
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitchable conformational changes of DNA nanogel shells containing disulfide–DNA hybrids for controlled drug release and efficient anticancer action
Wioletta Liwinskaa,
Iwona Stanislawskab,
Marek Lypb,
Zbigniew Stojeka and
Ewelina Zabost *a
*a
aFaculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland. E-mail: ezabost@chem.uw.edu.pl
bCollege of Rehabilitation, Kasprzaka 49, 01-234 Warsaw, Poland
First published on 3rd May 2019
Abstract
Oligonucleotide strands containing dithiol (–SS–) groups were used as the co-crosslinkers in PNIPA–AAc based nanogels (NGs). They hybridized with PEG–oligonucleotides introduced into the gels. The specific DNA hybrid formed in the nanogel/nanocarrier was involved in highly efficient accumulation of intercalators. The presence of –SS– groups/bridges improved the storing efficiency of doxorubicin (Dox) in DNA hybrids by 53, 40 and 20% compared to regular, single stranded and regular double stranded DNA crosslinkers, respectively. The explicit arrangement of the hybrids in the carrier enabled their reduction by glutathione and an effective cancer treatment while the side toxicity could be reduced. Compared to the NGs with traditional crosslinkers and those containing typical dsDNA-based hybrids, an improved, switchable and controlled drug release occurred in the novel NGs. Since the novel NGs can release the oligonucleotide strands during their degradation, this gives an opportunity for a combined drug-gene therapy.
1. Introduction
Some recent efforts in medicinal fields are directed towards the design of drug-release controlling systems. Therapeutic strategies that are characterized by high specificity, high efficacy and low adverse effects are urgently needed. A very promising strategy for effective cancer treatment is the design of localized treatment approaches, which combine strong targeted delivery aspects and controlled drug redistribution to cancer tissues. The main challenge in the design of the locally-operating delivery systems is to keep the drug distribution continuous in cancer tissue during the entire treatment cycle and to avoid frequent injections of the chemotherapy drug. Moreover, maintaining the minimum therapeutic concentration during the treatment time and effectively overcoming the risk of drug resistance in the cancer cells should be taken into account. Therefore, a single-injection delivery of a nanoplatform that simultaneously acts as a high-capacity drug reservoir and a prolonged, sustained- or controlled burst drug release system would be a great advantage.Rapid progress in the design of “smart”, environmental sensitive and multi-stimuli responsive nanocarriers dedicated for improved drug delivery and release has been seen.1,2 Polymeric-based systems incorporating biomolecules are promising carriers that gain great potential in biomedicine, due to: improved drug efficiency, targetability, biocompatibility, degradability and nontoxicity. Natural biopolymers that are mainly tested for the construction of polymeric-based hybrids are: PLA (polylactic acid), microbial polyesters, polysaccharides (e.g. chitosan, celluloses, hemicelluloses, lignins), starch-based polymers, proteins and lipids. Other bio-based polymers applied for the construction of drug delivery carriers are those chemically synthetized: PLA, PCL (polycaprolactone), polyacrylonitrile, polycarbonate, polyphenol, polydibutyl succinate, PNIPAM (poly(N-isopropylacrylamide)), PNIPAM–AAc.1–4
Recently, oligonucleotide-based nanotechnology has become a very promising approach for construction of drug carriers. Compared with synthetic polymers, oligonucleotides possess such features as: possibility of self-assembly and molecular recognition, material programmability, predictable nanoscale structure, response to stimuli, the ability of self-powering and self-propelling, and biocompatibility.5–9 The dynamic oligonucleotide-based networks have a potential to mimic natural networks that possess ability to lead to adaptive compositions and emerging new functions.10 Unique geometries can be formed by controlling the assembling of complementary single-stranded DNA molecules.11,12 The base-pairing effect of DNA was found useful in the design of macro- and micro-sized hydrogel networks for several biomedical applications.13–18
With respect to triggered drug delivery, DNA nanogels possess characteristic structural features such as: nanosize, porosity, stimuli responsiveness and high encapsulation ability. They can be made of pure DNA19–22 or DNA combined with other components.23–27 It has been shown, that DNA nanogels can reveal the resistance to nuclease degradation, enhanced cellular uptake, and more effective drug- or siRNA delivery to cancer cells. Recently, it was shown, that CpG-rich oligonucleotide layers merged with nanoparticles act as a nanoimmunotherapeutic and introduce a robust antitumor immunity through a multi-pronged mechanism of neuroblastoma.28 A possibility of triggering the structural transitions in DNA nanogels by means of an environmental stimuli, e.g. thermal, pH, photonic and chemical, leads to a range of possibilities for an increase in accumulation efficiency and more complete release of drugs.29,30
Apparently, the logic of reversibility of biomimetic DNA structural changes in nanogels is important for highly efficient controlled guest/drug release in DNA-based nanogels. Structural changes of grafted aptamers were studied on solid- and polyelectrolyte microcapsules and linear polymeric hydrogels, where the single and duplex DNA species can be separated and can specifically interact with drug molecules.27 Thanks to the presence of DNA crosslinkers in the soft nanogel network it was possible to get modulation of reversible phase transition of the hydrogel lattices for the achievement of selective drug binding and prolonged and controlled release of drugs.31,32
Herein, we report on the development of a biocompatible, multi-responsive DNA nanogel containing specific disulfide oligonucleotide-based co-crosslinkers for highly efficient loading and triggered, switchable drug release. Under physiological conditions the novel nanogel existed as a core–shell structure; it contained a polymeric core and an oligonucleotide-based shell. The modification of the DNA strands with disulfide groups can facilitate the release of the drug in the presence of a redox substance, e.g. glutathione tripeptide (GSH). A specific, controlled drug release based on a combination of breaking of disulfide bonds and DNA conformational change is proposed. The copolymerization of NIPA, AAc, and PEG with particular oligonucleotide allowed the implementation of pH-, temperature- and redox sensitivities to the gel NPs and an increase in the volume-phase-transition temperature. The presented type of nanogel was biocompatible and was well tolerated in the intracellular environment.
2. Materials and methods
2.1. Materials
Nanogel constituents: N-isopropyloacrylamide (NIPA 97.00%), N,N′-methylenebisacrylamide (BIS, 99.05%), potassium persulfate (KPS, 99.99%), acrylic acid (AAc, 99.00%) and tetramethylethylenediamine (TEMED) were purchased from Sigma Aldrich. NIPA monomer was recrystallized twice from benzene and hexane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) to remove inhibiting substances before polymerization of the nanogels. Other chemicals: (a) buffer constituents: sodium hydroxide (NaOH), sodium chloride (NaCl), potassium chloride (KCl), monosodium phosphate (NaH2PO4) and disodium phosphate (Na2HPO4), (b) cell culture medium constituents: fetal bovine serum (FBS), horse serum (HS), trypsin and dimethylforamide (DMF), were also purchased from Sigma Aldrich. Other culture cell chemicals, penicillin–streptomycin (10000 U mL−1), Gibco Fungizone®, DMEM, GlutaMAX™ media and MTT kits were provided by Thermo Fisher Scientific. The oligonucleotides modified with PEG2000 groups were synthesized by Friz Biochem (Neuried, Germany). Other oligonucleotides were prepared by Integrated DNA Technologies (IDT, Caralville, IA, USA). The sequences of used oligonucleotides, their molecular weights and melting temperatures are given in Table 1. The solutions of primary ssDNA (oligo1; c = 100 μM) and complementary ssDNA (oligo2 or oligo3; c = 100 μM) strands were prepared separately in 0.1 M PBS buffer, pH = 7.4, with 0.15 M NaCl and 0.002 M KCl. A model intercalator drug, doxorubicin hydrochloride (Dox), was purchased from LC Laboratories (Woburn, MA, USA). Concentration of Dox in the stock solution (0.1 M PBS, pH 7.4; 1 mL DMSO) was determined from UV-Vis measurements; (CDox ε485 nm = 10410 L mol−1 cm−1 (ref. 33)). For the preparation of all solutions, deionized water (Milli-Q, Millipore) with conductivity of 0.056 μS cm−1 was used.
:9) to remove inhibiting substances before polymerization of the nanogels. Other chemicals: (a) buffer constituents: sodium hydroxide (NaOH), sodium chloride (NaCl), potassium chloride (KCl), monosodium phosphate (NaH2PO4) and disodium phosphate (Na2HPO4), (b) cell culture medium constituents: fetal bovine serum (FBS), horse serum (HS), trypsin and dimethylforamide (DMF), were also purchased from Sigma Aldrich. Other culture cell chemicals, penicillin–streptomycin (10000 U mL−1), Gibco Fungizone®, DMEM, GlutaMAX™ media and MTT kits were provided by Thermo Fisher Scientific. The oligonucleotides modified with PEG2000 groups were synthesized by Friz Biochem (Neuried, Germany). Other oligonucleotides were prepared by Integrated DNA Technologies (IDT, Caralville, IA, USA). The sequences of used oligonucleotides, their molecular weights and melting temperatures are given in Table 1. The solutions of primary ssDNA (oligo1; c = 100 μM) and complementary ssDNA (oligo2 or oligo3; c = 100 μM) strands were prepared separately in 0.1 M PBS buffer, pH = 7.4, with 0.15 M NaCl and 0.002 M KCl. A model intercalator drug, doxorubicin hydrochloride (Dox), was purchased from LC Laboratories (Woburn, MA, USA). Concentration of Dox in the stock solution (0.1 M PBS, pH 7.4; 1 mL DMSO) was determined from UV-Vis measurements; (CDox ε485 nm = 10410 L mol−1 cm−1 (ref. 33)). For the preparation of all solutions, deionized water (Milli-Q, Millipore) with conductivity of 0.056 μS cm−1 was used.
| Name | Sequence type | Melting point temperature [Tm, °C] | Molecular weight [MW, g mol−1] |
|---|---|---|---|
| oligo1 | 5′-PEG2000-GGG GG-GC-TC-TT-GG-AA-CT-3′ | 57.6 | 7282.0 |
| oligo2 | 5′ AG-TT-CC AA-SS-GA-GC-CC-CCC 3′ | 57.3 | 5114.6 |
| oligo3 | 5′ AG-TT-CC-AA-GA-GC-CC-CCC 3′ | 57.3 | 5100.4 |
2.2. Synthesis of PNIPA–AAc–PEG–ssDNA NGs
NIPA and AAc monomers and BIS crosslinker were dissolved in 6 mL of deionized water. The total concentration of NIPA, AAc and BIS in the reaction mixture was 75 mM. The acrylic acid content in the mixture was 10%. A 100 μL sample of the initial solution of oligonucleotide (oligo1, c = 100 μM) was diluted to 3 mL with 0.1 M PBS (pH = 7.4) to achieve the final concentration of 3.35 μM. The prepared solution was added to the solution of monomers. Next, the mixture was stirred for circa 1 h at 1400 rpm and heated under argon atmosphere at 60 °C. That temperature was close to the melting point temperature of the used oligonucleotides and was higher than the LCST of the gel. Then, 1 mL of the KPS solution (10 mg mL−1) and 10 μL of TEMED were subsequently added to initiate the polymerization reaction. The polymerization was continued for 4 h under argon atmosphere at 1400 rpm stirring, and, at the end, the solution was slowly cooled down to room temperature. The purification of NGs was done by placing them in a dialysis bag (Spectra/Por RC, MCWO 8–10 kDa) and doing a 7 day dialysis process with triply-distilled water. Water was changed twice per day. The total concentration of oligonucleotides bound with NGs surfaces was estimated by measuring UV-Vis absorbance at 260 nm. The result was 2.173 μM. The extinction coefficient, ε260 m, was determined to be 184000 L mol−1 cm−1.
2.3. Hybridization process in PNIPA–AAc–PEG–ssDNA NGs
A 3 mL sample of PNIPA–AAc–PEG–ssDNA (oligo1) NPs solution was heated at 60 °C (temperature higher than Tm of the oligonucleotide) for 30 min, to make the nanogels shrink and to stretch the introduced ssDNA strands. Simultaneously, at the same temperature, the oligo2 complementary strand containing the SS-bridges, dissolved in 0.1 M PBS buffer with 0.15 M NaCl and 0.002 M KCl, was heated. Next, both solutions were mixed and incubated at 40 °C for 2 hours. The concentration of the oligo2 strand was by 10% higher than the oligo1 concentration in the nanogels. After heating the solution was cooled down in an ice bath and placed in a fridge for 24 h to complete the hybridization process. To remove the unbound strands the 1 week dialysis of NGs was applied. The total concentration of dsDNA in the NGs equaled 3.90 μM, it was determined spectrophotometrically. For a comparison purpose, we also hybridized oligo1 with complementary oligonucleotides without the –SS– bridges (oligo3).2.4. Loading of doxorubicin
A model intercalator, anthracycline drug doxorubicin (Dox), was selected for loading into nanogels. 2 mL of 200 μM initial solution of Dox were mixed with 2 mL of the suspension of one of available versions of PNIPA–AAc–PEG-based NGs. The mixture was kept for 3 h at 45 °C in the dark and was gently stirred. Next, the mixture was kept at room temperature for 24 h. The unbound Dox was separated by centrifugation (60000 rpm, 90 min). The obtained sediment was dissolved in the buffer solution (either 0.1 M PBS, pH = 7.4 or acetate buffer, pH = 5.0). The concentrations of Dox in individual NGs were determined spectrophotometrically (at 25 °C, A485 nm, ε485 nm = 10410 L mol−1 cm−1 (ref. 33)). The above procedure was applied for PNIPA–AAc–PEG, PNIPA–AAc–PEG–ssDNA-, PNIPA–AAc–PEG–dsDNA and PNIPA–AAc–PEG–SS–dsDNA NGs.
2.5. Doxorubicin release protocol
2 mL samples of solutions of particular NGs loaded with Dox were placed in dialysis bags (MWCO = 10 kDa). The bags were dialyzed against 20 mL of either 0.1 M PBS (pH = 7.4) or acetate buffer (pH = 5.0) with gentle stirring. The released Dox was determined in the solution outside the dialysis bag. The external solution was collected and measured at regular time intervals using electrochemical- (SVW) and spectroscopic (UV-Vis) methods. Generally, the measurements were carried out at physiological- (37 °C) and hyperthermia (45 °C) temperatures, in the absence and presence of 10 mM GSH. For the UV-Vis data, the cumulative level of released Dox was calculated using eqn (1):
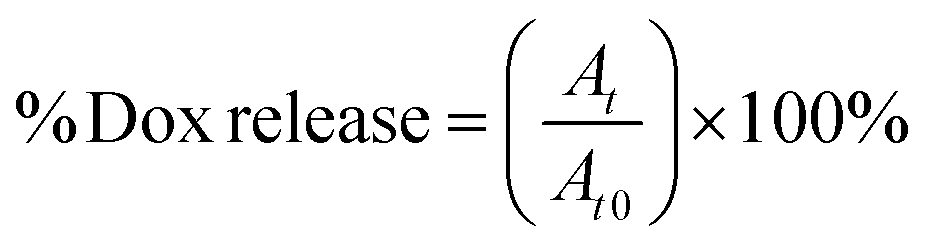 | (1) |
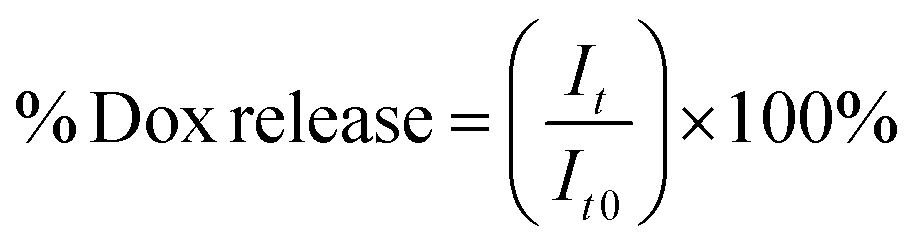 | (2) |
2.6. Cell-viability MTT assay
Human insulinoma β-TC3 cell line was cultured at 37 °C in a grow medium containing: Dulbecco's modified Eagle's medium (DMEM), high glucose GlutaMAX™, 12% HS, 3% FBS, 10 μg mL−1 fungizone, 100 μg mL−1 streptomycin and 100 U mL−1 penicillin in humidified atmosphere containing 5% CO2. The cytotoxicity measurements were performed with the use of MTT plate-based assay. We conducted measurements for: (a) reference Dox solution, (b) pure PNIPA–AAc–PEG–dsDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs, and (c) Dox loaded PNIPA–AAc–PEG–dsDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs. Before introducing particular NG to the cell cultures, the solutions were heated at 37 and 45 °C for 15 min and subsequently cooled down to 37 °C. Then, the solutions with released Dox were collected for the subsequent step of the investigation. Next, the cells were seeded onto 96 well plates. The density of β-TC3 cells was 3000 cells per 100 μL per well. The cells were incubated at 37 °C in 5% CO2 for 24 h to allow attachment of the cells. On the day of experiments, the cells were washed with warm PBS. Next, the cells were incubated with the reference Dox solution, the solution of pure NGs or Dox/PNIPA–AAc–PEG–dsDNA or PNIPA–AAc–PEG–SS–dsDNA NGs, and the solutions with released Dox. Dox concentration was in the range from 0.05 to 20 mg mL−1, while the nanogel concentration was constant: 7.8 mg mL−1. The cells were incubated for 30 min. This was followed by a 2 h interval at 37 °C. Then the cells were washed three times with 0.1 M PBS (100 μL) and a fresh growth medium was added. Finally, the cells were incubated for a total of 96 h. For MTT tetrazolium assay, 25 μL of 5 g mL−1 solution of MTT reagent were added and the incubation lasted 2 h. Next, 100 μL of lysis buffer (20% SDS, 50% DMF, pH 4.5) were introduced. After addition of the probes and the targets of particular assays, the fluorescence (fluorescence excitation and emission wavelengths were 480 and 520 nm, respectively) and absorbance (at 570 nm) were measured with a POLARStar Omega plate reader (BMG Labtech, Aylesbury, UK). All experiments were repeated 3 times. The presented results were corrected for: the values obtained for the media without the cells, the media with and without dyes, and the values obtained for assays constituents after their interaction with Dox-unloaded particular oligonucleotide-based NGs.2.7. Instrumental examination
3. Results and discussion
3.1. Synthesis and physicochemical characterization of NGs
The two-step synthesis process was applied for the preparation of novel PNIPA–AAc–PEG–dsDNA NGs containing DNA-based disulphide (–SS–) co-crosslinkers, see Fig. 1. In the first step (Fig. 1A) the nanogels were synthesized using the one-pot, free-radical and surfactant-free polymerization of NIPA and AAc monomers and PEG groups modified with single DNA strands. The strands were of 12 + 5 b.p length and were named oligo1. During the polymerization process, oligo1 strands were incorporated into the main polymer chains with the help of the PEG groups. The PNIPA–AAc-based nanogels were pH sensitive. In the second step (see Fig. 1B), a complementary oligo2 sequences, containing the disulfide groups, were hybridized with the oligo1 strands. The hybridization process occurred in the swollen NGs structures. For the comparison purpose, the hybridization of oligo1 strands and oligo3 strands without –SS– groups was also done. As a result, novel multi stimuli-responsive polymeric NGs with easily, GSH-triggered, cleaved disulphide/DNA-based hybrid co-crosslinkers were obtained for high-level loading and specific switchable release of intercalating compounds, e.g. anticancer drugs. As it is known, the intercalating drugs can interact with dissolved- and bound to NGs double stranded DNA by two modes: (1) the intercalation between the planar pairs of nucleic bases, and (2) electrostatic binding of the positively charged molecules to the negatively charged phosphate groups present in dsDNA minor and major groves. As we expected, three types of structural changes influenced the discharge of selected model-intercalator drug Dox during its GSH-mediated release process at selected pH and temperatures: (a) conformational change of double-stranded parts of DNA co-crosslinkers, (b) phase transition (swelling–shrinking) of the polymer net, and (c) disintegration/degradation of the nanogels due to breaking of the –SS– groups in the DNA-based co-crosslinkers.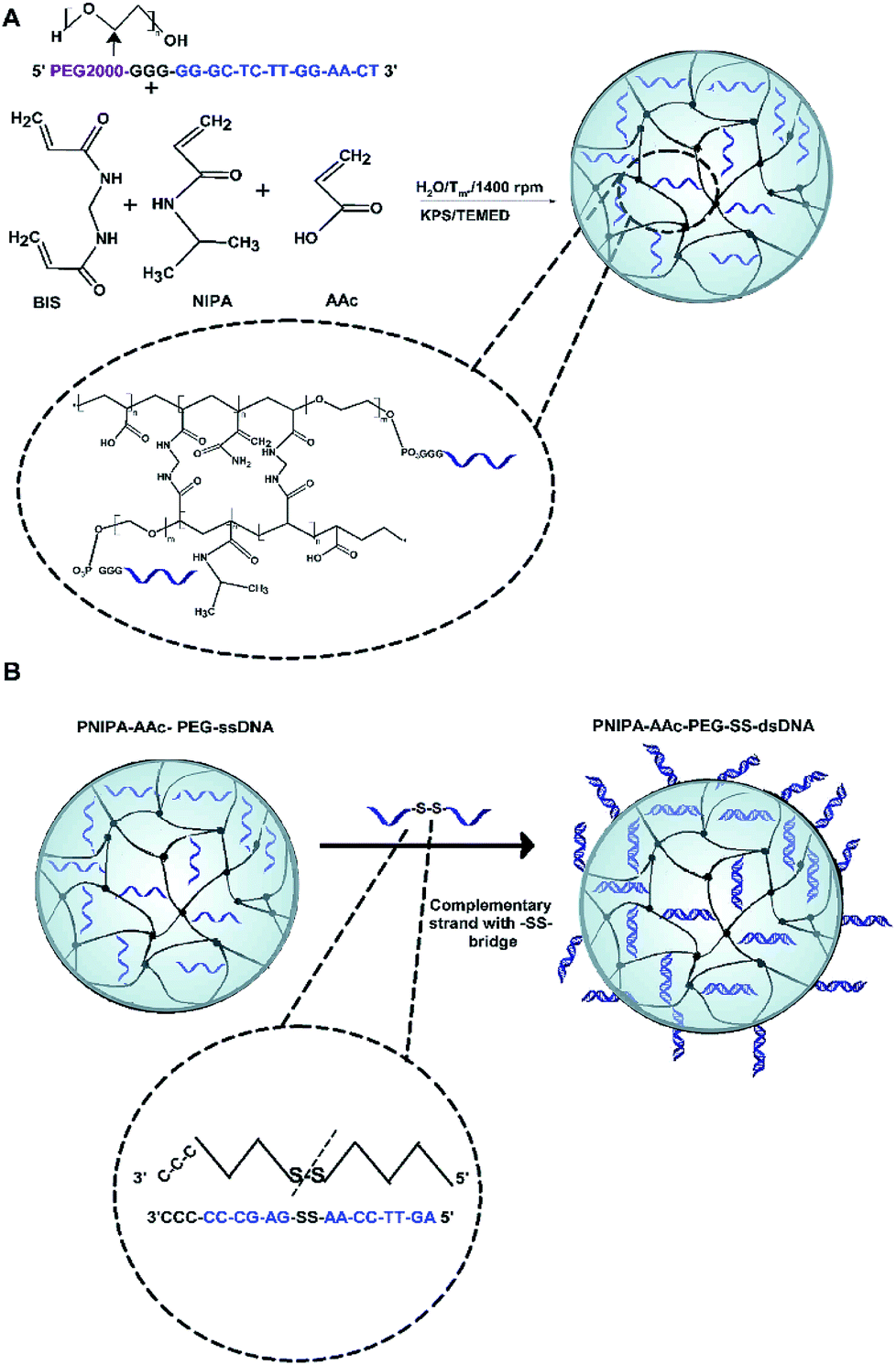 | ||
| Fig. 1 Two-step synthesis process of PNIPA–AAc–PEG–SS–dsDNA NGs. (A) Scheme of synthesis of PNIPA–AAc–PEG–ssDNA nanogels: covalent introduction of PEG–ssDNA oligos during NGs polymerization reaction. (B) Synthesis of PNIPA–AAc–PEG–SS–dsDNA NGs under conditions of swollen state: hybridization with complementary ssDNA strands containing internal –SS– bridges. | ||
A comparison of morphology of PNIPA–AAc-, PNIPA–AAc–PEG–ssDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs is visualized in Fig. 2A–D. Fig. 2A presents a typical TEM micrograph of neat and round PNIPA–AAc–PEG–ssDNA NGs. The hybridized PNIPA–AAc–PEG–dsDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs possessed also round shapes and similar sizes, circa 150–200 nm in diameter; however, their surface was slightly more irregular, see Fig. 2B–D. In fact, the sizes of the NGs with hybridized DNA and loaded with Dox were higher by circa 10%. As we used uranyl acetate (UA), and the uranyl cations possess high affinity to negatively charged phosphate groups in the DNA strands, the sun-type structures of hybridized NGs are well seen. It can be concluded, that while ssDNA strands introduced to NGs were partially placed inside the NGs nets, the hybridized dsDNA strands were rather positioned outside the NGs surfaces.
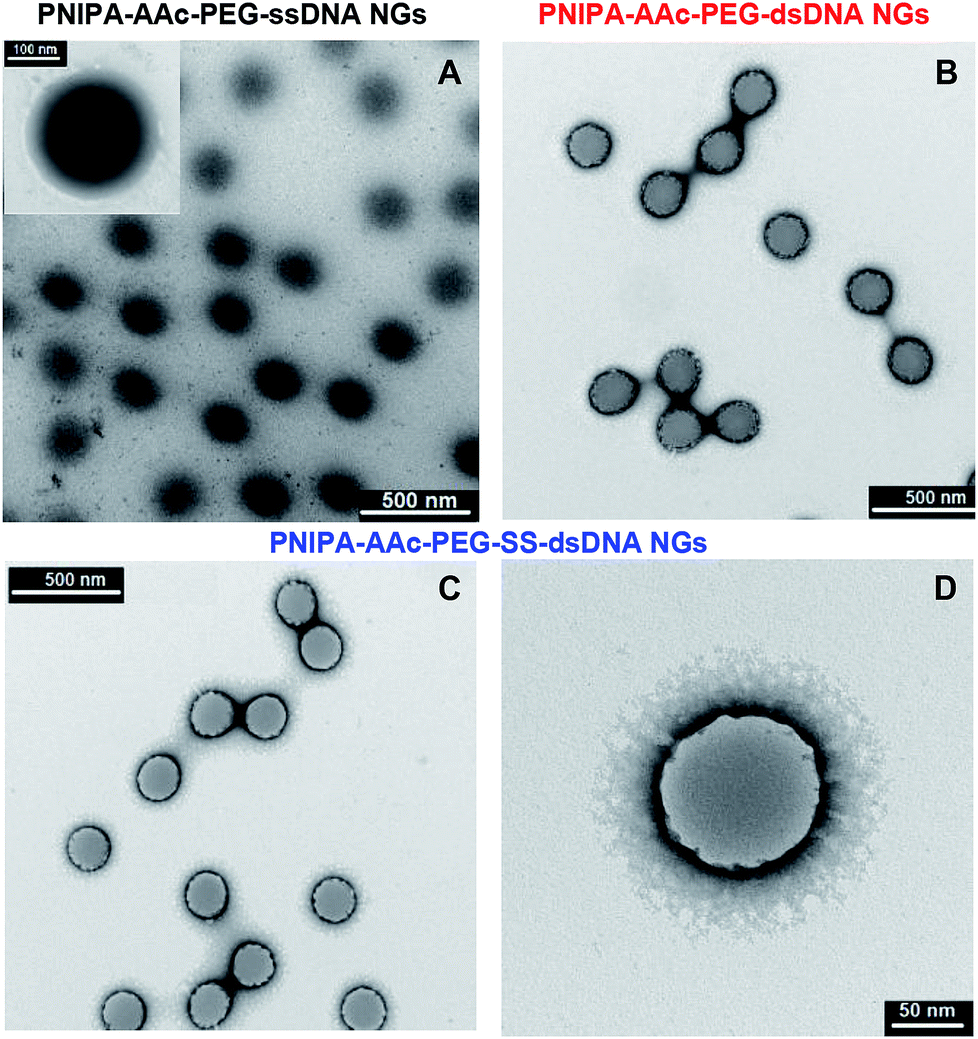 | ||
| Fig. 2 TEM micrographs of gently lyophilised and nondegraded NGs taken at 25 °C: (A) PNIPA–AAc–PEG–ssDNA- (B) PNIPA–AAc–PEG–dsDNA-, (C, D) PNIPA–AAc–PEG–SS–dsDNA. Uranyl acetate (UA) was applied as contrasting/staining agent. | ||
The presence of ssDNA (oligo1) in NGs and the yield of hybridization with the oligonucleotides either containing –SS– groups (oligo2) or without them (oligo3) were controlled by doing UV-VIS measurements, see Fig. 3A. Before the measurements we made sure that all unbound DNA molecules were removed from the NGs solutions. The characteristic band of dsDNA appeared at 260 nm. We assumed, in the calculations that typical and completed dsDNA hybridization (B conformation) leads to double decrease of the ssDNA oligonucleotide band intensity.35 The hybridization efficiency in NGs, a measure of which is absorbance, was compared with that occurring in the solution, see Fig. 3A. A comparison of dsDNA hybridization efficiency for the oligonucleotide strands dissolved in the solution and those present in the NGs was done. It appeared that the NGs with dsDNA hybrids containing –SS– bridges (oligo1 and oligo2) gave a slightly higher absorbance value than the unmodified dsDNA hybrids (oligo1 and oligo3). We could estimate the yield of DNA hybridization process. It equalled 76.9 and 85.3% for PNIPA–AAc–PEG–dsDNA and PNIPA–AAc–PEG–SS–dsDNA, respectively. Such quite small difference can be a result of various dsDNA hybrid spatial arrangements in particular NG nets.
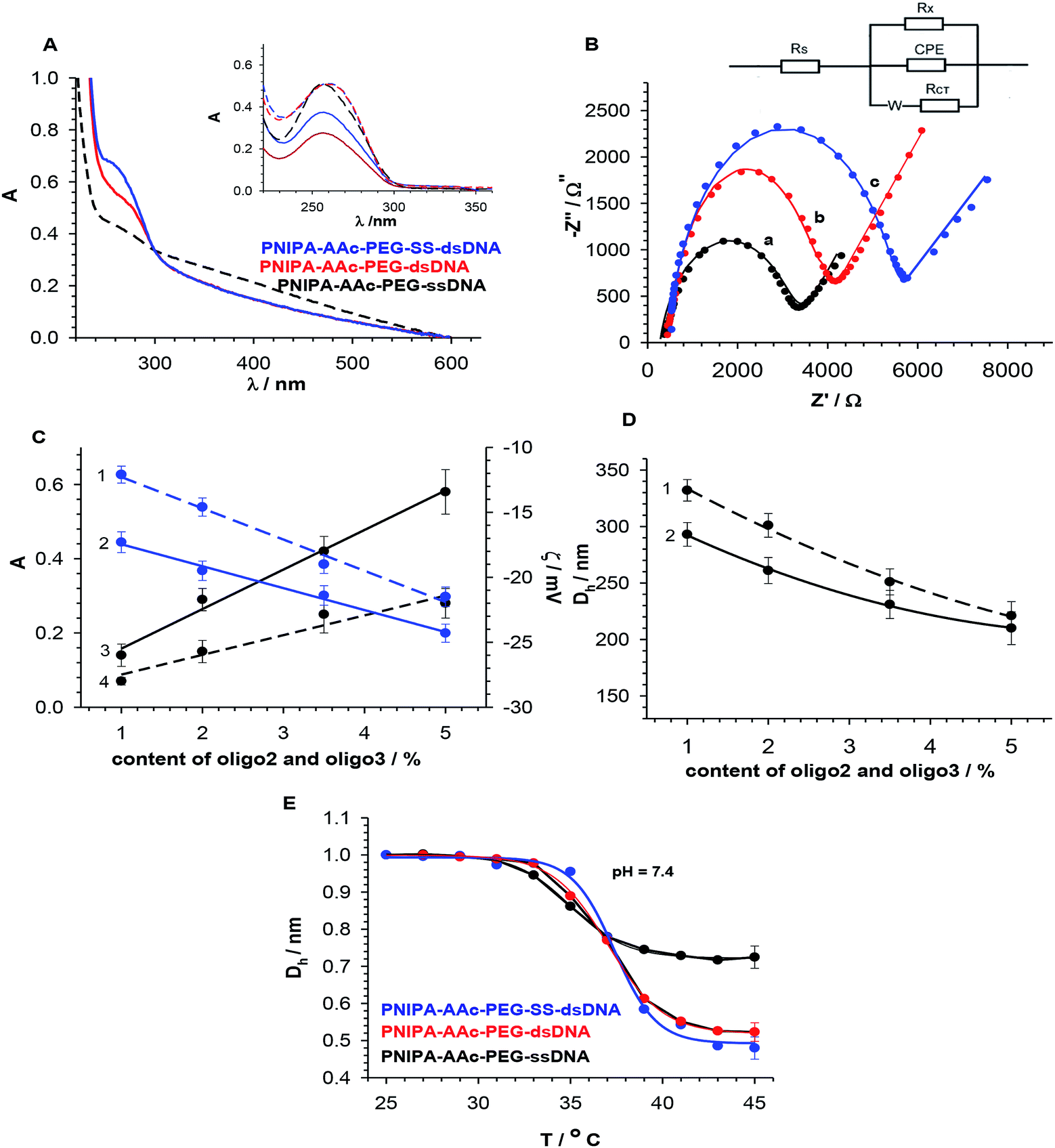 | ||
| Fig. 3 (A) UV-Vis spectra indicating hybridization efficiency in PNIPA–AAc–PEG–dsDNA (red line) and PNIPA–AAc–PEG–SS–dsDNA NGs (blue line). UV-Vis of PNIPA–AAc–PEG–ssDNA NGs (black line) is given as reference. For comparison absorbance spectra indicating hybridization efficiency in the solution are presented in inset. (B) EIS Nyquist plots obtained for 2 mM solution of [Fe(CN)6]3−/4− in 0.01 PBS, pH 7.4 for deposited NGs layers on GC electrode. (a) PNIPA–AAc–PEG–ssDNA- (black line), (b) PNIPA–AAc–PEG–dsDNA (red line) and (c) PNIPA–AAc–PEG–SS–dsDNA NGs (blue line), ϕ = 3 mm, T = 37 °C. Inset: modified Randles circuit used for fitting experimental data into simulated plots. Rs – solution resistance, Rx – additional resistance (DNA), Rct – charge transfer resistance, CPE – constant phase element and W – Warburg impedance. (C) Dependencies of Aλ = 260 nm and zeta (ζ) potential of NGs with hybridized dsDNA in PNIPA–AAc–PEG–dsDNA- (dashed lines) and PNIPA–AAc–PEG–SS–dsDNA (solid lines) NGs on content of introduced oligo2 (2,3) and oligo3 (1,4) crosslinkers. (D) Dependencies of hydrodynamic diameter, Dh, of PNIPA–AAc–PEG–dsDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs on content of introduced oligo2 (2) or oligo3 (1) crosslinkers. (E) Plots of hydrodynamic diameter, Dh, normalized vs. Dh at 25 °C, vs. temperature obtained for shrinking process of PNIPA–AAc–PEG–ssDNA- (black line), PNIPA–AAc–PEG–dsDNA (red line) and PNIPA–AAc–PEG–SS–dsDNA NGs (blue line), step time = 10 min. | ||
A more precise characterization of the dsDNA hybrids arrangement in the investigated NGs was possible with the electrochemical impedance spectroscopy (EIS), which is widely applied for detection of various DNA forms and tracking of the hybridization process.29,36 The EIS parameters were determined for layers of DNA attached directly to the electrode surface.37,38 In the fitting process the equivalent EIS electrical circuit developed by Randles was used,39 see inset in Fig. 3B. The selected circuit consists of solution resistance, Rs, charge transfer resistance, Rct, double-layer capacitance, C, Warburg impedance, W, and additional parallel resistance, Rx. For the nanogel layers that are in good contact with the electrode surface, a decrease in double layer capacitance should take place and the barrier for the interfacial electron transfer should increase. True, the existence of a complex system, e.g. conjugated π-system, could not be adequately described by the simple Randles circuit; however, the introduction of additional parallel resistance, Rx, improved the fit of the theoretical calculated spectra and the experimental data. The EIS results obtained for the PNIPA–AAc–PEG–ssDNA-, PNIPA–AAc–PEG–dsDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs are presented in Fig. 3B. All presented EIS spectra contained a semicircle and a linear component and were acquired at the formal potential of equimolar mixture of the [Fe(CN)6]3−/4− redox couple. The increase in Rct value obviously well illustrates the successful hybridisation process. Interestingly, the EIS spectra of PNIPA–AAc–PEG–SS–dsDNA NGs exhibited circa two times greater Rct value compared to the typical hybridization in PNIPA–AAc–PEG–dsDNA NGs. This indicates that in the case of –SS– the hybridization in the nanogels may have another form and may be incomplete. Taking into account other EIS parameters we noticed, that the solution resistance, Rs, had similar values for all measurements and was independent of the hybridization length of the oligonucleotides. The Warburg parameter, W, was nearly constant, thus, the mass transport of the redox probe to the electrode surface was not hindered in the NGs layers deposited on the electrode. The double layer capacitance decreased with an increase in length of the duplex and after introduction of the –SS– groups. Interestingly, the additional parallel resistance, Rx, increased for the hybridized DNA compared to ssDNA in the NGs, but decreased for PNIPA–AAc–PEG–SS–dsDNA NGs. This result can be explained by the fact that the spatial structures of dsDNA containing the –SS– group were slightly different than those without –SS– bonds.40,41
Fig. 3C and D present dependencies of absorbance, zeta potential (ζ) and hydrodynamic diameter (Dh) on percentage of introduced oligo1 or oligo2 strands in the swollen NGs. The absorbance and zeta potential increased and decreased linearly with content of both strands, respectively. In the case of hydrodynamic diameter we noticed a decrease in NG size with increasing co-crosslinker content. A comparison of hydrodynamic diameters and zeta potentials of NGs containing 3.5% of DNA-based co-crosslinkers and measured under conditions: (a) of drug loading (25 °C, pH 7.4 or 5.0), (b) physiological (37 °C, pH 7.4), (c) of cancer cells (37 °C, pH 5.0), and (d) of cancer hyperthermia treatment (45 °C, pH 7.4 or 5.0) is summarized in Table 2. In the DLS technique the information about the changes in the dynamics of the swollen polymeric NPs is extracted from the second-order autocorrelation function. The hydrodynamic diameter of a given particle is related to its diffusion coefficient.
| Nanogel type | PNIPA–AAc–PEG–ssDNA | PNIPA–AAc–PEG–dsDNA | PNIPA–AAc–PEG–SS–dsDNA |
|---|---|---|---|
| Size [Dh, nm], pH 7.4 | |||
| Dh/25 °C | 428.7 ± 6.2 | 381.6 ± 9.8 | 341.0 ± 8.1 |
| Dh/37 °C | 301.5 ± 7.5 | 251.0 ± 8.5 | 231.0 ± 9.5 |
| Dh/45 °C | 283.2 ± 4.8 | 170.5 ± 4.7 | 155.0 ± 7.5 |
|
|||
| Size [Dh, nm], pH 5.5 | |||
| Dh at 25 °C | 375.2 ± 5.1 | 327.0 ± 8.5 | 327.5 ± 7.1 |
| Dh at 37 °C | 285.5 ± 7.3 | 153.4 ± 7.7 | 105.5 ± 9.2 |
| Dh at 45 °C | 197.9 ± 5.2 | 143.6 ± 6.9 | 98.5 ± 6.7 |
|
|||
| Zeta potential [ζ, nm], pH 7.4 | |||
| ζ at 25 °C | −14.9 ± 1.6 | −16.6 ± 1.5 | −18.3 ± 1.2 |
| ζ at 37 °C | −16.8 ± 1.1 | −19.7 ± 1.4 | −21.4 ± 1.5 |
| ζ at 45 °C | −20.6 ± 1.4 | −21.6 ± 1.7 | −24.0 ± 1.3 |
|
|||
| Zeta potential [ζ, nm], pH 5.5 | |||
| ζ at 25 °C | −5.9 ± 1.7 | −9.6 ± 1.1 | −11.5 ± 1.2 |
| ζ at 37 °C | −13.2 ± 1.5 | −15.6 ± 1.3 | −18.2 ± 1.4 |
| ζ at 45 °C | −19.5 ± 1.2 | −21.2 ± 1.6 | −23.5 ± 1.7 |
|
|||
| Dox loading efficiency | |||
| Final CDox (μmol L−1) | 68.0 | 130.0 | 156.0 |
| Dox loading efficiency at 25 °C, pH 7.4 (%) | 34.0 | 63.0 | 78.3 |
These parameters are linked in the Stokes–Einstein eqn (3):
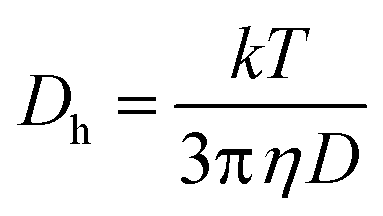 | (3) |
Hydrodynamic diameter of NGs containing 3.5% DNA-based co-crosslinkers, normalized vs. its value at 25 °C, is plotted as function of temperature in Fig. 3D. The initial sizes of the both effectively hybridized NGs were circa 20% lower than for NGs with ssDNA. A comparison of the plots in Fig. 3B and D led to a conclusion that the hybridization and the co-crosslinking process in the NGs was effective. In general, for PNIPA–AAc–PEG–SS–dsDNA NGs, after the hybridization process, the volume phase transition temperature (LCST) was increased by 2.7 degrees, the volume ratios of the swelling–shrinking process were bigger, and the transition occurred in a narrower temperature range compared to the ssDNA nanogels. This was caused by the presence of higher amounts of hydrophilic components attached to the nanogel network.
The impact of pH change on the volume phase transition of the PNIPA–AAc–PEG–SS–dsDNA NGs with 3.5% DNA-based crosslinkers was also investigated. Representative Ds vs. temperature plots obtained for different pH are presented in Fig. 4A. It can be seen, the volume phase transition temperature of the nanogels increased with increase in pH and dissociation of the carboxylic groups. At pH 7.0 (physiological pH) the VPT occurred at the physiological temperature (37 °C). From the above results, we can conclude that the way of the organization of disulfide/DNA co-crosslinkers in the NGs determined nanogel size and zeta potential for the work in particular physiological conditions.
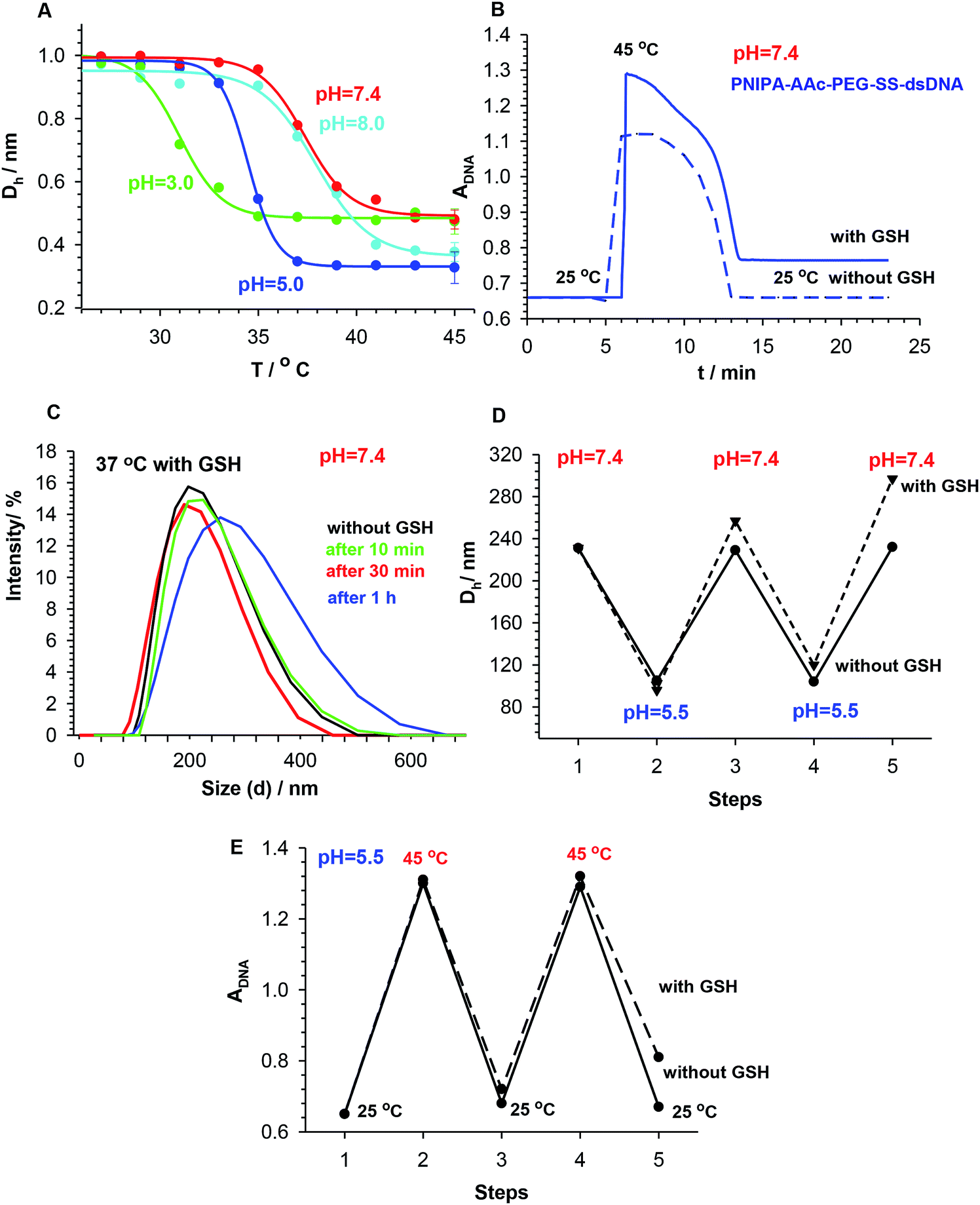 | ||
| Fig. 4 Analysis of structural changes and degradability of PNIPA–AAc–PEG–SS–dsDNA NGs: (A) plots of hydrodynamic diameter, Dh, normalized vs. Dh at 25 °C, vs. temperature for various pH, step time = 10 min. (B) AdsDNA, λ = 260 nm plotted vs. time for NGs heated from 25 °C to 45 °C and then cooled back to 25 °C. (C) Dh distribution change in time recorded for PNIPA–AAc–PEG–SS–dsDNA NGs after addition of 10 mM GSH, pH 7.4, 37 °C. (D) Oscillating changes of Dh measured for three-point pH system (7.4 to 5.5 and back to 7.4) and visualized for two consecutive cycles, for presence and absence of 10 mM GSH, step time = 10 min. (E) Oscillating changes of A measured for three-point temperature system (25 °C to 45 °C and back to 25 °C) and visualized for two consecutive cycles, for presence and absence of 10 mM GSH, step time = 10 min. | ||
Since the synthesized PNIPA–AAc–PEG–SS–dsDNA NGs contained redox-sensitive, cleavable disulfide bonds, they should be sensitive to GSH and disintegrate after addition of that reducing agent. In fact, the presence of substantially higher concentration of GSH (10 mM) was noticed in the cancer cells compared to the regular cells.42 By simple comparison of the absorbance of particular NGs at increased temperature and after addition of GSH, we could see that the addition of GSH resulted in the progressive disintegration of NGs, see Fig. 4B. The experiment was conducted by reversal heating of NGs: first under low-temperature hyperthermia conditions (45 °C) and then back at the physiological temperature (37 °C), in the presence and the absence of GSH. No significant activity of GSH towards the hybridized dsDNA co-crosslinkers at 25 °C was noticed.
The disintegration process of the NGs was also investigated by the DLS technique. Hydrodynamic diameter of the nanogels was measured over time after additions of 10 mM GSH, see Fig. 4C. It appeared that nanogel size increased in time due to relaxation of the polymer network associated with two processes: (a) the reduction of the –SS– bond and (b) the conformational changes of the DNA strands. After 1 hour of exposure to GSH, the distribution of size slightly widened. It can be concluded, that the availability of disulfide groups that are placed inside the oligonucleotide-based co-crosslinkers is limited for the interaction with GSH. This protection can lower the risk of premature drug release and can potentially diminish the side effects in cancer treatment.43
Fig. 4D confirms the previous conclusion that a departure from reversibility in the volume transitions of the NGs takes place; apparently the presence of GSH leads to a slow degradation of the NGs. Fig. 4E exhibits oscillations in DNA absorbance at pH 5.5 caused by repeated temperature changes from below to above LCST and back. A slow increase in absorbance is seen in the presence of GSH.
3.2. Loading efficiency and kinetics of release
The loading efficiency of selected intercalator Dox in the NGs was determined form UV-Vis spectra presented in Fig. 5A. The accumulation of Dox was done in a solution containing 200.00 μmol L−1 of Dox and 0.0078 mg L−1 of NGs. The percent of the initial Dox amount accumulated in particular NGs equaled 34, 65 and 78%, for PNIPA–AAc–PEG–ssDNA-, (b) PNIPA–AAc–PEG–dsDNA and (c) PNIPA–AAc–PEG–SS–dsDNA NGs, respectively. We can state that the introduction of –SS– bridges significantly increased the accumulation of Dox.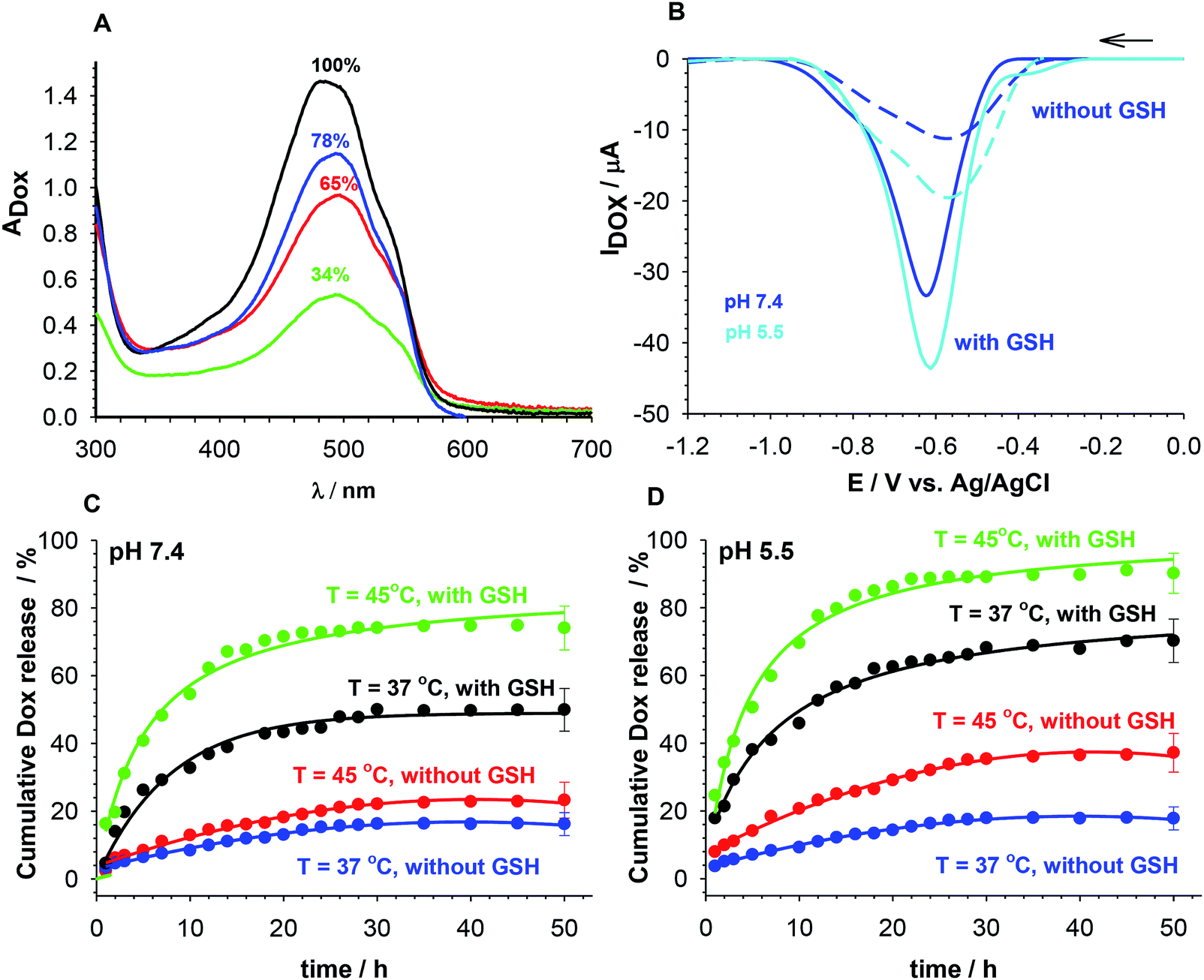 | ||
| Fig. 5 (A) UV-Vis spectra indicating loading levels of accumulated Dox in NGs. Initial, maximal Dox concentration in solution 200.0 μM; it was used for accumulation in NGs (black line), Dox accumulated in: PNIPA–AAc–PEG–SS–dsDNA- (blue line), PNIPA–AAc–PEG–dsDNA- (red line) and PNIPA–AAc–PEG–ssDNA NGs (green line). (B) Typical SW voltammograms recorded for released Dox from PNIPA–AAc–PEG–SS–dsDNA NGs after 10 h at 37 °C and at particular pH. (C) Release Dox profiles from PNIPA–AAc–PEG–SS–dsDNA NGs obtained at pH 7.4 and at 37 and 45 °C, with absence and presence of 10 mM GSH. (D) Release Dox profiles for PNIPA–AAc–PEG–SS–dsDNA NGs obtained at pH 5.5 and at 37 and 45 °C, with the absence and presence of 10 mM GSH. | ||
The next step of our investigation was to estimate the contribution of particular types of Dox bonding in the novel NGs. Two major ways of Dox binding to DNA strands should be considered: (a) electrostatic binding of positively charged molecules to negatively charged phosphate groups present in the minor- and major dsDNA groves, and (b) intercalation between the planar pairs of the bases. At different Dox/DNA concentration ratios and for various initial DNA structures either the first one or the second of the above DNA binding ways is preferred.44 Dox has also a high binding affinity to proteins containing –SS– groups. The McGhee and von Hippel model was applied to determine the binding parameters between oligonucleotides and Dox in the NGs. The working formula is given as eqn (4).
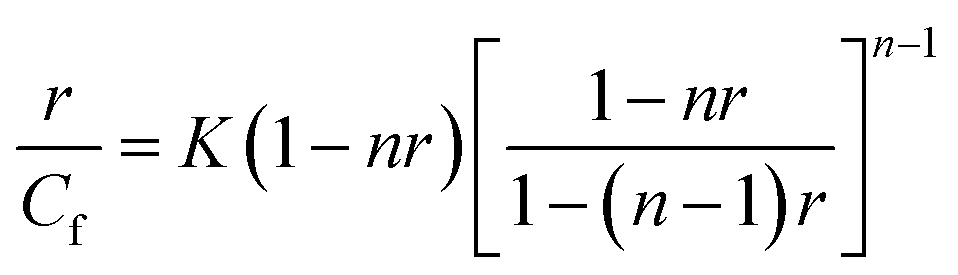 | (4) |
It is known, that among the two types of binding, the intercalation is the stronger one and the corresponding binding constant is bigger compared to that of the electrostatic binding.46 For the swollen PNIPA–AAc–PEG–SS–dsDNA NGs, the values of K and n determined at 25 °C (K1 and n1, K2 and n2) equaled (1.5 ± 0.2 × 104), (2.1 ± 0.1), (0.8 ± 0.1 × 104), (3.4 ± 0.3) for intercalation and electrostatic binding, respectively. A comparison of the K and n values with those given in our previous work25,26 led to two conclusions: (a) in the proposed NGs, the values of the binding constants were much higher than those in the NGs modified by oligonucleotides in a different way, (b) the contribution of electrostatic type of binding in the new NGs was also bigger.29,30 We expected from the above results, that the GSH-mediated release of Dox could be done in a controlled way and could have the “on–off” switchable character under particular conditions.
The kinetic profiles of GSH-mediated release of Dox from PNIPA–AAc–PEG–dsDNA and PNIPA–AAc–PEG–SS–dsDNA NGs at particular temperatures and pH were constructed from the square wave voltammetry (SWV) data. SWV appeared a very useful technique for quick determination of small concentrations of Dox.47,48 The dependence of SWV peak current vs. Dox concentration was found to be linear in a sufficiently wide range.35 Fig. 5B presents example SW voltammograms obtained after 10 h of release of Dox from PNIPA–AAc–PEG–SS–dsDNA NGs at 37 °C, at pH 5.5 and 7.4, and in presence and absence of GSH. Fig. 5C and D present prolonged Dox-release kinetic profiles recorded for PNIPA–AAc–PEG–SS–dsDNA NGs. The environment of health and cancer cells, pH 7.4 and 5.5, with- or without reducing agent GSH, and routine hyperthermia treatment of T = 45 °C, were adopted. At 37 °C, where there was no significant DNA structural change, the Dox release process was similar at both pH and reached 20% of the accumulated Dox. At 45 °C, were conformational dsDNA changes could take place, the Dox release process was again insignificantly dependent on pH and equaled 20–30% of the accumulated Dox. The addition of GSH significantly improved the efficiency of Dox release; it equaled from 50 to 70% and was by circa 20% higher at pH 5.5. The best Dox release results were obtained during the combination of hyperthermia treatment and addition of GSH. Then the amount of released Dox reached 78 and 95% at pH 7.4 and 5.5, respectively. Interestingly, after the 10 hour Dox release process the NGs could be regenerated and loaded with Dox to the previous initial concentration.
The process of Dox release from the NGs was analyzed using the Korsmeyer–Peppas model,49 see eqn (5):
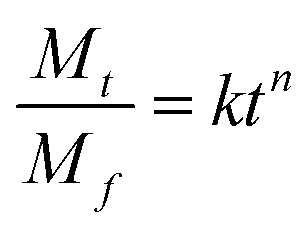 | (5) |
| Temperature | Peppas parameters of Dox release process | ||
|---|---|---|---|
| k | N | R | |
| Without GSH, pH 5.5 | |||
| 37 °C | 4.1 ± 0.3 | 0.4 ± 0.1 | 0.995 |
| 45 °C | 8.4 ± 0.6 | 0.6 ± 0.2 | 0.994 |
|
|||
| With GSH, pH 5.5 | |||
| 37 °C | 23.1 ± 1.6 | 0.4 ± 0.2 | 0.980 |
| 45 °C | 35.5 ± 3.0 | 0.3 ± 0.3 | 0.984 |
|
|||
| Without GSH, pH 7.4 | |||
| 37 °C | 3.7 ± 0.4 | 0.4 ± 0.1 | 0.992 |
| 45 °C | 4.9 ± 0.4 | 0.5 ± 0.1 | 0.990 |
|
|||
| With GSH, pH 7.4 | |||
| 37 °C | 14.4 ± 1.4 | 0.4 ± 0.2 | 0.983 |
| 45 °C | 26.4 ± 2.8 | 0.3 ± 0.3 | 0.986 |
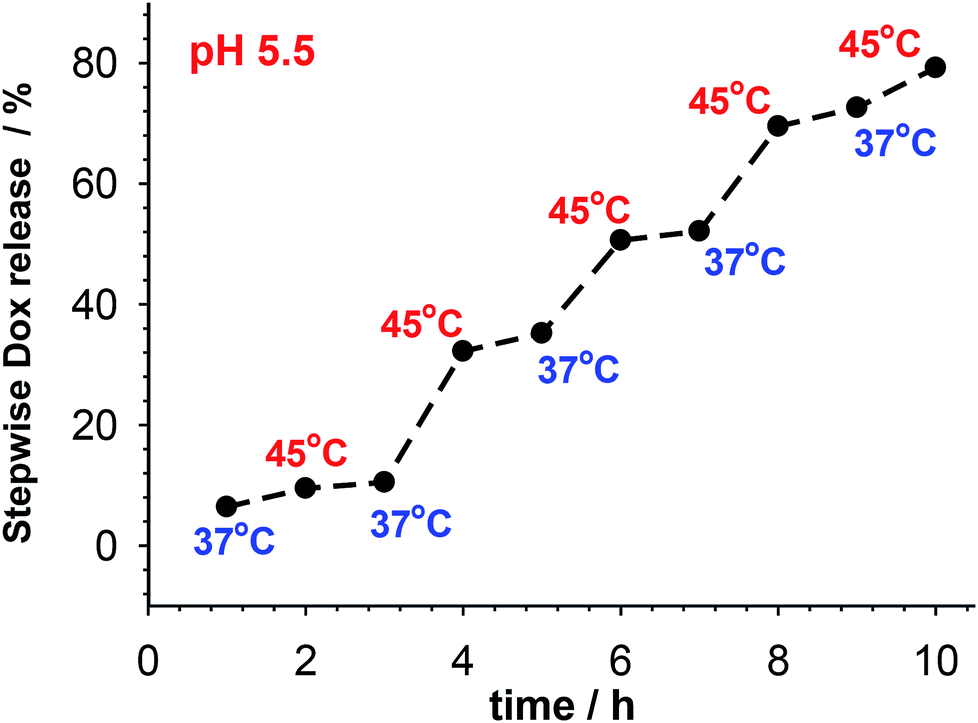 | ||
| Fig. 6 Pulsatile release of accumulated Dox from multi-responsive PNIPA–AAc–PEG–SS–dsDNA NGs at pH 5.5 and in presence of 10 mM GSH. Temperature was switched between 37 and 45 °C. | ||
3.3. Cell viability assays and uptake studies
As it is known, free Dox does not penetrate the cerebrospinal fluid. It binds, in 70–75%, to plasma proteins and circa 35% of them are irreversibly bound to tissues with microsomal proteins. Encapsulation of Dox in the NGs networks can minimize that negative effect. The cytotoxicity of the proposed NGs was examined with the MTT assay after 96 h accumulation with β-TC3 insulinoma cell lines. The measurements were conducted with pure NGs, the Dox-loaded NGs, and a reference Dox solution. It has been shown, that the major mechanism of doxorubicin toxicity in pancreatic β-cells is based on the DNA damage. This mechanism can be enhanced by redox cycling and production of H2O2.51 The results are presented in Fig. 7. Apparently, the presence of neat PNIPA–AAc–PEG–dsDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs did not limit the cell viability (see Fig. 7A). Fig. 7B presents the cytotoxicity profiles examined at pH 5.5, at 37 °C and 45 °C for: (a) reference Dox solution and (b) Dox loaded PNIPA–AAc–PEG–SS–dsDNA NGs in the presence of 10 mM and 1 mM GSH. It appeared that the viability of β-TC3 insulinoma in the presence of Dox loaded PNIPA–AAc–PEG–SS–dsDNA NGs and 10 mM GSH was significantly reduced compared to free Dox solution. Also, the mean% of viability reduction was circa 20% higher at 45 °C. As we expected, in the presence of low GSH level the viability of β-TC3 insulinoma cells was relatively high. We compared the cell viability results obtained from the fluorescence experiments with those from the UV-Vis measurements. They did not differ significantly, so the efficiency of Dox release was not significantly affected by the possible quenching-of-fluorescence effect after internalization of the nanogels to the cells.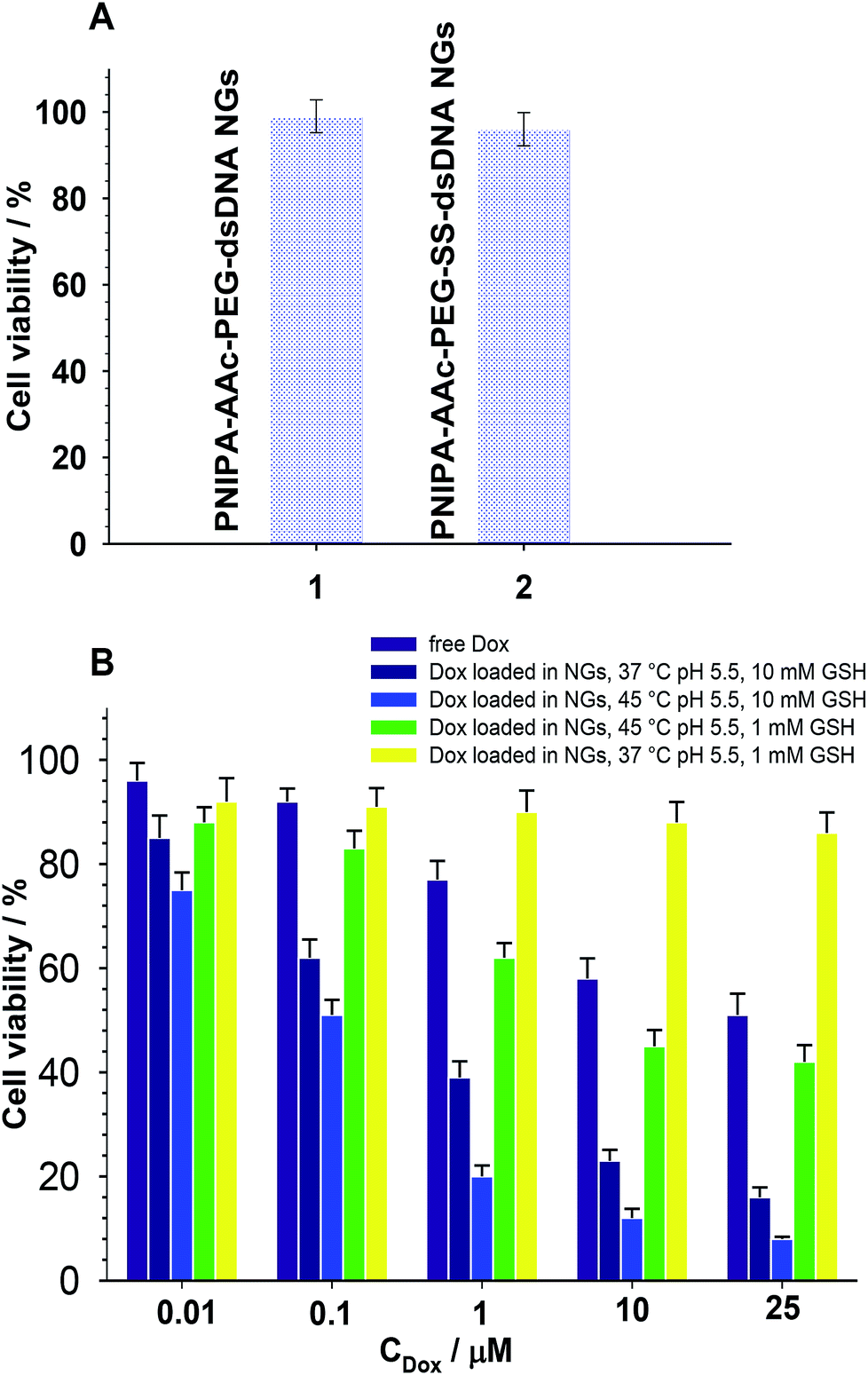 | ||
| Fig. 7 (A) Cell viability MTT assay of insulinoma cells measured after 48 h of their treatment with PNIPA–AAc–PEG–dsDNA- and PNIPA–AAc–PEG–SS–dsDNA NGs; CNGs = 7.8 mg mL−1, 37 °C, pH 7.4. (B) Cell viability MTT assay of insulinoma cells cultured with: free Dox (96 h, 37 °C, pH 5.5), pure PNIPA–AAc–PEG–SS–dsDNA NGs (CNGs = 7.8 mg mL−1, 48 h, 37 °C, pH 7.4), Dox loaded PNIPA–AAc–PEG–SS–dsDNA NGs (CNGs = 7.8 mg mL−1, measurement after 48 h, pH 5.5, 37 °C or 45 °C, GSH = 10 mM or GSH = 1 mM), β-TC3 cell lines; density 3000 cells per 100 μL per well, relative mean standard deviation – max. 6%. | ||
Cellular uptake of free Dox and Dox loaded in nanogels in β-TC3 insulinoma cells was quantified at different time intervals from 1 h to 60 h. Dox concentration in both forms equaled 1 mM. Fig. 8 illustrates the intracellular accumulation of free Dox in β-TC3 insulinoma cells. It should be stressed here, that Dox fluorescence is dramatically quenched upon its intercalation into dsDNA and the number of occupied DNA base pairs per one Dox molecule can vary depending on preferred mode of its interaction.31 As it was expected, the fluorescence intensity was lower for Dox accumulated in PNIPA–AAc–PEG–SS–dsDNA NGs. The decrease in Dox incubation in insulinoma cells might be also the effect of the increased detoxification capacity of the drug resistance mechanisms.52 When the β-TC3 insulinoma cells were incubated with Dox–PNIPA–AAc–PEG–SS–dsDNA NGs the fluorescence intensity increased linearly stronger, compared to free Dox. These results indicate that Dox loaded nanogels might not be detoxified as quickly as free Dox and the drug could be retained for a longer time in the resistant cells after internalization. The enhanced cellular uptake of Dox in nanogels also contributes to the overcoming of the drug resistance. The alternate conclusion is that the cells internalize the PNIPA–AAc–PEG–SS–dsDNA NGs slower than free Dox, and this might be due to the difference between endocytosis of the nanocarrier and the diffusion of a small-molecule drug.53
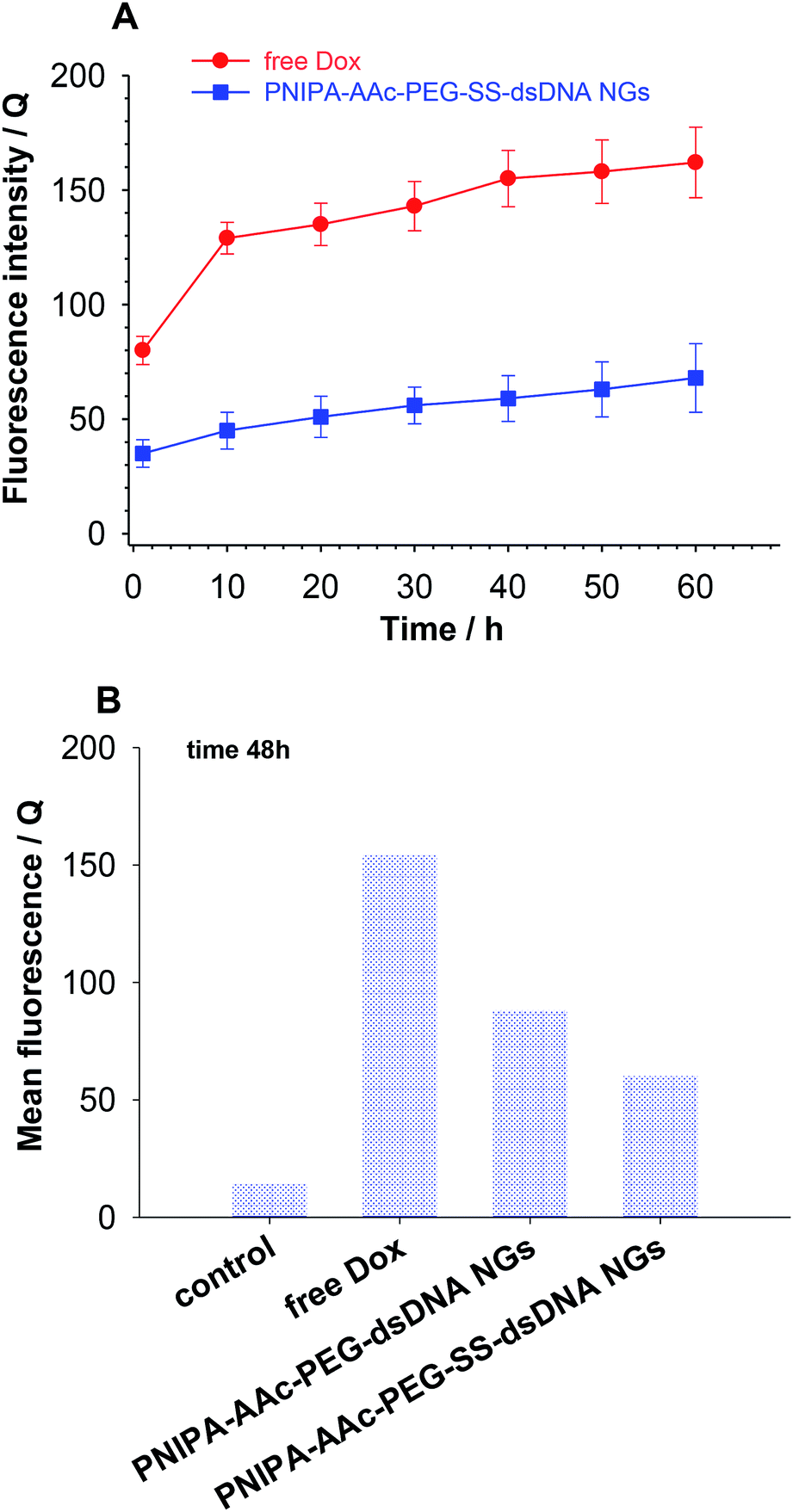 | ||
| Fig. 8 (A) Cell uptake of free Dox and Dox loaded in PNIPA–AAc–PEG–SS–dsDNA NGs by β-TC3 cells. (B) Quantitative comparison of Dox accumulation in β-TC3 cells after 48 h of incubation of free Dox and Dox loaded into nanogels. | ||
4. Conclusions
The introduction of oligonucleotide-based co-crosslinkers containing –SS– groups allowed us to develop multi responsive nanogels for prolonged and controlled release of drugs. Doxorubicin was selected for examination as the model drug. The DLS, spectroscopic and electrochemical experiments confirmed that switchable dynamical reorganization of DNA-based co-crosslinker shells took place in the proposed DNA nanogel structures. Finally, three processes that occurred in the nanogels appeared helpful in effective, controlled and prolonged drug release and nanogel degradation: (a) accumulation of the drug through intercalation into dsDNA, (b) reversible conformational changes and relaxation of the DNA hybrid crosslinkers and the hydrogel lattice and (c) reduction and breaking of disulfide groups. The cooperation of GSH-mediated cleavage with reversible conformational changes of oligonucleotides, which were involved in the effective accumulation and protection of the active form of the drug, resulted in enhanced efficiency of the pulsatile release process and promoted selective delivery to cancer cells. Moreover, the retarded degradation of the nanogel shells eliminated the risk of premature drug release and can potentially diminish the side effects in cancer treatment. The proposed complex mechanism of the drug release from the nanogels which possess reversible character under conditions of routine hyperthermia treatment, makes the nanogels potentially useful in conventional cancer therapies, as well as in advanced photothermal treatments. The non-loaded DNA nanogels containing –SS– groups in the co-crosslinkers appeared to exhibit no cytotoxicity, while the particles loaded with Dox possessed the ability of triggering the death response in insulinoma cancer cells. Moreover, the developed drug carrier can be regenerated. It appears it can serve as an evaluator of appropriate intercalation activity of the drug molecules and can be useful as a labelling agent in drug-route pharmacokinetic investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support of this work by NCN Preludium grant 2015/19/N/ST5/03672 is thankfully acknowledged.Notes and references
- M. Karbarz, M. Mackiewicz, K. Kaniewska, K. Marcisz and Z. Stojek, Applied Materials Today, 2017, 9, 516 CrossRef.
- S. Gao, G. Tang, D. Hua, R. Xiong, J. Han, S. Jiang, Q. Zhang and C. Huang, J. Mater. Chem. B, 2019, 7, 709 RSC.
- D. Hua, Z. Liu, F. Wang, B. Gao, F. Chen, Q. Zhang, R. Xiong, J. Han, S. K. Samal, S. C. De Smedt and C. Huang, Carbohydr. Polym., 2016, 151, 1240 CrossRef CAS PubMed; Z. Guo, G. Tanga, Y. Zhou, L. Shuwu, H. Hou, Z. Chen, J. Chen, C. Hu, F. Wang, S. C. De Smedt, R. Xiong and C. Huang, Carbohydr. Polym., 2017, 169, 198 CrossRef PubMed.
- Applications of Nanocomposite Materials in Drug Delivery, ed. I. A. M. Asiri and A. Mohammad, Woodhead Publishing, Elsevier, 2018 Search PubMed.
- U. Feldkamp and C. M. Niemeyer, Angew. Chem., 2006, 45, 1856 CrossRef CAS PubMed.
- S. H. Park, G. Finkelstein and T. H. LaBean, J. Am. Chem. Soc., 2008, 130, 40 CrossRef CAS PubMed.
- Y. He, T. Ye, M. Su, C. A. Zhang, E. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198 CrossRef CAS PubMed.
- A. M. Nowicka, E. Zabost, E. Gilant and Z. Stojek, Phys. Chem. Chem. Phys., 2011, 137, 500 Search PubMed.
- M. J. Campolongo, J. S. Kahn, W. Cheng, D. Yang, T. Gupton-Campolongo and D. Luo, J. Mater. Chem. B, 2011, 21, 6113 RSC.
- L. Yue, S. Wang, S. Wulf, S. Lilienthal, F. Remacle, R. D. Levine and I. Willner, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2843 CrossRef CAS PubMed.
- F. Wang, C. H. Lu and I. Willner, Chem. Rev., 2014, 114, 2881 CrossRef CAS PubMed.
- P. W. Rothemund, Nature, 2006, 16, 297 CrossRef PubMed.
- D. Wang, Y. Hu, L. P. Liu and D. Luo, Acc. Chem. Res., 2017, 50, 733 CrossRef CAS PubMed.
- J. S. Kahn, R. C. H. Ruiz, S. Sureka, S. Peng, T. L. Derrien and D. Luo, Biomacromolecules, 2016, 17, 2019 CrossRef CAS PubMed.
- S. R. Deshpande, R. Hammink, F. H. T. Nelissen, A. E. Rowan and H. A. Heus, Biomacromolecules, 2017, 18, 3310 CrossRef CAS PubMed.
- F. Huang, W. C. Liao, Y. S. Sohn, R. Nechushtai, C. H. Lu and I. Willner, J. Am. Chem. Soc., 2016, 138, 8936 CrossRef CAS PubMed.
- E. Zabost, W. Chmielowiec, T. Rapecki, M. Karbarz and Z. Stojek, Electrochem. Commun., 2014, 42, 50 CrossRef CAS.
- E. Zabost, W. Liwinska, M. Karbarz, E. Kurek, M. Lyp, M. Donten and Z. Stojek, Bioelectrochemistry, 2016, 106, 1 CrossRef PubMed.
- J. Li, C. Zheng, S. Cansiz, C. Wu, J. Xu, C. Cui, Y. Liu, W. Hou, Y. Wang, L. Zhang, I. T. Teng, H. H. Yang and W. Tan, J. Am. Chem. Soc., 2015, 137, 1412 CrossRef CAS PubMed.
- H. V. P. Thelu, S. K. Albert, M. Golla, N. Krishnan, D. Ram, S. M. Srinivasulab and R. Varghese, Nanoscale, 2018, 10, 222 RSC.
- Y. Ma, H. Liu, Q. Mou, D. Yan, X. Zhu and C. Zhang, Nanoscale, 2018, 10, 8367 RSC.
- C. Jiang, J. Song, X. Nangreave, L. Liu, D. Lin, Z. G. Qiu, G. Wang, X. Zou, H. Y. Liang and B. Ding, J. Am. Chem. Soc., 2012, 134, 13396 CrossRef PubMed.
- Y. Dayani and N. Malmstadt, Biomacromolecules., 2013, 14, 3380 CrossRef CAS PubMed.
- Y. Chi, Y. Ye and Y. Dayong, ACS Appl. Bio Mater., 2018, 1, 2012 CrossRef.
- F. Ding, Q. Mou, Y. Ma, G. Pan, Y. Guo, G. Tong, C. H. Choi, X. Zhu and C. Zhang, Angew. Chem., 2018, 57, 3064 CrossRef CAS PubMed.
- W. Sun, T. Jiang, Y. Lu, M. Reiff, R. Mo and Z. Gu, J. Am. Chem. Soc., 2014, 136, 14722 CrossRef CAS PubMed.
- B. Shi, M. Zheng, W. Tao, R. Chung, D. Jin, D. Ghaffari and O. C. Farokhzad, Biomacromolecules, 2017, 18, 2231 CrossRef CAS PubMed.
- J. Cano-Mejia, M. L. Bookstaver, E. E. Sweeney, C. M. Jewell and R. Fernandes, Biomater. Sci., 2019, 7, 1875 RSC.
- D. Banerjee, A. Bhatia, S. Saminathan, S. Chakraborty, S. Kar and Y. Krishnan, Angew. Chem., 2013, 52, 6854 CrossRef PubMed.
- C. H. Lu and I. Willner, Angew. Chem., 2015, 42, 12212 CrossRef PubMed.
- W. Liwinska, M. Symonowicz, I. Stanislawska, M. Łyp, Z. Stojek and E. Zabost, RSC Adv., 2016, 69, 1045 Search PubMed.
- W. Liwinska, I. Stanislawska, M. Lyp, M. Mackiewicz, Z. Stojek and E. Zabost, J. Mater. Chem. B, 2017, 5, 4713 RSC.
- Y. Tiam, L. Bromberg, S. N. Lin, T. A. Hatton and K. C. Tam, J. Controlled Release, 2007, 121, 137 CrossRef PubMed.
- I. Makra, P. Terejanszky and R. E. Gyurcsanyi, MethodsX, 2015, 2, 91 CrossRef PubMed.
- M. D'Abramo, C. L. Castellazzi, M. Orozco and A. Amadei, J. Phys. Chem. B, 2013, 117, 8697 CrossRef PubMed.
- R. Campos, J. Borme, G. R. Guerreiro, G. Machado, J. F. Cerqueira, D. Y. Petrovykh and P. Alpuim, ACS Sens., 2019, 4, 286 CrossRef CAS PubMed.
- C. Chen, Z. Lai, G. Wang and C. Wu, Biosens. Bioelectron., 2016, 77, 603 CrossRef CAS PubMed.
- B. Zhu, M. Booth, P. Shepherd and A. Sheppard, Biosens. Bioelectron., 2015, 64, 74 CrossRef CAS PubMed.
- A. Lasia, in Modern aspects of electrochemistry modeling and numerical simulations, ed. M. Schlesinger, Springer, New York, 2009 Search PubMed.
- N. Dolinnaya, V. Metelev, T. Oretskaya, D. Tabatadze and Z. Shabarova, FEBS Lett., 1999, 444, 285 CrossRef CAS.
- T. Takada and J. K. Barton, J. Am. Chem. Soc., 2005, 127, 12204 CrossRef CAS PubMed.
- G. Wu, Y. Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 482 Search PubMed; I. Stanisławska, B. Witek, M. Łyp, D. Rochon-Szmejchel, A. Wróbel, W. Fronczyk, A. Kamińska, A. Kołątaj and D. Załuski, Adv. Exp. Med. Biol., 2018, 81 CrossRef PubMed; B. Witek, D. Rochon-Szmejchel, I. Stanisławska, M. Łyp, K. Wróbel, A. Zapała, A. Kamińska and A. Kołątaj, Adv. Exp. Med. Biol., 2018, 73 Search PubMed.
- N. Drude, O. H. Winz, F. M. Mottaghy, M. Roller, H. Königs, M. Möller, S. Singh and A. Morgenroth, Small, 2018, 14, 1704093 CrossRef PubMed.
- C. Pérez-Arnaiz, N. Busto, J. Leal and B. García, J. Phys. Chem. B, 2014, 118, 1288 CrossRef PubMed.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469 CrossRef CAS PubMed.
- K. Nakamoto, M. Tsuboi and G. D. Strahan, Drug-DNA interactions: structures and spectra, John Wiley & Sons, Inc., Hoboken, New Jersey, 2008 Search PubMed.
- Y. Hahn and H. Y. Lee, Arch. Pharmacal Res., 2004, 27, 31 CrossRef CAS.
- L. Mora, K. Y. Chumbimuni-Torres, C. Clawson, L. Hernandez, L. Zhang and J. Wang, J. Controlled Release, 2009, 140, 69 CrossRef CAS PubMed.
- P. Ritger and N. Peppas, J. Controlled Release, 1987, 5, 23 CrossRef CAS.
- E. P. Holowka and S. K. Bhatia, Drug Delivery: Materials Design and Clinical Perspective, Springer Science & Business, New York, 2014 Search PubMed.
- E. A. Heart, S. Karandrea, X. Liang, M. E. Balke, P. A. Beringer, E. M. Bobczynski, D. Z. Burgos, T. Richardson and J. P. Gray, Toxicol. Sci., 2016, 152, 395 CrossRef CAS PubMed.
- K. Johansson, K. Ahlen, R. Rinaldi, K. Sahlander, A. Siritantikorn and R. Morgenstern, Carcinogenesis, 2007, 28, 465 CrossRef CAS PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |