
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly dispersed Ni nanoparticles on mesoporous silica nanospheres by melt infiltration for transfer hydrogenation of aryl ketones†
Hyemin Kweon‡
a,
Sanha Jang‡a,
Akerke Bereketovaa,
Ji Chan Park *b and
Kang Hyun Park*a
*b and
Kang Hyun Park*a
aDepartment of Chemistry, Pusan National University, Busan, 46241, Korea. E-mail: chemistry@pusan.ac.kr
bClean Fuel Laboratory, Korea Institute of Energy Research, Daejeon, 34129, Korea. E-mail: jcpark@kier.re.kr
First published on 7th May 2019
Abstract
Nickel-based catalysts have been applied to the catalytic reactions for transfer hydrogenation of carbonyl compounds. In the present work, highly dispersed nickel particles located at the pores of mesoporous silica spheres (Ni@mSiO2) were prepared via an optimized melt infiltration route. The nickel nanoparticles of 10 wt% in the Ni@mSiO2 catalyst could be uniformly loaded with high dispersion of 36.3%, resulting excellent performance for catalytic transfer hydrogenation of aryl ketones.
The reduction of carbonyl compounds has received steady interest as a common transformation in organic synthetic chemistry. Corresponding alcohols are important building blocks for the manufacture of chemicals, pharmaceuticals, and cosmetics.1,2 The transfer hydrogenation (TH) reaction of ketones has lots of advantages owing to its low cost, simple process, easy handling, and mild conditions.3 Isopropyl alcohol both as a solvent and hydrogen donor provides safe reaction conditions.4 Until now, many catalysts have been developed for TH reactions.5
Among the catalysts for TH reactions, nickel-based catalysts are representative, due to the metallic nickel surface can absorbs hydrogen and easily activates hydrogen in the atomic state.6–11 In addition, nickel compounds could be the significant catalyst candidate because of the low price, accessibility, and high reactivity.12,13 Therefore, several works used nickel nanocatalysts for TH reactions.14–17,44–51 However, developing the new supported catalyst with the high metal dispersion and narrow particle size distribution is still required, because it can enhance the reactant conversion and improve product yield.18 Recently, melt infiltration process has been exploited to prepare supported nanocatalysts as a fast and convenient route with no solvent use.19–21
The method which uses highly porous supports with regular porosity and hydrated metal salts with mild melting temperatures, allowed the metal salts to permeate inside the porous support via capillary forces. High performance nanocatalysts based on well-dispersed nanoparticles could be obtained through uniform infiltration of metal precursors and sequential thermal treatment.22 The obtained nanoparticles have narrow size distributions as well as small sizes, enlarging active sites.23,24
Mesoporous silica nanosphere (mSiO2) as a porous support can be an outstanding platform for the synthesis of metal nanoparticle, owing to the good thermal stability, high surface area, and large pore volume as well as uniform pore structures.25–29 Herein, we report a new catalyst with highly dispersed Ni nanoparticles at mesoporous silica sphere nanostructures (Ni@mSiO2) for catalytic TH reactions, which was synthesized through a melt infiltration of hydrated nickel salts and a sequential thermal reduction (Scheme 1). First, mSiO2 supports could be prepared by a sol–gel method based on a modified Stöber process. More details were described in the ESI.† The mesopores were formed in the silica nanospheres using the cationic surfactant C16TAB as a self-assembly template. The TEM images show mesoporous silica spheres with an average diameter of 417 ± 20 nm (Fig. 1a and b). Ordered mesoporous channels were generated by removing the long carbon chains of C16TAB by calcination at 500 °C. The corresponding Fourier-transform (FT) pattern showed a ring pattern of polycrystalline (inset of Fig. 1c). The inverse fast Fourier-transform (IFFT) image, calculated by Micrograph TM Gatan software, also demonstrated the existence of hexagonal lattice planes (Fig. 1c).
 | ||
| Scheme 1 A brief synthetic scheme of Ni@mSiO2 nanocatalyst. | ||
 | ||
| Fig. 1 (a and b) TEM images, (c) FT pattern for (b) (inset of c) and IFFT image, (d) HRTEM image, (e) N2 adsorption/desorption isotherms, and (f) pore size distribution diagrams of mSiO2. The bars represent 1 mm (a), 100 nm (b), and 20 nm (d). | ||
The brightest of the rings originated from a two-dimensional hexagonal (p6mm) structure with d100 spacing. The interplanar spacing computed from the brightest FFT diffraction ring was approximately 3 nm, corresponding to the pore sizes shown in the TEM image (Fig. 1d).
N2 sorption experiments for the mSiO2 nanosphere exhibited type IV adsorption–desorption hysteresis with H4 type hysteresis loop (Fig. 1e). The Brunauer–Emmett–Teller (BET) surface area and pore volume of the mSiO2 nanosphere were 1501 m2 g−1 and 0.70 cm3 g−1, respectively. The average pore size was estimated to be approximately 2 nm from the adsorption/desorption branches of N2 isotherms by using the Barrett–Joyner-Halenda (BJH) method (Fig. 1f).
Scheme 1 illustrated the simple procedure for the synthesis of the Ni@mSiO2 nanocatalyst. Based on the small pore size (3 nm) of mSiO2 nanospheres, very tiny nickel nanoparticles (∼2 nm) could be generated via a melt-infiltration process and thermal treatment under hydrogen flow. Using a 0.55 gnickel salt/gsilica support ratio, the hydrated nickel salt (melting point = 56.7 °C) was successfully incorporated in the mSiO2 nanosphere during the melt infiltration process, driven by the capillary forces. The Ni-loading content after the final thermal treatment was calculated to be nominally 10 wt% on the basis of Ni converted from the nickel nitrate salt. First, the hydrated nickel salt was melt-infiltrated into the mesoporous silica pores of the mSiO2 nanospheres by physical mixing at room temperature with subsequent aging at 60 °C for 24 h in a tumbling oven. Then, tiny nickel nanoparticles were generated by thermal reduction of the confined hydrated Ni salt in the small pores at 500 °C under hydrogen flow.
The low-resolution TEM images of the Ni@mSiO2 nanostructure show Ni nanoparticles as black dots (Fig. 2a and b). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image showed bright dots of a very small size (average 2.0 nm), which indicates uniform incorporation of the Ni nanoparticles in the porous silica (Fig. 2c and S1 in the ESI†). In the elemental mapping of silica (green color) and nickel (red color), high dispersion of nickel nanoparticles was observed (Fig. 2d). HRTEM analysis showed lattices of a single Ni nanoparticles in the mSiO2 pores (Fig. 2e). The inner particle size was observed to be around 2 nm. The Fourier-transform pattern represented a single crystal of metallic nickel with a distance of 0.2 nm between neighboring fringes, which corresponded to the (111) planes of face centered-cubic nickel (Fig. 2e).
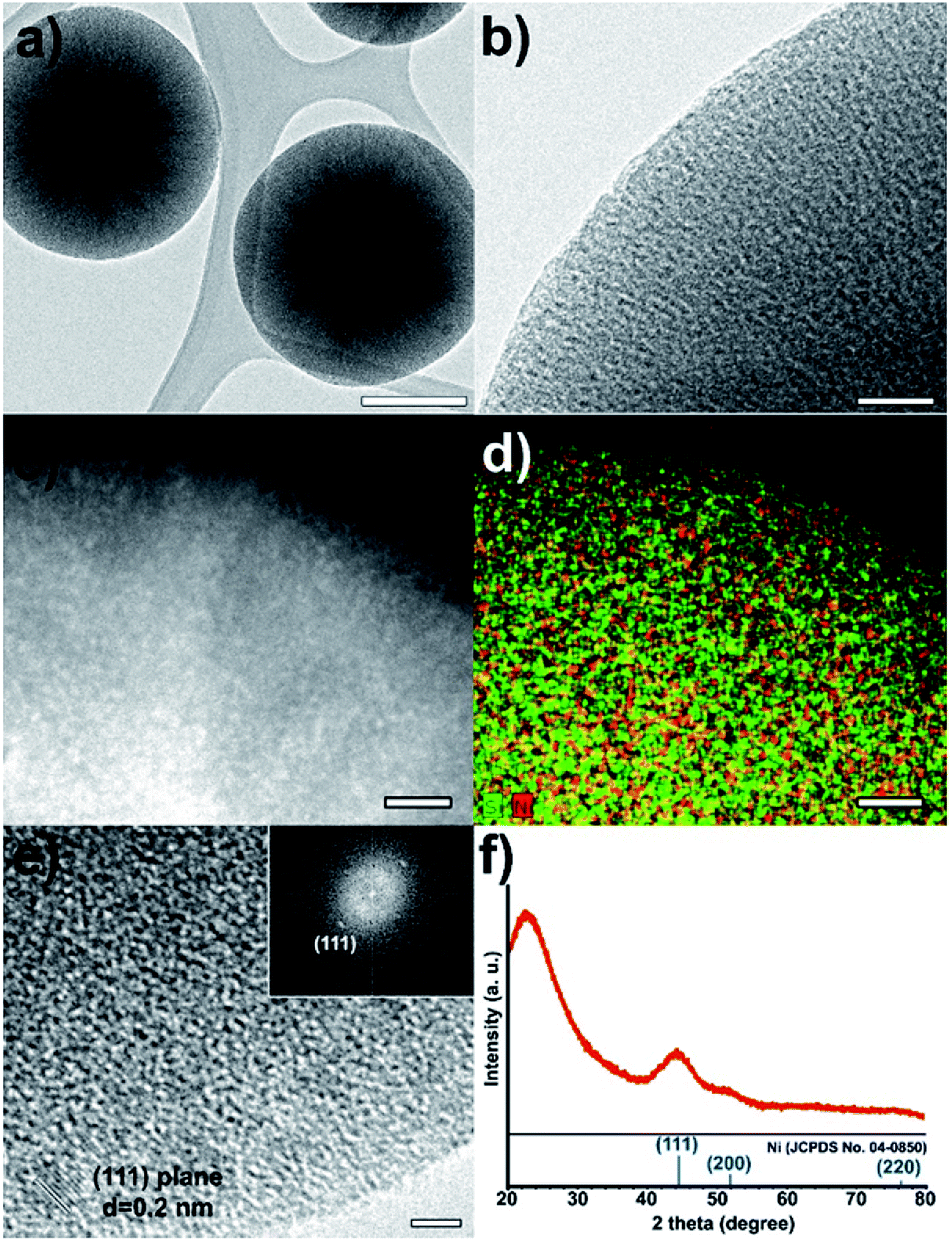 | ||
| Fig. 2 (a and b) TEM and (c) HAADF TEM images, (d) scanning TEM image with elemental mapping and (e) HRTEM images, and (f) XRD spectrum of Ni@mSiO2 nanostructure. The bars represent 200 nm (a), 20 nm (b–d), and 2 nm (e). | ||
The X-ray diffraction (XRD) spectrum of Ni@mSiO2 nanocatalyst has a broad peak at 2θ = 44.5°, which is assigned to the reflections of the (111) plane in the fcc-nickel phase (Fig. 2f, JCPDS No. 04-0850). The average crystal size of the nickel particle was estimated to be 1.8 nm, from the broadness of the (111) peak using the Debye–Scherrer equation, which is well-matched with that observed in the TEM images. The other intense peak around 23° of the Ni@mSiO2 nanocatalyst indicates the presence of amorphous silica.
Using a H2 chemisorption experiment, the active nickel surface area and average nickel particle size of the Ni@mSiO2 nanocatalyst could be analyzed, and measured 242 m2 g−1 and 2.78 nm, respectively. The obtained Ni dispersion was very high, and was calculated to be 36.3%.
The XPS spectrum analysis was carried out to probe the chemical states of Ni@mSiO2 (Fig. S2†). The Ni 2p peak of Ni@mSiO2 showed NiO and Ni peak. The four peaks at 853.9, 855.6, 860.7, and 863.5 eV are assigned to NiO, and the other peak at 852.1 eV is assigned metallic Ni peak. The metallic Ni surface of the small nanoparticles (∼2 nm) was easily oxidized under an ambient condition.
N2 sorption experiments for the Ni@mSiO2 nanocatalyst showed a type IV isotherm with type H4 hysteresis (Fig. 3a). The BET surface area was calculated to be 636 m2 g−1. The total pore volume was found to be 0.3 cm3 g−1, which is about 43% of the initial mSiO2 nanosphere (0.7 cm3 g−1). The significant decrease in pore volume was attributed to inner nickel nanoparticle occupancy. Applying the BJH method, small pore sizes were obtained by the adsorption/desorption branches (Fig. 3b). Because of the occupancy of tiny nickel particles in the pristine silica pores, the pore size of the Ni@mSiO2 nanocatalyst was also slightly decreased, and was observed to be 1.8 nm.
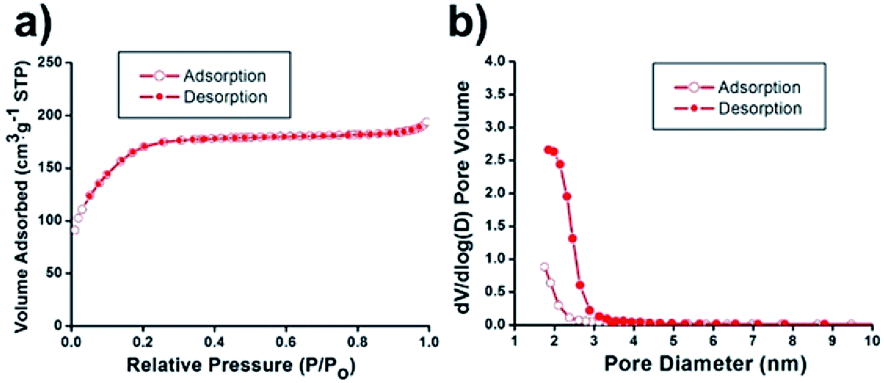 | ||
| Fig. 3 (a) N2 adsorption/desorption isotherms and (b) pore size distribution diagrams of Ni@mSiO2 nanocatalyst. | ||
The Ni@mSiO2 nanocatalyst was applied to the hydrogen-transfer reaction of acetophenone. Acetophenone is an ideal substrate in the hydrogen transfer reaction, and is generally used as a hydrogen acceptor.30 Among the various products of acetophenone reduction, only 1-phenylethanol resulted from the catalyzed reaction. The metallic Ni on the catalyst surface is the active species and the reaction was promoted by base.17 The dihydride species referred in this catalyst system to make alcohol from the transfer of the two hydrogen atoms of the donor to the surface of the metal.31 However, small amount of various byproduct including hemiacetals and condensation products which is occurred between ketone and alcohol were produced due to basic conditions.32 For optimization, the reaction parameters, such as amount of catalyst and base, temperature, and time, are adjusted. The TH reaction results of acetophenone are summarized in Table 1.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Temp. (°C) | Time (min) | Base (eq.) | Conv.b (%) | Yieldb (%) | TON |
| a Rxn. condition: acetophenone (2 mmol), i-PrOH (solvent, 10 mL), base (NaOH).b Determined by GC-MS spectroscopy.c nickel-aluminium alloy purchased from Lancaster (10034177) was applied. | |||||||
| 1 | 1 | 110 | 45 | 1 | 100 | 95 | 95 |
| 2 | 1 | 110 | 60 | 1 | 76 | 73 | 73 |
| 3 | 1 | 100 | 60 | 2 | 100 | 99 | 99 |
| 4 | 1 | 100 | 60 | 1 | 100 | 92 | 92 |
| 5 | 1 | 100 | 60 | 0.5 | 62 | 59 | 59 |
| 6 | 1 | 90 | 60 | 1 | 100 | 96 | 96 |
| 7 | 1 | 80 | 45 | 1 | 58 | 55 | 55 |
| 8 | 1 | 80 | 60 | 1 | 72 | 68 | 68 |
| 9 | 1 | 80 | 75 | 1 | 100 | 97 | 97 |
| 10 | 1 | 80 | 90 | 1 | 67 | 65 | 65 |
| 11 | 0.5 | 80 | 75 | 1 | 100 | 96 | 192 |
| 12 | 0.25 | 80 | 75 | 1 | 10 | 95 | 380 |
| 13 | 0.1 | 80 | 75 | 1 | 62 | 60 | 603 |
| 14 | 0.05 | 100 | 60 | 2 | 80 | 62 | 62 |
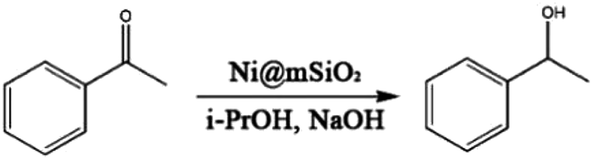
First, we studied the effect of the base and determined the amount of base required (Entries 3–6, Table 1). The reduction of acetophenone can be promoted by inorganic bases as the essential co-catalyst.
Alkaline bases enable increase in the concentration of the alkoxide ion and the deprotonation of the isopropyl alcohol coordinated to metal, which promotes the overall reaction.33,34 Although no reaction was observed without bases, a small amount of base was sufficient to trigger the reaction (Entry 5, Table 1). Subsequently, more than 1 equivalent of the base provided complete conversion efficiency of 1-phenylethanol (Entries 3, 4, Table 1) and 1 equivalent of base was sufficient to obtain high yield. Commercial nickel-aluminium alloy as a comparison catalyst showed lower conversion than that of Ni@mSiO2 nanocatalyst (Entry 15, Table 1). The highly dispersed nickel nanoparticles of Ni@mSiO2 nanocatalyst led to the increased activity.
The reaction temperature and time were also investigated. The reaction temperature was found to influence the reaction time dramatically (Entries 1, 2, 4, 6–10, Table 1). The rate of conversion increased as the temperature was increased. When applying same reaction time (60 min), the Ni@mSiO2 catalyst showed completed conversion at 90 and 100 °C (Entries 2, 4, 6, 8, Table 1). Above 90 °C, over 90% 1-phenylethanol was obtained within 1 h (Entries 1, 4, 6, Table 1). However, the conversion was decreased when the reaction was carried out at 110 °C from 45 to 60 min, and 80 °C from 75 to 90 min (Entries 1, 2, 9, 10, Table 1). At 80 °C, the progress of the reaction was monitored at every 15 min time interval (Entries 7–10, Table 1). The optimum time was found to be 75 min (Entry 10, Table 1).
Furthermore, the amount of catalyst was optimized. The amount of catalyst was 0.25 mol% and above all, acetophenone was fully converted to 1-phenylethanol with 1 equivalent of base for 75 min (Entries 9, 11, 12, Table 1). Under 60% of yield, lower conversions were attained with the addition of a small amount of Ni@mSiO2 (Entries 13, 14, Table 1). Regardless of the slight difference of selectivity of 1-phenylethanol, no further increase in the yield after 0.25 mol% was observed. Thus, it is concluded that 0.25 mol% is the optimum amount of catalyst (Entry 12, Table 1).
It was already mentioned that under the optimum conditions, the Ni@mSiO2 system provided an outstanding yield of 95%, following a good TON of 380. It exhibits higher TON compared to other heterogeneous nickel nanocatalysts including our previous article.6,15,35 The optimized conditions were applied to extend the scope.
To verify the general applicability of Ni@mSiO2, various aromatic ketones were tested (Table 2). Compared to acetophenone, most of the compounds were reduced to alcohol deficiently. Ni@mSiO2 was a highly selective catalyst to acetophenone. Thus we adjusted the reaction time longer excluding benzaldehyde (Entry 1, Table 2). Benzaldehyde was reduced to benzyl alcohol with only 65% yield under the optimized condition, although full conversion was attained. Benzaldehyde, which has higher reducibility, needed a shorter time to obtain higher yield (30 min). The low yield resulted from low selectivity and the fact that aldehyde easily reacts with alcohols, like a nucleophilic aromatic addition. The result of propiophenone was opposite to that of benzaldehyde (Entry 2, Table 2). Owing to electronic and steric interaction, propiophenone exhibited lower conversion compared to benzaldehyde. However, it showed outstanding selectivity with increasing time. A similar behavior was exhibited by 2-acetonaphthone; however, the gap in selectivity made a big difference yield (Entry 3, Table 2). Next, we investigated the effect of additional functional groups on the aromatic ring. According to the electron-donating or withdrawing character, we achieved unique outcomes. The aromatic ketone containing electron-donating groups disfavored the reaction in low conversion under standard reaction conditions (Entries 5–7, Table 2). On the other hand, they had superior selectivity. The substituted acetophenone with electron-withdrawing groups continued with a better conversion, although subordinate selectivity, compared to those with electron-donating groups (Entries 8–13, Table 2). Among the activated acetophenones, 4-methoxyacetophenone exhibited the lowest conversion and showed no further increase in yield via high temperature (Entry 5, Table 2). Together with the para-iodo compound, bromoacetophenone derivatives successfully received hydrogen atoms from isopropyl alcohol in a 96% yield for 3 h (Entries 10, 11, 13, Table 2). The steric factor of functional groups was also considered in this case. Because of its bulkiness, the ortho-substituted acetophenone had the lowest conversion (Entry 9, Table 2). Cyclohexanone, a sort of aliphatic ring, reduced to the corresponding alcohols, reaching up to 50% yield within 3 h (Entry 14, Table 2). Moreover, Ni@mSiO2 nanocatalyst shows highly catalytic activity compared with other Ni- and Ru-based catalysts previously reported (Table S1†).36–43
| Entry | Compound | Conv. (%) | Sel. (%) | Yield (%) |
|---|---|---|---|---|
| a Cat. (0.25 mol%), base NaOH (1 eq.), rxn. temp. 80 °C, rxn. time 75 min, determined by GC-MS spectroscopy.b Rxn. time 15 min.c Rxn. time 30 min.d Rxn. time 2 h.e Rxn. time 3 h.f Rxn. temp. 100 °C. | ||||
| 1 | 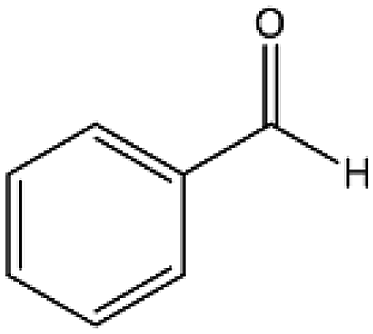 |
78b, 100c, 100 | 33b, 75c, 65 | 26b, 75c, 65 |
| 2 |  |
54, 68d, 44e | 92, 97d, 100e | 50, 66d, 44e |
| 3 |  |
55, 64d | 57, 55d | 31, 35d |
| 4 |  |
73 | 88 | 64 |
| 5 |  |
8, 35f | 88, 97f | 7, 34f |
| 6 |  |
40, 45e | 97, 93e | 39, 42e |
| 7 |  |
34, 56d | 93, 93d | 32, 52d |
| 8 |  |
55, 67e | 76, 57e | 42, 38e |
| 9 |  |
55 | 61 | 34 |
| 10 |  |
78, 99e | 96, 97e | 75, 96e |
| 11 |  |
69, 93e | 95, 100e | 66, 96e |
| 12 |  |
75, 95e | 64, 84e | 48, 79e |
| 13 |  |
71, 95e | 78, 97e | 56, 92e |
| 14 |  |
63, 91e | 64, 53e | 40, 48e |
In the TEM image of the recovered Ni@mSiO2 nanocatalyst shown totally collapsed silica structure and sintering nickel nanoparticles (Fig. S5a and b†). In the stability of structure, silica was occurred procreated due to the steam during the reaction. In the XRD data of the recovered Ni@mSiO2 nanocatalyst showed sharp peaks (Fig. S5c†). It means reflected the increased crystal size. In addition, metallic nickel phase changed nickel silicide and nickel carbide during the reaction.
The recycle reaction of the recovered Ni@mSiO2 catalyst showed a low conversion (79%) compared high conversion (100%) of fresh Ni@mSiO2 for 1 h. In the recycle reaction, the catalyst exhibited lower performance due to metallic nickel was changed no active site catalyst such as nickel carbide and nickel silicide.
Conclusions
In conclusion, we could newly synthesize highly dispersed nickel nanoparticles on mesoporous silica nanospheres, using melt infiltration process and subsequent thermal reduction. Based on the high particle dispersion of 36.3%, the Ni@mSiO2 nanocatalyst showed superior catalytic performance compared to commercial nickel-aluminium alloy for transfer hydrogenation reactions of acetophenone. Furthermore, the optimized catalytic system by Ni@mSiO2 catalyst can tolerate various ranges of functional groups.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2017R1A4A1015533 and NRF-2017R1D1A1B03036303). J. C. Park was supported by the Research and Development Program of the Korea Institute of Energy Research (KIER) (No. B9-2461-01).Notes and references
- D. A. Evans, S. G. Nelson, M. R. Gagne and A. R. Muci, J. Am. Chem. Soc., 1993, 115, 9800–9801 CrossRef CAS.
- E. J. Corey and C. J. Helal, Tetrahedron Lett., 1996, 37, 5675–5678 CrossRef CAS.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- F. Alonso, Nickel Nanoparticles in the Transfer Hydrogenation of Functional Groups, in Met. Nanoparticles Catal. Adv. Appl., The Royal Society of Chemistry, 2014, ch. 5, pp. 83–98 Search PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- J. Xiong, H. Shen, J. Mao, X. Qin, P. Xiao, X. Wang, Q. Wu and Z. Hu, J. Mater. Chem., 2012, 22, 11927 RSC.
- R. C. Mebane, K. L. Holte and B. H. Gross, Synth. Commun., 2007, 37, 2787–2791 CrossRef CAS.
- G. P. Boldrini, D. Savoia, E. Tagliavini, C. Trombini and A. Umani-Ronchi, J. Org. Chem., 1985, 50, 3082–3086 CrossRef CAS.
- G. Xanthopoulou, O. Thoda, E. D. Metaxa, G. Vekinis and A. Chroneos, J. Catal., 2017, 348, 9–21 CrossRef CAS.
- Y. Du, H. Chen, R. Chen and N. Xu, Appl. Catal., A, 2004, 277, 259–264 CrossRef CAS.
- P. Fouilloux, Appl. Catal., 1983, 8, 1–42 CrossRef CAS.
- F. Alonso, P. Riente and M. Yus, Tetrahedron, 2008, 64, 1847–1852 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- F. Alonso, P. Riente, J. A. Sirvent and M. Yus, Appl. Catal., A, 2010, 378, 42–51 CrossRef CAS.
- D. Kim, H. Kang, H. Park, S. Park, J. C. Park and K. H. Park, Eur. J. Inorg. Chem., 2016, 3469–3473 CrossRef CAS.
- J. C. Park, H. J. Lee, J. Y. Kim, K. H. Park and H. Song, J. Phys. Chem. C, 2010, 114, 6381–6388 CrossRef CAS.
- K. Shimura and K. Shimizu, Green Chem., 2012, 14, 2983–2985 RSC.
- H. Woo, K. Lee, J. C. Park and K. H. Park, New J. Chem., 2014, 38, 5626–5632 RSC.
- P. E. de Jongh and T. M. Eggenhuisen, Adv. Mater., 2013, 25, 6672–6690 CrossRef CAS PubMed.
- T. M. Eggenhuisen, J. P. den Breejen, D. Verdoes, P. E. de Jongh and K. P. de Jong, J. Am. Chem. Soc., 2010, 132, 18318–18325 CrossRef CAS PubMed.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS PubMed.
- J. I. Kwon, T. Kim, J. C. Park, J.-I. Yang and K. Y. Lee, J. Nanosci. Nanotechnol., 2016, 16, 1787–1792 CrossRef CAS PubMed.
- X. Liu, J. G. Khinast and B. J. Glasser, Ind. Eng. Chem. Res., 2014, 53, 5792–5800 CrossRef CAS.
- F.-S. Xiao, S. Zheng, J. Sun, R. Yu, S. Qiu and R. Xu, J. Catal., 1998, 176, 474–487 CrossRef CAS.
- M. Kim, J. C. Park, A. Kim, K. H. Park and H. Song, Langmuir, 2012, 28, 6441–6447 CrossRef CAS PubMed.
- B. G. Johnson, S. Raynor, D. Shephard, J. Thomas and M. Mantle, Chem. Commun., 1999, 1167–1168 RSC.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159 CrossRef CAS.
- H. Takahashi, B. Li, T. Sasaki, C. Miyazaki, T. Kajino and S. Inagaki, Microporous Mesoporous Mater., 2001, 44, 755–762 CrossRef.
- Y. Wang, D. Wang, M. Tan, B. Jiang, J. Zheng, N. Tsubaki and M. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 26767–26775 CrossRef CAS PubMed.
- J. S. M. Samec, J.-E. Backvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- F. Alonso, P. Riente and M. Yus, Tetrahedron Lett., 2008, 49, 1939–1942 CrossRef CAS.
- J. Clayden, N. Greeves, S. Warren and P. Wothers, “Organic Chemistry”, 2001, (n.d.) Search PubMed.
- M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466–1478 CrossRef CAS.
- R. L. Chowdhury and J.-E. Backvall, J. Chem. Soc., Chem. Commun., 1991, 1063–1064 RSC.
- A. F. Trasarti, N. M. Bertero, C. R. Apesteguía and A. J. Marchi, Appl. Catal., A, 2014, 475, 282–291 CrossRef CAS.
- N. Catellanos-Blanco, A. Arévalo and J. J. García, Dalton Trans., 2016, 45, 13604 RSC.
- Y. Li, S. Yu, W. Shen and J. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS PubMed.
- P. Yang, L. H. Lim, P. Chuanprasit, H. Hirao and J. S. Zhou, Angew. Chem., Int. Ed., 2016, 55, 12083–12087 CrossRef CAS PubMed.
- B. J. Simmons, M. Hoffmann, J. Hwang, M. K. Jackl and N. K. Grag, Org. Lett., 2017, 19, 1910–1913 CrossRef CAS PubMed.
- S. Guo, P. Yang and J. S. Zhou, Chem. Commun., 2015, 51, 12115–12117 RSC.
- R. Liu, T. Cheng, L. Kong, C. Chen, G. Liu and H. Li, J. Catal., 2013, 307, 55–61 CrossRef CAS.
- Z. Cao, H. Qiao and F. Zeng, Organometallics, 2019, 38, 797–804 CrossRef CAS.
- S. Guillarme, T. X. M. Nguyen and C. Saluzzo, Tetrahedron: Asymmetry, 2008, 19, 1450–1454 CrossRef CAS.
- S. Iyer and A. Sattar, Synth. Commun., 1998, 28, 1721–1725 CrossRef CAS.
- V. Polshettiwar, B. Baruwati and R. S. Varma, Green Chem., 2009, 11, 127–131 RSC.
- M. D. Le Page and B. R. James, Chem. Commun., 2000, 1647–1648 RSC.
- S. Iyer and J. P. Varghese, J. Chem. Soc., Chem. Commun., 1995, 4, 465–466 RSC.
- W. N. O. Wylei, A. J. Lough and R. H. Morris, Organometallics, 2009, 28, 6755–6761 CrossRef.
- B. Saes, D. G. A. Verhoeven, M. Lutz, R. J. M. Gebbink and M.-E. Moret, Organometallics, 2015, 34, 2710–2713 CrossRef CAS.
- P. Phukan and A. Sudalai, Synth. Commun., 2015, 30, 2401–2405 CrossRef.
- F. Foubelo, C. Nájera and M. Yus, Tetrahedron: Asymmetry, 2015, 26(15–16), 769–790 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01608b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |