydrogen bonded dimer of the complex. The gradient cut-off is s = 0.35 au, and the color scale is −0.04 < ρ < 0.04 au. | ||
3.4 Spectral and magnetic study
Strong and sharp band at 1634 cm−1 due to azomethine (CN) group has been routinely noticed in the IR spectrum of the complex.43 Existence of strong bands around 934 cm−1 and 825 cm−1 may be assigned to asymmetric and symmetric (OVO) vibrations of cis-VO2 groups.44 Sharp band at 3148 cm−1 may be assigned to (N–H) stretching vibration45 whereas a broad band at 3393 cm−1 indicates the presence of hydrogen bonded alcoholic group in the crystal structure of the complex.46 IR spectrum of the complex is shown in Fig. S1, ESI.† Electronic spectrum (Fig. S2, ESI†) of the complex in CH3CN displays an absorption band at 388 nm, which is indicative of the ligand-to-metal charge transfer (LMCT) transition originating from the pπ orbital on the phenolate oxygen to the empty d orbitals of vanadium(V).47
Magnetic susceptibility measurement shows that the complex is diamagnetic, which is quite obvious for any vanadium(V) complex with no unpaired electrons.48
3.5 Hirshfeld surface analysis
Hirshfeld surfaces and 2D fingerprint plots of the complex are shown in Fig. 5 and 6 respectively, where inter-molecular interactions appear as discrete spikes. The surfaces are shown as transparent to allow visualization of the molecule, in a similar orientation for all structures, around which they were calculated. | ||
| Fig. 5 Hirshfeld surfaces mapped with dnorm (left), shape index (middle) and curvedness (right) of the complex. | ||
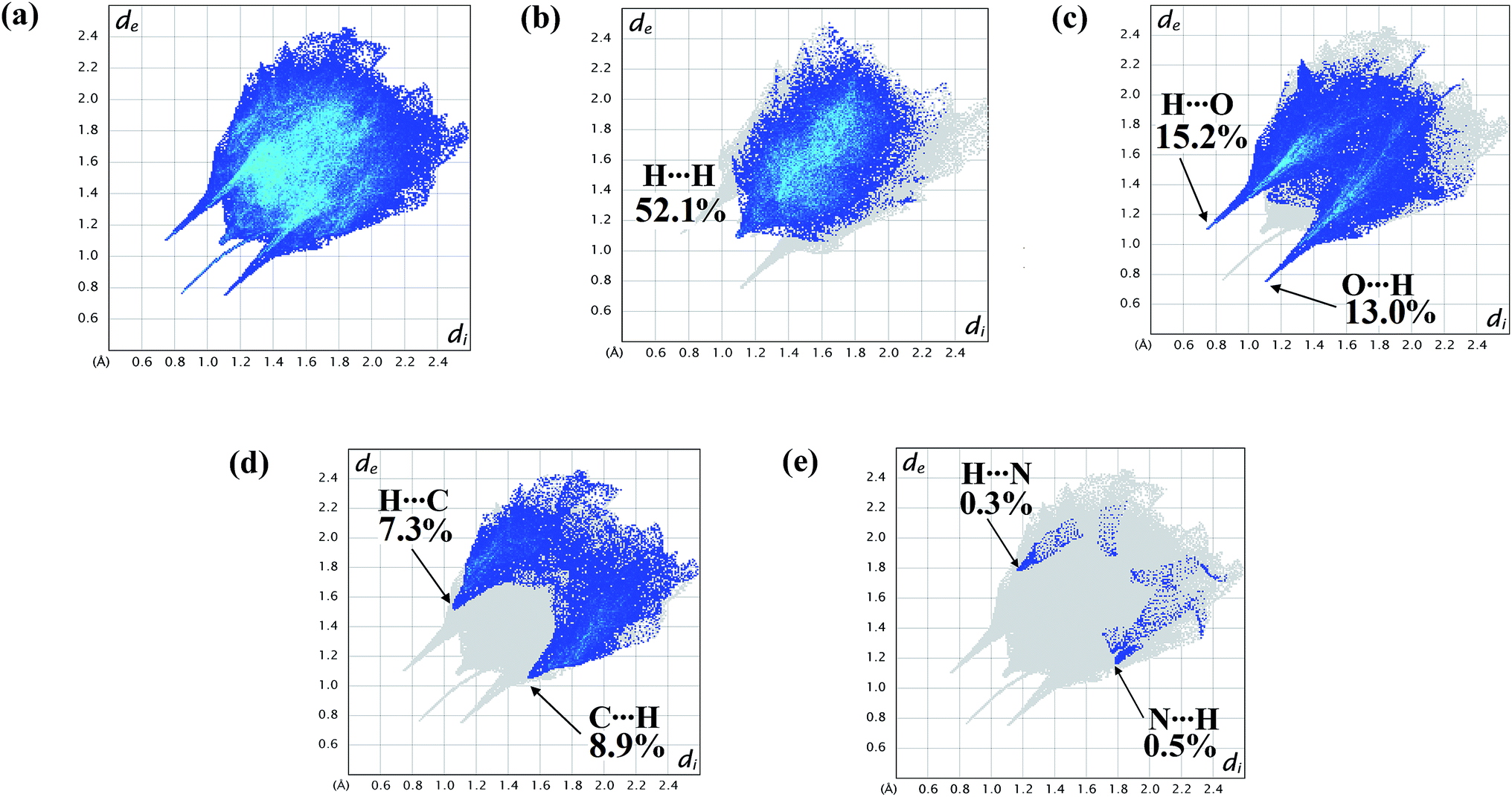 | ||
| Fig. 6 2D fingerprint plot of the complex showing (a) total surface area; proportions of (b) H⋯H; (c) O⋯H/H⋯O; (d) C⋯H/H⋯C; (e) N⋯H/H⋯N interactions. | ||
The fingerprint plots are also analyzed to illustrate particular atoms pair close contacts. This decomposition of fingerprint plots enables separation of contributions from different interaction types, which overlap in the full fingerprint. 2D fingerprint plot of the complex shows proportions of O⋯H/H⋯O and C⋯H/H⋯C interactions comprise 28.2% and 16.2% of total surface of the complex. The N⋯H/H⋯N interaction is found to be very low i.e. 0.8%. The H⋯H interaction constitutes significant amount of interaction, i.e. 52.1% of the total Hirshfeld surface (Fig. 7).
 | ||
| Fig. 7 Relative proportions of interactions in the total Hirshfeld surface area of the complex, represented in a bar diagram. | ||
The lower spike (di = 1.12, de = 0.76 Å) of Fig. 6c indicates O⋯H interaction and the upper spike (di = 0.76, de = 1.12 Å) indicates H⋯O interaction. Similarly C⋯H interaction is also represented by lower spike (di = 1.56, de = 1.06 Å) and H⋯C interaction is represented by upper spike (di = 1.06, de = 1.56 Å) (Fig. 6d).
4. Conclusion
A new dinuclear vanadium(V) Schiff base complex, (μ-O)2[V(O)(L)]2, has been synthesized and structurally characterized. The complex crystallizes in triclinic space group P. It consists of two independent dinuclear subunits. Hydrogen bonding interactions in two subunits are different. DFT calculations have been used to estimate the energy associated to the hydrogen bonding interactions present in the complex. NCI plot index computational tool further assists relevant theoretical findings. The competition between inter- and intra-molecular OH⋯O2V+ hydrogen bonds and their influence in the solid state architecture of this complex has been studied accordingly.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. T. thanks the DST, India, for awarding a Junior Research Fellowship (IF160359). A. Frontera and A. Franconetti gratefully acknowledge the financial support of this work by the MINECO/AEI of Spain (project CTQ2017-85821-R, FEDER funds). In addition, AF thanks the MINECO/AEI from Spain for a “Juan de la Cierva” contract. They thank the CTI (UIB) for free allocation of computer time. M. G. B. D. thanks the University of Reading and the EPSRC (U. K.) for funds for the diffractometer.Notes and references
- (a) G. Licini, V. Conte, A. Coletti, M. Mba and C. Zonta, Coord. Chem. Rev., 2011, 255, 2345–2357 CrossRef CAS; (b) Y. Zhang and R. H. Holm, Inorg. Chem., 1990, 29, 911–917 CrossRef CAS; (c) P. Adão, J. C. Pessoa, R. T. Henriques, M. L. Kuznetsov, F. Avecilla, M. R. Maurya, U. Kumar and I. Correia, Inorg. Chem., 2009, 48, 3542–3561 CrossRef; (d) S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott, A. D. Sutton and D. L. Thorn, J. Am. Chem. Soc., 2009, 131, 428–429 CrossRef CAS; (e) G. Santoni, G. Licini and D. Rehder, Chem.–Eur. J., 2003, 9, 4700–4708 CrossRef CAS PubMed.
- (a) P. Knopp, K. Wieghardt, B. Nuber, J. Weiss and W. S. Sheldrick, Inorg. Chem., 1990, 29, 363–371 CrossRef CAS; (b) J. J. H. Edema, W. Stauthamer, E. V. Bolhuis, S. Gambarotta, W. J. J. Smeets and A. L. Spek, Inorg. Chem., 1990, 29, 1302–1306 CrossRef CAS; (c) G. S. Fink, L. G. Cuervo, B. Therrien, H. S. Evans and G. B. Shul'pin, Inorg. Chim. Acta, 2004, 357, 475–484 CrossRef; (d) K. Jonas, B. Gabor, R. Mynott, K. Angermund, O. Heinemann and C. Kriiger, Angew. Chem., Int. Ed. Engl., 1997, 36, 1712–1714 CrossRef CAS; (e) S. L. Castro, W. E. Streib, J.-S. Sun and G. Christou, Inorg. Chem., 1996, 35, 4462–4468 CrossRef CAS; (f) H. Zhu, Q. Liu, Y. Deng, T. Wen, C. Chen and D. Wu, Inorg. Chim. Acta, 1999, 286, 7–13 CrossRef CAS; (g) X.-M. Zhang, M.-L. Tong, H. K. Lee and X.-M. Chen, J. Solid State Chem., 2001, 160, 118–122 CrossRef CAS; (h) A. Z. E. Sonbati, A. A. M. Belal, M. A. Diab and M. Z. Balboula, Spectrochim. Acta, Part A, 2011, 78, 1119–1125 CrossRef.
- (a) L. M. Mokry and C. J. Carrano, Inorg. Chem., 1993, 32, 6119–6121 CrossRef CAS; (b) S.-S. Qian, Z.-L. You, Y.-N. Li, X. Hu, M. Zhang and H.-L. Zhu, J. Coord. Chem., 2013, 66, 1311–1319 CrossRef CAS.
- (a) A. Escande, L. Guénée, K.-L. Buchwalder and C. Piguet, Inorg. Chem., 2009, 48, 1132–1147 CrossRef CAS; (b) L. Saghatforoush, Main Group Chem., 2015, 14, 115–125 CAS; (c) M. Chen, M. Hu, H. Zhao, J.-Y. Tian and C.-S. Liu, Z. Anorg. Allg. Chem., 2016, 642, 778–784 CrossRef CAS; (d) G. Mahmoudi, A. A. Khandar, J. K. Zaręba, M. J. Białek, M. S. Gargari, M. Abedi, G. Barandika, D. V. Derveer, J. Mague and A. Masoumi, Inorg. Chim. Acta, 2015, 429, 1–14 CrossRef CAS; (e) A. K. Cheetham, G. Kieslich and H. H.-M. Yeung, Acc. Chem. Res., 2018, 51, 659–667 CrossRef CAS; (f) M. Chen, Z.-W. Wang, H. Zhao and C.-S. Liu, Inorg. Chem. Commun., 2014, 45, 84–88 CrossRef CAS; (g) D. P. Malenov, G. V. Janjić, V. B. Medaković, M. B. Hall and S. D. Zaric, Coord. Chem. Rev., 2017, 345, 318–341 CrossRef CAS.
- (a) E. R. Johnson, S. Keinan, P. M. Sánchez, J. C. García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS; (b) S. Fang, S. Y.-L. Leung, Y. Li and V. W.-W. Yam, Chem.–Eur. J., 2018, 24, 15596–15602 CrossRef CAS; (c) Z. P. Shields, J. S. Murray and P. Politzer, Int. J. Quantum Chem., 2010, 110, 2823–2832 CrossRef CAS.
- (a) C. Schmuck and W. Wienand, Angew. Chem., Int. Ed., 2001, 40, 4363–4369 CrossRef CAS; (b) H. Guo and M. Karplus, J. Phys. Chem., 1994, 98, 7104–7105 CrossRef CAS; (c) L.-S. Teo, C.-Y. Chen and J.-F. Kuo, Macromolecules, 1997, 30, 1793–1799 CrossRef CAS.
- (a) L. J. Barbour, G. W. Orr and J. L. Atwood, Nature, 1998, 393, 671–673 CrossRef CAS; (b) H. W. Roesky and M. Andruh, Coord. Chem. Rev., 2003, 236, 91–119 CrossRef CAS; (c) F. Wang, M. A. J. Gillissen, P. J. M. Stals, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2012, 18, 11761–11770 CrossRef CAS.
- (a) A. Bouchet, J. Klyne, S.-I. Ishiuchi, O. Dopfer, M. Fujii and A. Zehnacker, Phys. Chem. Chem. Phys., 2018, 20, 12430–12443 RSC; (b) L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CrossRef CAS PubMed; (c) C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS.
- F. D. Rochon, M. Andruh and R. Melanson, Can. J. Chem., 1998, 76, 1564–1570 CAS.
- R. R. Fenton, L. F. Lindoy, R. C. Luckay, F. R. Turville and G. Wei, Aust. J. Chem., 2002, 54, 59–62 CrossRef.
- (a) M. Vicente, R. Bastida, C. Lodeiro, A. Macías, A. J. Parola, L. Valencia and S. E. Spey, Inorg. Chem., 2003, 42, 6768–6779 CrossRef CAS; (b) M. Woods, D. E. Woessner, P. Zhao, A. Pasha, M.-Y. Yang, C.-H. Huang, O. Vasalitiy, J. R. Morrow and A. D. Sherry, J. Am. Chem. Soc., 2006, 128, 10155–10162 CrossRef CAS; (c) T. Mani, A. C. L. Opina, P. Zhao, O. M. Evbuomwan, N. Milburn, G. Tircso, C. Kumas and A. D. Sherry, J. Biol. Inorg Chem., 2014, 19, 161–171 CrossRef CAS PubMed; (d) C. Stockheim, L. Hater, T. Weyhermiiller, K. Wieghardt and B. Nuber, J. Chem. Soc., Dalton Trans., 1996, 4409–4416 RSC; (e) K. S. Min and M. P. Suh, Eur. J. Inorg. Chem., 2001, 449–455 CrossRef CAS.
- H. Aneetha, Y.-H. Lai, S.-C. Lin, K. Panneerselvam, T.-H. Lu and C.-S. Chung, J. Chem. Soc., Dalton Trans., 1999, 2885–2892 RSC.
- M. K. C. T. Nagata, P. S. Brauchle, S. Wang, S. K. Briggs, Y. S. Hong, D. W. Laorenza, A. G. Lee and T. D. Westmoreland, Polyhedron, 2016, 114, 299–305 CrossRef CAS.
- (a) K. Chattopadhyay, G. A. Craig, A. Kundu, V. Bertolasi, M. Murrie and D. Ray, Inorg. Chem., 2016, 55, 10783–10792 CrossRef CAS; (b) W. J. Lian, X.-T. Wang, C.-Z. Xie, H. Tian, X.-Q. Song, H.-T. Pan, X. Qiao and J.-Y. Xu, Dalton Trans., 2016, 45, 9073–9087 RSC.
- (a) X. Li, M. S. Lah and V. L. Pecoraro, Inorg. Chem., 1988, 27, 4657–4664 CrossRef CAS; (b) C. R. Cornman, G. J. Colpas, J. D. Hoeschele, J. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1992, 114, 9925–9933 CrossRef CAS; (c) S.-Y. Liu and Y.-P. Ma, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 603–607 CrossRef CAS; (d) K.-H. Yang, Transition Met. Chem., 2014, 39, 469–475 CrossRef CAS.
- D. Sanna, J. Palomba, G. Lubinu, P. Buglyó, S. Nagy, F. Perdih and E. Garribba, J. Med. Chem., 2019, 62, 654–664 CrossRef CAS.
- (a) D. C. Crans, J. T. Koehn, S. M. Petry, C. M. Glover, A. Wijetunga, R. Kaur, A. Levina and P. A. Lay, Dalton Trans., 2019, 48, 6383–6395 RSC; (b) Z. Kazemi, H. A. Rudbari, V. Mirkhani, M. Sahihi, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork, A. A. Kajani and G. Azimi, Eur. J. Med. Chem., 2017, 135, 230–240 CrossRef CAS PubMed.
- O. A. Chavesa, M. C. C. de Oliveira, C. M. C. de Salles, F. M. Martins, B. A. Iglesiase and D. F. Back, J. Inorg. Biochem., 2019, 200, 110800 CrossRef.
- L.-Y. He, X.-Y. Qiu, J.-Y. Cheng, S.-J. Liu and S.-M. Wu, Polyhedron, 2018, 156, 105–110 CrossRef CAS.
- D. Dragancea, N. Talmaci, S. Shova, G. Novitchi, D. Darvasiová, P. Rapta, M. Breza, M. Galanski, J. Kozǐśěk, N. M. R. Martins, L. M. D. R. S. Martins, A. J. L. Pombeiro and V. B. Arion, Inorg. Chem., 2016, 55, 9187–9203 CrossRef CAS.
- P. Adak, B. Ghosh, B. Pakhira, R. Sekiya, R. Kuroda and S. K. Chattopadhyay, Polyhedron, 2017, 127, 135–143 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- J. L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- M. N. Burnett and C. K. Johnson, ORTEP-3: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Report ORNL-6895, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1996 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. R. Monge, R. Taylor, J. V. D. Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) S. Ghosh, S. Biswas, A. Bauzá, M. Barceló-Oliver, A. Frontera and A. Ghosh, Inorg. Chem., 2013, 52, 7508–7523 CrossRef CAS; (b) J. Adhikary, P. Chakraborty, S. Das, T. Chattopadhyay, A. Bauzá, S. K. Chattopadhyay, B. Ghosh, F. A. Mautner, A. Frontera and D. Das, Inorg. Chem., 2013, 52, 13442–13452 CrossRef CAS; (c) M. S. Gargari, V. Stilinović, A. Bauzá, A. Frontera, P. McArdle, D. Van Derveer, S. W. Ng and G. Mahmoudi, Chem.–Eur. J., 2015, 21, 17951–17958 CrossRef PubMed; (d) M. Mirzaei, H. Eshtiagh-Hosseini, Z. Bolouri, Z. Rahmati, A. Esmaeilzadeh, A. Hassanpoor, A. Bauzá, P. Ballester, M. Barceló-Oliver, J. T. Mague, B. Notash and A. Frontera, Cryst. Growth Des., 2015, 15, 1351–1361 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. C. García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- (a) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC; (b) F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS; (c) H. F. Clausen, M. S. Chevallier, M. A. Spackman and B. B. Iversen, New J. Chem., 2010, 34, 193–199 RSC.
- (a) A. L. Rohl, M. Moret, W. Kaminsky, K. Claborn, J. J. McKinnon and B. Kahr, Cryst. Growth Des., 2008, 8, 4517–4525 CrossRef CAS; (b) A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Crystal Explorer 2.0, University of Western Australia, Perth, Australia, 2007, http://hirshfeldsurfacenet.blogspot.com Search PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef.
- S. Ghosh, N. Hari, D. Pinkowicz, M. Fitta and S. Mohanta, New J. Chem., 2018, 42, 15917–15929 RSC.
- (a) A. Sarkar and S. Pal, Polyhedron, 2007, 26, 1205–1210 CrossRef CAS; (b) S. Pal and S. Pal, J. Chem. Crystallogr., 2000, 30, 329–333 CrossRef CAS.
- G. J. Colpas, B. J. Hamstra, J. W. Kampf and V. L. Pecoraro, Inorg. Chem., 1994, 33, 4669–4675 CrossRef CAS.
- S. Rayati, N. Sadeghzadeh and H. R. Khavasi, Inorg. Chem. Commun., 2007, 10, 1545–1548 CrossRef CAS.
- C. J. Carrano, C. M. Nunn, R. Quan, J. A. Bonadies and V. L. Pecoraro, Inorg. Chem., 1990, 29, 944–951 CrossRef CAS.
- (a) D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS; (b) D. Cremer, Acta Crystallogr., 1984, B40, 498–500 CrossRef CAS.
- A. D. French, Advances in Carbohydrate Chemistry and Biochemistry, Academic Press, Science, 2012, vol. 67, p. 24 Search PubMed.
- (a) M.-J. Xie, Y.-F. Niu, X.-D. Yang, W.-P. Liu, L. Li, L.-H. Gao, S.-P. Yan and Z.-H. Meng, Eur. J. Med. Chem., 2010, 45, 6077–6084 CrossRef CAS; (b) Z.-L. You, D.-H. Shi, J.-C. Zhang, Y.-P. Maa, C. Wang and K. Li, Inorg. Chim. Acta, 2012, 384, 54–61 CrossRef CAS; (c) K.-H. Yang, Transition Met. Chem., 2014, 39, 469–475 CrossRef CAS; (d) S.-Y. Liu and Y.-P. Ma, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 603–607 CrossRef CAS; (e) F. Xu, L.-W. Xue and C.-X. Zhang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2015, 45, 1678–1682 CrossRef CAS; (f) M. Ghorbani, A. D. Khalaji, N. Feizi, A. Akbari, V. Eigner and M. Dusek, J. Mol. Struct., 2017, 1130, 442–446 CrossRef CAS.
- S. Chattopadhyay, M. S. Ray, S. Chaudhuri, G. Mukhopadhyay, G. Bocelli, A. Cantoni and A. Ghosh, Inorg. Chim. Acta, 2006, 359, 1367–1375 CrossRef CAS.
- Y. Abe, A. Iyoda, K. Seto, A. Moriguchi, T. Tanase and H. Yokoyama, Eur. J. Inorg. Chem., 2008, 2148–2157 CrossRef CAS.
- (a) L. Sacconi and I. Bertini, J. Am. Chem. Soc., 1966, 88, 5180–5185 CrossRef CAS; (b) N. Raman, Y. P. Raja and A. Kulandaisamy, J. Chem. Sci., 2001, 113, 183–189 CrossRef CAS.
- G. Cros and J.-P. Laurent, Inorg. Chim. Acta, 1985, 105, 63–67 CrossRef CAS.
- B. Mondal, T. Ghosh, M. Sutradhar, G. Mukherjee, M. G. B. Drew and T. Ghosh, Polyhedron, 2008, 27, 2193–2201 CrossRef CAS.
- A. Niemann, U. Bossek, G. Haselhorst, K. Wieghardt and B. Nuber, Inorg. Chem., 1996, 35, 906–915 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1879621 and 1956388 contain the supplementary crystallographic data for the two complexes. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07006k |
| This journal is © The Royal Society of Chemistry 2019 |