
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile controlled synthesis of Ag3PO4 with various morphologies for enhanced photocatalytic oxygen evolution from water splitting†
Guiwei He,
Wanliang Yang *,
Wei Zheng,
Li Gong,
Xinghui Wang,
Yan An and
Mengkui Tian*
*,
Wei Zheng,
Li Gong,
Xinghui Wang,
Yan An and
Mengkui Tian*
School of Chemistry and Chemical Engineering, Guizhou University, Guiyang, Guizhou Province 550025, PR China. E-mail: yangwanlianghhhh@163.com; tianmk78@126.com; Tel: +86 15985159596 Tel: +86 18085027366
First published on 11th June 2019
Abstract
A facile and green hydrothermal method has been developed for the synthesis of Ag3PO4 with a variety of morphologies, including cubic, rhombic dodecahedral, spherical and roughly spherical, by using Ag4P2O7 as a sacrificial precursor. The as-prepared catalysts were characterized by carrying out X-ray diffraction (XRD), scanning electron microscopy (SEM), UV-visible diffuse reflectance spectroscopy (UV-Vis DRS), Fourier transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS). The morphology of Ag3PO4 was controlled by simply adjusting the hydrothermal reaction temperature and time, without adding any templates and organic additives. Kinetics studies and characterization results revealed that the transformation from P2O74− to a PO43− radical was a rate-determining step, and influenced the morphology of Ag3PO4. Different oxygen evolution rates were observed for samples subjected to different hydrothermal reaction times, and the highest initial rate of O2 evolution achieved was 582.55 μmol h−1 g−1. Furthermore, for the samples prepared using a hydrothermal reaction time of 96 h, as the hydrothermal reaction temperature was increased, the oxygen evolution rate of the resulting sample decreased first and then increased, and the highest initial rate of O2 evolution was 856.06 μmol h−1 g−1, about twice the 418.34 μmol h−1 g−1 value for the sample prepared using the coprecipitation method. A possible mechanism has been proposed to explain how the hydrothermal reaction temperature and time influenced the Ag3PO4 morphology. Our method provides a guiding hydrothermal strategy for the synthesis of insoluble electrolytes with various morphologies from relatively soluble electrolytes without the need to use templates and organic additives.
Introduction
Semiconductor photocatalysis has garnered tremendous research interest in recent years, especially due to the developing shortage of fossil fuels and the accumulation of industrial sewage.1–4 Multifarious traditional semiconductors such as TiO2 have been confirmed to be outstanding photocatalysts for water-splitting and resulting H2 production and for photodegradation.5–7 Nevertheless, these wide-band photocatalysts only respond to ultraviolet light, which makes up less than 4% of the entire solar spectrum. In order to make the best use of solar energy, the exploitation of photocatalysts that can respond to visible light has become particularly important to produce electron–holes in a more efficient way.8,9 Since silver phosphate (Ag3PO4) was first discovered by Ye in 2010, it has attracted great attention in the field of photocatalysis due to its proper band gap structure.10 Many studies have been carried out to improve its photocatalytic activity and stability. Controlling the morphology of a photocatalyst is deemed to be one of the best methods to improve its photocatalytic activity.11–13 For silver phosphate crystal catalysts, interfacial properties of the catalysts and reactants, such as specific surface area, exposed facets and photocurrent density, are determined by the sizes and morphologies of the crystals, which are the important parameters affecting photocatalytic activity.11,14,15In recent years, many investigations have been devoted to further optimizing the photocatalytic activity and stability of Ag3PO4. Of these investigations, the morphology-controlled synthesis of Ag3PO4 has made notable progress, including towards the control of facets and the construction of multidimensional structures.16,17 Photocatalysts with structures differing from those of their bulk counterparts have attracted considerable attention owing to their special chemical, electronic and optical properties.18 The morphology of a photocatalyst is intimately related to the exposed facets, which directly influence the activity of photocatalyst. According to the crystal growth kinetic theory, the growth rate of seeds and the diffusion rate of precursors jointly determine the growth habit of crystals.19,20 The growth habit can be adjusted by inorganic/organic additives, templates, reaction media and internal/external operating conditions, and such an adjustment ultimately results in changing the structural properties. Various Ag3PO4 nanostructures including spheres,10,21 rhombic dodecahedrons,22 concave trisoctahedrons,23 cubes,24 and tetrapods17 with controlled particle sizes have been designed and synthesized to improve the photocatalytic properties further. For example, Ag3PO4 samples with different morphologies were synthesized by using three different precipitating agents (Na3PO4, Na2HPO4, and NaH2PO4) whose hydrolyses resulted in diverse solution pH values.25,26 They have also been synthesized by using different organic additives, with these syntheses apparently proceeding according to the oriented aggregation mechanism: organic compounds containing Ag and a ligand complex were formed, followed by their having reacted with other compounds, then crystal nuclei having formed gradually and growing further, and finally with self-assembly and oriented aggregation having been realized. Different sizes and morphologies were produced using different templates such as polycarbonate membranes,27 vinyl pyrrolidone (PVP),28,29 and polyhedral Ag3PO4 microcrystals.30 The prepared parameters including reaction components,25,26 temperature, and time,31 usually play a vital role in tailoring the morphologies and structures of Ag3PO4 crystals. In general, the area of reactive facets often decreases owing to the reduction of surface energy during the crystal growth process, which creates low-facet-energy crystals. Nevertheless, high-energy facets can be maintained under certain synthesis situations. Engineering the morphology as well as facets of the Ag3PO4 crystal have a great impact on its photocatalytic activity. There have been many reports about the synthesis of various forms of Ag3PO4. Unfortunately, organic solvents and/or capping agents are currently essential for adjusting the sizes and shapes of Ag3PO4 particles. As a result, there is an urgent need to develop a simple and environmentally friendly way to synthesize Ag3PO4 particles with various morphologies. Moreover, as far as we know, the use of related compounds such as Ag4P2O7 in the synthesis of Ag3PO4 has hardly been studied so far.
In the current work, we explored a facile and green hydrothermal method without any templates and organic additives to carry out a shape-controlled synthesis of Ag3PO4 particles, with the procedure having them being transformed from rhombic dodecahedrons to spheres. The preparation was mainly divided into two stages. First, we made use of the synergetic reaction between silver cations and pyrophosphate anions to prepare Ag4P2O7 nanoparticles. Secondly, the as-prepared silver pyrophosphate was used as a sacrificial precursor to prepare Ag3PO4 nanoparticles of various morphologies only by adjusting hydrothermal reaction time and temperature. The formation of these structures was systematically studied. The formation of rhombic dodecahedral Ag3PO4 carried out at a hydrothermal reaction temperature of 100 °C was indicated to involve a transformation from Ag4P2O7 to Ag3PO4. The rhombic dodecahedral nanocrystals were transformed into spherical particles upon increasing the hydrothermal reaction temperature from 100 to 180 °C. A possible mechanism for the shape-controlled syntheses of Ag3PO4 was also proposed. In photocatalytic activity experiments that we carried out, the roughly spherical Ag3PO4 particles covered with small particles on their surfaces displayed better photocatalytic activity than did the other morphologies of Ag3PO4.
Experimental
Materials
All chemical agents used in the experiments were of analytical grade. Silver nitrate (AgNO3) and disodium hydrogen phosphate (Na2HPO4, A.R.) was obtained from Sinopharm Chemical Reagent Co., Ltd. Sodium pyrophosphate (Na4P2O7) was from Chengdu Jinshan Chemical Reagent Factory. Deionized water with a resistivity of 18.2 MΩ cm was used for aqueous solutions and photocatalytic activity tests.Synthesis of Ag3PO4 using the coprecipitation method
A typical synthesis of Ag3PO4 using the coprecipitation method was carried out as follows. A mass of 2.7 g of AgNO3 was scattered in 40 mL of deionized water, to which 40 mL of a solution of 0.2 M Na2HPO4 was then added. The resulting mixture was vigorously stirred for 5 h at 40 °C and then a yellow precipitate was obtained using filtration, washed with deionized water several times, and dried at 60 °C in air. This sample was marked as APO.Synthesis of Ag4P2O7
In the process used to synthesize Ag4P2O7, first a mass of 2.7 g of AgNO3 was dispersed in 40 mL deionized water, to which 40 mL of a solution of 0.1 M Na4P2O7 was then added. The resulting mixture was vigorously stirred at 40 °C for 5 h, and then white precipitate was obtained using filtration, washed with deionized water several times, and dried at 60 °C in air.Preparation of Ag3PO4 particles with different morphologies
The Ag3PO4 photocatalyst was prepared by using a simple hydrothermal method. In a typical process, a mass of 2.7 g of AgNO3 was dissolved in 40 mL of deionized water, to which 40 mL of a solution of 0.1 M Na4P2O7 was then added. The resulting mixture was vigorously stirred for 5 h at 40 °C, then transferred into a Teflon-lined autoclave and heated at 100 °C under autogenous pressure for 24 h, followed by being allowed to naturally cool to ambient temperature. The resulting product was separated using filtration, washed with deionized water three times and dried at 60 °C in air. Some Ag3PO4 samples were prepared by using different hydrothermal reaction times (24 h, 48 h, 72 h, 96 h) at 100 °C and were denoted as Ag3PO4 (t), and other Ag3PO4 samples were prepared by using different hydrothermal reaction temperatures (100, 120, 140, 160 and 180 °C) for 96 h, and were denoted as Ag3PO4 (T).Characterizations
X-ray diffraction (XRD) patterns of the samples were recorded from a D8 Advance Focus diffractometer (Bruker) using Cu-Kα radiation (λ = 0.15405 nm), in a 2θ angular range of 5–90°. The morphologies and sizes of the as-prepared products were characterized by using a field-emission scanning electron microscope (SEM) (S-3400N, HITACHI). The light absorption spectra of the samples were acquired when performing UV-visible diffuse reflection tests (UV-Vis, UV-3600 Plus, Shimadzu, Japan). The elemental composition and valence states of the samples were analyzed by carrying out X-ray photoelectron spectroscopy (XPS) (ThermoFisher K-Alpha, America). FTIR was applied to ascertain the chemical groups in Ag3PO4 photocatalysts. FTIR measurements were taken on a Nicolet IS50 infrared spectrometer after the sample was mixed with 300 mg of spectroscopic-grade KBr and pressed into thin slices.Assessment of photocatalytic performance
The photocatalytic activities of the prepared productions were estimated by measuring the photocatalytic O2 evolution under visible-light irradiation. The photocatalytic reaction was conducted in a Pyrex glass reactor at ambient temperature. A 300 W Xe arc lamp with a UV cut-off filter (λ ≥ 400 nm) was used as the light source. A mass of 0.425 g of AgNO3 was added into 110 mL of deionized water to form the mother solution used for the activity test, and then a mass of 0.1 g of as-prepared catalyst was dispersed into the above mixture as a sacrificial reagent. Then the above solution was subjected to ultrasonic dispersion for 3 min. The air in the reactor was replaced with Ar gas, which was irradiated by a 300 W Xe lamp with a cut-off filter (λ ≥ 400 nm) under 15 °C circulating condensed water. A gas chromatograph was acquired (using an Agilent, 7820A, GC system) to detect the oxygen produced.Results and discussion
We have developed a facile and green hydrothermal method to prepare Ag3PO4 with various morphologies by using silver pyrophosphate as a sacrificial precursor. The compositions and phase structures of all of the synthesized materials were determined from XRD measurements, and are shown in Fig. 1 and S1.† As shown in Fig. 1a, Ag4P2O7 was obtained by using AgNO3 and Na4P2O7 as raw materials. Only one main peak (2θ = 32.4°) appeared in the XRD pattern of the Ag4P2O7 of JCPDS card no. 37-0187, because this sample was not mineralized at the 100 °C temperature used. In our experiments, when the hydrothermal reaction temperature was 100 °C and the hydrothermal reaction time was 24 h or 48 h, three strong peaks at 27.14°, 28.79°, and 32.4° were observed (Fig. 1a), and indicated the presence of Ag4P2O7. The clear XRD pattern (see Fig. S1†) shows that the P2O74– was present in the Ag3PO4 lattice. A peak was observed at 33.293°, which indicated that Ag3PO4 formed (JPCDS no. 06-0505), and hence that Ag4P2O7 partly converted to Ag3PO4. The diffraction peaks corresponding to Ag4P2O7 became weaker as the hydrothermal reaction time was increased, and disappeared after the reaction time was prolonged to 96 h. Here, no other peaks beside those corresponding to Ag3PO4 were observed (Fig. 1a, 96 h), indicating that a pure phase of the Ag3PO4 photocatalyst was obtained. It is notable that the peak intensity of Ag3PO4 became sharper and stronger with the increasing hydrothermal reaction time, which showed that Ag4P2O7 was slowly converted to Ag3PO4. Furthermore, the peaks of the sample prepared for 96 h corresponded to the diffraction peak of Ag3PO4 (shown in Fig. 1b). The crystallinity of the as-prepared samples increased first as the hydrothermal reaction temperature was increased from 100 to 140 °C, and then decreased as the temperature was increased further to 180 °C (Fig. 1b), which may have been due to the destruction of crystals caused by the excessively high temperature.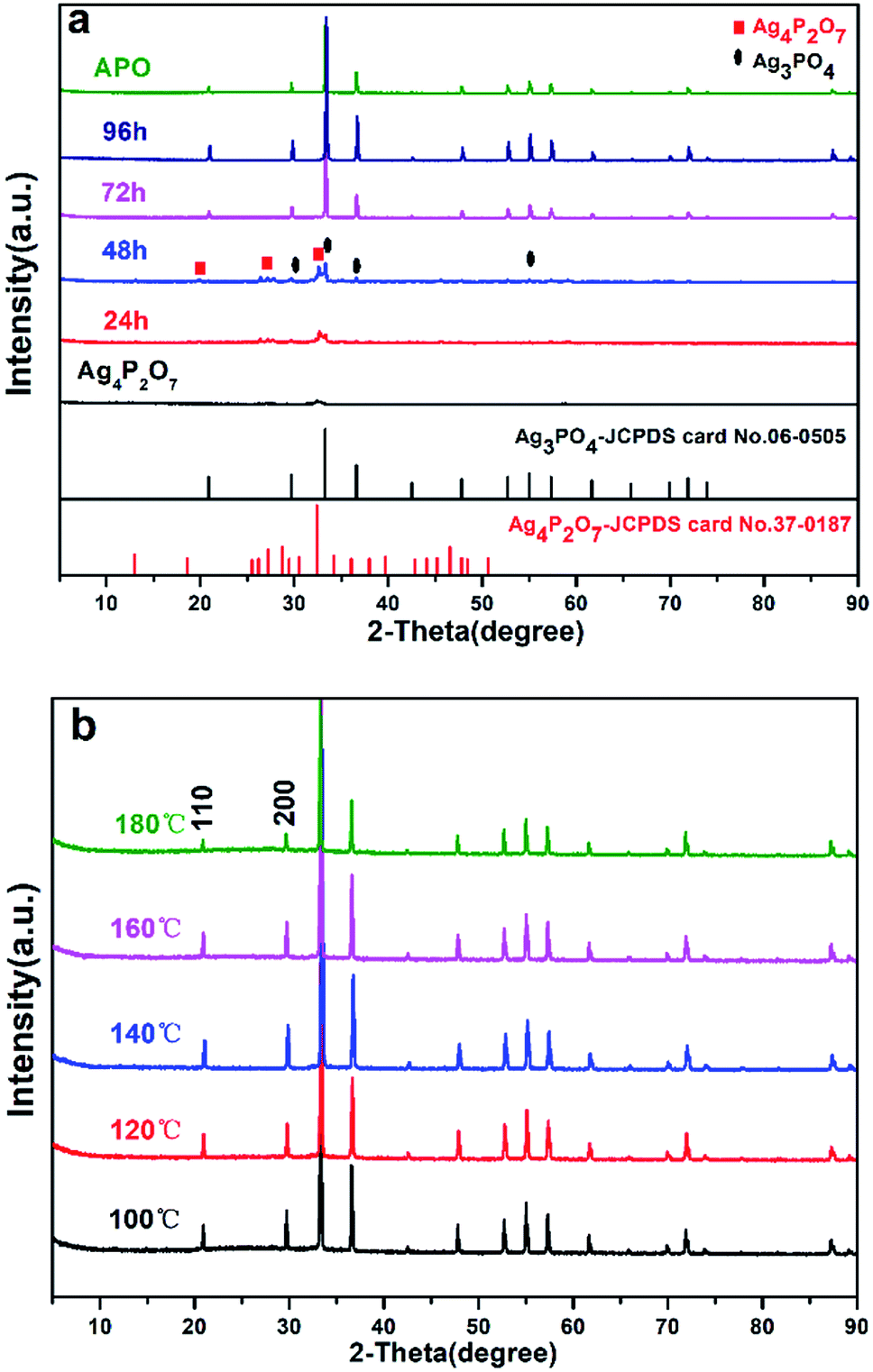 | ||
| Fig. 1 XRD patterns of samples (a) at 100 °C and different hydrothermal reaction times: 24 h, 48 h, 72 h and 96 h, and (b) for 96 h and different hydrothermal reaction temperatures: 100 °C, 120 °C, 140 °C, 160 °C and 180 °C. | ||
The changes in the morphology and crystallinity of the synthesized Ag3PO4 were examined using SEM. The APO sample synthesized using the coprecipitation method presented as a spherical structure but whose particle size was not uniform (200–700 nm), as shown in Fig. 2-APO. As shown in Fig. 2 and S2,† while Na4P2O7 was used as a precursor, the generated silver pyrophosphate formed a flaky polyhedron with an edge length of 100–300 nm and thickness of 30–40 nm (Fig. 2-Ag4P2O7). For a hydrothermal reaction time of 24 h, Ag3PO4 cubes with approximately 600 nm-long edges were clearly observed, as shown in Fig. 3-24 h (Fig. S2-24 h†), and Ag3PO4 coexisted with Ag4P2O7. With an increase of the reaction time to 48 h, the Ag4P2O7 still was present, but the Ag3PO4 cubes converted into rhombic dodecahedrons (Fig. 2-48 h and S2-48 h†). As shown in Fig. 2-72 h (Fig. S2-72 h†), no flaky silver pyrophosphate was observed when the hydrothermal reaction time was increased further to 72 h, and many Ag3PO4 rhombic dodecahedrons appeared. And for a reaction time of 96 h, the Ag3PO4 rhombic dodecahedrons with edge lengths of 300–600 nm were clearly observed (Fig. 2-96 h and S2-96 h†). The edges and corners of the pentagonal dodecahedrons were very clear, while the small flaky polyhedrons completely disappeared. The SEM data was consistent with the results of XRD (Fig. 1). The results taken together showed that, as the hydrothermal reaction time was increased, the Ag4P2O7 dissolved, the P2O74− converted gradually into PO43− and then the silver phosphate was formed. The Ag4P2O7 gradually converted to silver phosphate, but the diameters of the Ag3PO4 particles did not change significantly with changes in hydrothermal reaction time.10,18,33,34 Furthermore, when the hydrothermal reaction time was set at 96 h, the edges and corners of the Ag3PO4 rhombic dodecahedrons gradually disappeared as the hydrothermal reaction temperature was increased from 100 °C to 180 °C (Fig. 3 and S3†). For example, when 120 °C was used, the edges and corners appeared somewhat blurry. And when a hydrothermal reaction temperature of 140 °C was used, hardly any complete surface of an Ag3PO4 dodecahedron was seen (Fig. 3-140 °C). Perhaps, at the higher hydrothermal reaction temperatures, the high specific surface energy of the fine particles predominated and caused the formation of roughly spherical particles.35–37 In fact, spherical Ag3PO4 particles (3–5 μm) were clearly observed when a hydrothermal reaction temperature of 160 °C was used, and the surfaces of the silver phosphate particles became rough (Fig. 3-160 °C). This observation may be ascribed to the high hydrothermal reaction temperature having resulted in the small polyhedral silver phosphate particles falling onto the surface of the large particles of silver phosphate. When the hydrothermal reaction temperature was increased to 180 °C, the surfaces of the Ag3PO4 particles became even rougher (Fig. 3-180 °C). Such roughness may enhance their photocatalytic activity, because the small particles on the surface, being Ag3PO4, would increase the density of active sites on the surface and be beneficial to the contact of water molecules with the surface.38–40 The FTIR spectra acquired from Ag4P2O7, Ag4P2O7/Ag3PO4 (48 h), and Ag3PO4 (96 h) are illustrated in Fig. S4.† Absorption bands at 908 and 1118 cm−1 were clearly observed, and ascribed to the symmetric stretching vibration of the P–O–P group of Ag4P2O7, and another band was observed at 1108 cm−1, and assigned to the presence of P–O. Also, three absorption bands appeared in Ag4P2O7/Ag3PO4 (48 h), which resulted from the cleavage of some P–O–P bonds and the formation of P–O bonds. These results provided additional evidence that segmental Ag4P2O7 converted to Ag3PO4, in accordance with the mechanism mentioned previously.
| Na4P2O7 + 4AgNO3 → Ag4P2O7↓ + 4NaNO3 | (1) |
 | (2) |
 | (3) |
 | (4) |
 | ||
| Fig. 2 SEM images of Ag4P2O7, Ag3PO4 synthesized using the coprecipitation method (APO), and samples prepared at 100 °C but using different hydrothermal reaction times: 24 h, 48 h, 72 h and 96 h. | ||
 | ||
| Fig. 3 SEM images of Ag3PO4 samples synthesized for 96 h at different hydrothermal temperatures: 100 °C, 120 °C, 140 °C, 160 °C and 180 °C. | ||
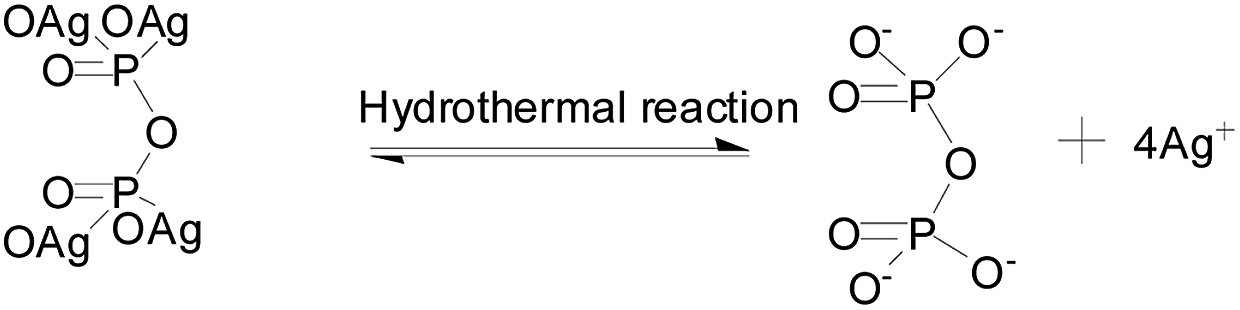
The above results clearly showed that Ag3PO4 materials with different morphologies can be successfully fabricated using the facile and green hydrothermal method. The morphology can be controlled by simply tuning the hydrothermal reaction temperature and time without adding any templates and organic additives. We came up with a possible mechanism, shown in Fig. 4, to explain the formation of Ag3PO4 with various morphologies from the use of Ag4P2O7 as a sacrificial precursor. Precipitation of Ag4P2O7 finished instantly after the silver ions and pyrophosphate anions were mixed. The transformation of P2O74− to a PO43− radical has been indicated, using kinetics principles, to be a rate-determining step, which would result in different morphologies of Ag3PO4.
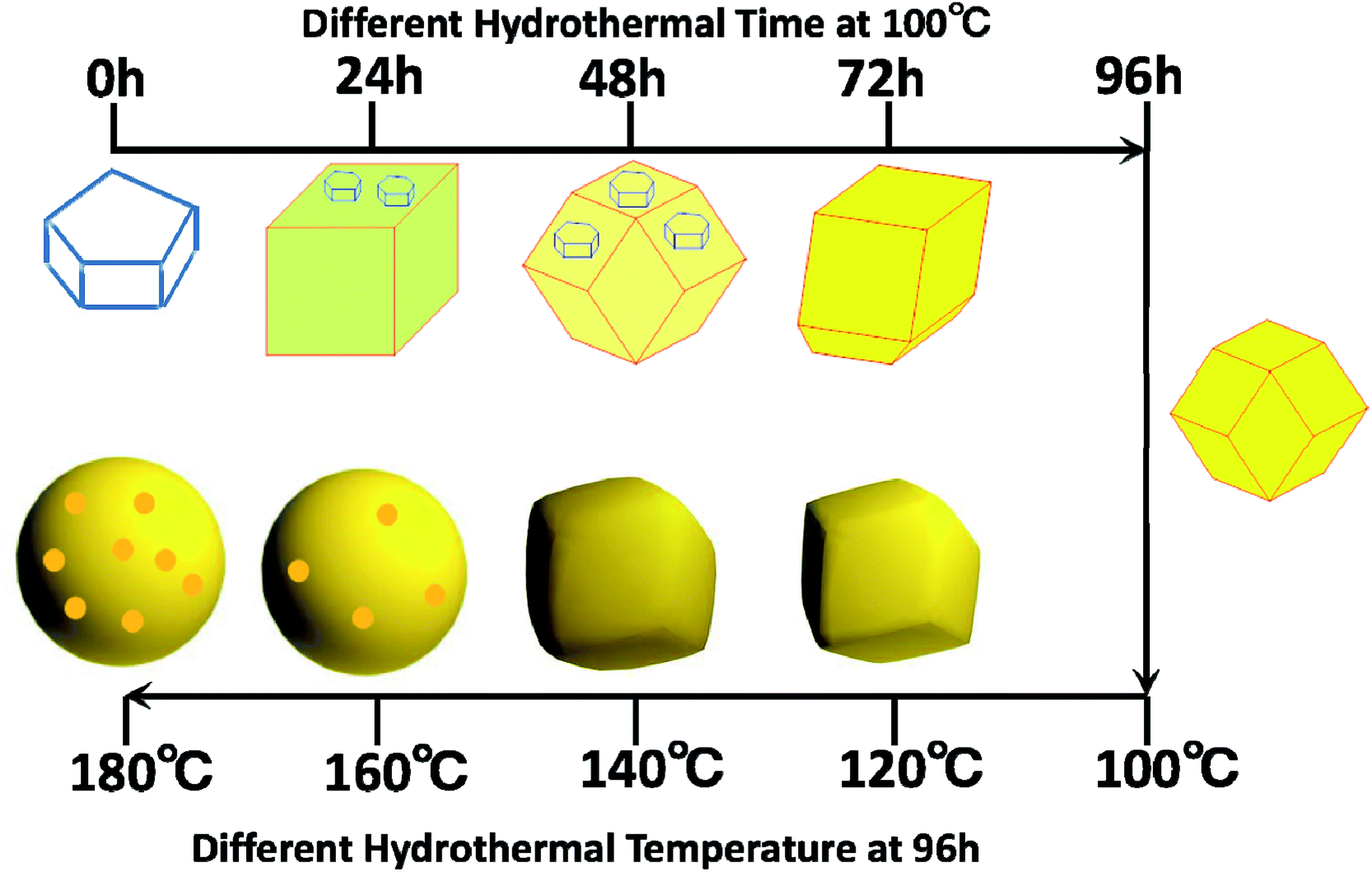 | ||
| Fig. 4 Mechanism of formation of the samples with different morphologies at the various hydrothermal reaction temperatures and times. | ||
According to the proposed reaction mechanism for the formation of Ag3PO4, first Ag4P2O7 formed when AgNO3 was simply mixed with Na4P2O7 at room temperature (eqn (1)). Then, the Ag4P2O7 decomposed into pyrophosphate anions and silver ions to achieve equilibrium at a given temperature (eqn (2)). Eqn (3) shows a P2O74− hydrolysis process in which PO43− anions formed at a certain temperature. At last, silver phosphate formed when the hydrolyzed PO43− anions combined with Ag+ (eqn (4)).
To realize the transformation of the morphologies of Ag3PO4 from cubes to rhombic dodecahedrons, nearly equimolar quantities (four moles worth) of silver ions and pyrophosphate anions were mixed, and precipitates of silver pyrophosphate immediately formed. The obtained mixture was then transferred into a Teflon-lined autoclave and kept at 100 °C under autogenous pressure for different hydrothermal reaction times to synthesize Ag3PO4 particles with different morphologies. Because the conversion from P2O74− to PO43− in the solution proceeded extremely slowly, silver pyrophosphate converted into silver phosphate and the silver phosphate particles converted from flaky polyhedrons to rhombic dodecahedrons as the hydrothermal reaction time was increased. When the hydrothermal reaction temperature was increased gradually, the silver phosphate particles transformed from the rhombic dodecahedrons to spheres, and then to rough spheres.
The proposed mechanism of the growth of the Ag3PO4 particles with various morphologies is shown in Fig. 4. To begin with, precipitates of silver pyrophosphate immediately formed when silver ions and pyrophosphate anions were mixed at room temperature, and the silver pyrophosphate particles were observed to be flaky polyhedrons. When the hydrothermal reaction time was set at 24 h, some of the Ag4P2O7 polyhedrons gradually transformed into Ag3PO4 cubes, which resulted in the simultaneous appearance of silver phosphate and silver pyrophosphate. As the hydrothermal reaction time was increased, much of the silver pyrophosphate gradually converted to cubic silver phosphate and then to rhombic dodecahedrons. With a further extension of the reaction time, silver pyrophosphate converted completely: that is, the transformation of the remaining silver pyrophosphate into rhombic dodecahedrons completed when the hydrothermal reaction time was increased to 96 h, and all the silver phosphate completely turned into rhombic dodecahedrons as well. All of the processes indicated that hydrothermal reaction time played a major role in the conversion of silver pyrophosphate to silver phosphate. The results also confirmed eqn (3) to be a rate-determining step in the transformation of Ag4P2O7 to Ag3PO4. Furthermore, as the hydrothermal reaction temperature was raised from 100 to 180 °C, the rhombic dodecahedral silver phosphate gradually evolved into spheres, i.e., the edges and corners disappeared. With a further increase of the hydrothermal reaction temperature, the surface of the sphere became rough, which indicated that the hydrothermal reaction temperature also functioned to change the morphology of the silver phosphate particle.31,32
To further confirm the proposed mechanism, we carried out XPS elemental analysis of Ag4P2O7, Ag4P2O7/Ag3PO4 (48 h at 100 °C), and Ag3PO4 (96 h at 100 °C), as shown in Fig. 5 and S5.† As Ag4P2O7 transformed to Ag3PO4 with the increase of hydrothermal reaction time, the energy level of the 2p orbital of P shifted from 133.08 eV in Ag4P2O7 to 132.58 eV in Ag3PO4 (Fig. 5a). Meanwhile, the binding energies of 1s orbitals of O varied from 530.88 eV to 530.48 eV (Fig. 5b). In contrast to these results for the Ag4P2O7 and Ag3PO4 samples, the energy levels of the P 2p and O 1s peaks of the sample prepared for 48 h at 100 °C were observed at positions (Fig. 5c and d) between those observed for the Ag4P2O7 and Ag3PO4 samples, suggesting that the sample prepared for 48 h at 100 °C contained both silver phosphate and silver pyrophosphate. The silver pyrophosphate molecule has, in addition to P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–O bonds also found in Ag3PO4, a P–O–P bond not found in Ag3PO4. This difference apparently led to a difference in the molecular environments and measured binding energy levels of P and O between these two molecules. However, the binding energies of the Ag 3d orbitals were measured to be almost the same (Fig. S5†), although their peak intensities differed, attributed to the different amounts on the particle surface.23,41,42 These conclusion are identical to the conclusions we arrived at as described above.
O and P–O bonds also found in Ag3PO4, a P–O–P bond not found in Ag3PO4. This difference apparently led to a difference in the molecular environments and measured binding energy levels of P and O between these two molecules. However, the binding energies of the Ag 3d orbitals were measured to be almost the same (Fig. S5†), although their peak intensities differed, attributed to the different amounts on the particle surface.23,41,42 These conclusion are identical to the conclusions we arrived at as described above.
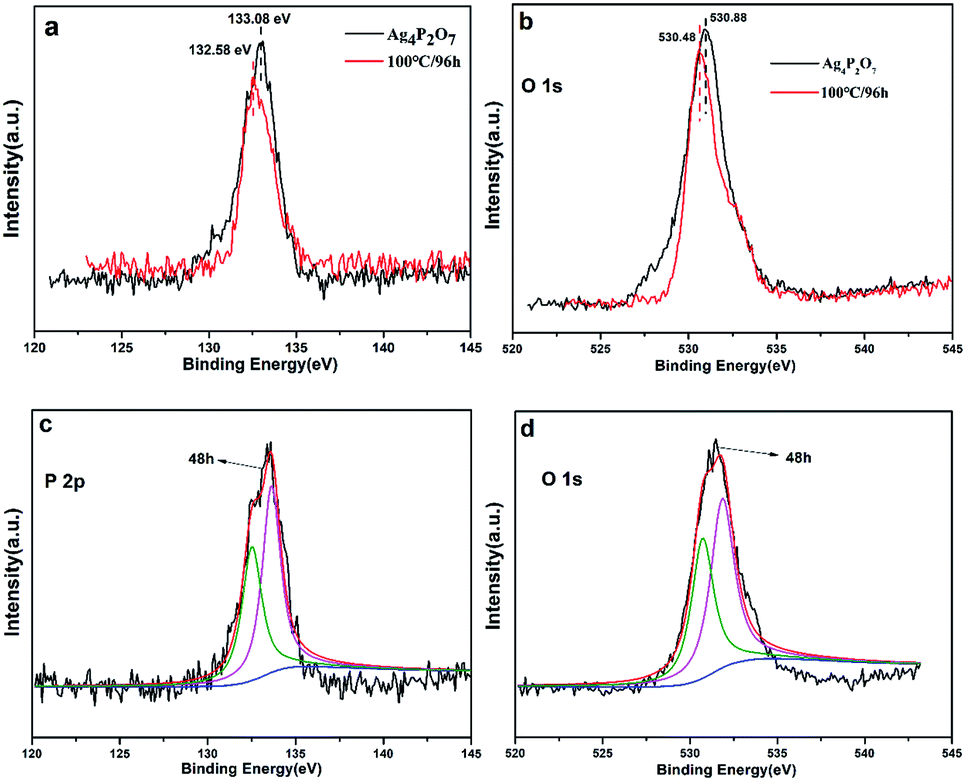 | ||
| Fig. 5 XPS spectra of Ag4P2O7, Ag4P2O7/Ag3PO4 (48 h at 100 °C) and Ag3PO4 (96 h at 100 °C) samples: P 2p (a) and O 1s (b) regions for Ag4P2O7 and Ag3PO4 (96 h at 100 °C), respectively; P 2p (c) and O 1s (d) regions for Ag4P2O7/Ag3PO4 (48 h at 100 °C). | ||
The UV-Vis analyses of the catalysts prepared with a hydrothermal reaction temperature set at 100 °C showed that the absorbance of the catalysts obviously red shifted as the hydrothermal reaction time was increased (Fig. 6a). Note that XRD results for a reaction time of 24 h indicated that the sample contained Ag3PO4, providing an explanation for the red shift of the sample in the UV-Vis spectrum. The absorbance intensities of Ag3PO4 prepared with reaction times of 72 h and 96 h were obviously greater than that of Ag4P2O7 and the mixture phase in the 300–500 nm region. While in the 500–700 nm region, as the hydrothermal reaction time was increased, the catalyst showed an increased ability to absorb these wavelengths of light. When the hydrothermal reaction temperature was set at 100 °C, an obvious red shift in the UV-Vis spectrum was observed with the increase of hydrothermal reaction time during the transformation process from Ag4P2O7 to Ag3PO4. Specifically, when the hydrothermal reaction times were 24 h and 48 h, there were two absorption band edges in the UV-Vis spectrum, which indicated that two different substances were present. Based on these results, combined with the above-described XRD (Fig. 1a) and SEM (Fig. 2) results, this phenomenon was concluded to have been caused by the coexistence of silver phosphate and silver pyrophosphate in the sample.32 When the hydrothermal reaction times were 72 h and 96 h, the absorption band edge of Ag4P2O7 was not obviously present, which indirectly indicated that all of the Ag4P2O7 was converted to Ag3PO4 when the hydrothermal reaction time was 72 h. However, in the 500–700 nm wavelength absorption region, the intensity of the absorbance for the 96 h catalyst sample was significantly greater than those of other catalysts. As mentioned above, this result was attributed to the obviously better absorbance displayed by the Ag3PO4 rhombic dodecahedron than by other catalysts with different morphologies.10,43 In addition, Ag3PO4 is an indirect semiconductor.44 And as can be seen from the UV-Vis spectra (Fig. 6), this indicated that no other impurity peaks formed. As shown in Fig. 6a and S6,† as the hydrothermal reaction time was increased, the absorption band edge was observed to increase from 357 nm to 533 nm, 546 nm, 536 nm, and finally to 540 nm; this result was ascribed to Ag4P2O7 having converted to Ag3PO4 under these conditions. This transformation resulted in the red shift of the absorption band, thereby increasing the visible light response. According to the formula Eg = 1240/λg (eV), where λg represents the threshold of the absorption wavelength, the corresponding band gaps were calculated to be 3.47 eV, 2.23 eV, 2.27 eV, 2.31 eV, 2.30 eV.45,46
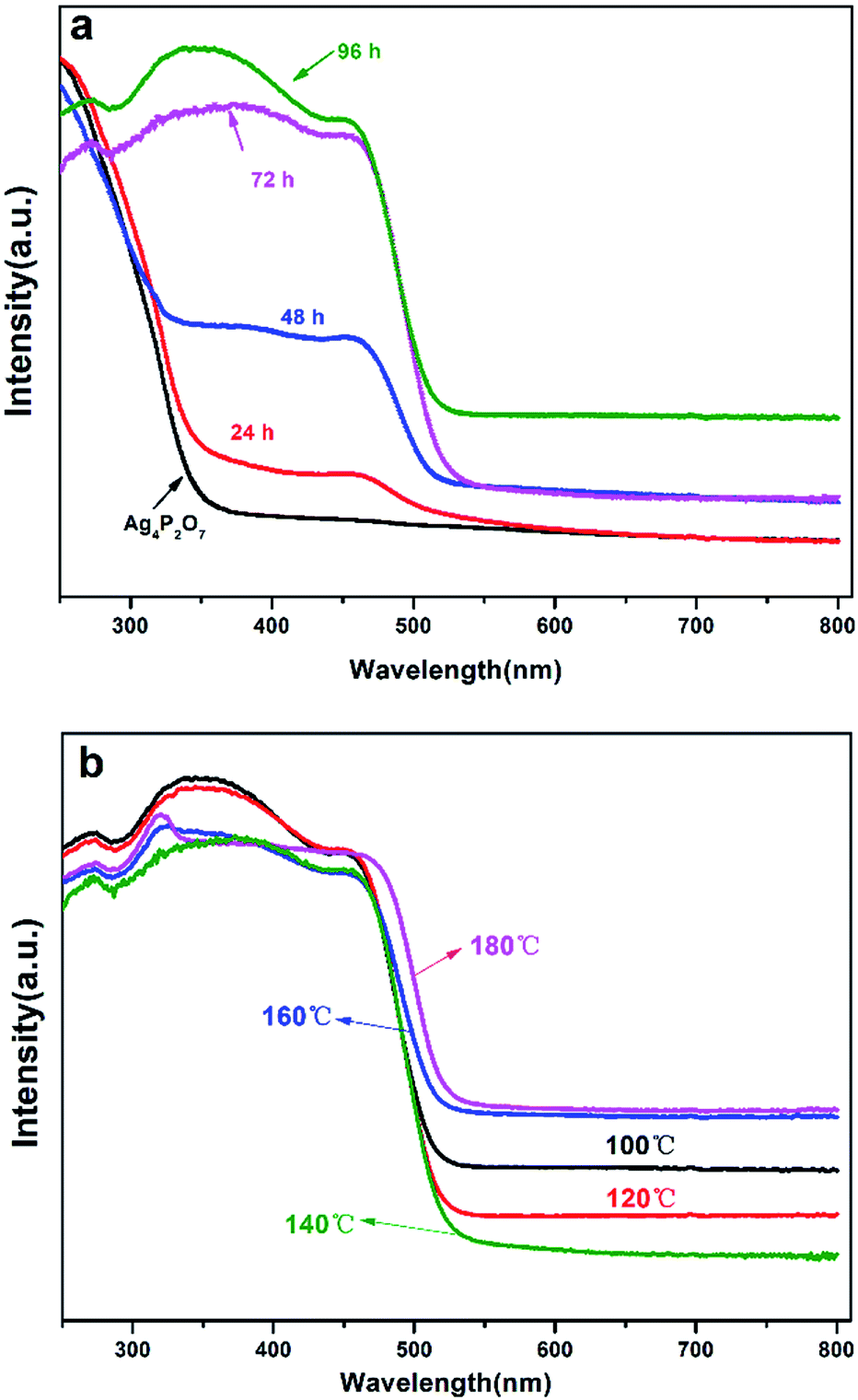 | ||
| Fig. 6 Ultraviolet-visible diffusive reflectance spectra of the various catalyst samples prepared (a) at 100 °C for different hydrothermal reaction times, of 24 h, 48 h, 72 h and 96 h, and (b) for 96 h at different hydrothermal reaction temperatures, of 100 °C, 120 °C, 140 °C, 160 °C and 180 °C. | ||
As shown in Fig. 6b, controlled the hydrothermal time at 96 hours, when the temperature is lower than 140 °C, the absorption band edge of the sample is almost unchanged, all of which are around 533 nm, which fully illustrates only Ag3PO4, no other impurities exists in the catalyst.43 However, when the temperature is higher than 160 °C, the surface of Ag3PO4 becomes rough, which results in partial red shift of its absorption edge. However, it can be seen that the intensity of the absorbance first decreased and then increased gradually with the increase of the hydrothermal reaction temperature in the region of 500–700 nm. Especially when the hydrothermal reaction temperature was below 140 °C, the intensity of the absorbance in this region tended to decrease with the increasing reaction time, and this result may have been due to the transformation from rhombic dodecahedral Ag3PO4 particles into spherical Ag3PO4 particles. When the hydrothermal reaction temperature was higher than 140 °C, the intensity of the absorbance in this region tended to increase with the increasing reaction time, and as mentioned above, this result may have been due to the surfaces of the spherical Ag3PO4 particles having become coarse.43
To assess the photocatalytic activities of the different Ag3PO4 samples, AgNO3 was used as a sacrificial reagent. For comparison, all of the performance tests were carried out with 0.425 g AgNO3 dissolved in 110 mL deionized water as a sacrificial reagent and added 0.1 g catalyst, and with other conditions remaining the same. Fig. 7a shows a diagram of the oxygen production of the photocatalysts prepared at 100 °C but with different hydrothermal reaction times. Here, the oxygen production levels of the samples clearly increased with increasing hydrothermal reaction time. The rate of oxygen evolution catalyzed by Ag3PO4 was 582.55 μmol h−1 g−1 when the hydrothermal reaction time was 96 h. This result was due to the generation of rhombic dodecahedral Ag3PO4 with excellent activity when the hydrothermal reaction time was increased to this level.22,31,47 Note that the oxygen production rate of APO was measured to be 418.34 μmol h−1 g−1, and the results discussed above illustrated that the photocatalytic activity of rhombic dodecahedral Ag3PO4 exceeded that of a sphere. As shown in Fig. 7b, when the hydrothermal reaction time was set at 96 h and the hydrothermal reaction temperature was increased, the oxygen production rate decreased first and then increased. The rates of oxygen production for Ag3PO4 (140 °C) and Ag3PO4 (180 °C) were 541.91 μmol h−1 g−1 and 856.06 μmol h−1 g−1 when the hydrothermal reaction temperatures were set at 140 °C and 180 °C, indicating that the oxygen production capacity of the Ag3PO4 sample made using a hydrothermal reaction temperature of 180 °C was obviously stronger than those of the samples made using other reaction temperatures. This result was attributed to the active facet with large Gibbs free energy on the surface gradually disappearing with the increase of hydrothermal reaction temperature. The exposed facet of the semiconductor will greatly affect the photocatalytic activity of the semiconductor; and especially for the {110} and {100} facets of silver phosphate, a higher facet activation energy results in better catalytic activity. The disappearance of the active facets of the rhombic dodecahedron caused the activity of the sample to decrease. As can be seen in Fig. 5, the spherical surface of the Ag3PO4 (180 °C) particles appeared rough, due to the deposition of small silver phosphate particles on the surfaces of the spherical silver phosphate particles.45,48 The decrease in the average size of the Ag3PO4 particles greatly increased the relative surface area of the particles and hence the quantity of active sites on the surfaces, features beneficial for the catalytic activity of spherical silver phosphate particles and that helped make the resulting oxygen production become more vigorous.41,42,49,50 In order to confirm the superiority of rhombic dodecahedral Ag3PO4 particle, it can be inferred that the decline of photocatalytic activity resulted from the disappearance of {110} when the hydrothermal reaction temperature was set at 120 °C (Fig. 8a). In addition, the photocatalytic activity increased along with the increase in surface roughness when the hydrothermal reaction temperature was set at 180 °C (Fig. 8b).
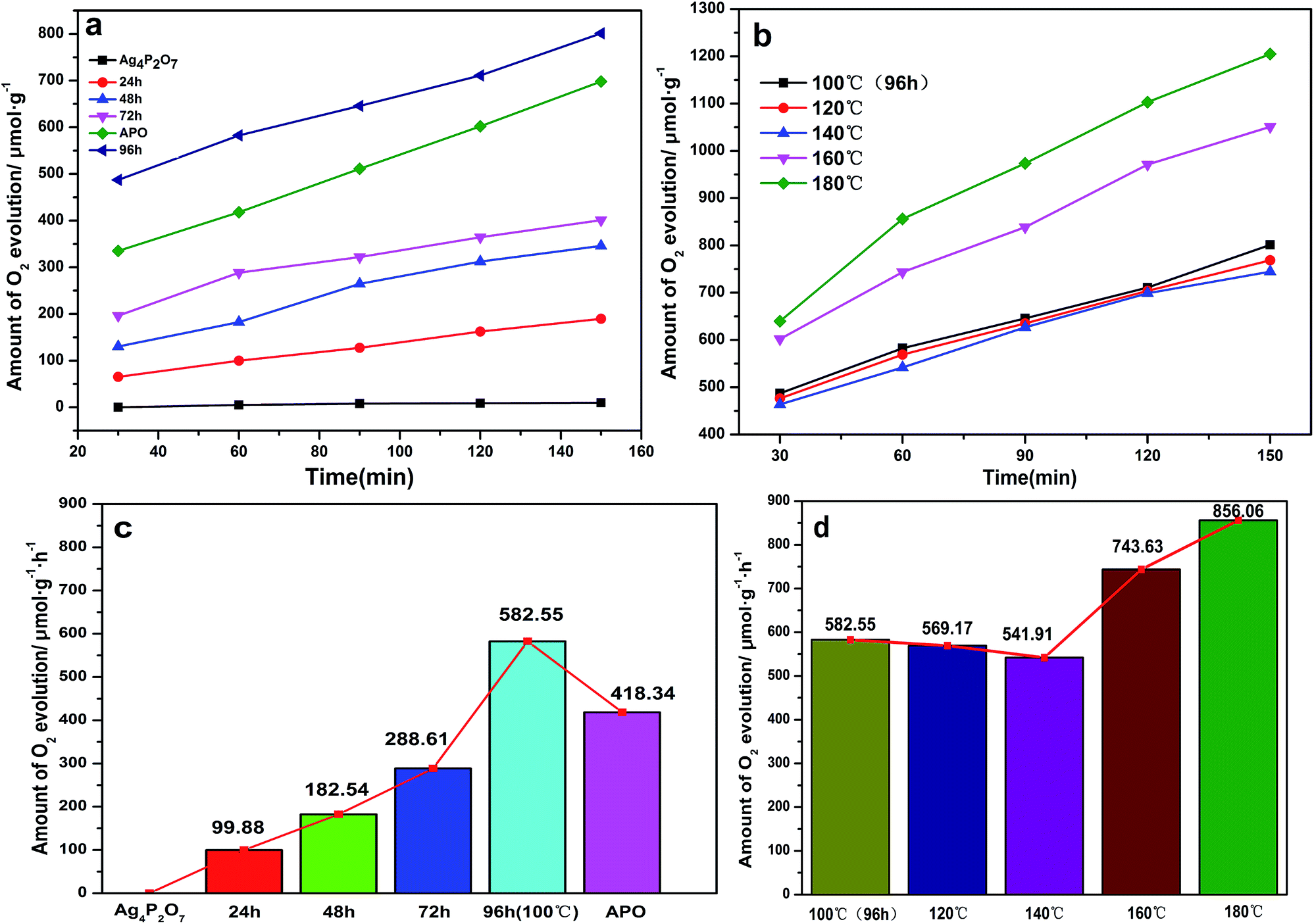 | ||
| Fig. 7 Photocatalytic activities of Ag3PO4 samples under visible light for the evolution of O2 from an aqueous AgNO3 solution. (a) Different hydrothermal reaction times for the 100 °C preparation. (b) Different hydrothermal reaction temperatures for the 96 h preparation. (c) Histogram of oxygen production of the 100 °C as-prepared samples. (d) Histogram of oxygen production of 96 h as-prepared samples. | ||
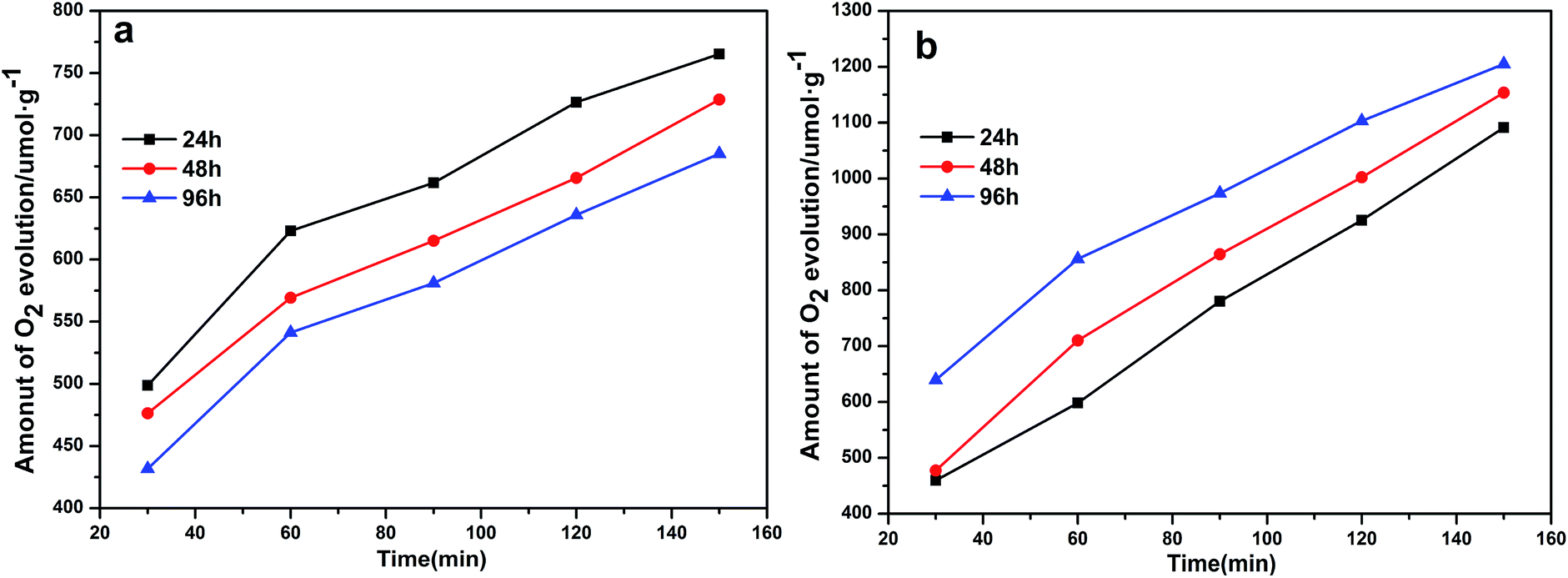 | ||
| Fig. 8 Photocatalytic activities of Ag3PO4 samples under visible light for O2 evolution from an AgNO3 aqueous solution. (a) Various hydrothermal reaction times for a hydrothermal reaction temperature of 120 °C. (b) Various hydrothermal reaction times for a hydrothermal reaction temperature of 180 °C. | ||
Pure Ag3PO4 exhibited a strong oxygen evolution ability according to the previous study10 (see Fig. 9). The valence band of Ag3PO4 was determined to be mainly composed of O 2p and Ag 4d, and the conduction band was determined to be mainly composed of Ag 5s, leading to the dispersion of many Ag s orbitals on the adjacent orbitals, in turn leading to the absence of Ag orbitals at the bottom of the conduction band, a feature also effective for dispersing Ag in s orbitals. The reduction of the total mass of effective electrons has been indicated to result in an increase in the separation effect of the photoelectrons. Moreover, the bottom of the conduction band of Ag3PO4 was shown to display an isotropic distribution, a feature also beneficial for the transfer of electrons and for increasing the photocatalytic activity.11–13,44,51–53 The strategy in which pyrophosphate was used as a precursor to prepare silver phosphate not only enriched the synthetic routes to Ag3PO4, but also provided a novel means to enhance the stability of Ag3PO4.
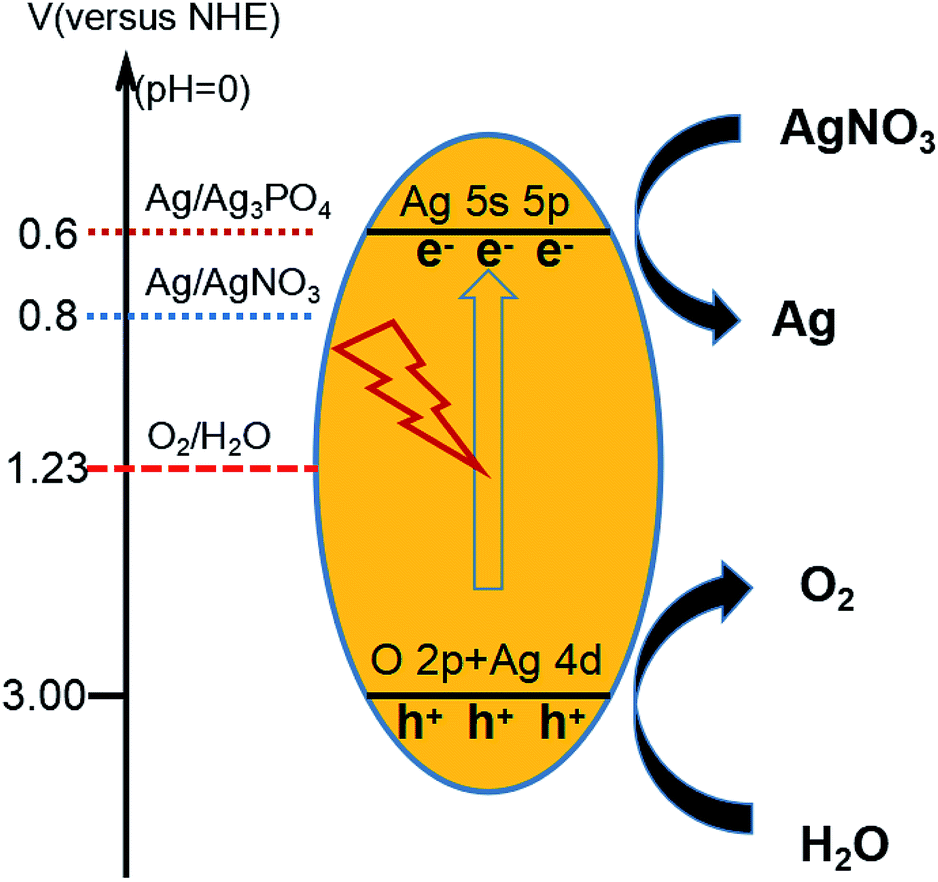 | ||
| Fig. 9 Schematic drawing of redox potentials of Ag3PO4 and the mechanism of O2 evolution. | ||
Conclusions
Ag3PO4 samples with different morphologies were prepared through the in situ conversion of Ag+ to Ag4P2O7 nanocrystals assisted by pyrophosphate anions, followed by the oriented attachment of the nanocrystals to form Ag3PO4 crystals with specific shapes determined by the hydrothermal reaction temperature and time used. The simple synthetic process used involved neither templates nor organic additives, and Ag4P2O7 nanocrystals functioned as not only sacrificial templates but also the source of Ag and phosphate anions for the generation of Ag3PO4 crystals. The hydrothermal reaction temperature and time were the predominant factors that were used to control the morphologies of the Ag3PO4 crystals, which in this way could be varied from nanoparticles to cubes, rhombic dodecahedrons, spheres and rough spheres. Photocatalytic oxygen production tests demonstrated that rough-spherical Ag3PO4 particles covered with small particles exhibited the best initial oxygen evolution rate of about 856 μmol h−1 g−1, and hence the most outstanding activity of all the photocatalyst samples tested. The second best Ag3PO4 catalytic activity was observed for spheres, third best for dodecahedrons, and lastly Ag4P2O7 nanocrystals. These observations can be explained by the visible light absorption capability and structural factors of these photocatalysts. This work has provided a new understanding of how to design highly efficient oxygen production photocatalysts displaying various morphologies. We believe that such a facile and green approach could also be used to prepare other insoluble electrolytes materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Planning Project of Guizhou Province [2017]5788-56, the Talent Introduction Program of Guizhou University (GDRJH2014-23 and GDRJH2014-21), Natural Science Foundation of China (No. 21663009), the Excellent Youth Fund of Guizhou Province [2017]5605 and Platform & Talent Program from Guizhou Province [2017]5788.Notes and references
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC
.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC
- S. Subudhi, D. Rath and K. M. Parida, Catal. Sci. Technol., 2018, 8, 679–696 RSC
- B. Zhu, R. Zou and Q. Xu, Adv. Energy Mater., 2018, 8, 1801193 CrossRef
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed
- L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC
- Y. Liu, L. Tian, X. Tan, X. Li and X. Chen, Sci. Bull., 2017, 62, 431–441 CrossRef CAS
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed
- X. Chen, Y. Dai and X. Wang, J. Alloys Compd., 2015, 649, 910–932 CrossRef CAS
- G. Huang, Z.-L. Ma, W.-Q. Huang, Y. Tian, C. Jiao, Z.-M. Yang, Z. Wan and A. Pan, J. Nanomater., 2013, 9, 1–8 Search PubMed
- D. J. Martin, G. Liu, S. J. A. Moniz, Y. Bi, A. M. Beale, J. Ye and J. Tang, Chem. Soc. Rev., 2015, 44, 7808–7828 RSC
- D. Wang, T. Hisatomi, T. Takata, C. Pan, M. Katayama, J. Kubota and K. Domen, Angew. Chem., Int. Ed., 2013, 52, 11252–11256 CrossRef CAS PubMed
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed
- M.-S. Hsieh, H.-J. Su, P.-L. Hsieh, Y.-W. Chiang and M. H. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 39086–39093 CrossRef CAS PubMed
- J. Wang, F. Teng, M. Chen, J. Xu, Y. Song and X. Zhou, CrystEngComm, 2013, 15, 39–42 RSC
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS PubMed
- R. Agarwal, Small, 2008, 4, 1872–1893 CrossRef CAS PubMed
- M. J. Bierman, Y. K. A. Lau, A. V. Kvit, A. L. Schmitt and S. Jin, Science, 2008, 320, 1060–1063 CrossRef CAS PubMed
- L. Wang, L. Wang, D. Chu, Z. Wang, Y. Zhang and J. Sun, Catal. Commun., 2017, 88, 53–55 CrossRef CAS
- Y. Bi, S. Ouyang, N. Umezawa, J. Cao and J. Ye, J. Am. Chem. Soc., 2011, 133, 6490–6492 CrossRef CAS PubMed
- Z. Jiao, Y. Zhang, H. Yu, G. Lu, J. Ye and Y. Bi, Chem. Commun., 2013, 49, 636–638 RSC
- Y. Bi, H. Hu, S. Ouyang, G. Lu, J. Cao and J. Ye, Chem. Commun., 2012, 48, 3748–3750 RSC
- P. Amornpitoksuk, K. Intarasuwan, S. Suwanboon and J. Baltrusaitis, Ind. Eng. Chem. Res., 2013, 52, 17369–17375 CrossRef CAS
- X. Song, R. Li, M. Xiang, S. Hong, K. Yao and Y. Huang, Ceram. Int., 2017, 43, 4692–4701 CrossRef CAS
- C.-T. Dinh, T.-D. Nguyen, F. Kleitz and T.-O. Do, Chem. Commun., 2011, 47, 7797–7799 RSC
- Y. Bi, H. Hu, Z. Jiao, H. Yu, G. Lu and J. Ye, Phys. Chem. Chem. Phys., 2012, 14, 14486–14488 RSC
- Q. Cao, J. Yu, K. Yuan, M. Zhong and J.-J. Delaunay, ACS Appl. Mater. Interfaces, 2017, 9, 19507–19512 CrossRef CAS PubMed
- P. Dong, Y. Wang, H. Li, H. Li, X. Ma and L. Han, J. Mater. Chem. A, 2013, 1, 4651–4656 RSC
- X. Guan, J. Shi and L. Guo, Int. J. Hydrogen Energy, 2013, 38, 11870–11877 CrossRef CAS
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef
- X. Yang, H. Cui, Y. Li, J. Qin, R. Zhang and H. Tang, ACS Catal., 2013, 3, 363–369 CrossRef CAS
- G. A. Somorjai and D. W. Blakely, Nature, 1975, 258, 580–583 CrossRef CAS
- H. Hu, Z. Jiao, H. Yu, G. Lu, J. Ye and Y. Bi, J. Mater. Chem. A, 2013, 1, 2387–2390 RSC
- D. J. Martin, N. Umezawa, X. Chen, J. Ye and J. Tang, Energy Environ. Sci., 2013, 6, 3380–3386 RSC
- G.-Y. Zhang, X.-M. Wei, X. Bai, C.-M. Liu, B.-Y. Wang and J.-W. Liu, Inorg. Chem. Front., 2018, 5, 951–961 RSC
- Y. Bi, H. Hu, S. Ouyang, Z. Jiao, G. Lu and J. Ye, Chem. - Eur. J., 2012, 18, 14272–14275 CrossRef CAS PubMed
- Y. Bi, H. Hu, S. Ouyang, Z. Jiao, G. Lu and J. Ye, J. Mater. Chem., 2012, 22, 14847–14850 RSC
- C. Li, P. Zhang, R. Lv, J. Lu, T. Wang, S. Wang, H. Wang and J. Gong, Small, 2013, 9, 3951–3956 CrossRef CAS PubMed
- Q. Guo, H. Li, Q. Zhang and Y. Zhang, Appl. Catal., B, 2018, 229, 192–203 CrossRef CAS
- J. Tian, T. Yan, Z. Qiao, L. Wang, W. Li, J. You and B. Huang, Appl. Catal., B, 2017, 209, 566–578 CrossRef CAS
- Q. Liang, W. Ma, Y. Shi, Z. Li and X. Yang, CrystEngComm, 2012, 14, 2966–2973 RSC
- N. Umezawa, O. Shuxin and J. Ye, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 035202–035210 CrossRef
- W. Zhang, R. Zhu, L. Ke, X. Liu, B. Liu and S. Ramakrishna, Small, 2010, 6, 2176–2182 CrossRef CAS PubMed
- F. Pang, X. T. Liu, M. Y. He and J. P. Ge, Nano Res., 2015, 8, 106–116 CrossRef CAS
- B. Zheng, X. Wang, C. Liu, K. Tan, Z. Xie and L. Zheng, J. Mater. Chem. A, 2013, 1, 12635–12640 RSC
- Z.-M. Yang, G.-F. Huang, W.-Q. Huang, J.-M. Wei, X.-G. Yan, Y.-Y. Liu, C. Jiao, Z. Wan and A. Pan, J. Mater. Chem. A, 2014, 2, 1750–1756 RSC
- X. Guan and L. Guo, ACS Catal., 2014, 4, 3020–3026 CrossRef CAS
- S. Huang, Y. Xu, Q. Liu, T. Zhou, Y. Zhao, L. Jing, H. Xu and H. Li, Appl. Catal., B, 2017, 218, 174–185 CrossRef CAS
- X. L. F. J. J. Liu, S. F. Chen and Y. F. Zhu, Appl. Phys. Lett., 2011, 99, 191903 CrossRef
- X. Ma, B. Lu, D. Li, R. Shi, C. Pan and Y. Zhu, J. Phys. Chem. C, 2011, 115, 4680–4687 CrossRef CAS
- P. Reunchan and N. Umezawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 245205–245210 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: More details of the SEM, FT-IR, XPS and UV-Vis of Ag3PO4 with various morphologies. See DOI: 10.1039/c9ra01306g |
| This journal is © The Royal Society of Chemistry 2019 |