
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide complexes combined with chiral salen ligands: application in the enantioselective epoxidation reaction of α,β-unsaturated ketones†
Xuexiu Xiaa,
Chengrong Lu*a,
Bei Zhao *ab and
Yingming Yaoab
*ab and
Yingming Yaoab
aCollege of Chemistry, Chemical Engineering and Materials Science, Dushu Lake Campus, Soochow University, Suzhou 215123, People's Republic of China. E-mail: zhaobei@suda.edu.cn
bKey Laboratory of Organic Synthesis of Jiangsu Province, People's Republic of China. E-mail: yaoym@suda.edu.cn; Fax: +86 51265880305; Tel: +86 512 65880305
First published on 3rd May 2019
Abstract
Readily available lanthanide amides Ln[N(SiMe3)2]3 (Ln = Nd (1), Sm (2), Eu (3), Yb (4), La (5)), combined with chiral salen ligands H2La ((S,S)-N,N′-di-(3,5-disubstituted-salicylidene)-1,2-cyclohexanediamine) and H2Lb ((S,S)-N,N′-di-(3,5-disubstituted-salicylidene)-1,2-diphenyl-1,2-ethanediamine) were employed in the enantioselective epoxidation of α,β-unsaturated ketones. It was found that the salen–La complex shows the highest efficiency and enantioselectivity. A relatively broad scope of α,β-unsaturated ketones was investigated, and excellent yields (up to 99%) and moderate to good enantioselectivities (37–87%) of the target molecules were achieved.
Introduction
Salen ligands are a particular class of Schiff bases, which are produced by condensation of two salicylaldehydes with a diamine. The use of salen metal complexes for a large variety of catalytic transformations has emerged, mainly because the salen ligands are easily available and highly tunable, and are capable of coordinating to many different metals to form various catalysts.1 In recent years, chiral salen ligands have become one of the most popular chiral ligands and a series of salen-transition metal catalysts have found application in enantioselective synthesis. In 2001, Kozlowski developed a set of modular bifunctional salen ligands, which showed enhanced reactivity in the asymmetric addition of diethylzinc to aldehydes and achieved excellent yield (up to 99%) with high enantioselectivity (up to 91%).2 In 2003, Jiang used salen-Ti complexes to catalyze the enantioselective addition of TMSCN to ketones.3 Rawal reported that the enantioselective carbonyl–ene reactions were catalyzed by a salen–Co complex, which generated chiral homoallylic alcohols in excellent yields, enantioselectivities and diastereoselectivities.4 In 2014, the asymmetric conia-ene-type cyclization of α-functionalized ketones was catalyzed by a chiral salen–Fe complex.5 Salen–Zr complexes catalyzed enantioselective α-hydroxylation of β-ketones esters using cumene hydroperoxide (CHP) as the oxidant in excellent yields and enantioselectivities.6 Epoxidation of non-functionalised alkenes catalyzed by Mn–salen was investigated by Vyas.7However, to the best of our knowledge, examples of highly active lanthanide complexes bearing chiral salen ligands are limited, and their applications are restricted to ring–opening reaction of epoxides,8 aminoalkene hydroamination/cyclisation,9 and nitro–mannich reaction.10 Shibasaki developed asymmetric epoxidation of the electron-deficient C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in enones, α,β-unsaturated amides and esters, using the combination of lanthanide-BINOL-Ph3AsO as catalyst.11 Some lanthanide–BINOL and its derivatives have also been investigated in such a transformation, which gave rise to high enantioselectivities in many cases.12 Feng employed Sc(OTf)3/N,N′-dioxide as catalyst to realize the asymmetric epoxidation of α,β-unsaturated ketones with excellent enantioselectivities.13 Recently, our group reported a series of rare-earth metal complexes together with phenoxy-functionalized chiral prolinols which are highly efficient catalysts in the epoxidation of α,β-unsaturated ketones. Both bisubstituted and trisubstituted chalcones produced the corresponding epoxides in excellent yields (up to 99%) and enantioselectivities (up to 99%) using tert-butylhydroperoxide (TBHP) as the oxidant.14 As a continuation of our research on the lanthanide-mediated asymmetric transformation, we herein report rare-earth metal complexes bearing chiral salen ligands and their catalytic potential in the enantioselective epoxidation of α,β-unsaturated ketones.
C bonds in enones, α,β-unsaturated amides and esters, using the combination of lanthanide-BINOL-Ph3AsO as catalyst.11 Some lanthanide–BINOL and its derivatives have also been investigated in such a transformation, which gave rise to high enantioselectivities in many cases.12 Feng employed Sc(OTf)3/N,N′-dioxide as catalyst to realize the asymmetric epoxidation of α,β-unsaturated ketones with excellent enantioselectivities.13 Recently, our group reported a series of rare-earth metal complexes together with phenoxy-functionalized chiral prolinols which are highly efficient catalysts in the epoxidation of α,β-unsaturated ketones. Both bisubstituted and trisubstituted chalcones produced the corresponding epoxides in excellent yields (up to 99%) and enantioselectivities (up to 99%) using tert-butylhydroperoxide (TBHP) as the oxidant.14 As a continuation of our research on the lanthanide-mediated asymmetric transformation, we herein report rare-earth metal complexes bearing chiral salen ligands and their catalytic potential in the enantioselective epoxidation of α,β-unsaturated ketones.
Results and discussion
To test the reactivity and selectivity of lanthanide amides in combination with chiral salen ligands, five amides Ln[N(SiMe3)2]3 (Ln = Nd (1), Sm (2), Eu (3), Yb (4), La (5)), two series of phenoxy-functionalized chiral salen ligands [(S,S)-N,N′-di-(3-R1-5-R2-salicylidene)-1,2-cyclohexanediamine, [R1 = R2 = Me (H2L1); R1 = R2 = tBu (H2L2); R1 = H, R2 = Cl (H2L3); R1 = H; R2 = tBu (H2L4); R1 = tBu, R2 = H (H2L5); R1 = Me, R2 = H (H2L6); R1 = tBu; R2 = 1-adam (H2L7)] and (S,S)-N,N′-(3-R1-5-R2-salicylidene)-1,2-diphenyl-1,2-ethanediamine [R1 = R2 = Me (H2L8); R1 = R2 = tBu (H2L9)] were synthesized according to the previous study.15 With the neodymium amide 1 and the salen ligand H2L1 in hand, the epoxidation reaction of chalcone was carried out in the presence of TBHP in THF. The results are listed in Table 1. The model reaction underwent quantitatively with unsatisfying enantioselectivity (99% yield and 31% ee). On the basis of this finding, the influence of the chiral salen ligands on enantioselectivity was carefully investigated via the modification of the phenol moiety. The yields of the corresponding epoxides remained excellent and the ee values varied from 11–31% (Table 1, entries 1–7). If the chiral linkage was changed to (S,S)-1,2-diphenyl-1,2-ethanediamine (H2L8 and H2L9), no significant improvement is observed (Table 1, entries 1–9).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Cat | n | Ligand | X | Solvent | T/°C | Yieldb (%) | eec (%) |
| a The reaction was performed with chalcone (0.3 mmol), TBHP (0.36 mmol) in 1 mL of solvent.b HPLC yield.c Determined by chiral HPLC analysis. | ||||||||
| 1 | Nd-1 | 10 | H2L1 | 12 | THF | rt | 99 | 31 |
| 2 | Nd-1 | 10 | H2L2 | 12 | THF | rt | 99 | 23 |
| 3 | Nd-1 | 10 | H2L3 | 12 | THF | rt | 86 | 31 |
| 4 | Nd-1 | 10 | H2L4 | 12 | THF | rt | 99 | 27 |
| 5 | Nd-1 | 10 | H2L5 | 12 | THF | rt | 99 | 30 |
| 6 | Nd-1 | 10 | H2L6 | 12 | THF | rt | 99 | 27 |
| 7 | Nd-1 | 10 | H2L7 | 12 | THF | rt | 86 | 11 |
| 8 | Nd-1 | 10 | H2L8 | 12 | THF | rt | 99 | 13 |
| 9 | Nd-1 | 10 | H2L9 | 12 | THF | rt | 99 | 23 |
| 10 | Sm-2 | 10 | H2L1 | 12 | THF | rt | 99 | 33 |
| 11 | Eu-3 | 10 | H2L1 | 12 | THF | rt | 89 | 35 |
| 12 | Yb-4 | 10 | H2L1 | 12 | THF | rt | 35 | 4 |
| 13 | La-5 | 10 | H2L1 | 12 | THF | rt | 99 | 41 |
| 14 | La-5 | 10 | H2L1 | 12 | Tol | rt | 99 | 5 |
| 15 | La-5 | 10 | H2L1 | 12 | Hex | rt | 87 | 37 |
| 16 | La-5 | 10 | H2L1 | 12 | DME | rt | 89 | 35 |
| 17 | La-5 | 10 | H2L1 | 12 | CH3CN | rt | 99 | 57 |
| 18 | La-5 | 10 | H2L1 | 12 | CH3CN | 0 | 99 | 70 |
| 19 | La-5 | 10 | H2L1 | 12 | CH3CN | -20 | 99 | 80 |
| 20 | La-5 | 10 | H2L1 | 12 | CH3CN | -40 | 82 | 56 |
| 21 | La-5 | 10 | H2L1 | 15 | CH3CN | -20 | 99 | 71 |
| 22 | La-5 | 10 | H2L1 | 20 | CH3CN | -20 | 99 | 75 |
| 23 | La-5 | 5 | H2L1 | 12 | CH3CN | -20 | 99 | 57 |
Thus, the chiral salen ligand H2L1 was the optimal choice for the model reaction considering both yield and enantioselectivity. Lanthanide amides of different metal centers were studied, and the lanthanum amide 5 gave the best result. The ee value of 7a reached 41% without sacrificing yield (Table 1, entries 1 and 10–13).
To further improve enantioselectivity, different solvents were screened. Fortunately, when acetonitrile was used as the solvent in the model reaction, the ee value increased to 57%, while the yield of 99% was maintained (Table 1, entries 13–17). Some successful cases in the asymmetric epoxidation reactions also achieved high ee values in acetonitrile.14a,16 Perhaps the weak coordination of acetonitrile to the central lanthanide metal changes the coordination environment. And it leads to better match for the catalyst and substrate, which may plays an important role in the metal-based catalytic asymmetric reaction. A relatively low temperature is usually helpful to improve the enantioselectivity of the asymmetric reaction. To our delight, attempts to lower the reaction temperature have a positive effect on the enantioselectivity. The ee value significantly increased when the reaction temperature decreased, and the 80% ee of the epoxide 7a was obtained at −20 °C (Table 1, entries 17–20). Finally, the ratio of the lanthanum amide 5 to the chiral salen ligand H2L1 was studied in CH3CN and the results showed that the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar ratio was optimal for the model reaction (Table 1, entries 19, 21–22). Thus, 10 mol% of La amide 5 in the combination of 12 mol% of chiral salen ligand H2L1 was an optimal catalytic system for the asymmetric epoxidation of chalcone with TBHP in CH3CN at −20 °C.
:1.2 molar ratio was optimal for the model reaction (Table 1, entries 19, 21–22). Thus, 10 mol% of La amide 5 in the combination of 12 mol% of chiral salen ligand H2L1 was an optimal catalytic system for the asymmetric epoxidation of chalcone with TBHP in CH3CN at −20 °C.
Subsequently, various α,β-unsaturated ketones were synthesized and investigated, and the results are summarized in Chart 1. Most of the disubstituted α,β-unsaturated ketones underwent the transformation to give epoxides in excellent yields (92–99%) and good to high enantioselectivities (57–83%), with the exception of 7h (37% ee) (Chart 1, 7a–7o). Delightedly, much better enantioselectivities, ranging from 80–87%, were observed in reactions of trisubstituted α,β-unsaturated ketones, which are in general bulky and challenging substrates (Chart 1, 7p–7y). It is noteworthy that these substrates are not only trisubstituted, but also cyclic with restricted mobility, hence more rigid transition states are supposed to generate, which may play a critical role in controlling the enantioselectivity. Relatively high results include the lanthanide–BINOL and its derivatives by Shibasaki, 11 Inanaga12a and Ding,12b and Sc(OTf)3/N,N′-dioxide catalyst by Feng,13 and rare-earth metal complexes together with phenoxy-functionalized chiral prolinols reported by us.14
 | ||
| Chart 1 Epoxidation of α,β-unsaturated ketones catalyzed by complex 5a. aReactions were performed with the substrate (0.3 mmol), TBHP (0.36 mmol) in 1 mL CH3CN at −20 °C for 10 h; isolated yield; ee value were determined by chiral HPLC analysis. b15 h. c48 h. | ||
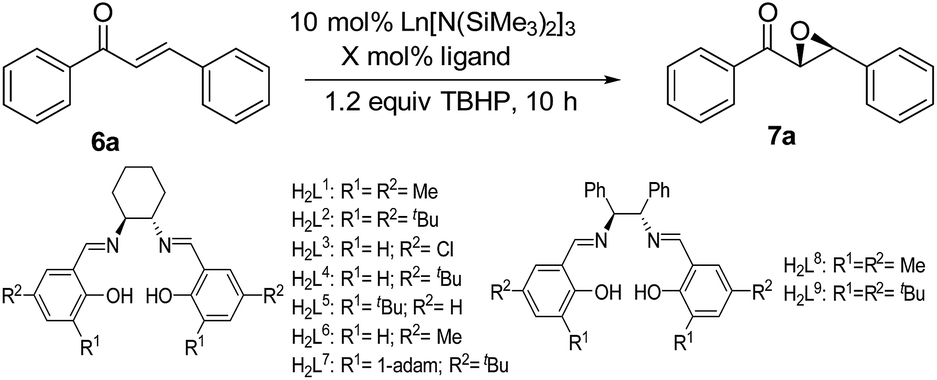
The real active species in the current system was investigated. La amide 5 was treated with 1.2 equiv. chiral salen ligand H2L1 in THF. After workup, complex 8 was isolated as yellow crystals (Scheme 1). Its 1H NMR spectrum shows absence of a peak at 0.1980 ppm, which is ascribed to the coordinated –N(SiMe3)2 group. Signals at 3.87–3.55, 1.60 and 1.02 ppm are assigned to cyclohexyl linkage. The peaks at 8.10, 7.91, 7.63, and 7.57 ppm are ascribed to the CH = N double bond. These observations prove the formation of complex 8 bearing the chiral salen ligand. Finally, the ratio of the lanthanum atom to chiral salen ligand is determined to be 2:3, based on results of elemental analysis and complex titration.
 | ||
| Scheme 1 The preparation of complex 8. | ||
To find out whether complex 8 is the active species, it was tested in the model asymmetric epoxidation of chalcone under standard conditions. However, neither the yield nor ee value was comparable with results of in situ generated catalyst. The target epoxide was obtained in 91% yield and only 70% ee (Table 2, entry 1). Comparing described catalyst system with complex 8, the difference is HN(SiMe3)2 generated in situ in the former, which may have the positive effects on the asymmetric transformation. To verify the hypothesis, complex 8 together with 20 mol% HN(SiMe3)2 were added to the model reaction. The outcome of 98% yield with 78% ee indicates that the addition of HN(SiMe3)2 indeed has a positive effect on the asymmetric catalytic process (Table 2, entry 2). Screening of basic additives, including 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU), Et3N, pyridine, and 2,2′-bipyridine, proved that HN(SiMe3)2 (20 mol%) is the optimal choice (Table 2, entries 2–9).
|
||||
|---|---|---|---|---|
| Entry | Additive | Y | Yeildb (%) | eec (%) |
| a Reactions were performed with chalcone (0.3 mmol), TBHP (0.36 mmol), 1 mL CH3CN.b HPLC yield.c Determined by chiral HPLC analysis. | ||||
| 1 | — | 20 | 91 | 70 |
| 2 | HN(SiMe3)2 | 20 | 98 | 78 |
| 3 | DBU | 20 | 93 | 15 |
| 4 | Pyridine | 20 | 88 | 59 |
| 5 | Et3N | 20 | 86 | 60 |
| 6 | 2,2-Bipyridine | 20 | 85 | 56 |
| 7 | HN(SiMe3)2 | 10 | 93 | 73 |
| 8 | HN(SiMe3)2 | 30 | 95 | 75 |
| 9 | HN(SiMe3)2 | 50 | 90 | 66 |

Finally, the outcome of the model reaction on a gram scale was satisfactory with an appropriate prolonging of the reaction time and the absolute configuration of the epoxides was determined by single crystal diffraction analysis, chiral HPLC and optical rotation analysis of a representative crystal 7e (Scheme 2).
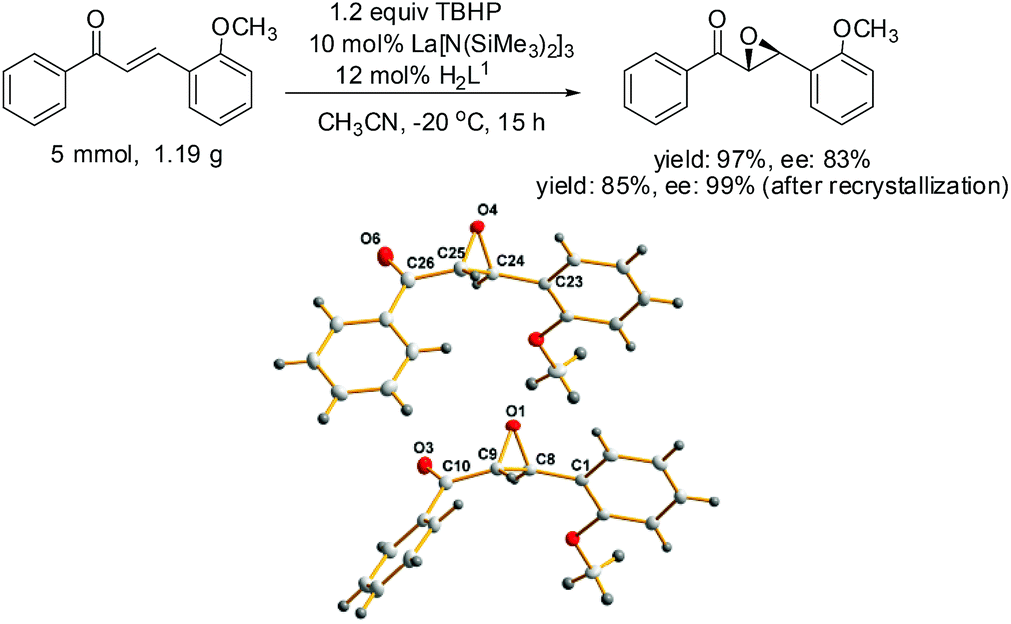 | ||
| Scheme 2 The synthesis and the molecular structure of compound 7e. | ||
The molecular structure of 7e showing 20% probability ellipsoids. Selected bond lengths (Å) and bond angles (°): O(1)–C(8) 1.4235(3), O(1)–C(9) 1.430(4), C(8)–C(9) 1.494(4), O(4)–C(24) 1.435(4), O(4)–C(25) 1.415(4), C24–C25 1.482(4), O(1)–C(8)–C(1) 116.6(2), O(1)–C(9)–C(10) 115.1(3), O(1)–O(8)–C(9) 58.37(18), O(1)–C(9)–C(8) 58.74(18), C(9)–O(1)–C(8) 62.89(19), O(4)–C(24)–C(23) 116.1(2), O(4)–C(25)–C(26) 113.8(3), O(4)–C(24)–C(25) 58.02(19), O(4)–C(25)–C(24) 59.33(19), C(24)–O(4)–C(25) 62.6(2).
Conclusions
In summary, the asymmetric epoxidation of α,β-unsaturated ketones was catalyzed by a new series of lanthanide amides with chiral salen ligands. After careful screening, 10 mol% of the La amide La[N(SiMe3)2]3 together with the chiral salen ligand (S,S)-N,N′-(3,5-dimethylsalicylidene)-1,2-cyclohexanediamine in a 1:1.2 molar ratio were found to be the optimal catalyst in CH3CN at −20 °C. For the disubstituted chalcone derivatives, the target epoxides were obtained in excellent yields (92–99%) with moderate to good ee values (57–83%), while epoxides from the trisubstituted α,β-unsaturated ketones were generated in high enantioselectivities (80–87%) and relative low yields (32–57%). Complex 8 resulting from reaction of lanthanum amide and salen ligand was isolated and characterized, which, together with HN(SiMe3)2 additive, gave comparable catalytic result with that of in situ generated catalyst. The absolute configuration of the target chiral epoxides was determined by single crystal diffraction analysis.
Experimental
General information
All manipulations and reactions involving air and water sensitive components were performed with the standard Schlenk techniques. Solvents, such as THF, toluene and hexane, were degassed and distilled from sodium benzophenone ketyl under argon before use. Analytical thin layer chromatography (TLC) was performed using F254 pre-coated silica gel plate (0.2 mm thickness). After elution, plates were detected using UV radiation (254 nm) on a UV lamp. Flash chromatography was performed using 200–300 mesh silica gel with freshly distilled solvents. Nuclear magnetic resonance spectra were obtained on a Bruker AV-400 apparatus (CDCl3 as solvent). High Resolution Mass (HRMS) spectra were obtained using Bruker ESI-TOF. Rare-earth metal analysis was performed by EDTA titration with a xylenol orange indicator and a hexamine buffer. Carbon, hydrogen and nitrogen analyses were performed by direct combustion with a Carlo-Erba EA-1110 instrument. The ee values determination was carried out using HPLC (Agilent Technologies 1200 Series) with Daicel chiralcel columns at room temperature. Optical rotation was measured using an Autopol IV polarimeter equipped with a sodium vapor lamp at 589 nm. The absolute configuration of 7e was determined by the detection of single crystal diffraction. Intensity data were collected with a Rigaku Mercury CCD area detector in ω scan mode using Cu–Kα radiation (λ = 1.54178 Å). Hence, the absolute configurations of 7a–7y were assigned by analogy, assuming the same reaction pathway and the same analysis.Synthesis of lanthanum complex 8
Under a standard Schlenk vacuum line, to a THF solution of 4 mmol La[N(SiMe3)2]3, 6 mmol H2L1 in 4 mL THF was added dropwise at room temperature. The mixture was continued to stir overnight. Then, removing the THF solvent in vacuo, the crude product was washed with hexane for three times and a faint yellow solid was obtained by centrifugal separation. Finally, complex 8 was purified by recrystallization in the mixed solvent of toluene and hexane.General procedure for the synthesis of the substituted epoxides 7a–7y
Under argon atmosphere, lanthanum amide La[N(SiMe3)2]3 (0.03 mmol, 18.65 mg) was added to a solution of H2L1 (0.036 mmol, 13.62 mg) in CH3CN (1 mL). After stirring for 30 min, unsaturated ketones (0.3 mmol) was added and stirred for further 30 min. After that, oxidant TBHP (0.36 mmol) was added to the mixture. The system was stirred for 10 h at −20 °C. Quenched by the saturated solution of Na2SO3, the crude product was extracted by ethyl acetate, then purified by column chromatography (ethyl acetate–petroleum ether, 1:10) to obtain the target epoxide. The enantiomeric excess of epoxide was determined by chiral HPLC analysis.
Spectroscopic data for ligands
Spectroscopic data for saymmetric epoxidation products
H), 4.44 (s, 1H, CH), 3.02 (m, 1H, CH2), 2.85 (m, 1H, CH2), 2.39 (m, 1H, CH2), 1.64 (m, 1H, CH2). HPLC: OD-H column, 97% hexanes, 3% iPrOH, 1.0 mL min−1, tr (minor) = 21.1 min, tr (major) = 19.5 min. HRMS (ESI, m/z) calcd for C17H13ClO2Na [M + Na]+: 307.0502, found: 307.0506. Mp: 132–134 °C.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant No. 21572151), the project of Scientific and Technologic Infrastructure of Suzhou (SZS201708), and PAPD.Notes and references
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- (a) M. Kitamura, S. Suga, K. Kawai and R. Noyori, J. Am. Chem. Soc., 1986, 108, 6071–6072 CrossRef CAS PubMed; (b) R. Noyori and M. Kitamura, Angew. Chem., Int. Ed. Engl., 1991, 30, 49–69 CrossRef; (c) M. Quirmbach, A. Kless, J. Holz, B. Tararov and A. Börner, Tetrahedron: Asymmetry, 1999, 10, 1803–1811 CrossRef CAS; (d) A. Kless, R. Kadyrov, A. Börner, J. Holz and H. B. Kagan, Tetrahedron Lett., 1995, 36, 4601–4602 CrossRef CAS; (e) E. F. Dimauro and M. C. Kozlowski, Org. Lett., 2001, 3(19), 3053–3056 CrossRef CAS PubMed.
- (a) E. F. DiMauro and M. C. Kozlowski, J. Am. Chem. Soc., 2002, 124, 12668–12670 CrossRef CAS PubMed; (b) C. Garcfa, L. K. LaRochelle and P. J. Walsh, J. Am. Chem. Soc., 2002, 124, 10970–10971 CrossRef; (c) Y. N. Belokon, B. Green, N. S. Ikonnikov, M. North and V. I. Tararov, Tetrahedron Lett., 1999, 40, 8147–8149 CrossRef CAS; (d) K. Yabu, S. Masumoto, M. Kanai, D. P. Curran and M. Shibasaki, Tetrahedron Lett., 2002, 43, 2923–2925 CrossRef CAS; (e) S. Masumoto, M. Suzuki, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2002, 43, 8647–8650 CrossRef CAS; (f) S. K. Tian and L. Deng, J. Am. Chem. Soc., 2001, 123, 6195–6197 CrossRef CAS PubMed; (g) H. Deng, M. P. Snapper and A. H. Hoveyda, Angew. Chem., Int. Ed., 2002, 41, 1009–1011 CrossRef CAS; (h) F. X. Chen, X. M. Feng, B. Qin, G. L. Zhang and Y. Z. Jiang, Org. Lett., 2003, 5(6), 949–952 CrossRef CAS PubMed.
- (a) D. A. Evans, C. S. Burgey, N. A. Paras, T. Vojkovsky and S. W. Tregay, J. Am. Chem. Soc., 1998, 120, 5824–5825 CrossRef CAS; (b) D. A. Evans, S. W. Tregay, C. S. Burgey, N. A. Paras and T. Vojkovsky, J. Am. Chem. Soc., 2000, 122, 7936–7943 CrossRef CAS; (c) Y. Yuan, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2003, 42, 5478–5480 CrossRef CAS PubMed; (d) D. A. Evans and J. Wu, J. Am. Chem. Soc., 2005, 127, 8006–8007 CrossRef CAS PubMed; (e) G. E. Huston, A. H. Dave and V. H. Rawal, Org. Lett., 2007, 9, 3869–3872 CrossRef PubMed.
- (a) F. D. Denes, A. Perez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366–2368 CrossRef CAS PubMed; (b) B. K. Corkey and F. D. Toste, J. Am. Chem. Soc., 2005, 125, 17168–17169 CrossRef PubMed; (c) T. Yang, A. Ferrali, F. Sladojevich, L. Campbell and D. Dixon, J. Am. Chem. Soc., 2009, 131, 9140–9141 CrossRef CAS PubMed; (d) A. Matsuzawa, T. Mashiko, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7616–7618 CrossRef CAS PubMed; (e) S. Shaw and J. D. White, J. Am. Chem. Soc., 2014, 136, 13578–13581 CrossRef CAS PubMed.
- (a) B. Gong, Q. Meng, T. Su, M. Lian, Q. Wang and Z. Gao, Synlett, 2009, 2009, 2659–2661 CrossRef; (b) Y. Cai, M. Lian, Z. Li and Q. Meng, Tetrahedron, 2012, 68, 7973–7978 CrossRef CAS; (c) Z. Li, M. Lian, F. Yang, Q. Meng and Z. Gao, Eur. J. Org. Chem., 2014, 2014, 3491–3495 CrossRef CAS; (d) M. Lian, Z. Li, J. Du, Q. Meng and Z. Gao, Eur. J. Org. Chem., 2010, 2010, 6525–6529 CrossRef; (e) F. Yang, J. N. Zhao, X. F. Tang, G. L. Zhou, W. Z. Song and Q. W. Meng, Org. Lett., 2017, 19, 448–451 CrossRef CAS PubMed.
- (a) R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. T. Patel and R. V. Jasra, Tetrahedron: Asymmetry, 2001, 12, 433–437 CrossRef CAS; (b) R. I. Kureshy, N. H. Khan, S. H. R. Abdi, I. Ahmed, S. Singh and R. V. Jasra, J. Mol. Catal. A: Chem., 2003, 219, 1–7 CAS; (c) R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. Singh, I. Ahmad, R. V. Jasra and A. P. Vyas, J. Catal., 2004, 224, 229–235 CrossRef CAS.
- S. Biswajit, L. Mei-Huey and T. V. RajanBabu, J. Org. Chem., 2007, 72, 8648–8655 CrossRef PubMed.
- P. N. O'Shaughnessy, D. K. Paul, M. Colin, M. G. Kevin and S. Peter, Chem. Commun., 2003, 1770–1771 RSC.
- S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 4900–4901 CrossRef CAS PubMed.
- (a) M. Bougauchi, S. Watanabe, T. Arai, H. Sasai and M. Shibasaki, J. Am. Chem. Soc., 1997, 119, 2329–2330 CrossRef CAS; (b) T. Nemoto, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 2725–2727 CrossRef CAS PubMed; (c) T. Ohshima, T. Nemoto, S.-y. Tosaki, H. Kakei, V. Gnanadesikan and M. Shibasaki, Tetrahedron, 2003, 59, 10485–10488 CrossRef CAS; (d) S. Matsunaga, T. Kinoshita, S. Okada, S. Harada and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 7559–7561 CrossRef CAS PubMed; (e) H. Kakei, R. Tsuji, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 8962–8963 CrossRef CAS PubMed.
- (a) K. Daikai, T. Hayano, R. Kino, H. Furuno, T. Kagawa and J. Inanaga, Chirality, 2003, 15, 83–88 CrossRef CAS PubMed; (b) X.-W. Wang, L. Shi, M.-X. Li and K.-L. Ding, Angew. Chem., Int. Ed., 2005, 44, 6362–6366 CrossRef CAS PubMed.
- Y. Y. Chu, X. H. Liu, W. Li, X. L. Hu, L. L. Lin and X. M. Feng, Chem. Sci., 2012, 3, 1996–1998 RSC.
- (a) Q. Qian, Y. Tan, B. Zhao, T. Feng, Q. Shen and Y. Yao, Org. Lett., 2014, 16, 4516–4519 CrossRef CAS PubMed; (b) C. Zeng, D. Yuan, B. Zhao and Y. Yao, Org. Lett., 2015, 17, 2242–2245 CrossRef CAS PubMed.
- (a) L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85–89 RSC; (b) R. Breinbauer and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 3604–3607 CrossRef CAS; (c) E. M. McMarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1566 CrossRef PubMed.
- (a) O. Cussó, I. Garcia-Bosch, X. Ribas, J. LIoret-Fillot and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871–14878 CrossRef PubMed; (b) R. V. Ottenbacher, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, ACS Catal., 2014, 1599–1606 CrossRef CAS; (c) D. Shen, B. Qin, D. Xu, C. Xia and W. Sun, Org. Lett., 2016, 18, 372–375 CrossRef CAS PubMed.
- W. Bin, W. Shoufang, X. Chungu and S. Wei, Chem.–Eur. J., 2012, 18, 7332–7335 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1888891 (for 7e) contains the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01529a |
| This journal is © The Royal Society of Chemistry 2019 |