
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase-specific bioactivity and altered Ostwald ripening pathways of calcium carbonate polymorphs in simulated body fluid†
Barbara Myszkaa,
Martina Schüßlerb,
Katrin Hurlec,
Benedikt Demmertb,
Rainer Detscha,
Aldo R. Boccaccini ad and
Stephan E. Wolf*bd
ad and
Stephan E. Wolf*bd
aInstitute of Biomaterials, Friedrich-Alexander-University of Erlangen-Nuremberg, Cauerstrasse 6, 91058 Erlangen, Germany
bInstitute for Glass and Ceramics, Friedrich-Alexander-University Erlangen-Nuremberg, Martensstraße 5, 91058 Erlangen, Germany. E-mail: stephan.e.wolf@fau.de
cGeoZentrum Nordbayern – Mineralogy, Friedrich-Alexander-University Erlangen-Nuremberg, Schlossgarten 5a, 91054 Erlangen, Germany
dInterdisciplinary Center for Functional Particle Systems (FPS), Friedrich-Alexander-University Erlangen-Nuremberg, 91058 Erlangen, Germany
First published on 24th June 2019
Abstract
Calcium carbonate is an abundant biomineral, and already archeological records demonstrate its bioactivity and applicability for osseo-integrative implants. Its solubility, which is generally higher than those of calcium phosphates, depends on its polymorph turning calcium carbonate into a promising biomaterial with tunable bioresorption rate. However, the phase-dependent bioactivity of calcium carbonate, i.e., its osteoconductivity, is still insufficiently characterized. In this study, we address this issue by monitoring the behavior of the four most important calcium carbonate phases, i.e., calcite, aragonite, vaterite, and amorphous calcium carbonate, in simulated body fluid solution at 37 °C. Our results demonstrate that the thermodynamically stable calcite phase is essentially inert. In contrast, the metastable phases aragonite and vaterite are bioactive, thus promoting the formation of calcium phosphate. Amorphous calcium carbonate (ACC) shows prominent bioactivity accompanied by pronounced redissolution processes. Mg-stabilized ACC was additionally tested since its increased stability eases formulation and handling in future applications. It is highly bioactive and, moreover, the additional release of Mg promotes cell viability. Overall, our results demonstrate that bioactivity of calcium carbonate is phase-dependent, allowing tailored response and bioactivity of future calcareous biomaterials. Our results also reveal that phosphate ions strongly interfere with Ostwald–Lussac step ripening of calcium carbonate, kinetically stabilizing metastable polymorphs such as vaterite and aragonite; this is a distinctive feature of the calcium carbonate mineral system which clearly has to be considered in future applications of calcium carbonate as a bioceramic.
Introduction
Calcium carbonate is an abundant biomineral able to form numerous morphologies and, as sediment, generates large geological deposits. Its three anhydrous and crystalline polymorphs predominate the bio- and geosphere. Beside rhombohedral calcite, orthorhombic aragonite, and hexagonal vaterite, an amorphous and unstable calcium carbonate (ACC) phase exists. Serving as a transient precursor phase to a crystalline material, ACC plays a dominant role in both biomineralization and synthetic crystallization processes, especially when crystallization inhibitors come into play.1–3 The presence of such crystallization inhibitors can trigger a range of so-called nonclassical pathways which allow the synthesis of a wide range of non-equilibrium morphologies, such as thin films on substrates or allow for the incorporation of organic matrices and foreign ions. Although calcium carbonate is a key model system for understanding mineral formation, featuring chemical distinctness which offers unparalleled synthetic and morpho-synthetic freedom, the use of calcium carbonate in biomedical applications is yet rare.It is thus surprising that already archaeological records demonstrate that calcium carbonate is bioactive and osseointegrative. The first evidence of the successful exploitation of a calcareous material in vivo was found in 1931 at Playa de los Muertos in the Ulloa valley, Honduras. The lower jaw of a Mayan skull, probably belonging to a female 20 years of age, bore three implants made from biogenic nacre. In 1972, Bobbio et al. evidenced by radiographic analysis of this peculiar ancient dental prosthesis that these implants were inserted ante mortem as compact bone formation and firm osseointegration could be demonstrated.4 Later work has shown that nacre indeed triggers calcium phosphate precipitation in simulated body fluids,5 and that nacre is able to induce bone formation by human osteoblasts in vitro.6 Subsequently, it was shown that aragonite is bioresorbable and that aragonite and other calcium carbonate polymorphs can serve as a drug release system.7–13 Despite the widespread occurrence of calcium carbonate in the biosphere, e.g., in human otoliths, it seems that the full potential of the calcium carbonate system is not yet tapped for the purposeful design of bioactive and bioresorbable biomaterials/bioceramics. Recent research also rose our awareness that calcium carbonate formation contributes to the bioactivity of bioglass.14
In fact, calcium carbonate is rarely used today as a bio-resorbable, osseointegrative, bioactive biomaterial compared to other pertinent mineral-based biomaterials such as calcium phosphate-based systems or bioactive glasses. A well-received example of the exploitation of a calcareous mineral for the generation of a bioactive bioceramic is given by the conversion of aragonite corals into apatitic calcium phosphate, which was first demonstrated in the early seventies using high temperature and pressures acid and basic phosphates during pseudomorphic conversion.15,16 This triggered a smaller series of detailed investigation on the formation mechanisms and on the exploitation of coralline calcium carbonate for biomedical applications.17–23 These contributions evidenced that coralline calcium carbonate is a potential bone filling material with good osteoconductivity.20,24–27 However, the use of biogenic and thus pre-formed porous ceramic matrices comes at a price which made this general approach less attractive: they typically pose problems due to their meager mechanical integrity and load-bearing capabilities which arise due to their ungovernable porosity. This also makes contouring and matching defect size challenging.28,29 Naturally, the scientific contributions on coralline calcium carbonate and aragonite nacre rose the awareness of the community that calcium carbonate might be a bioceramic of interest. In recent reports exploiting calcium carbonate as a mineral component of biomaterials, vaterite is nearly exclusively used, mostly in combination with hybrid systems or polymeric substrates.30–34 For instance, hybrid thin films containing calcium carbonate promoted the recovery of osteoblasts viability better than untreated titania surfaces, and polycaprolactone composite nanofibers can serve as functional guided bone regeneration membranes.31,32 In stark contrast, the purposeful use of the other polymorphs of calcium carbonate beside vaterite, e.g., aragonite or calcite, is only scarcely documented in the literature.7,35,36
Besides its clear biocompatibility, already evidenced by calcareous biominerals even in humans and its long-time usage in various contexts such as food additives or drug formulations, the polymorphism of calcium carbonate may be one of the key attributes which could render this mineral system a very attractive bioceramic if its reactivity profile is properly chartered. The different mineral phases, i.e., calcite, aragonite, vaterite and amorphous calcium carbonate (ACC), all exhibit a higher solubility than, for instance, hydroxyapatite (HAP) which fundamentally renders these calcareous minerals bioavailable and thus bioresorbable. Moreover, the different mineral phases exhibit different solubilities which may allow for the development of calcareous bioresorbable materials with attunable resorption rates. All of the polymorphs of calcium carbonate are synthetically accessible by direct precipitation from aqueous solutions. At standard conditions, calcite is the stable phase, whereas aragonite is known to be the polymorph stable at elevated temperature and/or pressure. Thermodynamically, vaterite is the least stable phase of the crystalline calcium carbonate mineral phases.37 Beside the crystalline phases also amorphous phases are known to play a central role in biomineralization processes of calcareous species. Amorphous calcium carbonate is a highly metastable phase, which is often highly hydrated (<1.6 moles of water per mole of CaCO3) and, after dehydration, transforms quickly to calcite, aragonite, or vaterite.38,39 A strict application of the Ostwald–Lussac law of stages would predict that ACC should transform into a thermodynamically stable state by obeying the energetically downhill sequence ACC → vaterite → aragonite → calcite.38 However, the transformation of vaterite to aragonite is rarely observed, and pure ACC usually transforms to calcite via vaterite intermediate at low temperatures (<30 °C) and via aragonite at higher temperatures (>60 °C).40 It seems clear that heterogeneous nucleation events on the surface of vaterite particles triggers the omission of the metastable aragonite phase at standard conditions.39 Already the presence of small-weight organic additives or foreign ions (e.g., Mg2+, Sr2+, aspartic acid, or citric acid) can radically affect the crystallization rates and pathways of ACC,41 which makes calcium carbonate a complicated system in non-pure environments. Rather than being a drawback, this responsive behaviour of the calcium carbonate system opens up a multitude of potential process parameters to steer and control the mineralization and phase transformation processes converting the CaCO3/H2O system into a potentially finely attunable bioceramic material or bone-filling material with adjustable reactivity.
In contrast to the yet untapped potential outlined above, the differences in bioactivity, i.e., its osteoconductivity, and phase transformation behaviour of the different calcium carbonate polymorphs were never addressed in the context of a potential usage as a bioceramic. Already the change in solubility and reactivity of calcium carbonate as a function of its mineral phase suggests that calcium carbonate polymorphs should behave distinctly different when subjected to in vivo conditions.
In this study, we map out the phase transformation and ripening processes of calcium carbonate powders in simulated body fluid (SBF, prepared according to Kokubo et al.42) in order to assess the potential bioactivity and reaction profiles for future exploitation of calcium carbonate as a biomaterial, be it in all-calcareous systems or as a minor/major component in biomaterial preparations. We demonstrate that the behaviour and bioactivity, with respect to biomimetic SBF solutions, is clearly polymorph-specific: calcite is relatively inert concerning CaP precipitation whereas all other calcium carbonate polymorphs show bioactivity. The bioactivity of the calcium carbonate polymorphs clearly scales with their solubility. The ripening behaviour in SBF of the investigated calcium carbonate phases is seemingly quite unexpected as the presence of phosphate ions in the solution can strongly suppress Ostwald–Lussac step ripening. This leads to the prolonged phase stability of the otherwise meta- and unstable mineral phases, e.g., aragonite and vaterite.
Materials and methods
Synthesis of phase-pure calcium carbonate polymorphs
Calcite at an analytical grade (purity > 99%) was purchased from Sigma-Aldrich (St. Louis, USA). Calcium carbonate in the form of aragonite, vaterite, or amorphous calcium carbonate is metastable and is not available commercially in sufficient purity; geological deposits are also impure and contain a wide and unpredictable range of foreign ions and organic inclusions.Phase-pure aragonite was prepared by a double decomposition by mixing solutions of calcium nitrate and sodium carbonate (purity > 99%; Sigma-Aldrich, St. Louis, USA) at elevated temperature. Both salts were dissolved in ultrapure water (Merck Milli-Q Direct 8 with UV photooxidation, 18.2 MΩ cm−1) at 0.1 mol L−1 and 1.0 mol L−1 concentration, respectively. The experiment was conducted at 75 °C, under a constant agitation speed of 400 rpm. Subsequently, 20 mL of calcium nitrate was added into 200 mL sodium carbonate at a rate of 5 mL min−1. Upon completed addition, the reaction was stirred further for additional 11 minutes to allow for ripening, yielding a total reaction time of 15 min. While the pH of initial sodium carbonate solution was 10.7 at 75 °C, it decreased to 7.2 during precipitation (HI-98140 Hannah Instruments, Vöhringen, Germany). The white precipitate was retrieved by vacuum filtration, thoroughly washed three times with ultrapure water, and was subsequently washed once with ethanol, then dried in a desiccator.
Phase-pure vaterite was prepared by double decomposition of calcium chloride and sodium carbonate at room temperature. For this, solutions with a concentration of 1.0 mol L−1 of sodium carbonate (purity > 99%; Sigma-Aldrich, St. Louis, USA) and of calcium chloride (purity > 99%; Fluka Analytical, Bucharest, Romania) were prepared. Under stirring at 400 rpm, 50 mL of 1.0 mol L−1 calcium chloride solution was added dropwise with a rate of 3.33 mL min−1 to 50 mL of 1.0 mol L−1 sodium carbonate at room temperature. The white precipitate was collected by vacuum filtration, thoroughly washed three times with ultrapure water, subsequently washed with ethanol, then dried in a desiccator. During precipitation, the pH of the solution dropped from 11.7 to 8.4 after completion of precipitation (HI-98140 Hannah Instruments, Vöhringen, Germany).
Phase-pure amorphous calcium carbonate (ACC) was prepared by double decomposition of calcium chloride and sodium carbonate at room temperature. For this, 25 mL of 40 mmol L−1 sodium carbonate was quickly added to 25 mL of 40 mmol L−1 calcium chloride under vigorous stirring at 800 rpm. After 5 seconds of reaction time, the precipitate was removed by vacuum filtration (MicronSep Nitrocellulose, 0.45 μm membrane disk, GVS Life Science, Findlay, USA). The powder was washed three times with dry ethanol and was then dried in a desiccator. During precipitation, the initial pH of 11.3 decreased to 10.2 (HI-98140 Hanna Instruments, Germany).
Phase-pure magnesium-stabilized ACC (Mg-ACC) was prepared by a direct-strike synthesis. First, a series of calcium chloride solutions with 10 wt%, 20 wt% and 40 wt% magnesium, in respect to the calcium content, was prepared by mixing 0.2 mol L−1 CaCl2 and 0.2 mol L−1 MgCl2 in the given proportions. The obtained solutions were rapidly mixed with 20 mL of 0.2 mol L−1 sodium carbonate solution under stirring at 750 rpm for 5 s, by adding sodium carbonate into the Ca/Mg solution. The suspension was filtered by vacuum filtration, washed with water and ethanol in sequence, and was dried at a temperature of 60 °C for 2 h. The incorporation of magnesium the ACC matrix was assured by inductively coupled plasma optical emission spectroscopy (Spectro Genesis FES, ICP-OES). For each ICP-OES measurement, 30 mg of powder was dissolved in nitric acid (1 mol L−1); every sample was measured at least in triplicate. Successful incorporation of Mg in the ACC matrix was assured, see Fig. D in the ESI.†
Incubation in simulated body fluid
Simulated body fluid (SBF) was prepared according to the protocol of Kokubo et al.;42 all reagents for SBF preparation were supplied by Sigma-Aldrich (St. Louis, Germany) or VWR (Radnor, USA) and the amount of the reagents was adjusted according to their purity. Each calcium carbonate polymorph was immersed in powder state as received from the synthesis given above; a constant ratio of 1.5 g L−1 between powder mass and SBF volume was obeyed. Samples were incubated in a rotational incubator (KS 400i control, IKA, Staufen, Germany) at 175 rpm at 37 °C temperature for varying times, from one day to 28 days. SBF solution was refreshed every three days. Upon completion, samples were collected and washed three times with 50 mL ethanol and centrifuged at 7000 rcf for 10 min (Centrifuge 5430, Eppendorf, Wesseling-Berzdorf, Germany). Finally, the samples were air-dried in a dust-protected container. As a control, samples were incubated in ultrapure water under otherwise identical conditions.Characterization of powders
X-ray diffraction (XRD) was carried out using a Siemens Kristalloflex D500 (Karlsruhe, Germany), equipped with a Cu Kα source; measurements were conducted over a 2θ range of 20° to 60° with a step size of 0.02° and dwell time 0.05 s. Phase analysis was accomplished using the software package MATCH! (Crystal Impact, Bonn, Germany). Fourier Transform IR (FTIR) spectra (IRAffinity-1S, Shimadzu, Japan, equipped with an ATR cell) were collected by gently grinding samples to ensure homogeneity. All spectra were obtained from 4000 to 400 cm−1; they are the average of two independent measurements with 32 scans each at a resolution of 4 cm−1. Raman spectroscopy analysis was done using a LabRAM HR800, Horiba spectrometer. Scanning electron microscopy (Carl Zeiss Auriga) was applied to follow the morphological evolution of powders; all samples were uncoated and observed at a working distance of 3.0 mm and an accelerating voltage of 1.0 kV. Simultaneous differential thermal analysis (DTA) and thermogravimetric analysis (TGA) were performed by means of an SDT 2960 DTA/TGA (TA Instruments, New Castle) using platinum crucibles; corundum powder was used as an inert reference. A heating rate of 5 °C min−1 was applied; samples were heated up to a maximum temperature of 1000 °C.Indirect cytotoxicity test of Mg-doped ACC
Cell culture experiments were performed by using a bone marrow stromal cell line (ST-2, Deutsche Sammlung für Mikroorganismen und Zellkultur, Germany). Cells were cultured in RPMO 1640 medium (Gibco, Germany) containing 10 vol% FBS (Sigma Aldrich, Germany) and 1 vol% penicillin/streptomycin (Sigma-Aldrich). ST-2 cells were seeded into 24-well plates in a concentration of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well (1 mL) for 24 hours. Simultaneously, 0.1 g granules were added to 10 mL culture medium (without cells) and incubated separately for 24 h at 37 °C. After that, the supernatant was extracted and diluted into different concentrations (0; 0.01; 0.1; 1 wt/vol) from all samples. The seeded ST-2 cells were washed with PBS, and the different suspensions of supernatant from the pre-incubated granules were transformed to the ST-2 cells for the next 48 h. The culture cells did not come in direct contact with the calcareous powder but with its release products.
000 cells per well (1 mL) for 24 hours. Simultaneously, 0.1 g granules were added to 10 mL culture medium (without cells) and incubated separately for 24 h at 37 °C. After that, the supernatant was extracted and diluted into different concentrations (0; 0.01; 0.1; 1 wt/vol) from all samples. The seeded ST-2 cells were washed with PBS, and the different suspensions of supernatant from the pre-incubated granules were transformed to the ST-2 cells for the next 48 h. The culture cells did not come in direct contact with the calcareous powder but with its release products.
Results
Synthesis and characterization of phase-pure calcium carbonate polymorphs
To assess the interaction of individual calcium carbonate phases with SBF solutions and to determine the phase-dependent impact on bioactivity, phase-pure synthetic calcite, aragonite, vaterite and amorphous calcium carbonate (ACC) were required at a high chemical purity since inorganic and organic impurities can strongly impact on crystallization and phase transformation behavior of calcium carbonate. Phase- and chemically pure calcite, the thermodynamically stable calcium carbonate phase, is commercially available. The purchased calcite is phase-pure, as demonstrated by XRD, FTIR, and Raman spectroscopy; and additional TGA/DSC analysis assured that no organic foreign material or polymorph was present (see Section A on calcite provided in the ESI†). Scanning electron microscopy revealed the classic and expected rhombohedral crystals of calcite (Fig. 1); their typical size is about 3 μm, and they show a minor number of stepped features on their crystal facets. The calcite crystals were not agglomerated, and the edges were well pronounced and sharp. The metastable phases of aragonite and vaterite are not commercially available, so both had to be synthesized by double decomposition using a reverse strike technique. Phase-purity was assessed by XRD, FTIR, and Raman; TGA/DSC assured that no inorganic components or second mineral phases were present (see Section B on aragonite and Section C on vaterite provided in the ESI†). Aragonite was obtained in a rod-like morphology, with a typical crystal size of 5–14 μm in length and 700–900 nm in width (Fig. 1). These rod-like structures were composed of smaller building units, i.e., microscale rods which assembled into structures which are reminiscent of mesocrystals reported by Cölfen and Antonietti.43 Zhou et al. synthesized aragonite rods with similar morphologies at comparable reaction conditions, i.e., by mixing calcium chloride with sodium carbonate at 95 °C.44 Phase-pure vaterite was obtained in form of polycrystalline and ellipsoidal aggregates which are built from even smaller nanocrystals with a mean length of 200 nm (Fig. 1). Finally, phase-pure amorphous calcium carbonate was also synthesized by a reverse-strike double-decomposition technique which yielded spheroidal and aggregated nanoparticles ranging in size from 120 to 300 nm (Fig. 1) which were shown to be X-ray amorphous (see Fig. D1-A in Section D on ACC provided in the ESI†). FTIR and Raman analyses further corroborated the absence of a crystalline phase; TGA/DTA assured that no foreign organic material was present and underlined the highly hydrated state of the synthesized ACC (see Section D on ACC provided in the ESI†). | ||
| Fig. 1 Scanning electron micrographs of the phase-pure calcium carbonate starting materials before incubation. | ||
Behaviour of calcium carbonate polymorphs exposed to simulated body fluid or water
In order to properly assess the distinct behavior of calcium carbonate polymorphs exposed to simulated body fluid, a control reaction performed in ultrapure water is needed by which standard crystal ripening processes, such as Ostwald–Lussac step ripening or Ostwald ripening due to the presence of fluid, can be identified. The Ostwald–Lussac rule of stages describes the typical behavior of a polymorphic system undergoing a series of phase transformations. Such systems are expected to transform into the thermodynamically stable phase via intermediate and transient stages of less stable polymorphs. From a homogenous solution, the less stable polymorph is expected to form first, more precisely at the highest rate, and then transforms into the next stable mineral phase, essentially feeding the growth of the subsequent polymorph. The direct transformation to the stable mineral phase (or to an intermediate kinetic product) is essentially possible, but the transformation rates are predicted to be distinctly lower due to a higher activation barrier of the direct pathway. The predictive power of the simplifying Ostwald–Lussac rule can already be seen in the synthesis protocols of the different calcium carbonate polymorphs whose conditions essentially reflect the step rule behavior. Elevated temperatures are needed to turn aragonite into the thermodynamically stable polymorph. Ostwald ripening describes the typical behavior of a dispersion in which smaller particles feed the growth of larger ones; the thermodynamic driving force of this coarsening process is essentially based on the reduction of interfacial energy and the relation of the surface tension of an interface to its curvature, given by the Ostwald–Freundlich equation. Overall a net mass flow is observed from particles with higher surface/volume ratio to particles with smaller surface/volume ratio of the same polymorphic phase; particles below the average will shrink or simply dissolve.Calcite changes its crystal morphology slightly upon immersion in water. The crystals still have well-defined edges, and the facets smoothen, but the crystals overall grow in size and start to agglomerate, either by accretion of touching crystals or by re-nucleation of calcite and growth on the already existing calcite crystals (Fig. 2-A). As calcite is the stable thermodynamic phase under the given conditions,45 no change in the polymorph composition is observed, as documented by XRD (see Fig. A2-A in Section A provided in the ESI†).
 | ||
| Fig. 2 Morphological evolution of calcite during incubation (A) in water and (B) in SBF, both at 37 °C. Calcite is stable in water and SBF. In SBF, no formation of calcium phosphate precipitates is observed even after an extended incubation period of 28 d. | ||
When immersed in SBF, the overall behaviour of calcite remains unaltered (Fig. 2-B). No deposition of calcium phosphate was observed, neither by XRD of the bulk (see Fig. A2-B in the ESI†) nor by visual inspection by scanning electron micrographs (Fig. 2-B). The absence of calcium phosphate was also corroborated by the lack of the characteristic phosphate bands at 1030 cm−1 in FTIR spectra (see Fig. A3 in the ESI†). This allows the conclusion that calcite is essentially inert against calcium phosphate deposition under SBF-conditions; even incubation for 28 d produced no calcium phosphate. Thus, in SBF solutions, calcite merely undergoes Ostwald ripening/growth processes identical to those observed in pure water.
Aragonite, upon immersion in water, starts to transform into the thermodynamically stable polymorph calcite slowly; the process is slow and not completed within 28 d of incubation. The phase transformation was evidenced by XRD analysis and FTIR (see Fig. B2-A and B3-A in Section B provided in the ESI†), and minor peaks of calcite appeared already after one day of immersion. Rietveld refinement gave a phase composition of 18 wt% of calcite and 82 wt% of aragonite after 28 d; a full list of refinements is given in Table B.4 in Section B (provided in the ESI†) which documents the slow phase transformation process. Apparently, calcite nucleates preferentially at the crystal surface or at surface defects of aragonite, e.g., at cleavages, fractures, and twin boundaries (Fig. 3-A).
 | ||
| Fig. 3 Morphological evolution of aragonite during incubation (A) in water and (B) in SBF, both at 37 °C. In water, the transformation to calcite takes more than 28 d to complete. In contrast, no calcite can be found in case of incubation in SBF, but calcium phosphate precipitates are present already after one day (marked with a red cross, the respective EDS given in Fig. B2-C in Section B on aragonite provided in the ESI†). | ||
In contrast to the behavior of aragonite in water, aragonite immersed into SBF at 37 °C for 1–28 days (Fig. 3-B) did not display any noticeable change in particle morphology from those before immersion in water. The aragonite precipitates showed a negligible change in their morphology after the first day of incubation, as some few spherical agglomerates have additionally formed on aragonite crystals. Such spheroidal aggregates composed of plate-like nanocrystals are a typical morphology of calcium phosphate.33 This interpretation is supported by EDS analysis, which demonstrated that these agglomerates are indeed composed of C, O, P, Ca (see Fig. B2-C in the ESI†). XRD analysis revealed that peaks from aragonite are present in the first 21 days; after 28 days of incubation, additional reflections attributed to calcium phosphate appear (see Fig. B2-B†). FTIR analysis corroborated these findings (see Fig. B3-B†); until 21 days of immersion, only bands characteristic of aragonite was present. However, the formation of calcium phosphate after 28 days produced absorption bands at 1029, 601 and 563 cm−1.46 It should be noted that CaP spheroids can be already spotted in SEM and EDS analysis well before 28 d of incubation; before it is undetectable by XRD, potentially because the formed calcium phosphate is amorphous or cryptocrystalline. It is also untraceable by FTIR; probably because its volume fraction is under the detection limit. Overall, these results clearly indicate that in SBF solution, Ostwald–Lussac step ripening of aragonite, which would result in the formation of calcite, is suppressed and, in contrast to calcite, aragonite allows for calcium phosphate formation, apparently heterogeneous nucleation.
Vaterite is thermodynamically metastable and, accordingly, the vaterite crystals undergo a fast transformation and re-dissolution after immersion in water (Fig. 4-A). Already after one day of immersion, rhombohedral crystals of calcite can be spotted. They accompany needle-like elongate crystals which are, according to XRD (see Fig. C2-A in the ESI†), probably the left-overs of the partially dissolved vaterite bundles. After 3 days of incubation, the elongated crystals are only sporadically present and disappear after 7 days. From day 7 on, only rhombohedral crystals of calcite are present, which then grew further in the course of the immersion time and develop their equilibrium habit after 21 days. Already after one day of immersion, reflections of calcite accompany those of vaterite (see Fig. C2-A†); at later stages, only calcite reflections are present. This is corroborated by FTIR analysis (Fig. C2-A†).
 | ||
| Fig. 4 Morphological evolution of vaterite during incubation (A) in water and (B) in SBF, both at 37 °C. Within one day, calcium precipitates nucleate on the vaterite particles (marked with a red cross) when incubated in SBF. | ||
Oppositely to these observations made in water, vaterite particles immersed into SBF at 37 °C did not display any clear change in particle morphology (Fig. 4-B). However, already after a single day of incubation, a few spheroidal agglomerates have been formed which, as determined by EDS analysis (see Fig. C2-C,† the characterized particle is marked in Fig. 4-B with a red cross), are composed of calcium phosphate.33 After 21 days, FTIR spectra corroborated the presence of phosphate since bands at 1029, 601 and 563 cm−1 appear which are attributed to calcium phosphate formation (see Fig. C3†).46 Probably due to the low crystallinity and relative low quantity of calcium phosphate, XRD reflection of calcium phosphate are not observed before 28 days (Fig. C1-B†). Most remarkably and quite similar to the aragonite case, XRD patterns show no other reflections beside those of vaterite until 21 days of incubation have passed by (see Fig. C2-B†). After 28 days, first reflections appear, which can be attributed to calcium phosphate. In contrast to the control experiments in water, in which vaterite already transforms after 3 d, no calcite can be found in case of incubation of vaterite in SBF. This underlines that vaterite did not transform to calcite even after 28 days of immersion in SBF at 37 °C. This again demonstrates that in SBF solutions, the Ostwald–Lussac rule of stages is not in effect for metastable calcium carbonate polymorphs; it seems like redissolution of calcium carbonate is suppressed allowing preservation of even metastable phases for long(er) periods of time. Similar to aragonite and in contrast to calcite, vaterite triggers calcium phosphate formation and, actually, in an even more pronounced fashion as it can be traced earlier and forms in larger amounts, according to the signal intensities.
Amorphous calcium carbonate (ACC), when brought into contact with water, transforms into calcite virtually immediately. This expected behaviour of ACC is demonstrated here both by XRD and FTIR analysis and scanning electron micrographs after 1 d of incubation (Fig. 5-A, see also Fig. D2-A in Section D provided in the ESI†). The morphology of calcite, whose growth is fed by the dissolving ACC, is very akin to those observed in case of calcite as a starting material.
 | ||
| Fig. 5 Morphological evolution of amorphous calcium carbonate (ACC) during in incubation (A) in water and (B) in SBF, both at 37 °C. | ||
In stark contrast to this, the transformation of ACC in SBF solution does not yield rhombohedral calcite; no rhombohedral or even no facetted crystals at all can be spotted, they are absent even after 28 days of incubation. Instead, large agglomerations of nano-sized spheres can be identified (Fig. 5-B). After 14 days of immersion, the precipitate develops morphologies best described as large but hollow spheres. EDS analysis clearly evidences the presence of phosphate in all of these structures, especially in the globular halo formed after 6 h of incubation (see Fig. D4 in the ESI†). Seemingly in contradiction with the observations made by SEM, XRD diffractograms reveal that ACC rapidly transforms via vaterite to calcite in a multistep process (see Fig. D2-B†), a small amount of vaterite is still present until up to 4 days of immersion. In line with these results, FTIR also suggests a cascade of transformations (see Fig. D3†), showing bands characteristic of calcite and vaterite in the first seven days. Additionally, bands which are associated with phosphate appear, i.e., anti-symmetric stretch vibration (ν3) at 1029 cm−1 and in-plane bending (ν4) at 601 and 563 cm−1, respectively. Moreover, the broad absorption bands between 3000 and 3600 cm−1 and the sharper band at 1632 cm−1 (O–H stretching), which are characteristic for ACC, disappear already after one day of immersion. In contrast to these analyses, a crystalline calcium phosphate phase was undetectable by X-ray diffraction, possibly because the forming calcium phosphate is low in crystallinity, cryptocrystalline or simply of amorphous character.47 The hollow-sphere morphology suggests that a thin coat of calcium phosphate forms on the transient ACC (or vaterite) spheres which act as sacrificial templates for the hollow sphere by dissolving in the later stages of the mineralization process. This intermediate stage can be well seen in Fig. 5-B, and also in Fig. D4 in Section D (provided in the ESI†), which show large calcium carbonate spheres which are already in the progress of sacrificial dissolution and carries a halo of calcium phosphate precipitates.
In order to corroborate this assumption and to check the role of calcium phosphate in the recrystallization of ACC, the test was repeated with simulated body fluid at a concentration tenfold increased. This modified simulated body fluid (mSBF) is, in comparison with the original SBF proposed by Kokubo et al., more stable due to the increased bicarbonate concentration so that no spontaneous precipitation of calcium phosphate is noticeable.48 After incubation of ACC in mSBF, the morphology of the particles was very similar to the one immersed into Kokubo's SBF. SEM micrographs show large agglomeration of nanosized spheres. The spheres are also initially covered with a thin layer composed of spherical particles which later is replaced by relatively large hollow spheres. In comparison to those observed in Kokubo's SBF, the CaP coating is much more pronounced, and the hollow spheres are better developed, the diameter of their mantle is distinctly thicker (see Fig. D5†). EDS analyses performed at various places on these agglomerates and after varying periods of incubation clearly indicated calcium phosphate as a component (see Fig. D5†). FTIR analysis gave already after one day of incubation the characteristic absorption bands of phosphate (see Fig. D6†). X-ray diffraction showed that, after one day, ACC recrystallized to vaterite and calcite, whereby vaterite reflections dominate in the first days and are no longer present after 3 days of incubation (see Fig. D6†). This contrasts with the experiment with the SBF solution; here, vaterite was still present until 14 days of incubation which essentially demonstrates that the reaction/supersaturation rates are increased in mSBF in comparison to Kokubo's SBF. Overall, the pronounced and more developed morphologies and, along with this, the specifically increased presence of CaP suggest that calcium phosphate formation plays a significant role in the transformation of amorphous calcium carbonate, creating a thin layer of calcium phosphate on the dissolving vaterite particles.
Mg-stabilized amorphous calcium carbonate (Mg-ACC) shows a reduced reactivity in contrast to pure, non-doped ACC. The extreme reactivity of non-doped ACC may pose a severe problem when ACC shall be used as a component in a formulation, e.g., a cement. We, therefore, extended our study to this ACC variant stabilized by the addition of magnesium ions during the precipitation. Magnesium is well-documented to stabilize an amorphous state of calcium carbonate, and biogenic calcium carbonate is typically remarkably rich in Mg. A series of Mg-doped ACC was prepared from different educts solutions containing 10 wt%, 20 wt%, or 40 wt% of magnesium. Due to a different partitioning of Mg and Ca between ACC and the solvent, magnesium is only partially incorporated in the ACC structure. Analyses by ICP-MS showed that starting solutions containing 10 wt%, 20 wt%, or 40 wt% of magnesium yielded 0.87 wt%, 2.6 wt%, and 5.7 wt% Mg in the obtained Mg-doped ACC power (see Table D.7†). The morphology of Mg-doped ACC is akin to that of pure ACC, and it is remarkably stable in its dry state as documented by consecutive XRD analyses; even after 1 month, the powders are X-ray amorphous (see Fig. D8†). Incubated in water, Mg-doped ACC retains its high reactivity and transforms quickly into a crystalline state. In case of 10 wt% Mg, distinct fractions of aragonite are detectable; a behavior which is expected since Mg is known to suppress calcite growth.49 At 20 wt% Mg, less aragonite is present; at 40 wt% Mg, only calcite is formed, and after 28 d also magnesite is formed. The morphological development is more complex than in the pure case since Mg is also a growth and habit modifier.49 In case of ACC prepared in the presence of 10 wt% Mg, aragonite needles can be clearly detected, whereas, at higher Mg content, spherical particles larger than the initial ACC particles form and dominate; the typical rhombohedral shape of calcite is not present (Fig. 6-A). Turning to SBF, we find a behavior of Mg-ACC, which is similar to pure ACC (Fig. 6-B). After the formation of large spheres (either vaterite or Ostwald-ripened ACC), these spheres are coated by a layer of CaP. The formation of CaP is also clearly detectable after 28 d at all Mg-concentrations. At this late stage, hollow spheres have been formed due to the redissolution of the calcareous sacrificial template. The stabilization of ACC by means of magnesium is not detrimental for a biomedical application, as demonstrated by in vitro cytotoxicity assays (Fig. 6-C). Indirect viability test of cells cultured in the presence of ACC doped with 10 wt%, 20 wt%, 40 wt% Mg and, for comparison, Mg-free ACC for 24 h revealed that the overall viability of the cells exposed to the extracts did not decrease when compared with the positive control. The cell viability in case of ACC prepared with 40 wt% Mg is comparable with the positive control and with pure ACC indicating that higher Mg content in the extract does not present any significant influence on cell viability. Turning to ACC with 10 wt% Mg and 20 wt% Mg, the cell test shows a statistically significant increase (p < 0.05) in viability, compared to the positive control and to pure ACC. Overall, ACC prepared in the presence of 20 wt% Mg extract shows the highest cell viability with an increase of ∼22%.
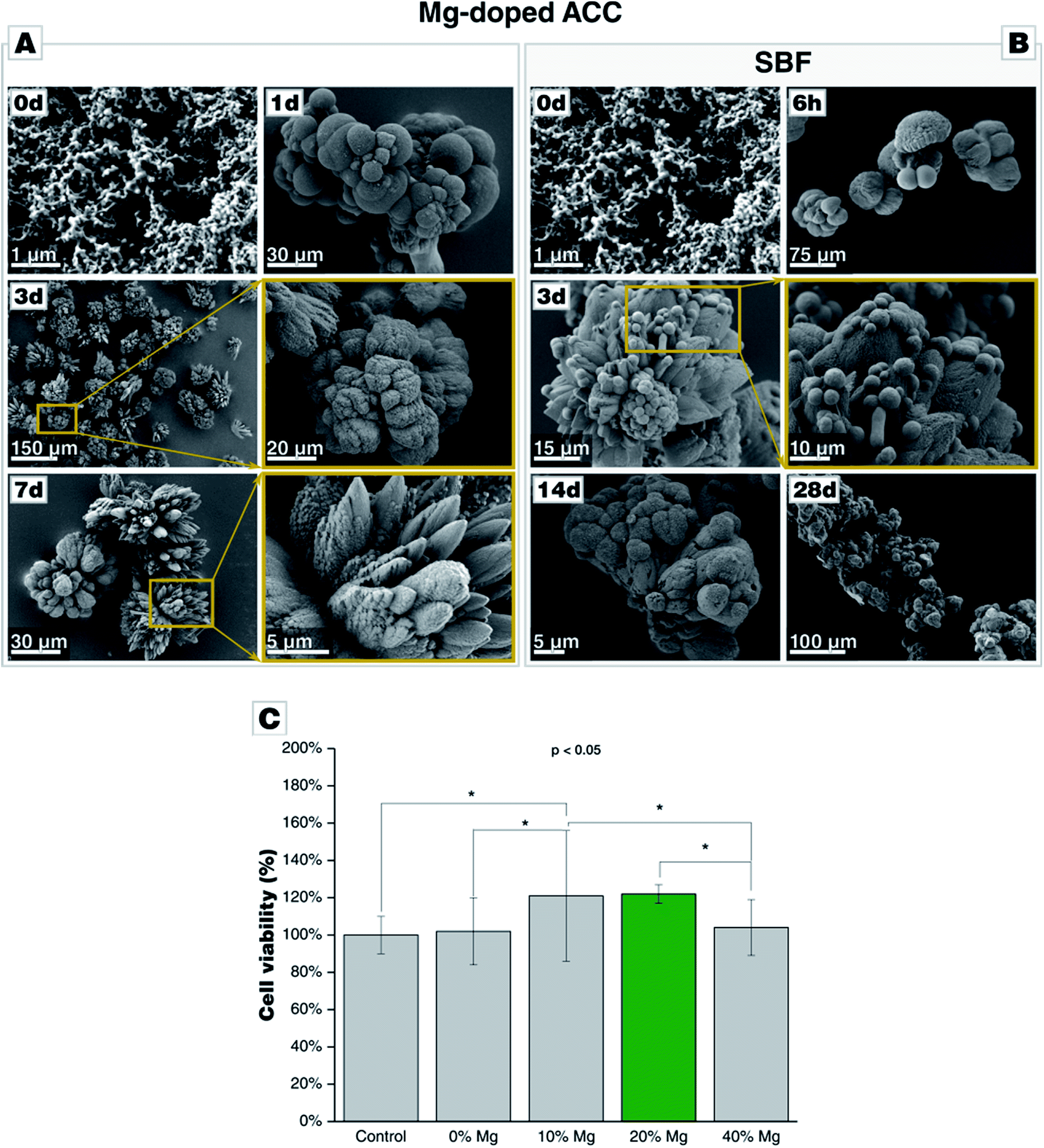 | ||
| Fig. 6 Morphological evolution of Mg-doped amorphous calcium carbonate (Mg-ACC) during incubation (A) in water and (B) in SBF, both at 37 °C. (C) Cytotoxicity assay of Mg-ACC on bone marrow stomal cell line (ST-2 cells) shows a significant increase in cell viability in presence of 10 wt% and 20 wt% Mg (p < 0.05, Anova). | ||
Discussion
Calcite is thermodynamically stable and, immersed in water, undergoes Ostwald ripening, which leads to the formation of agglomerated polycrystals. In SBF solutions, similar ripening is observed, and calcite appears to be essentially inert against calcium phosphate deposition under biomimetic conditions. We attribute this behavior to the low solubility of calcite, which is the lowest of all polymorphs at standard conditions. Changes in the calcium activity can be neglected, and the supersaturation of SBF with respect to calcium phosphate remains unaltered. SBF itself is supersaturated with respect to certain calcium phosphates (CaP), e.g., hydroxyapatite (HAP), which should form spontaneously. The absence of CaP crystallites on the surface of calcite lead us to the assumption that calcite is incapable of serving as a heterogeneous nucleator for CaP because the calcite {104} faces, which are expressed in thermodynamic equilibrium, are apparently incommensurable with those of hydroxyapatite (and potentially also with other calcium phosphates). Our observations correlate exceptionally well with in vivo study of calcite implanted into rabbit tibiae, demonstrating that pure calcite can bind to bone without the formation of a surface apatite layer.35Aragonite, metastable at standard conditions, transforms to calcite when brought into contact with water, obeying the Ostwald–Lussac step rule. In stark contrast to this, aragonite is stable in SBF, and no calcite appears during the first 28 d of incubation; in the framework of the Ostwald–Lussac step rule this means that the phase transformation of aragonite to calcite is impeded by increasing the barrier for this step, e.g., by crystal growth inhibitors suppressing the formation/growth of calcite. In SBF, only phosphate and sulphate might intervene with the crystallization of calcite/calcium carbonate (both forming sparely soluble calcium salts), and we assume that both or one of these ions blocks the growth of calcite, rendering aragonite kinetically stable. Phosphate has been repeatedly reported to inhibit calcite formation,50–52 whereas sulfate seems not to impede calcite growth.53 From this, we conclude that phosphate is capable to strongly interfere with the generic Ostwald–Lussac rule of calcium carbonate, remarkably raising the barrier for calcite formation/growth making calcite essentially inaccessible. In contrast to calcite, aragonite shows calcium phosphate precipitates forming on the crystallite surface, which indicates that the expressed aragonite face is capable of acting as a heterogeneous nucleator for CaP, and thus aragonite is bioactive. This is in line with an earlier report.7
Vaterite, thermodynamically metastable, undergoes expedient Ostwald–Lussac ripening yielding calcite when immersed in water; the omission of aragonite as intermediate corresponds with literature.38–40 Contrasting the water control experiment but congruent to the case of aragonite, vaterite is stable in SBF solutions and does not transform to calcite. Vaterite is more bioactive than aragonite and shows a more pronounced formation of CaP on its surface which is also corroborated by increased signal intensities in various characterization techniques; we attribute this behaviour to the higher solubility of vaterite leading to a higher efflux of calcium ions increasing the solution's supersaturation with respect to CaP formation.
Amorphous calcium carbonate (ACC) is unstable and, in the presence of water, rapidly transforms to calcite. The potential intermediate stage of vaterite is not captured in our experiments due to the coarse time resolution, but it is well documented in the literature.54 In SBF, ACC transforms to calcite probably via vaterite as traces of vaterite are detectable even after 14 d; in water, vaterite transforms within 3 d to calcite. ACC is similarly bioactive as vaterite; it produces, in contrast to vaterite, an amorphous or cryptocrystalline CaP untraceable by XRD. This might be due to the absence of a crystalline substrate providing the required stereochemistry allowing for heterogeneous nucleation. More probable is that the extreme high solubility of ACC might also cause a fast release of calcium ions into the solution which ramps up the supersaturation with respect to CaP, facilitating the precipitation of a disordered and amorphous CaP phase. The latter scenario also directly explains the peculiar hollow-sphere morphology; transient vaterite spheres serve as sacrificial templates for CaP precipitation.
The finding that ACC transforms in SBF solution to calcite and vaterite contradicts with the tests of vaterite aged in SBF solution, which remained stable for up to 28 d. This disagreement can be resolved by considering, again, the high release rates of calcium ions into SBF, fueled by the rapid redissolution of ACC, which is not observed in the vaterite case. This pronouncedly increases the solution's supersaturation with regard to calcite so that either the increased barrier of the vaterite-to-calcite transformation becomes more probable to be overcome or that calcite formation is directly triggered, without the involvement of intermediate stage. We assume that the latter aspect dominates. As we observed similar morphologies and transformation behavior in PBS buffer, which does not contain sulfate ions, it is clear that the behavior has to be attributed to the presence of phosphate in the system.
In order to prolong the stability of ACC, allowing for extended storage for weeks and in order to ease formulations based on ACC, we also prepared Mg-stabilized ACC which behaves in SBF very similar to pure ACC. The incorporation of Mg in the calcareous biomaterial is not detrimental for cell viability or bioactivity. In contrast to pure ACC, the presence of magnesium seems to facilitate the formation of a crystalline CaP phase, as demonstrated by XRD. Moreover, the presence of Mg suppressed the formation of vaterite and increased cell viability. In our experiments, the addition of magnesium in amounts of 10 and 20 wt% thus promoted the cell viability, indicating excellent biocompatibility and allows for further fine-tuning the phase transformation behavior in SBF solutions.
The SBF system has been criticized as an efficient and reliable in vitro test for bioactivity.55 It goes without saying that SBF tests are highly simplifying and even if they cannot provide the chemical complexity of an in vivo test, the reductionistic approach of SBF test gives valuable insights into the behavior of inorganic minerals in the presence of foreign ions, especially phosphate. Our key findings that (a) phosphate acts as an inhibitor in Ostwald–Lussac step ripening and that (b) calcite seems incapable to nucleate CaP in contrast to all other calcium carbonate polymorphs should also hold true in more complex systems. Our results for calcite are in exceptionally good agreement with the findings of Fujita et al. who showed calcite plates, when implanted in rabbit tibiae, to bind to bone tissue without the formation of surficial apatite layer.35 As the process of heterogeneous nucleation is a fundament of CaP formation on surfaces (leaving aside mere CaP deposition, e.g., by sedimentation), and as the thermodynamic concepts of phase transformation and their inhibition also valid in organisms, the observation made in SBF tests are valuable for a first assessment and a fundamental understanding of the behaviour of calcium carbonate. Moreover, good correlations between bioactivity in SBF tests and bioactivity in vivo have been established.42 Indeed, by SBF tests, inorganic materials proposed for bone substitution and bone regeneration can be compared.
Conclusions
For the first time, the four major polymorphs of calcium carbonate – calcite, aragonite, vaterite, and amorphous calcium carbonate (ACC) – have been systematically mapped out with respect to their bioactivity and phase transformation behavior in SBF solution. Reviewing our results, it becomes clear that the Ostwald–Lussac step rule is distinctly altered when turning away from pure water to SBF solutions mimicking the inorganic components of body fluids, such as blood. The results are summarized in Fig. 7, contrasting the phase transformation behavior of calcium carbonate in water with that in SBF, i.e., in the presence of phosphate ions. | ||
| Fig. 7 Schematic Ostwald–Lussac step ripening processes occurring upon immersion of calcium carbonate polymorphs in water and in SBF, respectively. The grayed-out arrows indicate those transformation which are typically not observed under the chosen conditions. The respective activation barriers are not to scale. | ||
Based on our results, it seems that calcite is not bioactive with respect to CaP formation in SBF solution. All other calcium carbonate polymorphs show distinct bioactivity, and the bioactivity increases with the polymorph's solubility. Further, the phase transformation and re-dissolution behavior are remarkably altered by the presence of phosphate, generating passivating coating and stabilizing metastable calcium carbonate polymorphs. In order to stabilize the highly unstable ACC, in order to ease its formulation and processing, we also generated and tested Mg-stabilized ACC. This Mg-doped ACC was clearly bioactive in SBF and, moreover, promoted cell viability. This observation evidences not only the excellent biocompatibility of this calcareous biomaterial. It also shows that the reactivity and bioactivity of calcareous biomaterials can be designed by adjusting its phase and ion/dopant composition. In future application, this feature of calcium carbonate-based biomaterials will allow to design and optimize the host's response in clinical applications. Overall, our findings pave the way for designing calcium carbonate-based bone replacement materials with adjustable bioactivity and bioresorption rate; they clearly demonstrate that calcium carbonate is indeed a bioceramic suitable for bone replacement material as its behavior can be tuned by crystal phase and composition.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. M. and A. R. B. received support from the European Horizon 2020 ITN “NanoHeal” (Grant Agreement # 642976). S. E. W. acknowledges financial support by an Emmy Noether starting grant issued by the German Research Foundation (DFG, no. WO1712/3-1).Notes and references
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- Z. Zou, I. Polishchuk, L. Bertinetti, B. Pokroy, Y. Politi, P. Fratzl and W. J. E. M. Habraken, J. Mater. Chem. B, 2018, 6, 449–457 RSC.
- D. Gebauer and S. E. Wolf, J. Am. Chem. Soc., 2019, 141(14), 4490–4504 CrossRef CAS PubMed.
- A. Bobbio, Bull. Hist. Dent., 1972, 20, 1–6 CAS.
- M. Ni and B. D. Ratner, Biomaterials, 2003, 24, 4323–4331 CrossRef CAS PubMed.
- E. Lopez, B. Vidal, S. Berland, S. Camprasse, G. Camprasse and C. Silve, Tissue Cell, 1992, 24, 667–679 CrossRef CAS PubMed.
- A. Lucas, J. Gaudé, C. Carel, J.-F. Michel and G. Cathelineau, Int. J. Inorg. Mater., 2001, 3, 87–94 CrossRef CAS.
- B. Ramalapa, O. Crasson, M. Vandevenne, A. Gibaud, E. Garcion, T. Cordonnier, M. Galleni and F. Boury, J. Mater. Chem. B, 2017, 5, 7360–7368 RSC.
- Y. Wu, W. Gu, J. Tang and Z. P. Xu, J. Mater. Chem. B, 2017, 5, 7194–7203 RSC.
- C. Wang, S. Chen, Q. Yu, F. Hu and H. Yuan, J. Mater. Chem. B, 2017, 5, 2068–2073 RSC.
- J. Jia, Q. Liu, T. Yang, L. Wang and G. Ma, J. Mater. Chem. B, 2017, 5, 1611–1623 RSC.
- F. Tewes, O. L. Gobbo, C. Ehrhardt and A. M. Healy, ACS Appl. Mater. Interfaces, 2016, 8, 1164–1175 CrossRef CAS PubMed.
- Y. Guo, W. Jia, H. Li, W. Shi, J. Zhang, J. Feng and L. Yang, J. Mater. Chem. B, 2016, 4, 5650–5653 RSC.
- M. Mozafari, S. Banijamali, F. Baino, S. Kargozar and R. G. Hill, Acta Biomater., 2019, 91, 35–47 CrossRef CAS PubMed.
- D. M. Roy, W. Eysel and D. Dinger, Mater. Res. Bull., 1974, 9, 35–39 CrossRef CAS.
- E. W. White, J. N. Weber, D. M. Roy, E. L. Owen, R. T. Chiroff and R. A. White, J. Biomed. Mater. Res., 1975, 9, 23–27 CrossRef CAS PubMed.
- R. A. Yukna and C. N. Yukna, J. Clin. Periodontol., 1998, 25, 1036–1040 CrossRef CAS PubMed.
- A. Piattelli, A. Scarano and M. Quaranta, Biomaterials, 1997, 18, 577–579 CrossRef CAS PubMed.
- S.-B. Nam, Y.-C. Bae, J.-S. Moon and Y.-S. Kang, Ann. Plast. Surg., 2006, 56, 263–267 CrossRef CAS PubMed.
- A. Piattelli, G. Podda and A. Scarano, Biomaterials, 1997, 18, 623–627 CrossRef CAS PubMed.
- C. M. Zaremba, D. E. Morse, S. Mann, P. K. Hansma and G. D. Stucky, Chem. Mater., 1998, 10, 3813–3824 CrossRef CAS.
- I. Sethmann, C. Luft and H.-J. Kleebe, J. Funct. Biomater., 2018, 9, 69 CrossRef PubMed.
- I. Sethmann, S. Völkel, F. Pfeifer and H.-J. Kleebe, J. Funct. Biomater., 2018, 9, 67 CrossRef CAS PubMed.
- W. R. Walsh, P. J. Chapman-Sheath, S. Cain, J. Debes, W. J. M. Bruce, M. J. Svehla and R. M. Gillies, J. Orthop. Res., 2003, 21, 655–661 CrossRef CAS PubMed.
- R. Vago, D. Plotquin, A. Bunin, I. Sinelnikov, D. Atar and D. Itzhak, J. Biochem. Biophys. Methods, 2002, 50, 253–259 CrossRef CAS PubMed.
- H. Liao, H. Mutvei, M. Sjöström, L. Hammarström and J. Li, Biomaterials, 2000, 21, 457–468 CrossRef CAS PubMed.
- G. Atlan, Biomaterials, 1999, 20, 1017–1022 CrossRef CAS PubMed.
- H. Elmazar, I. T. Jackson, D. Degner, T. Miyawaki, K. Barakat, L. Andrus and M. Bradford, Eur. J. Plast. Surg., 2003, 25, 362–368 CrossRef.
- F. Baino, Acta Biomater., 2011, 7, 3248–3266 CrossRef CAS PubMed.
- H. Maeda, V. Maquet, Q. Z. Chen, T. Kasuga, H. Jawad and A. R. Boccaccini, Mater. Sci. Eng., C, 2007, 27, 741–745 CrossRef CAS.
- K. Fujihara, M. Kotaki and S. Ramakrishna, Biomaterials, 2005, 26, 4139–4147 CrossRef CAS PubMed.
- M. A. E. Cruz, G. C. M. Ruiz, A. N. Faria, D. C. Zancanela, L. S. Pereira, P. Ciancaglini and A. P. Ramos, Appl. Surf. Sci., 2016, 370, 459–468 CrossRef CAS.
- S. Kim and C. B. Park, Biomaterials, 2010, 31, 6628–6634 CrossRef CAS PubMed.
- Z. Xu, G. Liang, L. Jin, Z. Wang, C. Xing, Q. Jiang and Z. Zhang, J. Cryst. Growth, 2014, 395, 116–122 CrossRef CAS.
- Y. Fujita, T. Yamamuro, T. Nakamura, S. Kotani, C. Ohtsuki and T. Kokubo, J. Biomed. Mater. Res., 1991, 25, 991–1003 CrossRef CAS PubMed.
- E. Tolba, W. E. G. Müller, B. M. Abd El-Hady, M. Neufurth, F. Wurm, S. Wang, H. C. Schröder and X. Wang, J. Mater. Chem. B, 2016, 4, 376–386 RSC.
- L. Brecevic and D. Kralj, Croat. Chem. Acta, 2007, 80, 467–484 CAS.
- A. V. Radha, T. Z. T. Z. Forbes, C. E. Killian, P. U. P. A. Gilbert and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16438–16443 CrossRef CAS PubMed.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265–271 RSC.
- J. D. Rodriguez-Blanco, K. K. Sand and L. G. Benning, New Perspectives on Mineral Nucleation and Growth, Springer, Switz., 2017 Search PubMed.
- D. J. Tobler, J. D. Rodriguez-Blanco, K. Dideriksen, N. Bovet, K. K. Sand and S. L. S. Stipp, Adv. Funct. Mater., 2015, 25, 3081–3090 CrossRef CAS.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef PubMed.
- G. T. Zhou, Q. Z. Yao, J. Ni and G. Jin, Am. Mineral., 2009, 94, 293–302 CrossRef CAS.
- S. L. S. Stipp and M. F. Hochella, Geochim. Cosmochim. Acta, 1991, 55, 1723–1736 CrossRef CAS.
- T. Ikoma, T. Tonegawa, H. Watanaba, G. Chen, J. Tanaka and Y. Mizushima, J. Nanosci. Nanotechnol., 2007, 7, 822–827 CrossRef CAS PubMed.
- C. Drouet, BioMed Res. Int., 2013, 4, 1–13 CrossRef PubMed.
- L. Müller and F. A. Müller, Acta Biomater., 2006, 2, 181–189 CrossRef PubMed.
- K. J. Davis, P. M. Dove and J. J. De Yoreo, Science, 2000, 290, 1134–1137 CrossRef CAS PubMed.
- E. A. Burton and L. M. Walter, Geochim. Cosmochim. Acta, 1990, 54, 797–808 CrossRef CAS.
- W. A. House, J. Colloid Interface Sci., 1987, 119, 505–511 CrossRef CAS.
- P. M. Dove and M. F. Hochella, Geochim. Cosmochim. Acta, 1993, 57, 705–714 CrossRef CAS.
- M. M. Reddy and G. H. Nancollas, J. Cryst. Growth, 1976, 35, 33–38 CrossRef CAS.
- J. Ihli, W. C. Wong, E. H. Noel, Y.-Y. Kim, A. N. Kulak, H. K. Christenson, M. J. Duer and F. C. Meldrum, Nat. Commun., 2014, 5, 3169 CrossRef PubMed.
- M. Bohner and J. Lemaitre, Biomaterials, 2009, 30, 2175–2179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Extended details on the characterization and phase transformation behaviour of the different polymorphs. See DOI: 10.1039/c9ra01473j |
| This journal is © The Royal Society of Chemistry 2019 |