
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrP (58–93) peptide from unstructured N-terminal domain of human prion protein forms amyloid-like fibrillar structures in the presence of Zn2+ ions†
Maciej Gielnik a,
Zuzanna Pietralika,
Igor Zhukovbc,
Aneta Szymańskad,
Wojciech M. Kwiateke and
Maciej Kozak*afg
a,
Zuzanna Pietralika,
Igor Zhukovbc,
Aneta Szymańskad,
Wojciech M. Kwiateke and
Maciej Kozak*afg
aDepartment of Macromolecular Physics, Faculty of Physics, Adam Mickiewicz University, Uniwersytetu Poznańskiego 2, PL 61-614 Poznań, Poland. E-mail: mkozak@amu.edu.pl
bInstitute of Biochemistry and Biophysics, Polish Academy of Sciences, PL 02-106 Warszawa, Poland
cNanoBioMedical Centre, Adam Mickiewicz University, PL 61-614 Poznań, Poland
dDepartment of Biomedical Chemistry, Faculty of Chemistry, Gdańsk University, PL 80-308 Gdańsk, Poland
eInstitute of Nuclear Physics Polish Academy of Sciences, PL 31-342 Krakow, Poland
fJoint Laboratory for SAXS Studies, Faculty of Physics, Adam Mickiewicz University, PL 61-614 Poznań, Poland
gNational Synchrotron Radiation Centre SOLARIS, Jagiellonian University, PL 30-392 Kraków, Poland
First published on 17th July 2019
Abstract
Many transition metal ions modulate the aggregation of different amyloid peptides. Substoichiometric zinc concentrations can inhibit aggregation, while an excess of zinc can accelerate the formation of cytotoxic fibrils. In this study, we report the fibrillization of the octarepeat domain to amyloid-like structures. Interestingly, this self-assembling process occurred only in the presence of Zn(II) ions. The formed peptide aggregates are able to bind amyloid specific dyes thioflavin T and Congo red. Atomic force microscopy and transmission electron microscopy revealed the formation of long, fibrillar structures. X-ray diffraction and Fourier transform infrared spectroscopy studies of the formed assemblies confirmed the presence of cross-β structure. Two-component analysis of synchrotron radiation SAXS data provided the evidence for a direct decrease in monomeric peptide species content and an increase in the fraction of aggregates as a function of Zn(II) concentration. These results could shed light on Zn(II) as a toxic agent and on the metal ion induced protein misfolding in prion diseases.
Introduction
Insoluble fibrillar polypeptide structures appear as a result of a misfolding process in numerous proteins or peptides and accompany the amyloid aggregation process.1–6 The most spectacular structures of this type are linked to the development of fatal neurodegenerative disorders such as Alzheimer's disease,7,8 Icelandic type amyloidosis9,10 or Creutzfeldt–Jakob disease.11,12 The latter belongs to the group of diseases called transmissible spongiform encephalopathies (TSE) in which a misfolding of human prion protein (PrP) is responsible for the formation of fibrillar aggregates in the brain.11–15Mature form of cellular human prion protein (PrPC) is 208 residues long glycosylphosphatidylinositol-anchored to outer membrane protein,16 located in the synaptic cleft and expressed at high levels in striatum, hippocampus, cortex and olfactory bulb.17 As shown by NMR studies, human PrPC protein is composed of two structurally different domains: unstructured N-terminal part of polypeptide chain containing residues 23–120 and structured, globular C-terminal domain composed of residues 121–231.18 For the development of TSE, a conformational transformation of the cellular form of the prion protein into the pathogenic and insoluble PrPSc is necessary. This transformation involves the formation of the intermolecular cross-β structure at the sacrifice of the helical structure of the PrP C-terminal domain.13,14 The molecular function of PrPC is not known, however, it has been suggested that it might be responsible for the upkeep of divalent transition metal ions and disruption of such homeostasis may facilitate the formation of PrPSc.19–21 Prion protein contains also two high-affinity metal binding sites: the octarepeat region (residues 60–91) and non-octarepeat region (residues 92–111), both located in unstructured N-terminal domain.22 Octarepeat region contains four tandem octapeptide repeats characterized by PHGGGWGQ sequence, which exhibits high binding affinity towards Cu(II)22–29 and also binds Zn(II) with lower affinity.22,30,31
Zinc is the second most abundant transition metal in living organisms and it is responsible for many important biological functions, including structure stabilization, catalysis of biochemical reactions and regulation of enzymatic activity.32 In the highest concentrations Zn(II) occurs in the brains, it is on average equal to 150 μM.33 Zinc plays a key role in synaptic transmission and during the synaptic excitation the local transient concentration of Zn(II) can reach values up to 300 μM.33,34 Assuming that the cellular function of PrPC is most likely connected with metal binding, it was proposed that PrPC is responsible for zinc homeostasis in the brain.21 Three main functions are attributed to PrPC: zinc sensor, transporter or sequester.21,35,36 PrPC enhances the rate of zinc uptake in neurons and neuronal cell lines.37 Zinc at concentration 100 μM also stimulates endocytosis of PrPC from the cell surface into endosomes and Golgi apparatus.38,39 At the molecular level, coordination of Zn(II) by octarepeat region of PrPC causes a tertiary contact between Zn(II) saturated octarepeat region and surface of helices 2 and 3 in C-terminal domain.40 Zinc may also play an important role during TSE. In prion diseases, zinc concentration in the brain decreases to 20%41 and inoculation of neuronal cell lines with scrapie disrupts zinc uptake.37 Also tertiary contacts between Zn(II) coordinated octarepeat domain and helices 2 and 3 in pathogenic mutants of PrPC are altered.40
Here we studied the aggregation of isolated metal binding PrP58–93 domain, naturally located in the unstructured N-terminal domain of PrPC in the presence of Zn(II).
We report for the very first time the fibrillization of the octarepeat domain to amyloid-like structures. This self-assembling process occurred in the presence of Zn2+ ions. The assemblies obtained were analyzed by various spectroscopic, microscopic and scattering techniques. The formed PrP58–93 fibrillar structures were characterized by the amyloid-specific assays (thioflavin T, Congo red) and were visualized using atomic force microscopy (AFM) and transmission electron microscopy (TEM). The presence of the cross-β-structure in the formed fibrils was verified by the X-ray diffraction pattern combined with Fourier transform infrared (FTIR) spectroscopy. SAXS experiments suggested that Zn(II) had a direct impact on PrP58–93 monomeric state. The presented studies may be crucial for better comprehension of the molecular etiology of TSE and other amyloid diseases.
Materials and methods
Peptide synthesis and purification
PrP58–93 was synthesized by standard Fmoc/tBu amino acid chemistry and purified to at least 98% purity by means of reversed-phase high-performance liquid chromatography. The molecular weight of the peptide was confirmed by the mass spectrometry method using an ESI-IT-TOF-LC-MS system (Shimadzu) with a C12 Jupiter Proteo column (150 × 2 mm, 4 μm, 90 Å; Phenomenex). The exact procedure of peptide synthesis and purification is described in ESI.† For determination of PrP58–93 concentration, we used extinction coefficient calculated in protparam42 equal 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.
000 M−1 cm−1.
Thioflavin T assay
Spectrofluorometric measurements of PrP58–93 were performed in a 1 cm cuvette using an FP-8300 spectrofluorimeter (Jasco, Tokyo, Japan). Excitation and emission wavelengths were 440 and 480 nm, respectively. 2 mM stock solution of Thioflavin T (ThT) was prepared in 10 mM NaH2PO4/Na2HPO4 buffer, pH 7.4 and filtered through 0.22 μm syringe filter. Samples for thioflavin T assay contained 40 μM of ThT and 10 μM of PrP58–93 peptide in 10 mM NaH2PO4/Na2HPO4, pH 7.4. Zinc(II) ions were added as ZnCl2 solution directly into the cuvette after recording the fluorescence intensity for 40 μM ThT with 10 μM peptide for a few minutes. All ThT experiments were performed in triplicate at room temperature and in quiescent conditions.Congo red binding assay
Congo red (CR) stock solution was prepared in 10 mM NaH2PO4/Na2HPO4 buffer, pH 7.4 with 10% (v/v) ethanol and filtered through an 0.22 μm syringe filter. Concentration of CR stock solution was determined in 1 mM NaH2PO4/Na2HPO4 buffer, pH 7.0 with 40% ethanol at 505 nm using the molar extinction coefficient ε = 59300 M−1 cm−1.43
Congo red binding assays were performed in a 1 cm cuvette (2500 μl volume). CR at concentration of 2 μM with 10 μM PrP58–93 peptide samples were incubated with or without 40 μM ZnCl2 for 12 hours in 10 mM NaH2PO4/Na2HPO4 buffer, pH 7.4. Spectra were recorded every 15 minutes over the spectral range from 300 to 800 nm on a V-650 UV-Vis spectrometer (Jasco, Tokyo, Japan), with bandwidth 1 nm, scan speed 200 nm min−1. All CR experiments were performed in room temperature and in quiescent conditions.
Fourier transform infrared spectroscopy
The absorption spectra within the Amide I bands region for PrP58–93 peptide solution (600 μM PrP58–93 dissolved in 10 mM NaH2PO4/Na2HPO4, pH 7.4) were collected with a resolution of 6 cm−1 at room temperature. A small drop of PrP58–93 peptide solution (20 μl) was placed on a horizontal single-reflection diamond crystal of PLATINUM ATR (Bruker, Billerica, USA) unit and then FTIR spectra (128 scans) were collected using TENSOR 27 (Bruker, Billerica, USA) equipped with MCT detector. After addition of 25 mM of ZnCl2 solution (2 μl), the FTIR spectra were collected every 5 or 10 minutes for up to 2 hours. The recorded FTIR spectra were corrected for buffer absorption and smoothed with 10 point Savitzky–Golay filter. Second-derivative analysis was performed as described by Yang et al.44Atomic force microscopy
The topographic images of peptide fibrils were collected using JPK Nanowizard 4 atomic microscope (JPK, Berlin, Germany) operated in air contact mode. The imaging was conducted using Tap150AL AFM cantilevers (Ted Pella, Inc., Redding, USA). Sample preparation procedure for AFM imaging of 10 μM PrP58–93 peptide (apo) and 10 μM peptide incubated for 24 h with 40 μM of ZnCl2 was identical: 10 μl of solution was placed on a freshly cleaved mica surface, adsorbed for 3 min, rinsed with a small amount of distilled water and dried at room temperature. The AFM images were analyzed using the Gwyddion 2.49 software.45Transmission electron microscopy
The PrP58–93 peptide solution at concentration of 10 μM was incubated with 40 μM of ZnCl2 in 10 mM NaH2PO4/Na2HPO4 buffer, pH 7.4 for 24 h at room temperature. After incubation, 5 μl of samples were placed on PELCO® 300 mesh copper grid (Ted Pella, Inc., Redding, USA) and adsorbed within 60 seconds on the grid. Then the sample was removed from the grid using filter paper strips. The peptide fibrils adsorbed on the grid were contrasted by the use of 0.5% uranyl formate solution for 60 seconds. TEM images were recorded using JEM-1400 transmission electron microscope (JEOL, Tokyo, Japan) operated at 120 kV.Synchrotron radiation small angle X-ray scattering (SR-SAXS)
SR-SAXS data were collected at P12 beamline, PETRA III storage ring (DESY, Hamburg), using a Pilatus 3X 6M pixel detector (DECTRIS Ltd. Baden-Daettwil, Switzerland) and synchrotron radiation (λ = 0.124 nm).46 The sample-to-detector distance was 3.0 m and the scattering vector (s) range was: 0.026 < s < 7.288 nm−1. In SR-SAXS experiments 0.8 mM PrP58–93 peptide was dissolved in 50 mM HEPES, pH 7.4, 100 mM NaCl and ZnCl2 (concentration from 0 to 0.8 mM). For each sample, thirty subsequent frames, 50 ms each, were recorded and radiation damage was inspected. Due to radiation damage only the first three frames were analyzed.Buffer subtraction and Guinier analysis were performed in PRIMUS47 from ATSAS 2.8 package.48 The pair distribution function (P(r)) was calculated using GNOM.49
Due to the polydispersity of sample after addition of ZnCl2, we performed two component analysis of SR-SAXS data in SASfit.50 The shape of apo PrP58–93 peptide can be reduced into a spheroid and the form factor can be calculated analytically:50
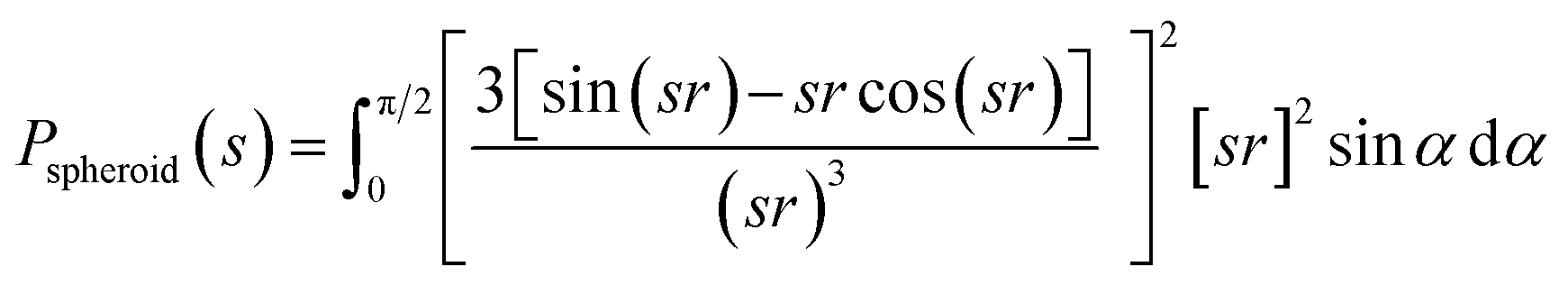 | (1) |
|
r = s[Rp2sin2(α) + Re2 cos2(α)]1/2
| (2) |
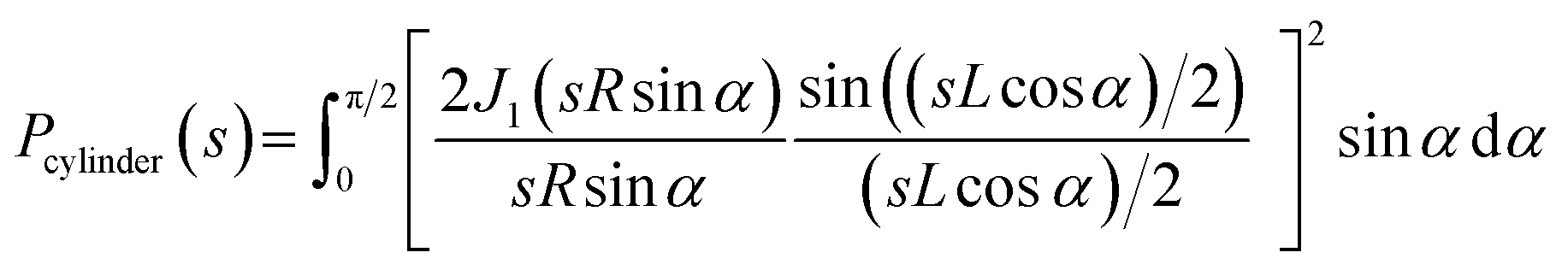 | (3) |
X-ray diffraction
X-ray fiber diffraction experiment of PrP58–93 fibrils was conducted on XEUSS 2.0 SAXS/WAXS system (XENOCS, Sassenage, France), using Ga Kα radiation from a MetalJet microfocus generator with a liquid-metal jet anode (Excillum AB, Kista, Sweden) and a Pilatus 3R 1M detector (DECTRIS Ltd. Baden-Daettwil, Switzerland). The sample-to-detector distance was calibrated by the use of silver behenate (reference q001 = 1.070 nm−1). 500 μl of 0.4 mM PrP58–93 peptide prepared in 50 mM HEPES, pH 7.4, 100 mM NaCl with 1.6 mM ZnCl2 was incubated at room temperature for 24 h. After incubation time, the aggregated deposits, visible on the walls of the Eppendorf test tube, were transferred into thin-walled borosilicate glass and the data were collected.Results and discussion
PrP58–93 peptide binds amyloid specific dyes only in the presence of Zn(II)
For the initial characterization of PrP58–93 peptide, we performed circular dichroism spectroscopy experiments (Fig. S1, ESI†). The CD spectrum of this peptide is well-documented in literature:23,26,27,51,52 and it shows a weak positive band at 225 nm, a strong negative band at 200 nm, which are interpreted to correspond to a mixture of random coil and polyproline-II helix – extended left-handed helix.51,52 Surprisingly, we found that the investigated peptide was very stable – 1 mM stock solution incubated over six months at room temperature and diluted into 5 μM concentration did not show any significant changes in the secondary structure (Fig. S1, ESI†). However, the addition of 20 μM ZnCl2 into the cuvette with 5 μM PrP58–93 peptide resulted in a rapid loss of the CD signal, which suggests precipitation or aggregation of the peptide (data not shown). In order to check the possible aggregation of PrP58–93 peptide in the presence of Zn(II) we performed ThT and CR assays.ThT is an amyloid-specific dye whose fluorescence increases upon binding via hydrophobic interaction with β sheets to amyloid fibrils, therefore it is commonly used to monitor the fibrillization kinetics.53,54 The fluorescence of 40 μM of ThT with 10 μM of apo PrP58–93 peptide showed a slight increase in intensity over time (Fig. 1a, black line), which can be attributed to rather slow fibril formation and subsequent ThT binding. To rule out a possible aggregation of apo PrP58–93 peptide, we conducted additional dynamic Congo red (CR) binding assay. Congo red is a diazo dye highly specific for binding a cross-β structure present in amyloid fibrils. When it binds to fibrils, CR absorption increases and shows a red shift from 490 to 540 nm.43 Time-resolved spectra obtained for PrP58–93 peptide revealed low and constant CR absorbance at 541 nm over time (Fig. 1b, black squares). This result, together with the ThT and CD data, strongly indicate that the PrP58–93 alone does not aggregate and its structure is stable in solution.
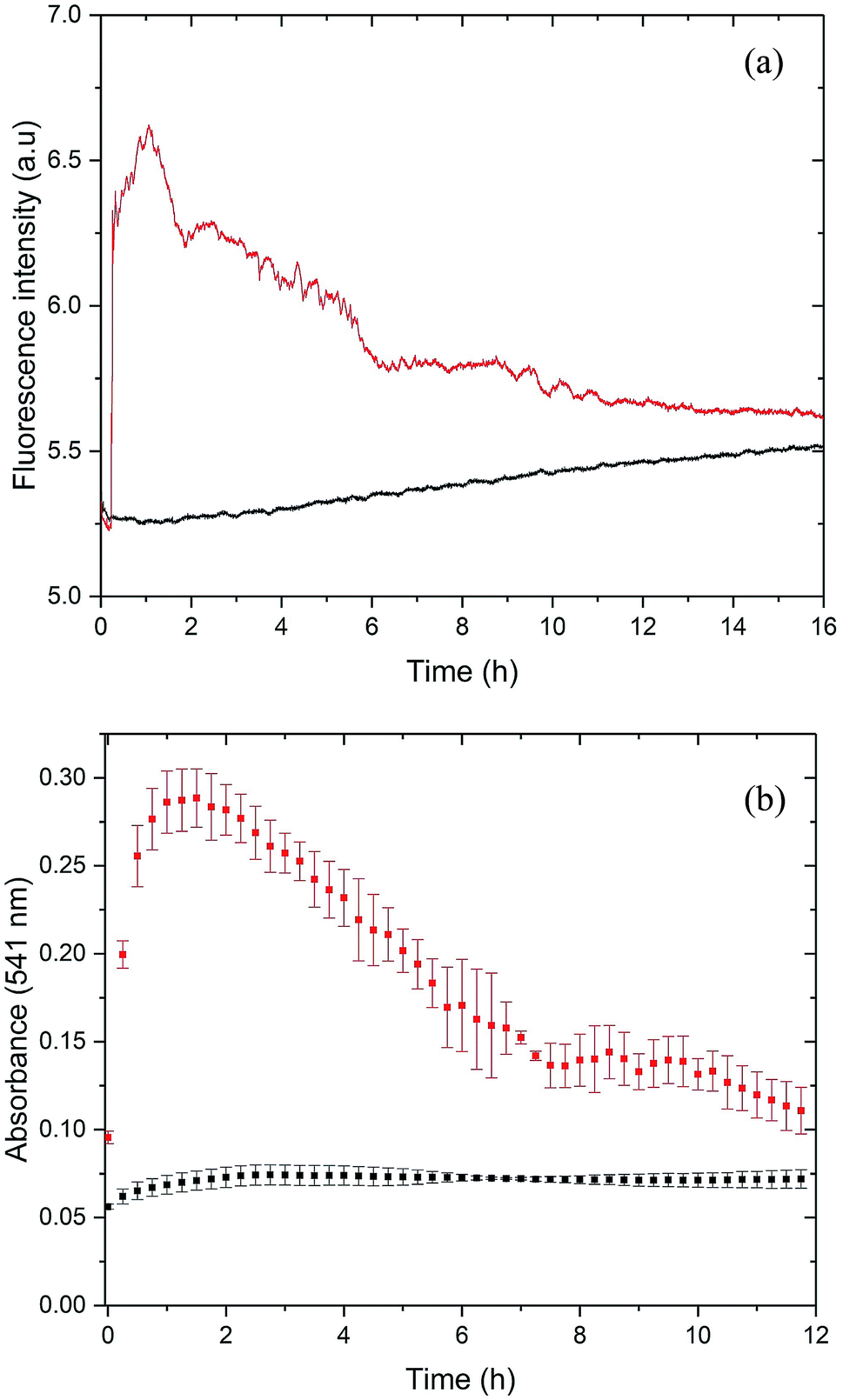 | ||
| Fig. 1 (a) ThT binding assay for 10 μM PrP58–93 peptide (black line) and for 10 μM PrP58–93 peptide with 40 μM of ZnCl2 (red line). (b) CR binding assay for 10 μM PrP58–93 peptide (black squares) and for 10 μM PrP58–93 peptide with 40 μM of ZnCl2 (red squares). Both experiments were performed in 10 mM NaH2PO4/Na2HPO4 buffer, pH 7.4. | ||
Upon addition of ZnCl2 (40 μM) to the 10 μM peptide sample, ThT fluorescence increased rapidly and PrP58–93 acquired the ability to bind ThT (Fig. 1a, red line). This observation suggests that upon interaction with Zn(II) PrP58–93 peptide forms amyloid-like structures. The kinetic ThT curve showed no lag phase, what suggests that the induced fibrillization is a very rapid process which quickly exhausts most of the monomeric peptide form. Indeed, the fluorescence intensity reached a plateau after ∼1 h of incubation. Longer incubation resulted in a decrease in the ThT fluorescence intensity and a final stabilization of the signal at a constant level after ∼11 h. The loss of ThT signal was most probably caused by sedimentation of the formed fibrils, visible as a yellowish precipitate at the bottom of the cuvette after 12 h.
For the cross-validation of the possible fibrillization of PrP58–93 in the presence of Zn(II), we applied the Congo red (CR) binding assay (Fig. 1b, red squares). In the presence of Zn(II), the absorbance of CR at 541 nm increased significantly from 0.07 to 0.27 very quickly after metal ion addition. This elevation in the absorbance is an evidence for the binding of CR to the β-sheets and suggests fibril formation by PrP58–93 peptide in the presence of Zn(II). The shape of the kinetic curve in the CR assay resembles the ThT curve (Fig. 1a red line). CR absorbance reached a plateau very fast, in about 1 h after addition of ZnCl2, which confirms a rapid fibril formation. Further incubation resulted in a decrease in CR absorbance in a fashion similar to the ThT fluorescence intensity for PrP58–93 peptide with Zn(II). At the end of CR assay, a reddish precipitate was visible at the bottom of the cuvette, therefore reduction of CR absorbance was probably caused by direct binding of CR to the formed fibrils and sedimentation of the aggregates. In conclusion, both ThT and CR assays indicated that PrP58–93 peptide might form amyloid-like structures in the presence of Zn(II) in contrast to the apo peptide that seemed to be stable over time.
Zn(II) induces β-sheet formation in PrP58–93
CD spectroscopy failed to monitor β-sheet formation, probably due to sedimentation of formed fibrillar structures. Therefore, in order to observe a transition in the secondary structure of PrP58–93 peptide after addition of Zn(II), we subjected the samples to ATR-FTIR spectroscopy. Amide I band for PrP58–93 peptide was broad and had a maximum of absorption at 1661 cm−1 (Fig. 2, dark green line). Second-derivative analysis suggested, that apart from the predominant absorption maximum at 1645 cm−1, corresponding to the random coil structure, the bands assigned to 310-helix, β-turns and β-sheets were also present (Fig. S2, ESI†).44 These bands, other than assigned to random coil, seemed to be a consequence of the inherence of PPII helix secondary structure in the PrP58–93 peptide.51,52 Theoretical and experimental amide I FTIR spectra of (PPG)10 collagen peptides, forming triple PPII helix suggest that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of PPII amide group gives rise to frequencies described in a standard secondary structure analysis as 310-helix, β-turns and β-sheets55 and match well with our data. As a consequence, the secondary structure of apo PrP58–93 peptide is a mixture of a random coil and PPII helix, as postulated in literature.51,52
O stretching vibration of PPII amide group gives rise to frequencies described in a standard secondary structure analysis as 310-helix, β-turns and β-sheets55 and match well with our data. As a consequence, the secondary structure of apo PrP58–93 peptide is a mixture of a random coil and PPII helix, as postulated in literature.51,52
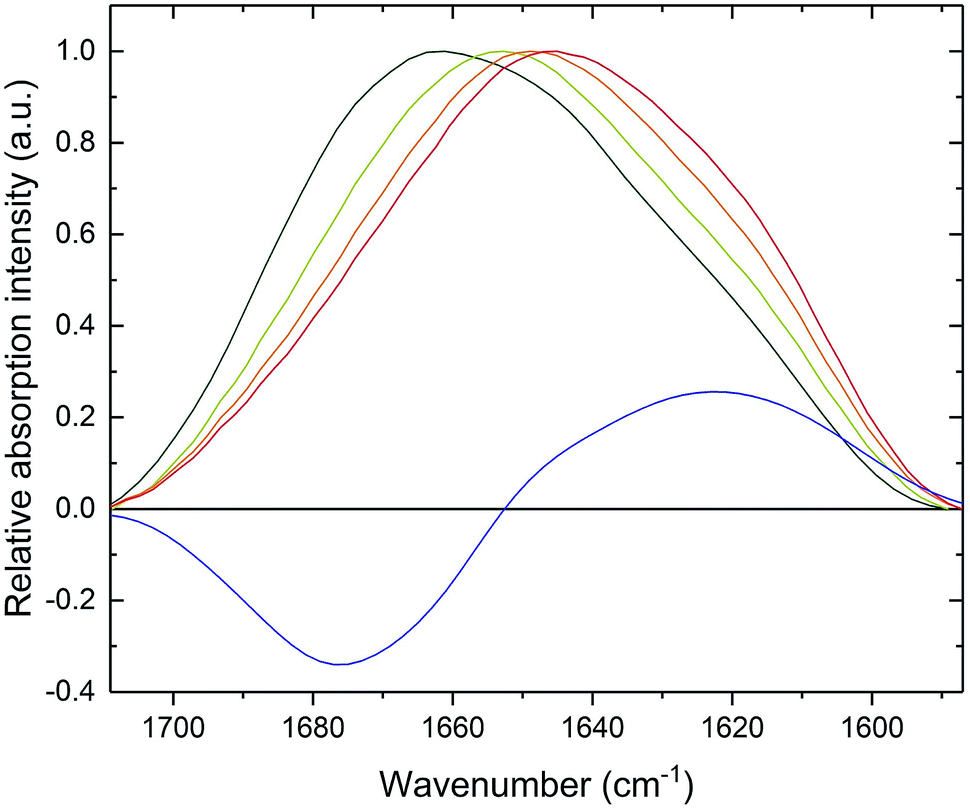 | ||
| Fig. 2 FTIR spectra of 600 μM PrP58–93 peptide (olive), 600 μM PrP58–93 peptide with 2.3 mM ZnCl2 (lime), 600 μM PrP58–93 peptide with 2.3 mM ZnCl2 after 15 minutes (orange), 600 μM PrP58–93 peptide with 2.3 mM ZnCl2 after 30 minutes (red), differential spectrum between 600 μM PrP58–93 peptide with 2.3 mM ZnCl2 after 30 minutes and 600 μM PrP58–93 peptide (blue). | ||
Addition of Zn(II) cations to the PrP58–93 sample resulted in a gradual shift of the absorption maximum up to 1645 cm−1 after 30 minutes (Fig. 2, red line). The accompanying decrease in amide I band intensity at 1675 cm−1 and the concurrent increase at 1622 cm−1 (Fig. 2, blue line) corresponded to a structural transition in a PrP58–93 peptide induced by Zn(II). These changes in FTIR spectrum indicated a reduction of β-turn content and formation of β-sheets, respectively.44 Further second-derivative analysis confirmed the formation of β-sheets, the disappearance of peaks that might be assigned to PPII helix, and an increase in random coil content (Fig. S3, ESI†). Zn(II) binding with PrP58–93 may cause the unfolding of the PPII helix what, accompanied by the lowering of the energetic barrier for nucleation and aggregation, might result in a conformational transition unavailable for apo PrP58–93. The FTIR data are consistent with the results from ThT and CR assays and provide further evidence for β-sheet formation in PrP58–93 peptide upon interaction with Zn(II).
PrP58–93 upon interaction with Zn(II) forms structured fibrils with cross-β structure
To further investigate the effect of Zn(II) on PrP58–93 peptide aggregation and possible fibril formation, we performed also microscopic studies. Fig. 3a presents the AFM image of monomeric 10 μM PrP58–93 peptide adsorbed on the mica surface and dried at room temperature. The measured height of dried peptide monomers varies from 1.5 to 2.5 nm. After 24 h incubation of 10 μM PrP58–93 peptide with 40 μM ZnCl2, some spherical oligomers, as well as single peptide fibrils were visible (Fig. 3b). PrP58–93 fibrils had a well-defined shape and quite uniform height in the range 1.5 to 1.8 nm. Moreover, the AFM images suggested that the formed fibrils had lateral branches, directed at right angles from the main fibril axis. The shape of the spherical oligomers was not well defined and their height ranged from 5 to 16 nm. Interestingly, the spherical oligomers were connected with the ends of the fibrils and therefore, the observed spherical oligomers might be centers of fibril nucleation.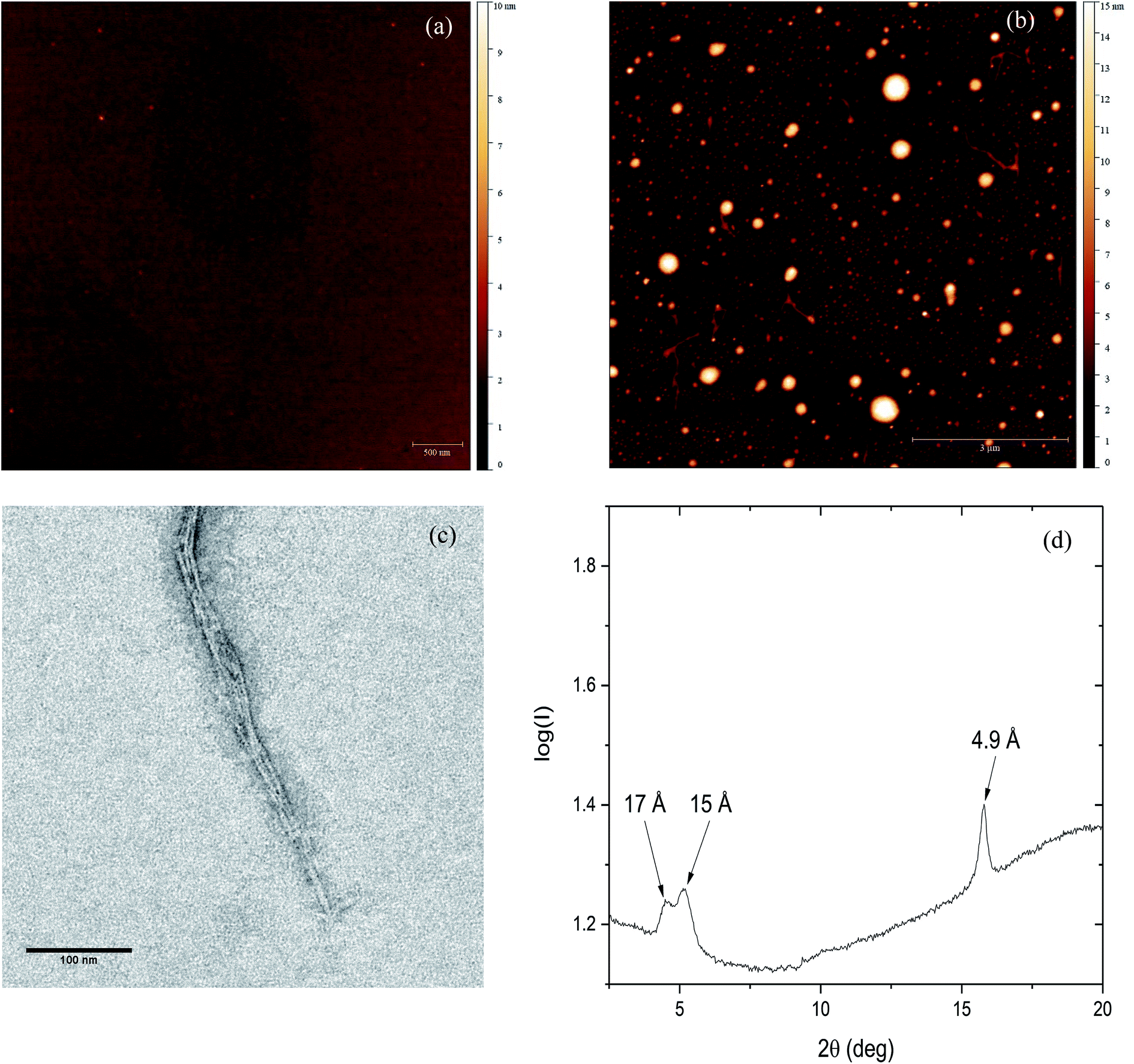 | ||
| Fig. 3 (a) AFM micrograph of monomeric PrP58–93 peptide. (b) AFM micrograph of formed PrP58–93 fibrils and oligomers in presence of ZnCl2. (c) TEM image of PrP58–93 fibrils formed in presence of ZnCl2. (d) X-ray diffraction pattern for PrP58–93 fibrils formed in presence of ZnCl2. Three reflections are visible: one meridional at 4.9 Å and two equatorial at ∼15 Å and ∼17 Å. | ||
The microstructure of PrP58–93 peptide fibrils with ZnCl2 was also verified by the transmission electron microscopy (TEM). The formed structures show a high tendency to cluster and form well-defined parallel bundles (Fig. 3c), nevertheless the diameters of individual fibrils could be assessed and ranged from 2.6 to 3 nm. Taking into account the grain size of uranyl formate (∼0.4 nm)56 the diameter of fibrils measured in TEM images is in good agreement with the height of single fibrils measured by AFM. Spherical oligomers were not visible in TEM micrographs probably because they could not be well stained with uranyl formate. One possible reason for that is a high content of random coil. Our FTIR spectra suggest that, upon the interaction between Zn(II) and PrP58–93, the content of random coil in peptide increases. Therefore, the spherical oligomers might be unstructured intermediates in fibril formation.
The cross-β structure of the formed peptide fibrils was confirmed by X-ray diffraction (Fig. 4d). The diffraction pattern shows an intense and sharp meridional reflection at 4.9 Å, that indicates a structural spacing of β-sheets along the fibril axis.8,57 Two weak and broad equatorial reflections at ∼15 Å and ∼17 Å are also visible (Fig. S4, ESI†). This result together with our previously presented spectroscopic and microscopic studies, indicates the existence of cross-β-structure for PrP58–93 fibrils formed in the presence of Zn(II).
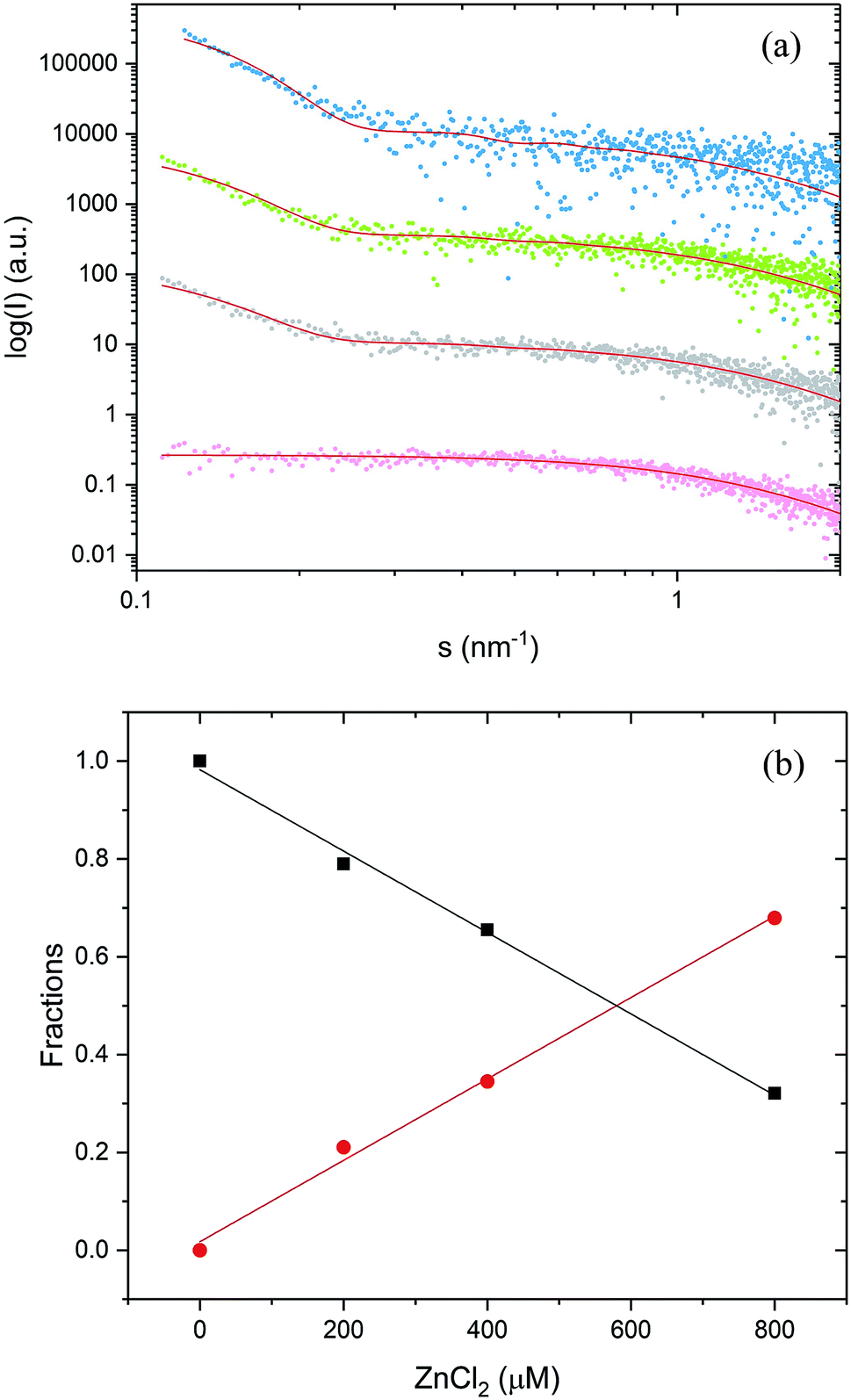 | ||
| Fig. 4 SAXS analysis of PrP58–93 peptide in solution. (a) SAXS data fitted in SASfit as a spheroid for 0.8 mM apo peptide (pink, MSWD 1.06) and as a mixture of spheroids and cylinders for 0.8 mM peptide with: 0.2 mM ZnCl2 (gray MSWD 1.14), 0.4 mM ZnCl2 (green, MSWD 1.09) and 0.8 mM ZnCl2 (blue, MSWD 1.16). SAXS curves were displaced along the vertical axis for clarity. (b) Fractions of monomeric PrP58–93 peptide (black squares) and fibrils (red dots) as a function of ZnCl2 concentration. | ||
Effect of Zn(II) on PrP58–93 peptide monomeric state
To gain structural information on the peptide structure in solution we subjected it to synchrotron radiation small angle X-ray scattering study. The initial apo PrP58–93 sample was monodisperse (DLS – Fig S5 and S6, ESI†). For PrP58–93 at a concentration of 0.8 mM, the peptide molecules were characterized by the radius of gyration (Rg) equal to 1.31 ± 0.03 nm (smaxRg < 1.3, fidelity 0.74; Fig. S7, ESI†). This value is significantly smaller than the theoretical Rg = 1.65 nm calculated for an intrinsically disordered protein of the same length,58 which suggests compactness and nonlinear shape of the peptide molecule. The shape of the calculated pair-distance distribution function suggests that the peptide molecule adopts a spherical shape with an elastic tail, characterized by Dmax = 4.3 nm (Fig. S8, ESI†).On the basis of our initial SAXS analysis, the behavior of PrP58–93 peptide seemed to be different than expected for an unstructured peptide. SAXS data alone can be used for a construction of a low-resolution model of a peptide structure59 therefore we performed molecular modeling with two different approaches. In the first approach, commonly used for structured proteins, molecular envelope of a peptide was reconstructed by ab initio bead-modeling in DAMMIN,60 and in the second, commonly used in the analysis of intrinsically disordered proteins,61 a large pool of random PrP58–93 peptide conformations was created and a final ensemble of models best matching the experimental data was chosen.62 Models generated by both approaches fitted well our data (Fig. S9, ESI,† χ2 = 1.013 and χ2 = 1.011 respectively). From the pool of random conformations, PrP58–93 peptide apparently adopted two equally distributed states: compact and extended. Even though each of the two states is highly dynamic the compact state seemed to form a loop and was similar to the NMR structure of PrP61–84 published by Zahn (PDB: 1OEI),63 where the extended conformation resembled the features of PPII helix (Fig. S10, ESI†).64 Both conformations could be superimposed with the calculated ab initio bead-model (Fig. S10, ESI†), which suggested that the molecular envelope may correspond to a volume available for all conformational states of PrP58–93.
Subsequent addition of ZnCl2 up to the equimolar concentration of the prion peptide (0.8 mM), resulted in an increase in the scattering intensity at the scattering vectors s < 0.3 nm−1, which can be attributed to peptide aggregation65 caused by zinc (Fig. 4a, gray, green and blue circles Fig. 4a). Because of the appearance of sample polydispersity after addition of ZnCl2, further analysis was performed in SASfit46 upon three assumptions: (1) the molecular shape of apo peptide can be simplified to a spheroid with the polar semi-axis = 2.86 nm, equatorial semi-axis = 0.98 nm and scattering length density = 0.33, (2) addition of Zn2+ results in the appearance of the second scattering component in SAXS curve and (3) only the number of scatters can change. The fitting procedure of the polydisperse SAXS curve is complex because both, the particle shape, as well as the particle size distribution of the second component can vary. The second scattering component was chosen arbitrarily as a cylinder of a radius 14.1 nm, length 36.7 nm, and scattering length density = 0.33, because the cylinder shape corresponds to the shape of fibrillar structures, observed by us in the TEM and AFM images. After careful model selection, scattering intensity weighted models were fitted well to experimental data (red lines Fig. 4a).
The proposed models fitted well our SAXS data with the highest MSWD = 1.16 for 0.8 mM PrP58–93 with 0.8 mM ZnCl2. Two-component analysis of SAXS data revealed that the addition of zinc into PrP58–93 sample causes a gradual decrease in the content of the monomer (spheroid) fraction in favor of the oligomer (cylinder) fraction (Fig. 4b). The reported dissociation constant for Zn(II)-octarepeat peptide complex differs from ∼0.4 μM to ∼200 μM,22,30 however in the experiment with 800 μM PrP58–93 concentration, the direct binding should give a linear response up to the stoichiometric point. In our SAXS experiments, the content of the monomer fraction changed linearly as a function of zinc ions concentration, which can be interpreted as a result of direct metal binding to the octarepeat peptide.
Many different metal ions modulate aggregation of amyloid peptides or proteins.66 Here we reported the discovery of amyloid-like structures formed by an isolated prion protein octarepeat domain PrP58–93. The fibrils formed from the unstructured PrP58–93 peptide in the presence of Zn2+ ions fulfill the fundamental criteria to be classified as amyloid structures: they form cross-β structure, bind two amyloid-specific dyes and form elongated structures, observed by AFM and TEM.5 Even though the outcome of our research is a model, we hypothesize, that such a phenomenon might occur in the synaptic cleft, where transient zinc concentration can reach millimolar values.36
The results of ThT, CR assays and SAXS experiments suggest heterogeneous primary nucleation of PrP58–93 peptide in the presence of Zn(II) cations.54 PrP58–93 starts to aggregate after the addition of zinc cations and the relative content of monomeric peptide drops gradually only in the presence of the metal ion. Nevertheless, branched fibrils visible in AFM micrographs indicate the existence of secondary nucleation.68 Spherical oligomers seem to be the centers of fibril nucleation, as fibrils appear to grow directly from them, nevertheless, they could be observed only in AFM images. The reason for the inability to observe these oligomers in TEM images is the apparent lack of structure, which can hamper staining with uranyl formate. Oligomers were also not visible on the SAXS curves, probably due to a concentration dependent shift of equilibrium towards fibrils rather than to oligomers.
On the basis of our data we propose a hypothetical scenario of PrP58–93 aggregation that requires several steps: (i) Zn2+ cation is bound by four deprotonated histidine residues of a single peptide molecule; (ii) direct binding induces structural rearrangements that are not available for peptide in the apo state; (iii) two newly formed Zn(II)-PrP58–93 species have low energetic barrier for nucleation and can form a dimer; (iv) elongation occurs by addition of other Zn(II)-PrP58–93 molecules as building bricks (but not apo PrP58–93 segments), (v) fibril termini contain solvent-exposed hydrophobic residues, therefore, in order to minimize energy, they can connect to already existing fibrils, forming perpendicular branches.
A similar heterogeneous primary nucleation has been observed for the four-repeat domain of Tau protein in the presence of heparin. In this case, two Tau protein molecules bound to the same heparin molecule can form aggregation-prone dimer and further fibril elongation occurs by the addition of monomers to the ends of the formed nucleation center.69 Our results are in contrast to the studies performed on the full-length mouse and human PrPC, in which the presence of Zn(II) cations inhibited the formation of amyloid fibrils.70,71 Nevertheless, these studies were performed under denaturing conditions, where different conformational states of the Zn(II)-occupied octarepeat domain are favored. Denaturing conditions may disrupt the coordination of Zn(II) by the octarepeat domain and therefore change the aggregation path. Other possible explanation of the formation of PrP58–93 fibrils in the presence of Zn2+ ions might be the lack of tertiary contact between the Zn(II)-occupied octarepeat domain and helices 2 and 3.40 Such a tertiary fold is altered in the E199K point mutant of mouse PrPC, which is responsible for familial Creutzfeldt–Jakob disease, and a lack of such interaction might be responsible for PrPC misfolding and formation of PrPSc.40
Similar studies have been also performed for amyloid β (Aβ), the peptide involved in the formation of amyloid aggregates in Alzheimer's disease. Dissociation constant reported for Zn(II)-Aβ40 is in low micromolar range,72,73 which is to some degree similar to the reported KD for PrPC,22,30,31 nevertheless, zinc seems to have a protective influence on Aβ aggregation. Stoichiometric concentrations of Zn(II) induced precipitation of Aβ oligomers,74,75 the form more cytotoxic than fibrillar structures.76 At sub-stoichiometric concentrations Zn(II) inhibited fibril formation77 and reduced toxicity in cultured primary hippocampal neurons.78 Prion protein was shown to interact with Aβ oligomers and fibrils,79–81 therefore it would be interesting to investigate Aβ fibrillization with PrP58–93 at different Zn(II) concentration.
The formation of fibrillar structures by various proteins or peptides accompanies the protein misfolding processes. Some of these proteins are characterized by the intrinsic flexibility of their three-dimensional structure. A good example of such flexible protein structure is human cystatin C (HCC) for which, in the native state, the domain swapping phenomenon, the formation of polymorphic structures, oligomers, fibrils, and aggregates have been observed.82–87
Conclusions
In our study we investigated the effect of Zn(II) on the aggregation of prion octarepeat peptide. Our results show that this monomeric peptide upon interaction with Zn(II) forms ThT and CR-specific fibrillar aggregates with cross-β structure. The formed fibrils fulfill the main criteria of classifications as amyloid fibrils. Moreover, we proposed a hypothetical model of Zn(II)-induced PrP58–93 aggregation, that might be verified by future kinetic or structural studies. Our results provide important clues on protein misfolding and Zn(II) homeostasis that are associated with prion diseases.PrP58–93 peptide fibrils formed are also a potential functional material.88 Therefore, from the bionanotechnological point of view, we can expect that this system could be very useful in the design of nanofibrils formed in the presence of divalent cations (e.g. Zn2+).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a research grant (2014/15/B/ST4/04839) from the National Science Centre (Poland). The synchrotron SAXS data were collected at beamline P12 operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany). We would like to thank the beamline staff for kind assistance in using the beamline. Authors thank Dr Barbara Peplinska from NanoBioMedical Centre (A. Mickiewicz University) for support in collection of TEM images.References
- N. Gour, P. C. Kanth, B. Koshti, V. Kshtriya, D. Shah, S. Patel, R. Agrawal-Rajput and M. K. Pandey, ACS Chem. Neurosci., 2019, 10, 1230–1239 CrossRef CAS PubMed.
- S. Shaham-Niv, P. Rehak, D. Zaguri, A. Levin, L. Adler-Abramovich, L. Vuković, P. Král and E. Gazit, Commun. Chem., 2018, 1, 25 CrossRef.
- W. Gospodarczyk and M. Kozak, RSC Adv., 2017, 7, 10973–10984 RSC.
- R. N. Rambaran and L. C. Serpell, Prion, 2008, 2, 112–117 CrossRef.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef.
- T. Stromer and L. C. Serpell, Microsc. Res. Tech., 2005, 67, 210–217 CrossRef CAS PubMed.
- M. Wahlbom, X. Wang, V. Lindström, E. Carlemalm, M. Jaskolski and A. Grubb, J. Biol. Chem., 2007, 282, 18318–18326 CrossRef CAS.
- A. Szymańska, E. Jankowska, M. Orlikowska, I. Behrendt, P. Czaplewska and S. Rodziewicz-Motowidło, Front. Mol. Neurosci., 2012, 5, 82 Search PubMed.
- P. Saá, D. A. Harris and L. Cervenakova, Expert Rev. Mol. Med., 2016, 18, e5 CrossRef PubMed.
- N. J. Cobb and W. K. Surewicz, Biochemistry, 2009, 48, 2574–2585 CrossRef CAS PubMed.
- D. W. Colby and S. B. Prusiner, Cold Spring Harbor Perspect. Biol., 2011, 3, a006833 Search PubMed.
- S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13363–13383 CrossRef CAS PubMed.
- N. Mabbott, Pathogens, 2017, 6, 60 CrossRef.
- N. Stahl, D. R. Borchelt, K. Hsiao and S. B. Prusiner, Cell, 1987, 51, 229–240 CrossRef CAS.
- N. Salès, K. Rodolfo, R. Hässig, B. Faucheux, L. Di Giamberardino and K. L. Moya, Eur. J. Neurosci., 1998, 10, 2464–2471 CrossRef.
- R. Zahn, A. Liu, T. Lührs, R. Riek, C. von Schroetter, F. López García, M. Billeter, L. Calzolai, G. Wider and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 145–150 CrossRef CAS.
- M.-A. Wulf, A. Senatore and A. Aguzzi, BMC Biol., 2017, 15, 34 CrossRef.
- A. R. Castle and A. C. Gill, Front. Mol. Biosci., 2017, 4, 19 Search PubMed.
- N. T. Watt and N. M. Hooper, Trends Biochem. Sci., 2003, 28, 406–410 CrossRef CAS.
- G. S. Jackson, I. Murray, L. L. Hosszu, N. Gibbs, J. P. Waltho, A. R. Clarke and J. Collinge, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8531–8535 CrossRef CAS.
- J. H. Viles, F. E. Cohen, S. B. Prusiner, D. B. Goodin, P. E. Wright and H. J. Dyson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2042–2047 CrossRef CAS.
- C. S. Burns, E. Aronoff-Spencer, C. M. Dunham, P. Lario, N. I. Avdievich, W. E. Antholine, M. M. Olmstead, A. Vrielink, G. J. Gerfen, J. Peisach, W. G. Scott and G. L. Millhauser, Biochemistry, 2002, 41, 3991–4001 CrossRef CAS.
- M. Chattopadhyay, E. D. Walter, D. J. Newell, P. J. Jackson, E. Aronoff-Spencer, J. Peisach, G. J. Gerfen, B. Bennett, W. E. Antholine and G. L. Millhauser, J. Am. Chem. Soc., 2005, 127, 12647–12656 CrossRef CAS.
- N. D. Younan, M. Klewpatinond, P. Davies, A. V. Ruban, D. R. Brown and J. H. Viles, J. Mol. Biol., 2011, 410, 369–382 CrossRef CAS.
- A. P. Garnett and J. H. Viles, J. Biol. Chem., 2003, 278, 6795–6802 CrossRef CAS.
- M. L. Kramer, H. D. Kratzin, B. Schmidt, A. Römer, O. Windl, S. Liemann, S. Hornemann and H. Kretzschmar, J. Biol. Chem., 2001, 276, 16711–16719 CrossRef CAS.
- M. A. Wells, C. Jelinska, L. L. P. Hosszu, C. J. Craven, A. R. Clarke, J. Collinge, J. P. Waltho and G. S. Jackson, Biochem. J., 2006, 400, 501–510 CrossRef CAS.
- E. D. Walter, D. J. Stevens, M. P. Visconte and G. L. Millhauser, J. Am. Chem. Soc., 2007, 129, 15440–15441 CrossRef CAS.
- P. Davies, F. Marken, S. Salter and D. R. Brown, Biochemistry, 2009, 48, 2610–2619 CrossRef CAS.
- B. L. Vallee and D. S. Auld, Biochemistry, 1990, 29, 5647–5659 CrossRef CAS.
- J. H. Weiss, S. L. Sensi and J. Y. Koh, Trends Pharmacol. Sci., 2000, 21, 395–401 CrossRef CAS.
- S. Y. Assaf and S. H. Chung, Nature, 1984, 308, 734–736 CrossRef CAS.
- S. Lehmann, Curr. Opin. Chem. Biol., 2002, 6, 187–192 CrossRef CAS.
- N. T. Watt, H. H. Griffiths and N. M. Hooper, Prion, 2013, 7, 203–208 CrossRef CAS.
- N. T. Watt, D. R. Taylor, T. L. Kerrigan, H. H. Griffiths, J. V. Rushworth, I. J. Whitehouse and N. M. Hooper, Nat. Commun., 2012, 3, 1134 CrossRef.
- L. R. Brown and D. A. Harris, J. Neurochem., 2003, 87, 353–363 CrossRef CAS PubMed.
- P. C. Pauly and D. A. Harris, J. Biol. Chem., 1998, 273, 33107–33110 CrossRef CAS PubMed.
- A. R. Spevacek, E. G. B. Evans, J. L. Miller, H. C. Meyer, J. G. Pelton and G. L. Millhauser, Structure, 2013, 21, 236–246 CrossRef CAS.
- B. S. Wong, D. R. Brown, T. Pan, M. Whiteman, T. Liu, X. Bu, R. Li, P. Gambetti, J. Olesik, R. Rubenstein and M. S. Sy, J. Neurochem., 2001, 79, 689–698 CrossRef CAS.
- M. R. Wilkins, E. Gasteiger, A. Bairoch, J. C. Sanchez, K. L. Williams, R. D. Appel and D. F. Hochstrasser, Methods Mol. Biol., 1999, 112, 531–552 CAS.
- W. E. Klunk, R. F. Jacob and R. P. Mason, Methods Enzymol., 1999, 309, 285–305 CAS.
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS.
- D. Nečas and P. Klapetek, Cent. Eur. J. Phys., 2012, 10, 181–188 Search PubMed.
- C. E. Blanchet, A. Spilotros, F. Schwemmer, M. A. Graewert, A. Kikhney, C. M. Jeffries, D. Franke, D. Mark, R. Zengerle, F. Cipriani, S. Fiedler, M. Roessle and D. I. Svergun, J. Appl. Crystallogr., 2015, 48, 431–443 CrossRef CAS.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- D. Franke, M. V. Petoukhov, P. V. Konarev, A. Panjkovich, A. Tuukkanen, H. D. T. Mertens, A. G. Kikhney, N. R. Hajizadeh, J. M. Franklin, C. M. Jeffries and D. I. Svergun, J. Appl. Crystallogr., 2017, 50, 1212–1225 CrossRef CAS.
- D. I. Svergun, J. Appl. Crystallogr., 1992, 25, 495–503 CrossRef CAS.
- I. Breßler, J. Kohlbrecher and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 1587–1598 CrossRef.
- C. J. Smith, A. F. Drake, B. A. Banfield, G. B. Bloomberg, M. S. Palmer, A. R. Clarke and J. Collinge, FEBS Lett., 1997, 405, 378–384 CrossRef CAS.
- G. Di Natale, G. Pappalardo, D. Milardi, M. F. M. Sciacca, F. Attanasio, D. La Mendola and E. Rizzarelli, J. Phys. Chem. B, 2010, 114, 13830–13838 CrossRef CAS.
- R. Khurana, C. Coleman, C. Ionescu-Zanetti, S. A. Carter, V. Krishna, R. K. Grover, R. Roy and S. Singh, J. Struct. Biol., 2005, 151, 229–238 CrossRef CAS.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1405–1412 CrossRef CAS PubMed.
- M. A. Bryan, J. W. Brauner, G. Anderle, C. R. Flach, B. Brodsky and R. Mendelsohn, J. Am. Chem. Soc., 2007, 129, 7877–7884 CrossRef CAS.
- C. A. Scarff, M. J. G. Fuller, R. F. Thompson and M. G. Iadaza, J. Visualized Exp., 2018, 132, 57199 Search PubMed.
- K. L. Morris and L. C. Serpell, Methods Mol. Biol., 2012, 849, 121–135 CrossRef CAS.
- P. Bernadó and D. I. Svergun, Mol. BioSyst., 2012, 8, 151–167 RSC.
- H. D. T. Mertens and D. I. Svergun, J. Struct. Biol., 2010, 172, 128–141 CrossRef CAS.
- D. I. Svergun, Biophys. J., 1999, 76, 2879–2886 CrossRef CAS.
- A. G. Kikhney and D. I. Svergun, FEBS Lett., 2015, 589, 2570–2577 CrossRef CAS.
- G. Tria, H. D. T. Mertens, M. Kachala and D. I. Svergun, IUCrJ, 2015, 2, 207–217 CrossRef CAS.
- R. Zahn, J. Mol. Biol., 2003, 334, 477–488 CrossRef CAS.
- A. A. Adzhubei, M. J. E. Sternberg and A. A. Makarov, J. Mol. Biol., 2013, 425, 2100–2132 CrossRef CAS.
- S. Skou, R. E. Gillilan and N. Ando, Nat. Protoc., 2014, 9, 1727–1739 CrossRef CAS.
- L. Breydo, J. M. Redington and V. N. Uversky, Int. Rev. Cell Mol. Biol., 2017, 329, 145–185 CAS.
- A. K. Buell, Int. Rev. Cell Mol. Biol., 2017, 329, 187–226 Search PubMed.
- S. Linse, Pure Appl. Chem., 2019, 91, 211–229 CAS.
- G. Ramachandran and J. B. Udgaonkar, J. Biol. Chem., 2011, 286, 38948–38959 CrossRef CAS.
- O. V. Bocharova, L. Breydo, V. V. Salnikov and I. V. Baskakov, Biochemistry, 2005, 44, 6776–6787 CrossRef CAS.
- K. Pan, C.-W. Yi, J. Chen and Y. Liang, Biochim. Biophys. Acta, Proteins Proteomics, 2015, 1854, 907–918 CrossRef CAS.
- A. Clements, D. Allsop, D. M. Walsh and C. H. Williams, J. Neurochem., 1996, 66, 740–747 CrossRef CAS.
- J. Danielsson, R. Pierattelli, L. Banci and A. Gräslund, FEBS J., 2007, 274, 46–59 CrossRef CAS PubMed.
- K. Garai, B. Sahoo, S. K. Kaushalya, R. Desai and S. Maiti, Biochemistry, 2007, 46, 10655–10663 CrossRef CAS.
- K. Garai, P. Sengupta, B. Sahoo and S. Maiti, Biochem. Biophys. Res. Commun., 2006, 345, 210–215 CrossRef CAS.
- M. Sakono and T. Zako, FEBS J., 2010, 277, 1348–1358 CrossRef CAS.
- A. Abelein, A. Gräslund and J. Danielsson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5407–5412 CrossRef CAS.
- M. A. Lovell, C. Xie and W. R. Markesbery, Brain Res., 1999, 823, 88–95 CrossRef CAS.
- K. Nieznanski, K. Surewicz, S. Chen, H. Nieznanska and W. K. Surewicz, ACS Chem. Neurosci., 2014, 5, 340–345 CrossRef CAS.
- J. Laurén, D. A. Gimbel, H. B. Nygaard, J. W. Gilbert and S. M. Strittmatter, Nature, 2009, 457, 1128–1132 CrossRef.
- D. A. Gimbel, H. B. Nygaard, E. E. Coffey, E. C. Gunther, J. Lauren, Z. A. Gimbel and S. M. Strittmatter, J. Neurosci., 2010, 30, 6367–6374 CrossRef CAS.
- R. Janowski, M. Kozak, E. Jankowska, Z. Grzonka, A. Grubb, M. Abrahamson and M. Jaskolski, Nat. Struct. Biol., 2001, 8, 316–320 CrossRef CAS PubMed.
- M. Kozak, E. Jankowska, R. Janowski, Z. Grzonka, A. Grubb, M. Alvarez Fernandez, M. Abrahamson and M. Jaskolski, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1939–1942 CrossRef CAS.
- R. Janowski, M. Kozak, M. Abrahamson, A. Grubb and M. Jaskolski, Proteins: Struct., Funct., Bioinf., 2005, 61, 570–578 CrossRef CAS.
- G. Östner, V. Lindström, P. Hjort Christensen, M. Kozak, M. Abrahamson and A. Grubb, J. Biol. Chem., 2013, 288, 16438–16450 CrossRef.
- M. Chrabąszczewska, M. Maszota-Zieleniak, Z. Pietralik, M. Taube, S. Rodziewicz-Motowidło, A. Szymańska, K. Szutkowski, D. Clemens, A. Grubb and M. Kozak, J. Appl. Phys., 2018, 123, 174701 CrossRef.
- M. Taube, Z. Pietralik, A. Szymanska, K. Szutkowski, D. Clemens, A. Grubb and M. Kozak, Sci. Rep., 2019, 9, 8548 CrossRef.
- C. Li, R. Qin, R. Liu, S. Miao and P. Yang, Biomater. Sci., 2018, 6, 462–472 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01510h |
| This journal is © The Royal Society of Chemistry 2019 |